Hydrogen bonding-driven highly stable homoduplexes formed by benzene/naphthalene amide oligomers†
Yun-Xiang
Xu
a,
Tian-Guang
Zhan
a,
Xin
Zhao
*a and
Zhan-Ting
Li
*ab
aShanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, China. E-mail: xzhao@mail.sioc.ac.cn; ztli@mail.sioc.ac.cn; Fax: +86 21 6416 6128; Tel: +86 21 5492 5122
bDepartment of Chemistry, Fudan University, 220 Handan Road, Shanghai 200433, China
First published on 20th December 2013
Abstract
This paper describes the dimerization of aromatic amide oligomers in chloroform. The oligomers were prepared by step-by-step coupling of benzene-m-dicarboxylic acid and naphthalene-2,7-diamine. Triethylene glycol chains were introduced into the 5-position of the benzene rings to provide solubility in organic solvents. 1H NMR investigations revealed that, although shorter oligomers did not exhibit good dimerization ability, longer oligomers with five or more amide units could form stable homoduplexes which were stabilized by intermolecular multiple N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonds. The association constants (Ka) of the 3-, 4- and 5-mer oligomers were determined using 1H NMR dilution experiments, while the Ka values of a 5-mer oligomer and a 7-mer oligomer, which were appended with two or one pyrene unit, were also evaluated using fluorescent dilution experiments. The results showed that the stability of the homoduplexes of the aromatic amide oligomers is nearly linearly related to the number of their amide units, and a Ka of 3.0 × 107 M−1 was determined for the 7-mer oligomer.
C hydrogen bonds. The association constants (Ka) of the 3-, 4- and 5-mer oligomers were determined using 1H NMR dilution experiments, while the Ka values of a 5-mer oligomer and a 7-mer oligomer, which were appended with two or one pyrene unit, were also evaluated using fluorescent dilution experiments. The results showed that the stability of the homoduplexes of the aromatic amide oligomers is nearly linearly related to the number of their amide units, and a Ka of 3.0 × 107 M−1 was determined for the 7-mer oligomer.
Introduction
Self-assembly of linear oligomers into double-stranded complexes is a common phenomenon in biology. In the past two decades, chemists have had considerable interest in constructing artificial duplexes to reveal the noncovalent rules that govern the natural structures and to develop useful recognition motifs or functional supramolecular systems.1 Among others, hydrogen bonding has been demonstrated to be the most versatile noncovalent force for this self-assembly purpose.2 By introducing multiple hydrogen bonds into a compact entity and/or preorganizing the backbone,3 very stable dimeric motifs can be created, which have found wide applications in generating more complicated supramolecular species.4 Incorporating preorganized aromatic amides into long sequences of aliphatic amides or ureas also enables the formation of extended duplexes through multiple N–H⋯OC hydrogen bonds.5–8 However, sequences that fully consist of unnatural aliphatic amino acid residues usually prefer to form helical conformations.9–11 For linear arylamide oligomers, poor solubility, due to too strong aromatic stacking and intermolecular N–H⋯OC hydrogen bonding,12 has prevented them from forming resolvable well-defined duplexes in an organic medium, although intramolecular hydrogen bonding-induced azaheterocycle-derived foldamers can readily self-assemble into double helices or more complicated aggregates.13,14
We recently prepared a series of arylamide oligomers through step-by-step coupling of benzene-m-dicarboxylic acid and benzene-1,3-diamine or naphthalene-2,7-diamine,15–17 which are highly soluble in organic solvents of both low and high polarity due to the introduction of long ethylene glycol chains. We found that these arylamide oligomers are able to strongly bind carboxylate anions in dimethylsulfoxide (DMSO) by folding into a helical conformation15,16 and to stack into vesicles in methanol.17 We herein report that this kind of linear molecule can self-assemble in chloroform to form highly stable homoduplexes which are stabilized by multiple intermolecular N–H⋯OC hydrogen bonds and aromatic stacking.
Results and discussion
Compounds 1a, 1b–e,16,171f, g, 2a–c, 3a, b, 4a16 and 4b were prepared to investigate their ability to form homodimers. The introduction of the ethylene glycol side chains provides them with solubility in common organic solvents, including chloroform. The synthetic routes for 1a, 1f, and 1g are shown in Scheme 1. Compound 1a was prepared from the reaction of 5 and di-tert-butyl dicarbonate in 75% yield, while 1f was prepared from the coupling of 617 and 717 in DMF in the presence of 1-[bis(di-methyl-amino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxide hexafluorophosphate (HATU) and N,N-diisopropylethyl-amine (DIEA). Under similar reaction conditions, 7 reacted with diamine 816 in DMF to give rise to 1g in 39% yield. The synthetic routes of 2a–c are provided in Scheme 2. Diacid 917 reacted with n-butylamine in dichloromethane in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) and hydroxybenzotriazole (HOBT) to produce 2a in 64% yield. By using a similar procedure, 11 was prepared in 76% yield from the reaction of 1017 and n-butylamine and then hydrolyzed with lithium hydroxide to give acid 12 in 98% yield. EDCI-mediated coupling of 12 with diamine 5 afforded 2b in 79% yield, while HATU-mediated coupling of 12 with longer diamine 6 produced 2c in 68% yield. By using similar coupling reactions, compounds 3a, b and 4b were prepared in moderate to good yields (Scheme 3). | ||
| Scheme 1 The synthesis of compounds 1a, 1f and 1g. | ||
 | ||
| Scheme 2 The synthesis of compounds 2a–c. | ||
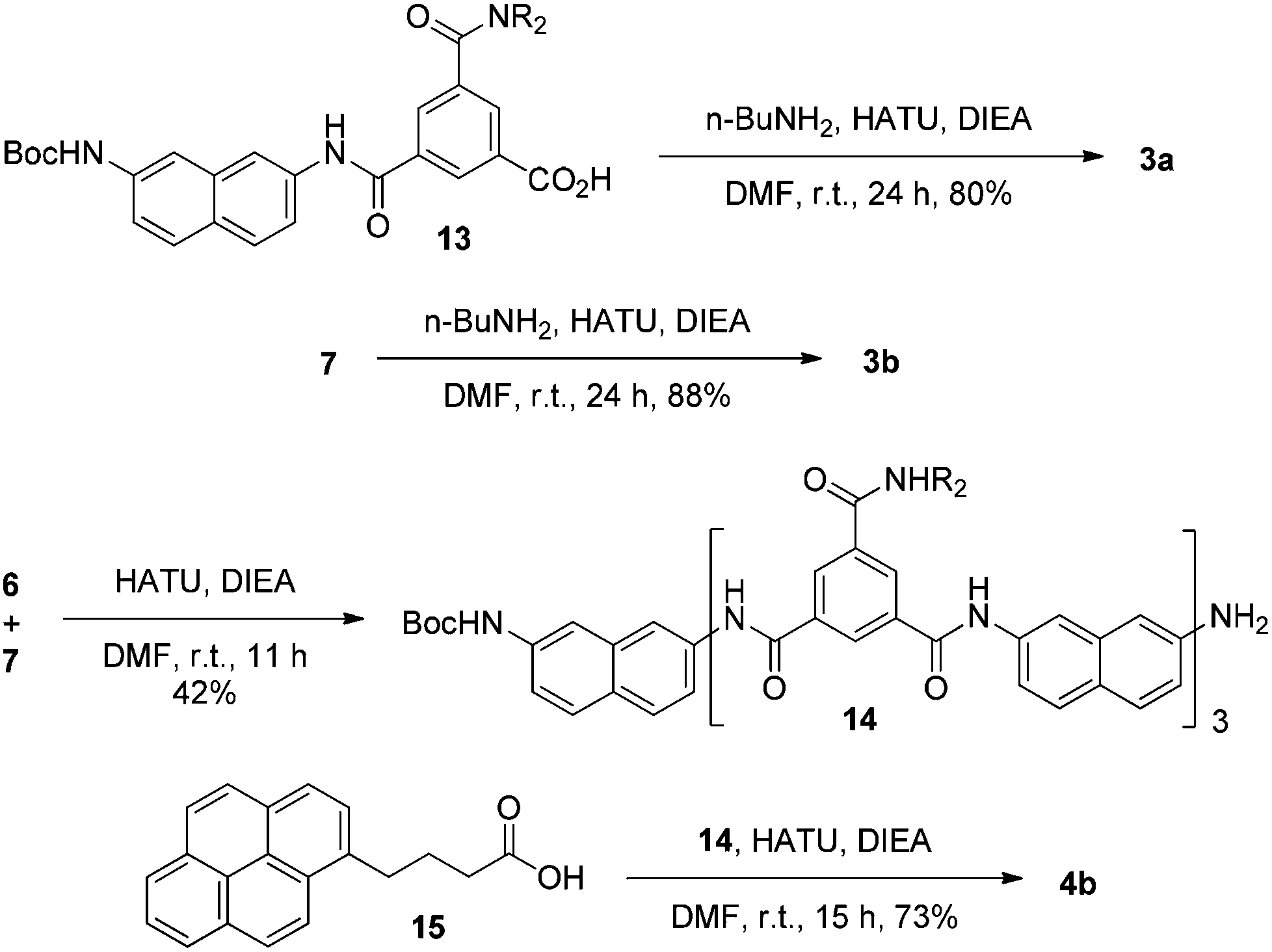 | ||
| Scheme 3 The synthesis of compounds 3a, b and 4b. | ||
The 1H NMR spectra of compounds 1a–g of the same concentration (6.0 mM) were recorded in both DMSO-d6 and CDCl3. The spectra in DMSO-d6 all exhibited signals of high resolution (Fig. S1, ESI†). The signal of the BocNH protons of all the compounds appeared around 9.59 ppm. In addition, diluting the solution to 0.20 mM did not cause important shifting (<0.03 ppm) for the signals in the downfield area, suggesting that these compounds were overwhelmingly in a single molecular state in this highly polar solvent. A comparison of the spectra of this series of oligomers in CDCl3 revealed important shifting of the NH signals. The spectra of 1b–d are shown in Fig. 1. Compared with that of 1a, whose BocNH signal appeared at 6.62 ppm, the BocNH signal of 1b–d shifted downfield by 0.18, 0.39, and 0.44 ppm (Fig. 1a–c), respectively. Moreover, the resolution of the spectra quickly decreased with the elongation of the backbone, and the long oligomers 1e–g exhibited only broad signals for the NH protons. A similar downfield shifting was also observed for the NH signals of 3b related to the corresponding signals of 3a (Fig. 1d and e). Actually, the BuNH (H-a) and BocNH (H-b) signals shifted by 0.61 and 0.41 ppm, respectively, whereas the NpNH signals shifted by 0.51, 0.69, and 0.80 ppm, respectively. Compared with the n-BuNH signal of 2a, the corresponding signals of 2b and 2c shifted downfield by 0.38 and 0.70 ppm (Fig. 2), respectively, and the two NpNH signals of 2c shifted downfield by 0.17 and 0.31 ppm, respectively, related to the signal of 2b. All these results show that the three series of oligomers formed intermolecular hydrogen bonding which was strengthened with the elongation of the backbone.
 | ||
| Fig. 1 Partial 1H NMR spectra (400 MHz) of (a) 1b, (b) 1c, (c) 1d, (d) 3a and (e) 3b in CDCl3 (6.0 mM) at 25 °C. | ||
 | ||
| Fig. 2 Partial 1H NMR spectra (400 MHz) of (a) 2a, (b) 2b, and (c) 2c in CDCl3 (6.0 mM) at 25 °C. | ||
The 1H NMR spectra of 1b and 1c were also recorded in binary CDCl3 and DMSO-d6. As expected, the NH signals shifted downfield with the increase of Φ, which represents the volume ratio of DMSO-d6 over the total volume of the two solvents. This shifting can be readily attributed to the more competitive hydrogen bonding between the NH and the DMSO-d6 oxygen. The aromatic signals also shifted downfield. For H-a, this downfield shifting reached the maximum (about 0.16 and 0.30 ppm, respectively) at Φ = ca. 0.15 (Fig. 3). Further increase of Φ (up to 0.4) did not cause the signal to shift. However, the same signal of 2a did not exhibit a similar downfield shifting (<0.03 ppm) when Φ was increased to 0.15. Thus, the pronounced downfield shifting observed for 1b and 1c should support the formation of the intermolecular N–H⋯OC hydrogen bonding in chloroform, which promoted the stacking of their aromatic units. The intermolecular hydrogen bonding was weakened and finally completely broken by the increasing DMSO, which led to the downfield shifting of the aromatic signal due to weakened π–π stacking.
 | ||
| Fig. 3 Chemical shift of the H-a of (a) 1b (●) and (b) 1c (■) (0.77 mM) versus Φ at 25 °C. | ||
1H NMR dilution experiments in CDCl3 were also carried out for 1c. The plot of the chemical shifts of the typical signals against concentration is provided in Fig. 4. It can be seen that, upon dilution from 50 mM to 0.1 mM, the three NH signals shifted upfield by −0.43, −0.58, and −0.69 ppm, respectively, indicating the weakening of the intermolecular N–H⋯OC hydrogen bonding. Accordingly, the signals of their aromatic protons all shifted downfield pronouncedly, reflecting the weakening of intermolecular stacking. This observation is consistent with the above 1H NMR result, supporting that the intermolecular hydrogen bonding promoted the stacking of the aromatic units in chloroform. Dilution experiments were also conducted for other oligomers that exhibited a good resolution in 1H NMR spectra (Fig. S2–6, ESI†), and a similar shifting was observed.
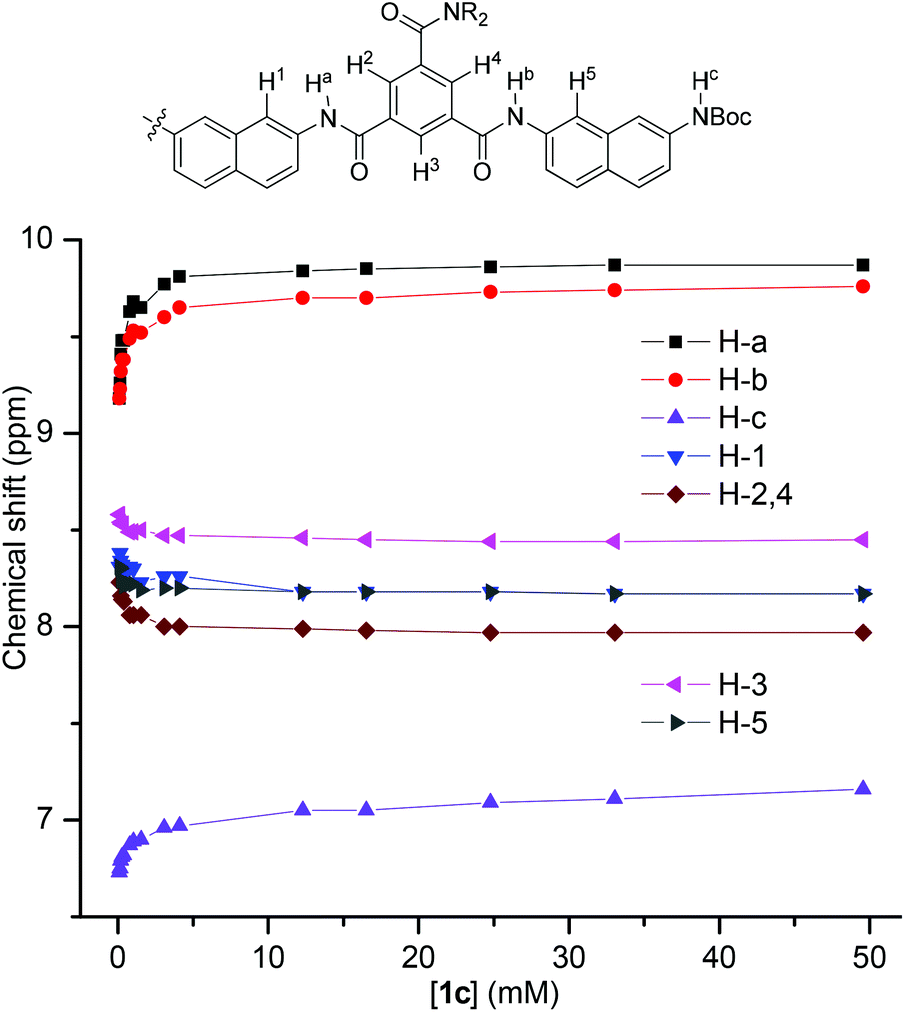 | ||
| Fig. 4 The plot of chemical shifts of the 1H NMR signals of 1cversus [1c] in CDCl3 at 25 °C. | ||
The UV spectra of 1b, 1d and 1g were further recorded in binary CHCl3 and DMSO. Fig. 5 provides the plot of the molar extinction coefficient of the absorption at 323 nm, which was produced by the naphthalene unit, versus Φ. It can be seen that, with the increase of DMSO, the absorption of the oligomers all displayed an obvious hyperchromic effect at the initial stage and reached the maximum at Φ = ca. 0.15. This observation was also consistent with the above 1H NMR result, further evidencing that the increase of DMSO weakened their intermolecular hydrogen bonding and thus the stacking of their aromatic units, which usually produces a hypochromic effect for aromatic structures.18 The increased hyperchromicity from 1b to 1d and then to 1g also qualitatively supports that the longer the oligomer was, the more stable the intermolecular hydrogen bonding would be.
 | ||
| Fig. 5 The apparent molar extinction coefficient ε of the absorption at 323 nm of 1b (■), 1d (●), and 1 g (▲) versus Φ at 25 °C. [Np] = 30 μM. | ||
To get more insight into the binding of the oligomers, NOESY experiments were carried out for compounds 1c, 2c and 3b in CDCl3 (Fig. S7–S11). Pentamer 1c displayed NOE contacts between H-5 and H-4 and H-6, H-c and H-12, and H-10 and H-12 (Fig. 6). In contrast, no similar NOE contacts were observed for the shorter analogue 1b of the same concentration. For pentamer 2c, intermolecular NOEs were observed between H-1 and H-2, H-9 and H-11, and H-10 and H-11 (Fig. 6). However, similar contacts were not found for the shorter analogue 2b of the same concentration. Tetramer 3b also displayed intermolecular NOEs between H-1 and H-2, H-1 and H-3, H-2 and H-3, H-4 and H-6, and H-5 and H-6, whereas the shorter dimer 3a of the same concentration did not show any of these contacts. Under similar conditions, pentamer 16,16 which may form four intermolecular N–H⋯OC hydrogen bonds, also gave rise to NOEs between H-1 and H-3 and H-2 and H-3 (Fig. 6, Fig. S10, ESI†). All these observations support that these longer oligomers formed stable duplexes which were driven by multiple intermolecular N–H⋯OC hydrogen bonds. Due to low resolution, no NOESY experiments were performed for even longer oligomers 1d–g. However, we may expect that they could form more stable dimers as a result of the increased number of intermolecular hydrogen bonds.
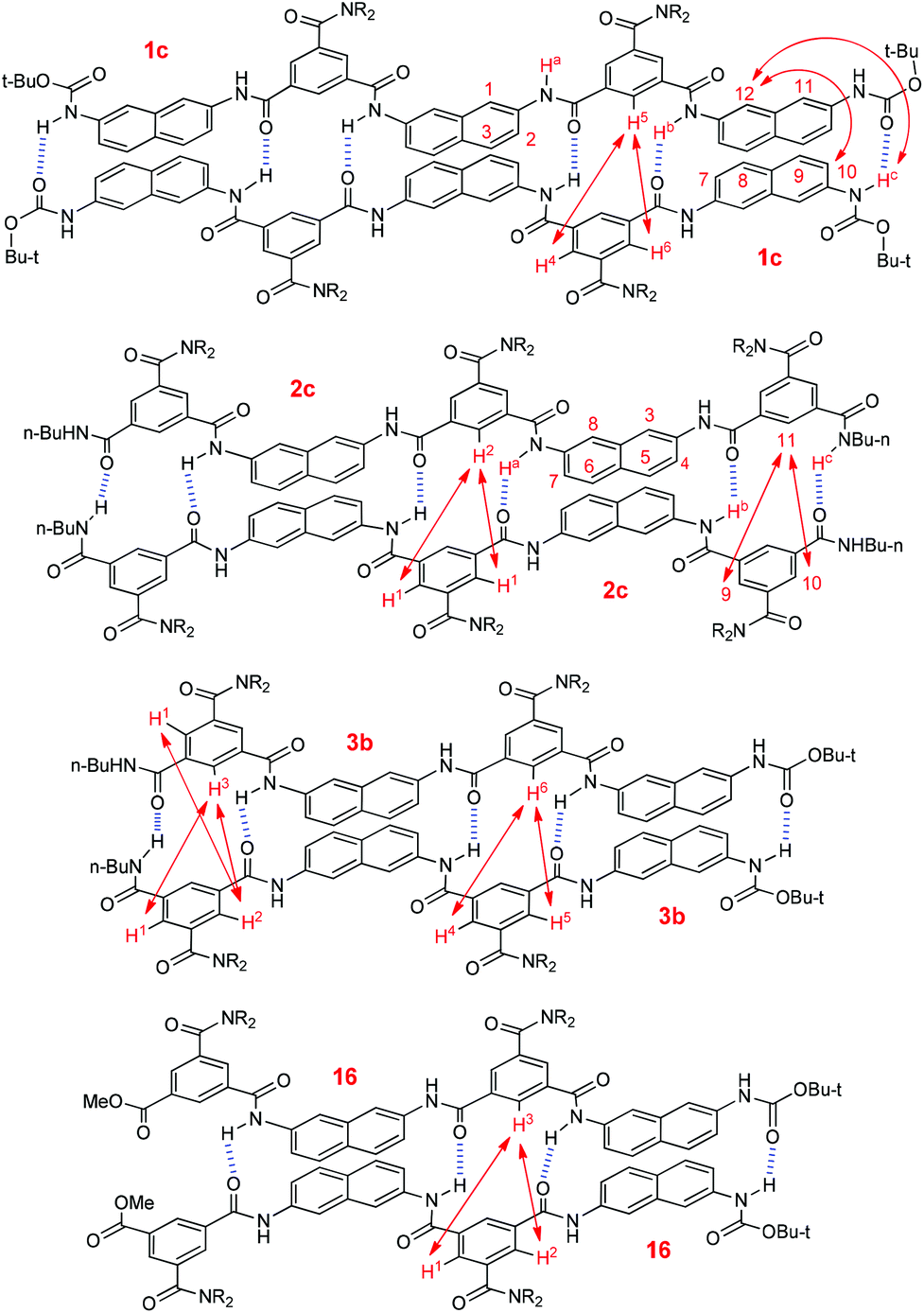 | ||
| Fig. 6 Intermolecular NOE connections of compounds 1c, 2c, 3b and 16 displayed by NOESY spectra in CDCl3 (6.0 mM), which supports the formation of homoduplexes. | ||
1H NMR dilution experiments were then performed for the shorter oligomers in CDCl3. By assuming a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding stoichiometry and on the basis of the concentration-dependent shifting of the amide protons,19 we could determine the association constants (Ka) of the new hydrogen bonded homoduplexes, which are listed in Table 1. Because of the low resolution of their 1H NMR spectra in CDCl3, the Ka values of longer oligomers 1d–g could not be evaluated using the 1H NMR dilution method. We thus further prepared compounds 4a and 4b, which were appended with two or one pyrene unit from the side as a fluorescent probe. Diluting their solution in chloroform caused a decrease of the excimer emission of pyrene centred at 479 nm (Fig. S11, ESI†), which could be produced only through dimerization of the aromatic backbone. We thus could determine the Ka value (Table 1) of their homoduplexes using the fluorescent dilution method.20
:1 binding stoichiometry and on the basis of the concentration-dependent shifting of the amide protons,19 we could determine the association constants (Ka) of the new hydrogen bonded homoduplexes, which are listed in Table 1. Because of the low resolution of their 1H NMR spectra in CDCl3, the Ka values of longer oligomers 1d–g could not be evaluated using the 1H NMR dilution method. We thus further prepared compounds 4a and 4b, which were appended with two or one pyrene unit from the side as a fluorescent probe. Diluting their solution in chloroform caused a decrease of the excimer emission of pyrene centred at 479 nm (Fig. S11, ESI†), which could be produced only through dimerization of the aromatic backbone. We thus could determine the Ka value (Table 1) of their homoduplexes using the fluorescent dilution method.20
| Homoduplex | K a (M−1) | −ΔG (kJ mol−1) | Homoduplex | K a (M−1) | −ΔG (kJ mol−1) |
|---|---|---|---|---|---|
| a Too small to be evaluated using 1H NMR dilution experiments. b Determined using fluorescent dilution experiments. | |||||
| 1a·1aa | — | — | 16·16 | 1.6(±0.1) × 102 | 12.6 |
| 2a·2aa | — | — | 3b·3b | 7.1(±0.2) × 102 | 16.3 |
| 3a·3a | 7 ± 2 | 4.8 | 1c·1c | 5.5(±0.4) × 103 | 21.3 |
| 1b·1b | 64 ± 6 | 10.3 | 2c·2c | 2.0(±0.1) × 103 | 18.8 |
| 2b·2b | 57 ± 4 | 10.0 | 4a·4ab | 1.8(±0.2) × 106 | 35.6 |
| 4b·4bb | 3.0(±0.5) × 107 | 42.7 | |||
It can be seen that both 1a and 2a did not dimerize, although they bear two amide units, while 3a, which contains three amide units, formed a dimer with a very small Ka. The stability of the dimer of 1b and 2b, both of which have four amide units, was increased pronouncedly. With the elongation of the backbone, compounds 16, 3b, 1c and 2c exhibited increasing dimerization ability. The Ka values of 1c and 2c, which bear six amide units, are notably lower than those formed by other partially or fully preorganized aromatic amide oligomers.21,22 This is not unexpected because preorganization can remarkably reduce the flexibility of the backbones and thus enhances dimerization of the corresponding monomers. Clearly, the longer oligomers generated more stable homoduplexes due to the increased number of amide units by forming more hydrogen bonds. Although no Ka values were obtained for longer analogues 1d–g, we may expect that they also formed homoduplexes of a comparable or higher stability.
For every oligomer, the maximum number of possibly formed intermolecular hydrogen bonds is the number of the amide units. The plot of the binding free energies listed in Table 1 against the number of amide units of the oligomers is provided in Fig. 7. It can be found that for oligomers with the same number of aromatic units, the 1 series is more robust than the 2 series and the binding free energy of both series is nearly linearly increased with the increase of the repeat aromatic units. This observation suggests that their intermolecular hydrogen bonds did not produce a positive or negative cooperativity. The Ka of 4a was remarkably higher than that of 1c and 2c of the same number of amide units, and the Ka of 4b was also much higher than the value expected by extending the linearity of both 1 and 2 series. These large differences implied that the formation of the excimer of the pyrene unit through π–π stacking facilitated the intermolecular hydrogen bonding of the aromatic amide backbones, even though the peripheral amide and t-butoxycarbonylamino units might contribute differently to the formation of the duplexes.
 | ||
| Fig. 7 Free energy of dimerization versus the number of amide units of the oligomer. | ||
To further evaluate the stability of the homoduplexes and also detect the possibility of forming other more complicated aggregates, vapour pressure osmometry (VPO) experiments were performed for the solution of 5-mer 1c and 7-mer 1d in chloroform,23 which gave rise to a number-average molecular weight (Mn) of 2980 (±300) and 4640 (±460) for the two oligomers. The two values are consistent with the calculated molecular weight of their respective homoduplex (3211 and 4459, respectively), again supporting the high stability of the homoduplexes.
Conclusions
We have demonstrated that soluble aromatic amide oligomers can self-assemble into homoduplexes in chloroform. The stability of the homoduplexes of this series of linear amides is revealed to be nearly linearly related to the length of the backbones and thus long oligomers with five or more amide units can form very stable duplexes, with association constants being as high as 106–107 M−1, which are driven by multiple intermolecular hydrogen bonds. Different from previously reported partially or fully preorganized amide oligomers, this new family of intramolecular hydrogen bonding-free aromatic amide oligomers does not exhibit a positive cooperativity in dimerizing. Therefore, even a very long 13-mer oligomer does not dimerize in competitive DMSO. The high stability of the homoduplexes of long sequences bodes well for future design of stable chiral homoduplexes by simply introducing the chirality group into the end(s) of the backbones, and soluble polymeric sequences may exhibit a new unique duplex–helix conversion which can be tuned by anion guests.References
- M. Albrecht, Chem. Rev., 2001, 101, 345 CrossRef CAS PubMed; V. Berl, I. Huc, R. G. Khoury and J.-M. Lehn, Chem.–Eur. J., 2001, 7, 2810 CrossRef; C. Baldoli, P. Cerea, C. Giannini, E. Licandro, C. Rigamonti and S. Maiorana, Synlett, 2005, 1984 Search PubMed; M. Barboiu and J.-M. Lehn, Rev. Roum. Chim., 2006, 51, 581 Search PubMed; G. H. Clever, C. Kaul and T. Carell, Angew. Chem., Int. Ed., 2007, 46, 6226 CrossRef PubMed; Y. Furusho and E. Yashima, Chem. Rec., 2007, 7, 1 CrossRef PubMed; Y. Takezawa and M. Shionoya, Acc. Chem. Res., 2012, 45, 2066 CrossRef PubMed; G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933 RSC.
- A. P. Bisson, F. J. Carver, D. S. Eggleston, R. C. Haltiwanger, C. A. Hunter, D. L. Livingstone, J. F. McCabe, C. Rotger and A. E. Rowan, J. Am. Chem. Soc., 2000, 122, 8856 CrossRef CAS; E. A. Archer, A. E. Sochia and M. J. Krische, Chem.–Eur. J., 2001, 7, 2059 CrossRef.
- S. C. Zimmerman and P. S. Corbin, Struct. Bonding, 2000, 96, 63 CrossRef CAS; R. P. Sijbesma and E. W. Meijer, Chem. Commun., 2003, 5 RSC; X. Zhao, X.-Z. Wang, X.-K. Jiang, Y.-Q. Chen, Z.-T. Li and G.-J. Chen, J. Am. Chem. Soc., 2003, 125, 15128 CrossRef PubMed; J. Taubitz and U. Lüning, Aust. J. Chem., 2009, 62, 1550 CrossRef; B. A. Blight, C. A. Hunter, D. A. Leigh, H. McNab and P. T. Thomson, Nat. Chem., 2011, 3, 244 CrossRef PubMed.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed; S. K. Yang and S. C. Zimmerman, Isr. J. Chem., 2013, 53, 511 CrossRef.
- H. Zeng, R. S. Miller, R. A. Flowers and B. Gong, J. Am. Chem. Soc., 2000, 122, 2635 CrossRef CAS; B. Gong, Polym. Int., 2007, 56, 436 CrossRef; B. Gong, Acc. Chem. Res., 2012, 45, 2077 CrossRef PubMed.
- Y. Yang, J.-F. Xiang and C.-F. Chen, Org. Lett., 2007, 9, 4355 CrossRef CAS PubMed; W.-J. Chu, Y. Yang and C.-F. Chen, Org. Lett., 2010, 12, 3156 CrossRef PubMed; Y. Yang, W.-J. Chu, J.-W. Liu and C.-F. Chen, Curr. Org. Chem., 2011, 15, 1302 CrossRef.
- J. S. Nowick, Acc. Chem. Res., 2008, 41, 1319 CrossRef CAS PubMed.
- P.-H. Zhang, H.-Z. Chu, X.-H. Li, W. Feng, P.-C. Deng, L.-H. Yuan and B. Gong, Org. Lett., 2011, 13, 54 CrossRef CAS PubMed; X. Li, Y. Jia, Y. Ren, Y. Wang, J. Hu, T. Ma, W. Feng and L. Yuan, Org. Biomol. Chem., 2013, 11, 6975 Search PubMed.
- D. Seebach and J. L. Matthews, Chem. Commun., 1997, 2015 RSC.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173 CrossRef CAS.
- Y.-D. Wu, W. Han, D.-P. Wang, Y. Gao and Y.-L. Zhao, Acc. Chem. Res., 2008, 41, 1418 CrossRef CAS PubMed.
- D. Tanner, J. A. Fitzgerald and B. R. Phillips, Angew. Chem., Int. Ed. Engl., 1989, 28, 649 CrossRef CAS; H. G. Chae and S. Kumar, J. Appl. Polym. Sci., 2006, 100, 791 CrossRef; J. M. Garcia, F. C. Garcia, F. Serna and J. L. de la Peña, Prog. Polym. Sci., 2010, 35, 623 CrossRef PubMed.
- V. Berl, I. Huc, R. G. Khoury and J.-M. Lehn, Chem.–Eur. J., 2001, 7, 2810 CrossRef CAS; D. Haldar, H. Jiang, J.-M. Léger and I. Huc, Angew. Chem., Int. Ed., 2006, 45, 5483 CrossRef PubMed; Q. Gan, Y. Ferrand, N. Chandramouli, B. Kauffmann, C. Aube, D. Dubreuil and I. Huc, J. Am. Chem. Soc., 2012, 134, 15656 CrossRef PubMed.
- Q. Gan, C. Bao, B. Kauffmann, A. Grelard, J. Xiang, S. Liu, I. Huc and H. Jiang, Angew. Chem., Int. Ed., 2008, 47, 1715 CrossRef CAS PubMed; Q. Gan, J. Shang, B. Kauffmann, Y. Wang, F. Bie and H. Jiang, Tetrahedron, 2012, 68, 4479 CrossRef PubMed; Q. Gan, Y. Ferrand, C. Bao, B. Kauffmann, A. Grelard, H. Jiang and I. Huc, Science, 2011, 331, 1172 CrossRef PubMed; Q. Gan, F. Li, G. Li, B. Kauffmann, J. Xiang, I. Huc and H. Jiang, Chem. Commun., 2010, 46, 297 RSC.
- Z.-M. Shi, S.-G. Chen, X. Zhao, X.-K. Jiang and Z.-T. Li, Org. Biomol. Chem., 2011, 9, 8122 Search PubMed; Z.-M. Shi, C.-F. Wu, T.-Y. Zhou, D.-W. Zhang, X. Zhao and Z.-T. Li, Chem. Commun., 2013, 49, 2673 RSC.
- Y.-X. Xu, X. Zhao, X.-K. Jiang and Z.-T. Li, J. Org. Chem., 2009, 74, 7267 CrossRef CAS PubMed.
- Y.-X. Xu, G.-T. Wang, X. Zhao, X.-K. Jiang and Z.-T. Li, Langmuir, 2009, 25, 2684 CrossRef CAS PubMed.
- J. C. Nelson, J. G. Saven, J. S. Moore and P. G. Wolynes, Science, 1997, 277, 1793 CrossRef CAS.
- K. A. Conners, Binding Constants the Measurement of Molecular Complex Stability, Wiley, New York, 1987 Search PubMed; C. S. Wilcox, in Frontiers in Supramolecular Organic Chemistry and Photochemistry, ed. H.-J. Schneider and H. Dürr, VCH, New York, 1991, p. 123 Search PubMed.
- S. H. M. Sontjens, R. P. Sijbesma, M. H. P. van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487 CrossRef CAS; P. S. Corbin, L. J. Lawless, Z. Li, Y. Ma, M. Witmer and S. C. Zimmerman, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5099 CrossRef PubMed.
- R. Cao, J. Zhou, W. Wang, W. Feng, X. Li, P. Zhang, P. Deng, L. Yuan and B. Gong, Org. Lett., 2010, 12, 2958 CrossRef CAS PubMed.
- J. Zhu, J.-B. Lin, Y.-X. Xu, X.-B. Shao, X.-K. Jiang and Z.-T. Li, J. Am. Chem. Soc., 2006, 128, 12307 CrossRef CAS PubMed.
- J. P. Mathias, C. T. Seto, E. E. Simanek and G. M. Whitesides, J. Am. Chem. Soc., 1994, 116, 1725 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of new compounds, 1H NMR dilution and NOESY spectra, and fluorescent spectra. See DOI: 10.1039/c3qo00032j |
| This journal is © the Partner Organisations 2014 |