Palladium-catalyzed base-accelerated direct C–H bond alkenylation of phenols to synthesize coumarin derivatives†
Xi-Sha
Zhang‡
a,
Zhao-Wei
Li‡
a and
Zhang-Jie
Shi
*ab
aBeijing National Laboratory of Molecular Sciences (BNLMS) and Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry and Molecular Engineering and Green Chemistry Center, Peking University, Beijing, 100871, China. E-mail: zshi@pku.edu.cn
bState Key Laboratory of Organometallic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
First published on 20th December 2013
Abstract
A palladium-catalyzed base-accelerated ortho-selective C–H alkenylation of phenols to synthesize bioactive coumarin derivatives was developed. The reaction condition was mild and the substrate scope was broad with both electron-neutral and electron-deficient phenols, which is complementary to the previous methods to synthesize electron-rich coumarins. Several bioactive molecules were functionalized and several coumarins with bioactive properties were synthesized. Mechanistic studies showed that this reaction underwent C–H bond activation via direct metallation rather than the Friedel–Crafts pathway.
Phenols are important motifs in natural products and drugs1 and the direct functionalization of phenols is useful for organic synthesis. Although the hydroxyl group has been used as an efficient directing group in C–H bond activation,2 phenols cannot form five- or six-membered metallacycles, which are important intermediates for directing strategies.3 As a result, they are usually transformed to their derivatives with proper directing groups, like carbamate,4 ester,5 amide,6 carboxylic acid,7N-heterocycle8 and silyl groups.9 For unprotected phenols, the selective ortho-functionalization requires specific structures (2-phenylphenol10 and naphthalen-1-ol11), acceleration by phosphines12 or other methods.13 For the transition metal-catalyzed direct ortho-functionalization of phenols, only limited examples were reported, including the additions to alkynes and alkenes,14 oxidative couplings,15 and others.16
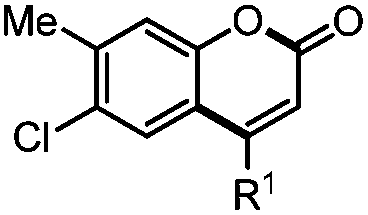
Coumarin derivatives17 are one kind of important natural products in plants and display good biological and pharmacological activity, including antitumor,18 anti-HIV,19 antioxidant,20 antibacterial,21 and anti-inflammatory22 activity. In addition, they also have good optical properties and are widely used in laser devices, light-emitting diodes, as fluorescent probes and in other areas23 (Scheme 1).
 | ||
| Scheme 1 Selected coumarin derivatives with bio-activities and photochromic properties. | ||
Traditional synthesis of coumarin derivatives is based on condensation reactions, such as Pechmann,20 Perkin, Knoevenagel, Wittig,24 Ponndorf, Horner–Wittig, Houben–Hoesch, and so on.25 Although these methods are widely used and efficient, harsh conditions and stoichiometric quantities of strong Lewis or Brønsted acids are usually required, and the regioselectivity is not ideal and leads to mixtures. Transition metal-catalyzed intermolecular or intramolecular coupling reactions (Heck reaction, carbonylation, etc.) partially solved this problem.26 However, prefunctionalization of the starting materials (halides and organometallic reagents) made these reactions non-economic. Transition metal-catalyzed direct C–H bond addition to alkynes developed by Trost and Fujiwara is more atom economic and has seen wide application in recent years14d,e,27 (Scheme 2, (2)). However, this reaction is generally limited to electron-rich phenols and a Friedel–Crafts pathway rather than a C–H activation pathway cannot be ruled out clearly. Direct C–H bond alkenylation provide another strategy for the synthesis of coumarins. This reaction was first realized in 2005 by Pd catalysis with TFA (trifluoroacetic acid) as solvent, although only electron-rich phenols can be adopted28 (Scheme 2, (3)). Very recently, a tandem reaction starting from cyclohexanone with phenol as intermediate was reported.29 Again, the reaction was conducted in acid and the Friedel–Crafts pathway rather than C–H activation cannot be ruled out clearly. As we were preparing our manuscript, a Pd-catalyzed alkenylation of phenols to synthesis benzofurans and coumarins was reported by Maiti.30 Here, we report a base-accelerated alkenylation reaction of phenol to synthesize coumarins under mild conditions (without acid as solvent) and good functional group tolerance (Scheme 2).
 | ||
| Scheme 2 Synthesis of coumarins by transition metal catalysis. | ||
Initially, we selected 4-(tert-butyl)phenol (1a) and methyl acrylate (2a) as model substrates to screen the conditions (Table 1). When Pd(OAc)2 was used as catalyst and Cu(OAc)2 as oxidant, the alkenylation/cyclization product 6-(tert-butyl)-2H-chromen-2-one (3a) was obtained in 11% NMR yield (Table 1, entry 1). Screening of other palladium catalysts showed that Pd(CH3CN)4(BF4)2 gave the best yield (Table 1, entry 3). We proposed that addition of proper base may promote the formation of phenolic anion, which will coordinate with the palladium catalyst more readily and thus promote the ortho-C–H bond activation. Screening of a series of bases showed that many of the bases could accelerate the reaction to different degrees, and NaOPiv and KOPiv showed the highest activity (Table 1, entries 6 to 12). Changing the solvent from DCE to mesitylene further promoted the yield to 67% (Table 1, entry 14). Increasing or decreasing the temperature showed inferior results (Table 1, entries 15 and 16). No product was obtained in the absence of the catalyst (Table 1, entry 17). Replacement of the palladium catalyst by Lewis acids, Cu(OTf)2 or Sc(OTf)3, all showed no reaction, ruling out the Friedel–Crafts reaction pathway (Table 1, entries 18 and 19).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (10 mol%) | Solvent | Additive | Yieldb (%) |
| a Reaction conditions: 1a (0.20 mmol, 1.0 equiv.), methyl acrylate 2a (0.40 mmol, 2.0 equiv.), Pd catalyst (0.020 mmol, 10 mol%), Cu(OAc)2 (0.40 mmol, 2.0 equiv.) and additive were reacted in solvent (2.0 mL) at 120 °C for 20 h under N2 atmosphere. DCE (1,2-dichloroethane). b NMR yield with CH2Br2 as internal standard. c Isolated yield. d 140 °C. e 90 °C. | ||||
| 1 | Pd(OAc)2 | DCE | None | 11 |
| 2 | Pd(OTFA)2 | DCE | None | <10 |
| 3 | Pd(CH3CN)4(BF4)2 | DCE | None | 14 |
| 4 | Pd(CH3CN)2(OTs)2 | DCE | None | 8 |
| 5 | Pd(dba)2 | DCE | None | <5 |
| 6 | Pd(CH3CN)4(BF4)2 | DCE | Na2CO3 (100 mol%) | 19 |
| 7 | Pd(CH3CN)4(BF4)2 | DCE | K2CO3 (100 mol%) | 19 |
| 8 | Pd(CH3CN)4(BF4)2 | DCE | Cs2CO3 (100 mol%) | 15 |
| 9 | Pd(CH3CN)4(BF4)2 | DCE | K3PO4 (100 mol%) | 9 |
| 10 | Pd(CH3CN)4(BF4)2 | DCE | NaOPiv (100 mol%) | 30 |
| 11 | Pd(CH3CN)4(BF4)2 | DCE | KOPiv (100 mol%) | 30 |
| 12 | Pd(CH3CN)4(BF4)2 | DCE | NaOPiv (80 mol%) | 33 |
| 13 | Pd(CH3CN)4(BF4)2 | Decalin | NaOPiv (80 mol%) | 53 |
| 14 | Pd(CH3CN)4(BF4)2 | Mesitylene | NaOPiv (80 mol%) | 67 (70)c |
| 15d | Pd(CH3CN)4(BF4)2 | Mesitylene | NaOPiv (80 mol%) | 48 |
| 16e | Pd(CH3CN)4(BF4)2 | Mesitylene | NaOPiv (80 mol%) | 38 |
| 17 | None | Mesitylene | NaOPiv (80 mol%) | 0 |
| 18 | Cu(OTf)2 | Mesitylene | NaOPiv (80 mol%) | 0 |
| 19 | Sc(OTf)3 | Mesitylene | NaOPiv (80 mol%) | 0 |
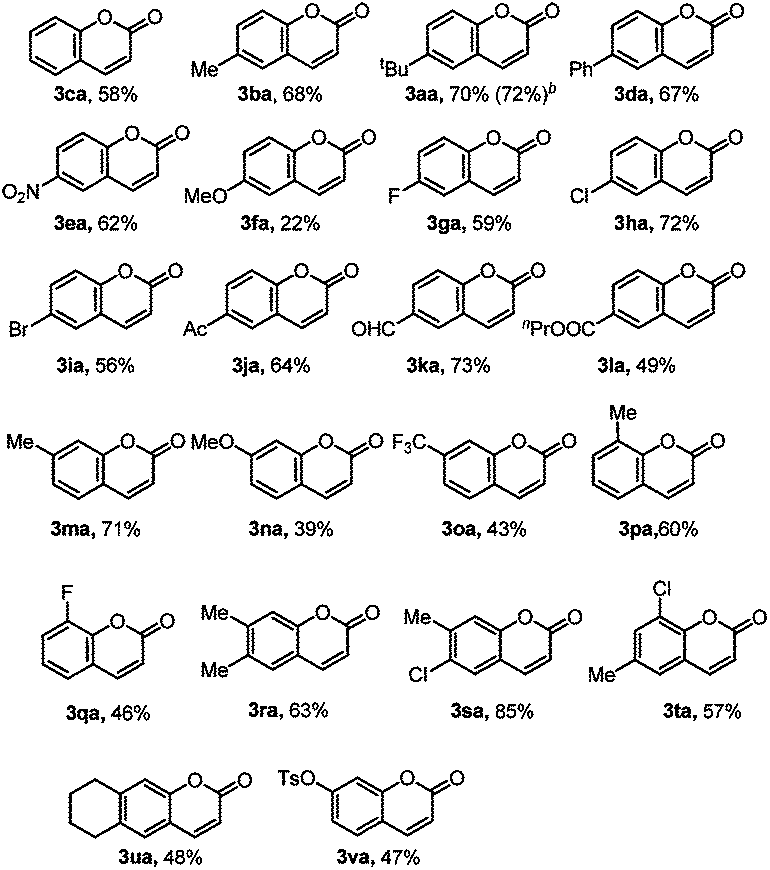
With the reaction conditions in hand, we first investigated the substrate scope of phenol derivatives (Table 2). Both electron-rich and electron-deficient phenols could be tolerated, and electron-deficient ones (3ea, 3ga, 3ka, 3ja, 3la, 3oa, 3qa) are better than electron-rich ones (3fa, 3na). This kind of electronic effect complements the developed methods, which makes this transformation synthetically useful.14e,27b,31 Apart from alkyl (3aa, 3ba, 3ma) and aryl (3da) substituents, functional groups like nitro (3ea), halide (3ga to 3ia), acetyl (3ja), formyl (3ka), and ester (3la) could all be well tolerated, providing access to synthesize more complex molecules by further transformation of these functional groups.32Meta-substituted phenols underwent this reaction selectively at the less sterically hindered position (3ma to 3oa), avoiding the difficult separation of regioisomers.27b,e Di-substituted phenols also reacted smoothly to give moderate to good yields (3ra to 3ua). The tolerance of the OTs group (3va) also provided the possibility for further cross-coupling reactions.26a,33 It should be noted that this reaction can even be conducted on a 1.0 mmol scale without a decrease in yield (3aa), further suggesting its synthetic applications.
|
|
|---|
| a Reaction conditions: 1 (0.20 mmol, 1.0 equiv.), methyl acrylate (0.40 mmol, 2.0 equiv.), [Pd(CH3CN)4](BF4)2 (0.020 mmol, 10 mol%), Cu(OAc)2 (0.40 mmol, 2.0 equiv.) and NaOPiv (0.16 mmol, 0.80 equiv.) were reacted in mesitylene (2.0 mL) at 120 °C for 20 h under N2 atmosphere. Isolated yields were reported. b 1.0 mmol scale in 10.0 mL of mesitylene. |
|
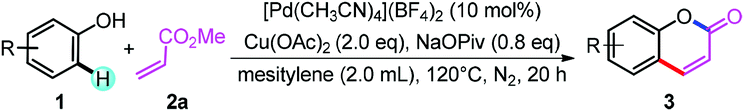
Next, we began to investigate the substrate scope of the alkenes (Table 3). Variation of the protecting groups of the ester showed a reduced activity of more sterically hindered acrylate (Table 3, entries 1 to 6). Apart from mono-substituted alkenes, 1,2-di-substituted alkenes can also show acceptable to moderate yields (Table 3, entries 7 to 9). The reactivity of methyl cinnamate (2g) provided a method to synthesize 4-aryl coumarins with bioactivity.34 However, the 1,1-di-substitued alkenes showed no reactivity (Table 3, entry 10).
|
|
|||
|---|---|---|---|
| Entry | 2 | 3 | Yield |
| a Reaction conditions: 1 (0.20 mmol, 1.0 equiv.), alkene (0.40 mmol, 2.0 equiv.), [Pd(CH3CN)4](BF4)2 (0.020 mmol, 10 mol%), Cu(OAc)2 (0.40 mmol, 2.0 equiv.) and NaOPiv (0.16 mmol, 0.80 equiv.) were reacted in mesitylene (2.0 mL) at 120 °C for 20 h under N2 atmosphere. Isolated yields were reported. b NMR yield. | |||
| 1–6 |

|
|
R = Me, 70%b; Bu, 52%b |
| R = Et, 32%b; Ph, 5%b | |||
| R = tBu, 20%b; Bn, 36%b | |||
| 7 |
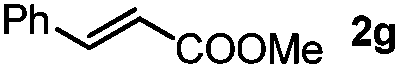
|
|
R1 = Ph, 3sg, 57% |
| 8 |
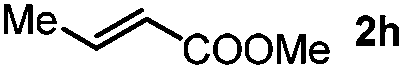
|
R1 = Me, 3sh, 37% | |
| 9 |
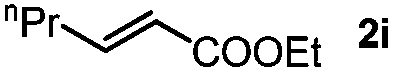
|
R1 = nPr, 3si, 21%b | |
| 10 |
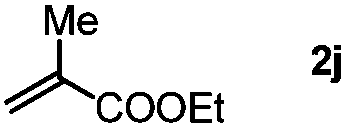
|
|
3sj, Trace |
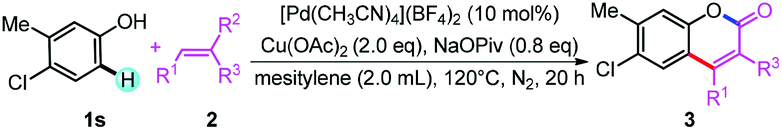
When [1,1′-biphenyl]-4,4′-diol (4) was selected as the substrate, di-functionalization occurred and the product 5 with more complexity was obtained in 34% yield (Scheme 3, Eq. 1). Interestingly, for 2-phenylphenol (6) as substrate, the compound 8 was produced via an oxidative alkenylation/oxidative cyclization sequence rather than the alkenylation/Michael addition product reported in the literature (Scheme 3, Eq. 2).35
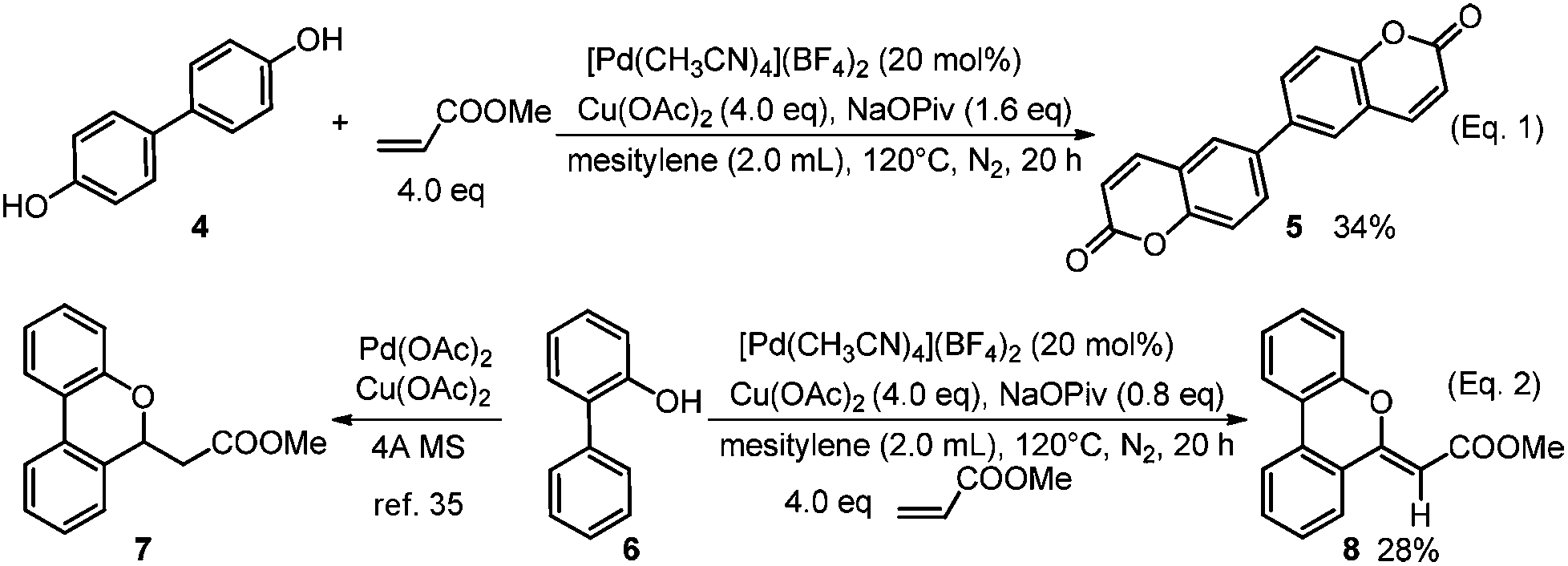 | ||
| Scheme 3 Synthesis of complex and interesting molecules. | ||
To verify whether this reaction was initiated by alkenylation or esterification, we conducted the reaction with phenyl acrylate (9) or (E)-methyl 3-(2-hydroxyphenyl)acrylate (10) as starting materials in parallel with the standard reaction (Scheme 4, Eq. 3–5). (E)-Methyl-3-(2-hydroxyphenyl)acrylate (10) can be converted to the product in comparable yield with the standard reaction while only a trace amount of product can be obtained from phenyl acrylate, indicating the C–H bond alkenylation occurs first. A tandem process of C–H activation, olefin insertion, proto-demetalation, condensation and oxidation was also ruled out by using 11 (chroman-2-one) as the starting material under the standard conditions (Scheme 4, Eq. 6).36
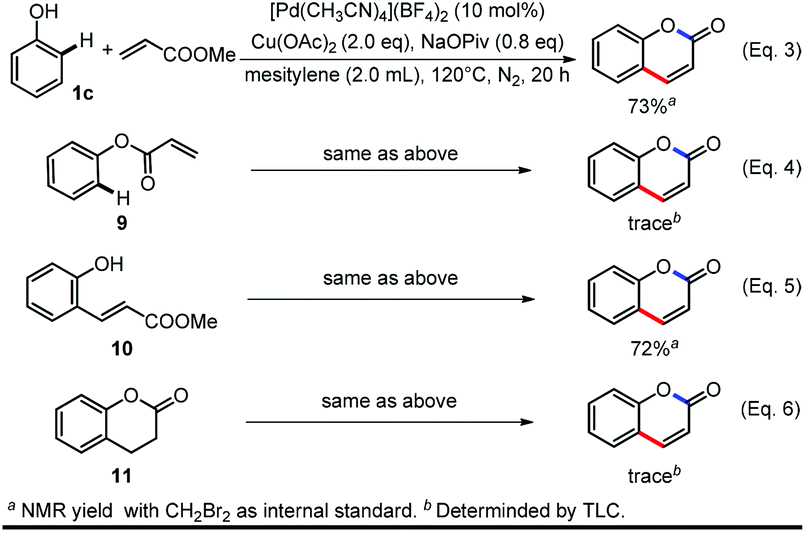 | ||
| Scheme 4 Control experiments to identify the reaction pathway. | ||
An intramolecular deuterium labeling experiment showed a kinetic isotopic effect (KIE) value of 1.9 (Scheme 5, Eq. 7), showing that the C–H activation was involved in the rate-determining step. When 4-methylphenol (1b) was conducted under the conditions in the presence of 4.0 equivalents of D2O but in the absence of alkene, no deuterium exchange we observed in the recovered starting material (Scheme 5, Eq. 8).
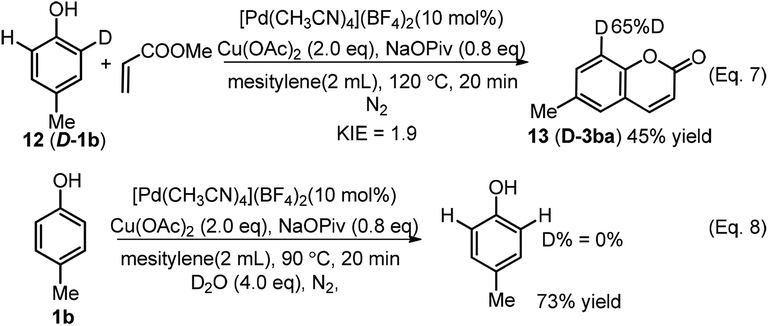 | ||
| Scheme 5 Deuterium labeling experiments. | ||
Competing experiments between phenols with different electronic factors showed that electron-rich phenols reacted faster than electron-deficient ones (Scheme 6).
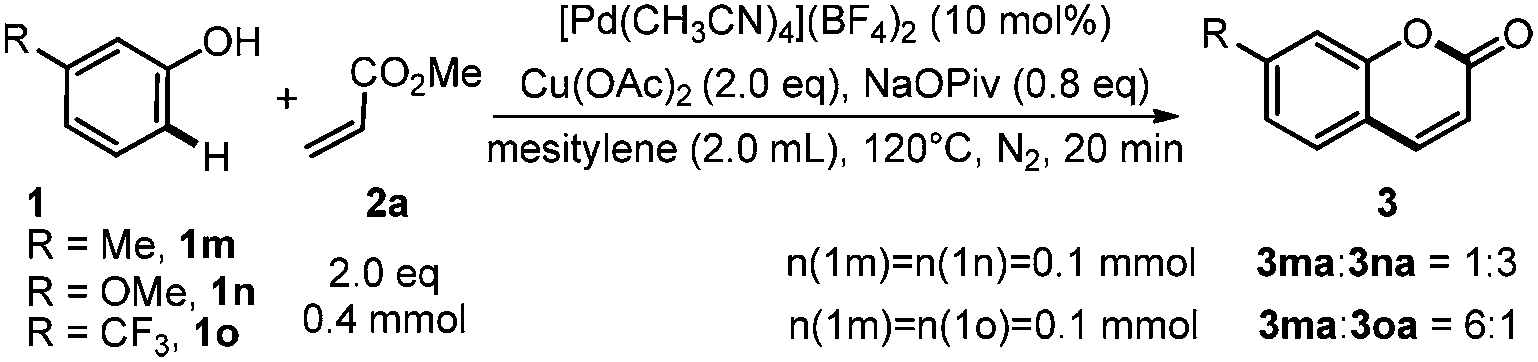 | ||
| Scheme 6 Competing reaction. | ||
Based on these results and the literature reports,13,14c,37 we proposed the mechanism as shown in Scheme 7. In the presence of base, phenol was transformed to its sodium salt I, which coordinated with the palladium catalyst to form intermediate II. Palladium 1,3-migration viaIII generated the C–H bond metalation intermediate IV. Alkene insertion and β-H elimination released the alkenylated product and the PdH species VI, which was oxidized to the Pd(II) species to furnish the catalytic cycle. Intramolecular esterification produced the final product.
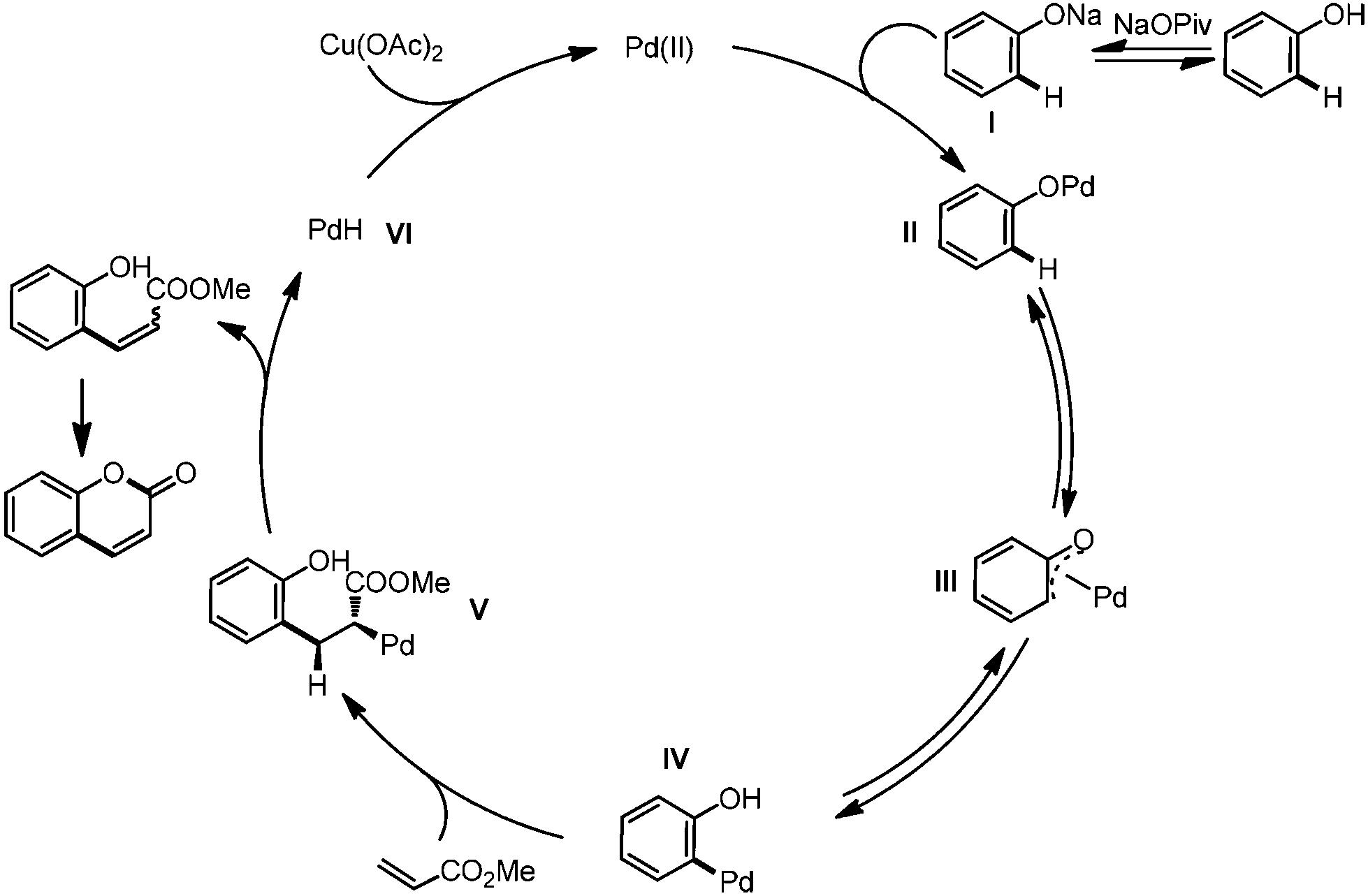 | ||
| Scheme 7 Proposed catalytic cycle. | ||
With our method, several bioactive molecules can also be functionalized to synthesize more complex structures (Scheme 8). Estrone (1,3,5(10)-estratrien-3-ol-17-one)38 and Phth-Me-D-tyrosine39 all reacted smoothly under the standard conditions to afford moderate yields.40 In addition, with this methodology, several bio-active molecules can also be constructed (Scheme 9). Apart from the herniarin (7-methoxy-2H-chromen-2-one, 3na) with anti-inflammatory activity,41 deprotection of 2-oxo-2H-chromen-7-yl-4-methylbenzenesulfonate (3va) can also generate the natural umbelliferone molecule with anti-oxidant activity.17m,42
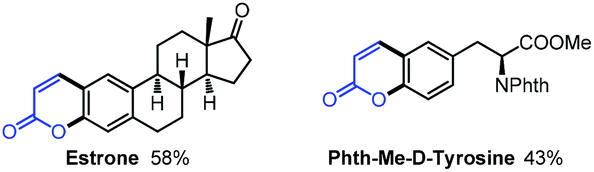 | ||
| Scheme 8 Functionalization of bio-active molecules and natural products. | ||
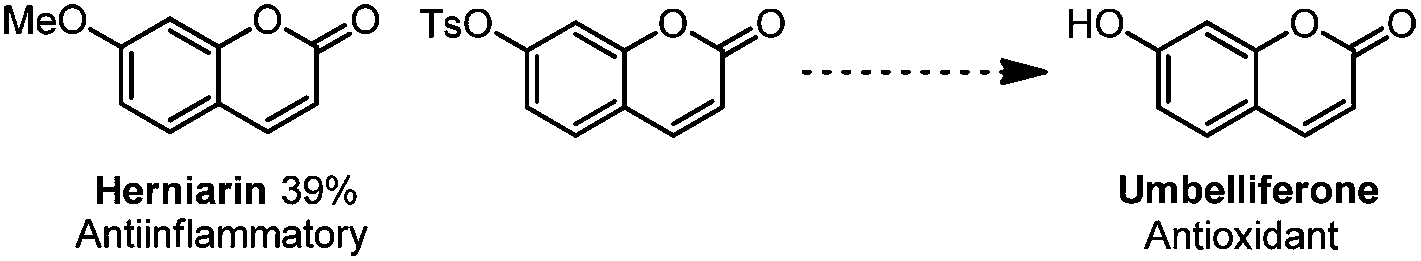 | ||
| Scheme 9 Synthesis of natural products with bioactivity. | ||
In summary, a palladium-catalyzed base-accelerated ortho-selective phenol C–H alkenylation reaction was developed without acid as solvent. The substrate scope for the phenol was very wide and both electron neutral and electron-deficient phenols were well tolerated, which is complementary to the previous methods to synthesize electron-rich coumarins. Several bioactive molecules were functionalized and several coumarins with bioactive or fluorescent properties were synthesized. Mechanistic studies showed that this reaction underwent C–H bond activation via metallation but not the Friedel–Crafts pathway. Expansion of this chemistry to synthesize other useful molecules is undergoing in our laboratory.
Experimental section
General procedures
To a 50 mL of Wattecs (a Chinese company making chemical apparatuses) reaction tube with a stirring bar was added 4-(tert-butyl)phenol 1a (0.2 mmol, 1.0 equiv., 30.0 mg), Cu(OAc)2 (0.4 mmol, 2.0 equiv., 72.5 mg) and NaOPiv (0.16 mmol, 0.8 equiv., 19.9 mg) under air atmosphere. After adding Pd(CH3CN)4(BF4)2 (0.02 mmol, 8.9 mg) in a glove box, methyl acrylate 2a (0.4 mmol, 2.0 equiv., 34.4 mg) and solvent (mesitylene) were added and the tube was sealed and heated to 120 °C for 12 h. After cooling to room temperature, the system was purified by column chromatography with hexane–EtOAc (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to afford the product as a white solid in 70% yield (28.5 mg).
:1) to afford the product as a white solid in 70% yield (28.5 mg).
Acknowledgements
Support of this work by the “973” Project from the MOST of China (2009CB825300) and NSFC (Nos. 20925207 and 21002001) is gratefully acknowledged.Notes and references
- B. N. Shyamala, M. M. Naidu, G. Sulochanamma and P. Srinivas, J. Agric. Food Chem., 2007, 55, 7738–7743 CrossRef CAS PubMed.
- X. Wang, Y. Lu, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 12203–12205 CrossRef CAS PubMed.
- (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (b) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed.
- (a) A. John and K. M. Nicholas, J. Org. Chem., 2012, 77, 5600–5605 CrossRef CAS PubMed; (b) T.-J. Gong, B. Xiao, Z.-J. Liu, J. Wan, J. Xu, D.-F. Luo, Y. Fu and L. Liu, Org. Lett., 2011, 13, 3235–3237 CrossRef CAS PubMed; (c) C. Feng and T.-P. Loh, Chem. Commun., 2011, 47, 10458–10460 RSC; (d) X. Zhao, C. S. Yeung and V. M. Dong, J. Am. Chem. Soc., 2010, 132, 5837–5844 CrossRef CAS PubMed.
- B. Xiao, Y. Fu, J. Xu, T.-J. Gong, J.-J. Dai, J. Yi and L. Liu, J. Am. Chem. Soc., 2010, 132, 468–469 CrossRef CAS PubMed.
- Y. Shen, G. Liu, Z. Zhou and X. Lu, Org. Lett., 2013, 15, 3366–3369 CrossRef CAS PubMed.
- H.-X. Dai, G. Li, X.-G. Zhang, A. F. Stepan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 7567–7571 CrossRef CAS PubMed.
- L. Ackermann, E. Diers and A. Manvar, Org. Lett., 2012, 14, 1154–1157 CrossRef CAS PubMed.
- (a) C.-H. Huang, B. Chattopadhyay and V. Gevorgyan, J. Am. Chem. Soc., 2011, 133, 12406–12409 CrossRef CAS PubMed; (b) C. Huang, N. Ghavtadze, B. Chattopadhyay and V. Gevorgyan, J. Am. Chem. Soc., 2011, 133, 17630–17633 CrossRef CAS PubMed; (c) C. Huang and V. Gevorgyan, J. Am. Chem. Soc., 2009, 131, 10844–10845 CrossRef CAS PubMed; (d) T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 7534–7535 CrossRef CAS PubMed.
- (a) S. Luo, F.-X. Luo, X.-S. Zhang and Z.-J. Shi, Angew. Chem., Int. Ed., 2013, 52, 10598–10601 CrossRef CAS PubMed; (b) J. Zhao, Y. Wang, Y. He, L. Liu and Q. Zhu, Org. Lett., 2012, 14, 1078–1081 CrossRef CAS PubMed; (c) B. Xiao, T.-J. Gong, Z.-J. Liu, J.-H. Liu, D.-F. Luo, J. Xu and L. Liu, J. Am. Chem. Soc., 2011, 133, 9250–9253 CrossRef CAS PubMed; (d) Y. Wei and N. Yoshikai, Org. Lett., 2011, 13, 5504–5507 CrossRef CAS PubMed; (e) T. Satoh, Y. Kawamura, M. Miura and M. Nomura, Angew. Chem., Int. Ed. Engl., 1997, 36, 1740–1742 CrossRef CAS; (f) Q. Zhu, J. Zhao and Y. Wang, Synthesis, 2012, 44, 1551–1555 CrossRef PubMed.
- (a) J. D. Dooley, C. S. Reddy and H. W. Lam, J. Am. Chem. Soc., 2013, 135, 10829–10836 CrossRef CAS PubMed; (b) V. S. Thirunavukkarasu, M. Donati and L. Ackermann, Org. Lett., 2012, 14, 3416–3419 CrossRef CAS PubMed; (c) S. Mochida, M. Shimizu, K. Hirano, T. Satoh and M. Miura, Chem.–Asian J., 2010, 5, 847–851 CrossRef CAS PubMed; (d) Y. Nishinaka, T. Satoh, M. Miura, H. Morisaka, M. Nomura, H. Matsui and C. Yamaguchi, Bull. Chem. Soc. Jpn., 2001, 74, 1727–1735 CrossRef CAS.
- (a) R. B. Bedford, S. J. Coles, M. B. Hursthouse and M. E. Limmert, Angew. Chem., Int. Ed., 2003, 42, 112–114 CrossRef CAS; (b) S. Oi, S.-I. Watanabe, S. Fukita and Y. Inoue, Tetrahedron Lett., 2003, 44, 8665–8668 CrossRef CAS PubMed; (c) L. N. Lewis and J. F. Smith, J. Am. Chem. Soc., 1986, 108, 2728–2735 CrossRef CAS.
- D. D. Hennings, S. Iwasa and V. H. Rawal, J. Org. Chem., 1997, 62, 2–3 CrossRef CAS.
- (a) Z. Huang, L. Jin, Y. Feng, P. Peng, H. Yi and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 7151–7155 CrossRef CAS PubMed; (b) R. Zhu, J. Wei and Z. Shi, Chem. Sci., 2013, 4, 3706–3711 RSC; (c) M. R. Kuram, M. Bhanuchandra and A. K. Sahoo, Angew. Chem., Int. Ed., 2013, 52, 4607–4612 CrossRef CAS PubMed; (d) C. Jia, D. Piao, J. Oyamada, W. Lu, T. Kitamura and Y. Fujiwara, Science, 2000, 287, 1992–1995 CrossRef CAS; (e) B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1996, 118, 6305–6306 CrossRef CAS.
- X. Guo, R. Yu, H. Li and Z. Li, J. Am. Chem. Soc., 2009, 131, 17387–17393 CrossRef CAS PubMed.
- D.-H. Lee, K.-H. Kwon and C. S. Yi, J. Am. Chem. Soc., 2012, 134, 7325–7328 CrossRef CAS PubMed.
- (a) K. N. Venugopala, V. Rashmi and B. Odhav, BioMed Res. Int., 2013, 963248 CAS; (b) A. Y. Fedorov, A. V. Nyuchev and I. P. Beletskaya, Chem. Heterocycl. Compd., 2012, 48, 166–178 CrossRef CAS; (c) A. Jerezano, F. Jimenez, M. d. C. Cruz, L. E. Montiel, F. Delgado and J. Tamariz, Helv. Chim. Acta, 2011, 94, 185–198 CrossRef CAS; (d) M. E. Riveiro, K. N. De, A. Moglioni, R. Vazquez, F. Monczor, C. Shayo and C. Davio, Curr. Med. Chem., 2010, 17, 1325–1338 CrossRef CAS; (e) I. Kostova, Curr. Med. Chem.: Anti-Cancer Agents, 2005, 5, 29–46 CrossRef CAS; (f) M. M. Garazd, Y. L. Garazd and V. P. Khilya, Chem. Nat. Compd., 2005, 41, 245–271 CrossRef CAS PubMed; (g) F. Borges, F. Roleira, N. Milhazes, L. Santana and E. Uriarte, Curr. Med. Chem., 2005, 12, 887–916 CrossRef CAS; (h) A. Lacy and R. O'Kennedy, Curr. Pharm. Des., 2004, 10, 3797–3811 CrossRef CAS; (i) K. C. Fylaktakidou, D. J. Hadjipavlou-Litina, K. E. Litinas and D. N. Nicolaides, Curr. Pharm. Des., 2004, 10, 3813–3833 CrossRef CAS; (j) S. Sardari, S. Nishibe and M. Daneshtalab, Stud. Nat. Prod. Chem., 2000, 23, 335–393 CAS; (k) V. M. Malikov and A. I. Saidkhodzhaev, Chem. Nat. Compd., 1998, 34, 202–264 CrossRef CAS; (l) A. Estevez-Braun and A. G. Gonzalez, Nat. Prod. Rep., 1997, 14, 465–475 RSC; (m) J. R. S. Hoult and M. Paya, Gen. Pharmacol., 1996, 27, 713–722 CrossRef CAS; (n) R. D. H. Murray, Nat. Prod. Rep., 1995, 12, 477–505 RSC; (o) R. D. H. Murray, Prog. Chem. Org. Nat. Prod., 1991, 58, 83–316 CAS; (p) S. M. Sethna and N. M. Shah, Chem. Rev., 1945, 36, 1–62 CrossRef CAS.
- H. Zhao, A. C. Donnelly, B. R. Kusuma, G. E. L. Brandt, D. Brown, R. A. Rajewski, G. Vielhauer, J. Holzbeierlein, M. S. Cohen and B. S. J. Blagg, J. Med. Chem., 2011, 54, 3839–3853 CrossRef CAS PubMed.
- (a) R. Dayam, R. Gundla, L. Q. Al-Mawsawi and N. Neamati, Med. Res. Rev., 2008, 28, 118–154 CrossRef CAS PubMed; (b) L. Xie, Y. Takeuchi, L. M. Cosentino and K.-H. Lee, J. Med. Chem., 1999, 42, 2662–2672 CrossRef CAS PubMed.
- T. Symeonidis, M. Chamilos, D. J. Hadjipavlou-Litina, M. Kallitsakis and K. E. Litinas, Bioorg. Med. Chem. Lett., 2009, 19, 1139–1142 CrossRef CAS PubMed.
- E. Melliou, P. Magiatis, S. Mitaku, A.-L. Skaltsounis, E. Chinou and I. Chinou, J. Nat. Prod., 2005, 68, 78–82 CrossRef CAS PubMed.
- (a) F. Gosselin, R. A. Britton, I. W. Davies, S. J. Dolman, D. Gauvreau, R. S. Hoerrner, G. Hughes, J. Janey, S. Lau, C. Molinaro, C. Nadeau, P. D. O'Shea, M. Palucki and R. Sidler, J. Org. Chem., 2010, 75, 4154–4160 CrossRef CAS PubMed; (b) C. A. Kontogiorgis and D. J. Hadjipavlou-Litina, J. Med. Chem., 2005, 48, 6400–6408 CrossRef CAS PubMed.
- (a) R. Sheng, P. Wang, Y. Gao, Y. Wu, W. Liu, J. Ma, H. Li and S. Wu, Org. Lett., 2008, 10, 5015–5018 CrossRef CAS PubMed; (b) R. S. Koefod and K. R. Mann, Inorg. Chem., 1989, 28, 2285–2290 CrossRef CAS; (c) X. Xu, X. Hu and J. Wang, Beilstein J. Org. Chem., 2013, 9, 254–259 CrossRef CAS PubMed; (d) Y. Zhou, K. Liu, J.-Y. Li, Y. Fang, T.-C. Zhao and C. Yao, Org. Lett., 2011, 13, 1290–1293 CrossRef CAS PubMed.
- D. Audisio, S. Messaoudi, J.-D. Brion and M. Alami, Eur. J. Org. Chem., 2010, 1046–1051 CrossRef CAS.
- (a) E. Fillion, A. M. Dumas, B. A. Kuropatwa, N. R. Malhotra and T. C. Sitler, J. Org. Chem., 2006, 71, 409–412 CrossRef CAS PubMed; (b) M. S. Reddy, N. Thirupathi and M. Haribabu, Beilstein J. Org. Chem., 2013, 9, 180–184 CrossRef CAS PubMed.
- (a) S. Valente and G. Kirsch, Tetrahedron Lett., 2011, 52, 3429–3432 CrossRef CAS PubMed; (b) D. Giguere, P. Cloutier and R. Roy, J. Org. Chem., 2009, 74, 8480–8483 CrossRef CAS PubMed; (c) Y. Yamamoto and N. Kirai, Org. Lett., 2008, 10, 5513–5516 CrossRef CAS PubMed; (d) M. Catellani, G. P. Chiusoli, G. Marzolini and E. Rossi, J. Organomet. Chem., 1996, 525, 65–69 CrossRef CAS; (e) K. Sasano, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2013, 135, 10954–10957 CrossRef CAS PubMed; (f) J. Ferguson, F. Zeng and H. Alper, Org. Lett., 2012, 14, 5602–5605 CrossRef CAS PubMed.
- (a) C. Jia, W. Lu, J. Oyamada, T. Kitamura, K. Matsuda, M. Irie and Y. Fujiwara, J. Am. Chem. Soc., 2000, 122, 7252–7263 CrossRef CAS; (b) B. M. Trost, F. D. Toste and K. Greenman, J. Am. Chem. Soc., 2003, 125, 4518–4526 CrossRef CAS PubMed; (c) Z. Shi and C. He, J. Org. Chem., 2004, 69, 3669–3671 CrossRef CAS PubMed; (d) H. A. Wegner, S. Ahles and M. Neuburger, Chem.–Eur. J., 2008, 14, 11310–11313 CrossRef CAS PubMed; (e) J.-L. Cao, S.-L. Shen, P. Yang and J. Qu, Org. Lett., 2013, 15, 3856–3859 CrossRef CAS PubMed.
- S. Aoki, J. Oyamada and T. Kitamura, Bull. Chem. Soc. Jpn., 2005, 78, 468–472 CrossRef CAS.
- D. Kim, M. Min and S. Hong, Chem. Commun., 2013, 49, 4021–4023 RSC.
- U. Sharma, T. Naveen, A. Maji, S. Manna and D. Maiti, Angew. Chem., Int. Ed., 2013, 52, 12669–12673 CrossRef CAS PubMed.
- T. Kitamura, K. Yamamoto, M. Kotani, J. Oyamada, C. Jia and Y. Fujiwara, Bull. Chem. Soc. Jpn., 2003, 76, 1889–1895 CrossRef CAS.
- (a) B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500–1511 CrossRef CAS PubMed; (b) A. Zapf and M. Beller, Chem.–Eur. J., 2000, 6, 1830–1833 CrossRef CAS; (c) S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1439–1439 CrossRef CAS PubMed; (d) N. S. Sitnikov, A. S. Shavyrin, G. K. Fukin, I. P. Beletskaya, S. Combes and A. Y. Fedorov, Russ. Chem. Bull., 2010, 59, 626–631 CrossRef CAS PubMed; (e) M. I. Naumov, A. V. Nuchev, N. S. Sitnikov, Y. B. Malysheva, A. S. Shavyrin, I. P. Beletskaya, A. E. Gavryushin, S. Combes and A. Y. Fedorov, Synthesis, 2009, 1673–1682 CAS.
- B.-J. Li, D.-G. Yu, C.-L. Sun and Z.-J. Shi, Chem.–Eur. J., 2011, 17, 1728–1759 CrossRef CAS PubMed.
- (a) S. Combes, P. Barbier, S. Douillard, A. McLeer-Florin, V. Bourgarel-Rey, J.-T. Pierson, A. Y. Fedorov, J.-P. Finet, J. Boutonnat and V. Peyrot, J. Med. Chem., 2011, 54, 3153–3162 CrossRef CAS PubMed; (b) C. Bailly, C. Bal, P. Barbier, S. Combes, J.-P. Finet, M.-P. Hildebrand, V. Peyrot and N. Wattez, J. Med. Chem., 2003, 46, 5437–5444 CrossRef CAS PubMed.
- M. Miura, T. Tsuda, T. Satoh and M. Nomura, Chem. Lett., 1997, 1103–1104 CrossRef CAS.
- Z. Shi, C. Grohmann and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 5393–5397 CrossRef CAS PubMed.
- (a) J. A. Tunge and L. N. Foresee, Organometallics, 2005, 24, 6440–6444 CrossRef CAS; (b) M. Ochiai, K. Fukui, S. Iwatsuki, K. Ishihara and K. Matsumoto, Organometallics, 2005, 24, 5528–5536 CrossRef CAS; (c) D. D. Hennings, S. Iwasa and V. H. Rawal, Tetrahedron Lett., 1997, 38, 6379–6382 CrossRef CAS.
- N. M. Howarth, A. Purohit, M. J. Reed and B. V. L. Potter, J. Med. Chem., 1994, 37, 219–221 CrossRef CAS.
- (a) S. Papst, A. F. M. Noisier, M. A. Brimble, Y. Yang and G. W. Krissansen, Bioorg. Med. Chem., 2012, 20, 5139–5149 CrossRef CAS PubMed; (b) M. Gynther, K. Laine, J. Ropponen, J. Leppänen, A. Mannila, T. Nevalainen, J. Savolainen, T. Järvinen and J. Rautio, J. Med. Chem., 2008, 51, 932–936 CrossRef CAS PubMed.
- J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed.
- A. M. Silván, M. J. Abad, P. Bermejo, M. Sollhuber and A. Villar, J. Nat. Prod., 1996, 59, 1183–1185 CrossRef PubMed.
- S. P. Pillai, S. R. Menon, L. A. Mitscher, C. A. Pillai and D. M. Shankel, J. Nat. Prod., 1999, 62, 1358–1362 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3qo00010a |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2014 |