
Synthesis, polymorphism, and electronic structures of Sr3Sn2As4†
Xiao-Cun
Liu
,
Ming-Yan
Pan
,
Xin
Li
,
Sheng-Qing
Xia
* and
Xu-Tang
Tao
State Key Laboratory of Crystal Materials, Institute of Crystal Materials, Shandong University, Jinan, Shandong 250100, People's Republic of China. E-mail: shqxia@sdu.edu.cn
First published on 12th September 2014
Abstract
Two polymorphs of Sr3Sn2As4 were synthesized from the Sn-flux reactions, and their structures were determined by the single-crystal X-ray diffraction technique. α-Sr3Sn2As4 adopts the Sr3Sn2P4 structure type with the orthorhombic space group Cmca (a = 25.798(2) Å, b = 12.8883(11) Å, c = 19.1244(16) Å, V = 6358.8(9) Å3, Z = 24), whereas β-Sr3Sn2As4 belongs to Ca3Si2As4 structure type and crystallizes in the monoclinic crystal system P21/c (a = 7.7049(13), b = 19.118(3), c = 7.6877(13), β = 112.003(2)°, V = 1049.9(3), Z = 4). Despite the obvious structural differences, the polyanion units of both compounds feature similar [Sn2As6] octahedra. Differential thermal analysis and thermogravimetry measurements indicate that α-Sr3Sn2As4 has good thermal stability and melts at 1185 K, while β-Sr3Sn2As4 starts to decompose above 800 K. Diffuse reflectance spectral measurements proved that both compounds had a band gap of about 0.9 eV, supported by the density functional calculations.
Introduction
Zintl phases are valence precise compounds formed among metals with very different electronegativities, such as alkali metals, alkali earth metals, and group 13–15 elements.1,2 Recently, Zintl phases have become appealing to more and more scientists due to their diverse structures and rich physical properties, including superconductivity,3–5 magnetoresistance,6–8 mixed valence,9,10 thermoelectricity11–13 and so on. Numerous studies on the crystal and electronic structures, as well as related properties have been reported in the last decade.14 Surprisingly, although so many compounds have been synthesized, the variety of such phases seems far from being exhausted.For Zintl compounds with the general formula A3M2Pn4 (A = alkaline-earth metals, Yb or Eu; M = group 12, 13 or 14 metals; Pn = pnictogen elements), various polyanion structures have been discovered, i.e. two-dimensional [Cd2Sb4]6− layers in Ba3Cd2Sb4,15 one-dimensional [In2P4]6− chains in Eu3In2P4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16 and zero-dimensional [Sn12P24]36− clusters in Sr3Sn2P4,17 which can all be rationalized using the Zintl–Klemm concept. The variety of the 3-2-4 family has aroused many studies involving the correlation between structures and properties. As part of such investigations, the A3Tt2Pn4 (Tt = Si, Ge or Sn) system is of special interest due to its structural diversity. The structures of A3Tt2Pn4 generally feature polyanionic rings, such as four-membered rings in Sr3Si2As4,18 five-membered rings in Ca3Si2As419 and six-membered rings in Sr3Sn2P4.17 More interestingly, all these different rings are composed of the same Tt2Pn6 octahedral units.
16 and zero-dimensional [Sn12P24]36− clusters in Sr3Sn2P4,17 which can all be rationalized using the Zintl–Klemm concept. The variety of the 3-2-4 family has aroused many studies involving the correlation between structures and properties. As part of such investigations, the A3Tt2Pn4 (Tt = Si, Ge or Sn) system is of special interest due to its structural diversity. The structures of A3Tt2Pn4 generally feature polyanionic rings, such as four-membered rings in Sr3Si2As4,18 five-membered rings in Ca3Si2As419 and six-membered rings in Sr3Sn2P4.17 More interestingly, all these different rings are composed of the same Tt2Pn6 octahedral units.
Herein, we present the flux synthesis of two new ternary Zintl polymorphs, α-Sr3Sn2As4 and β-Sr3Sn2As4, which belong to the Sr3Sn2P417 and Ca3Si2As419 structure types, respectively. Compounds with the Ca3Si2As4 structure type are very common and more than half of the A3Tt2Pn4 series adopt this structure. However, for the Sr3Sn2P4 structure type, only one phosphide, Sr3Sn2P4, has been reported so far. By single-crystal X-ray diffraction methods, the crystal structures of α-Sr3Sn2As4 and β-Sr3Sn2As4 were determined accurately, and in cooperation with the density functional theoretical calculations their electronic structures were studied as well. The theoretical results indicated the semiconducting properties of both compounds, which were consistent with the measured optical diffuse reflectance spectral data. Thermal stability studies were performed with the aid of differential thermal analysis and thermogravimetry measurements.
Results and discussion
Structure description
Compound α-Sr3Sn2As4 crystallizes in the orthorhombic Sr3Sn2P4 structure type (Cmca), whereas β-Sr3Sn2As4 crystallizes in the monoclinic Ca3Si2As4 structure type (P21/c). Since the structures of both compounds have been well discussed in previous studies, only some important structural features will be discussed here. Information on data collection, structure solution and refinement is summarized in Table 1. Positional and equivalent isotropic displacement parameters, and selected bond distances are all listed in Tables S1 and S2 (ESI†).| Formula | α-Sr3Sn2As4 | β-Sr3Sn2As4 |
|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|; wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2, and w = 1/[σ2Fo2 + (AP)2 + BP], P = (Fo2 + 2Fc2)/3; A and B are weight coefficients. | ||
| Fw/g mol−1 | 799.92 | 799.92 |
| T/K | 296(2) | 296(2) |
| Radiation, wavelength | Mo-Kα, 0.71073 Å | |
| Crystal system | Orthorhombic | Monoclinic |
| Space group, Z | Cmca, 24 | P21/c, 4 |
| Unit cell dimensions | ||
| a/Å | 25.798(2) | 7.7049(13) |
| b/Å | 12.8883(11) | 19.118(3) |
| c/Å | 19.1244(16) | 7.6877(13) |
| β/° | — | 112.003(2) |
| V/Å3 | 6358.8(9) | 1049.9(3) |
| ρ calc/g cm−3 | 5.013 | 5.060 |
| μ Mo Kα/mm−1 | 31.968 | 32.268 |
| Extinction coefficient | 0.000069(2) | — |
| GOF | 1.019 | 1.050 |
| Final R indicesa [I > 2σ(I)] | R 1 = 0.0329 | R 1 = 0.0324 |
| wR2 = 0.0513 | wR2 = 0.0795 | |
| Final R indicesa [all data] | R 1 = 0.0610 | R 1 = 0.0408 |
| wR2 = 0.0667 | wR2 = 0.0831 | |
A structure comparison between α-Sr3Sn2As4 and β-Sr3Sn2As4 is provided in Fig. 1. As shown in Fig. 1, the anion structure of α-Sr3Sn2As4 features an interesting [Sn12As24]36− cluster, which encapsulates a Sr2+ cation in the center. Each [Sn12As24]36− cluster can be viewed as composed of six [Sn2As6] octahedra, connected in the edge-sharing manner. Such an ethane-like [Sn2As6] fragment is very common in pnictide Zintl phases and similar examples have been frequently reported as in Sr3Si2As4,18 Ca3Si2As419 and ACdGeAs2 (A = K, Rb).20 However, it is really interesting if this structure is compared with its polymorph β-Sr3Sn2As4, in which the polyanion structure turns out to be a completely different infinite chain. In addition, such one-dimensional chains are also built of the same [Sn2As6] octahedra as in α-Sr3Sn2As4 and the connection of these [Sn2As6] octahedral units still embodies the edge-sharing fashion. A better illustration of the difference in the anion structures of these two polymorphs is presented in Fig. 2, shown by both polyhedral and ball-and-stick fashions. In α-Sr3Sn2As4, the edge-sharing connection between two neighbouring [Sn2As6] octahedra takes place via two As atoms shared by four different [SnAs3] triangular pyramids. This connection pattern results in a unique [Sn4As2] six-membered ring, which helps build the “circle-like” anion clusters in α-Sr3Sn2As4. While for β-Sr3Sn2As4 the situation is a little different: these two As atoms are shared by three [SnAs3] triangular pyramids, which are still from two different [Sn2As6] octahedral and in this way, polyanion chains are formed featuring new [Sn3As2] five-membered rings.
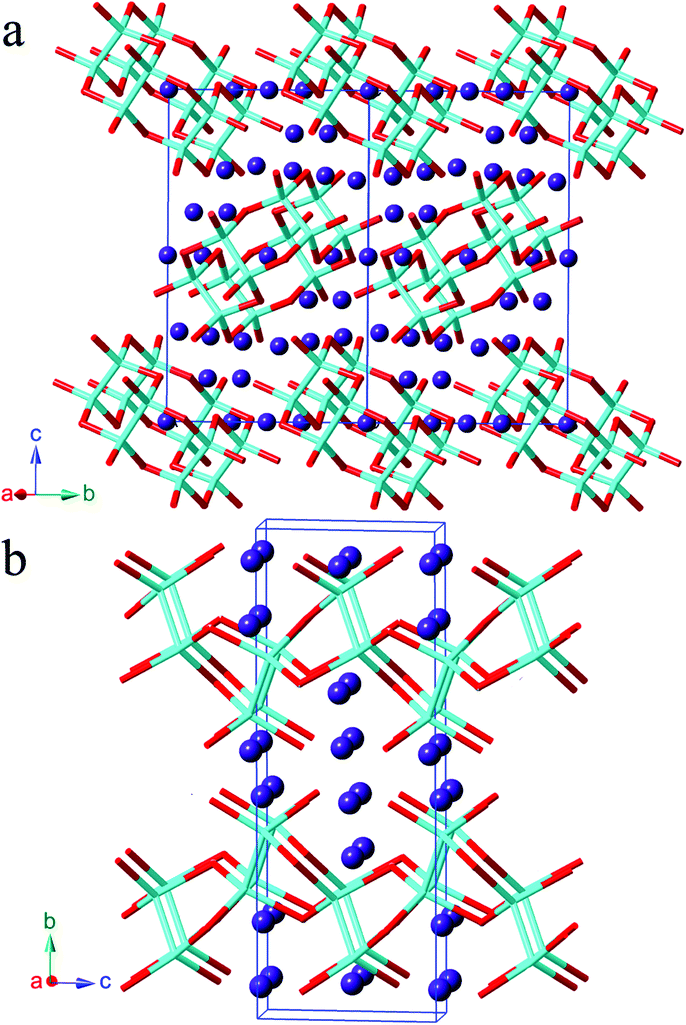 | ||
| Fig. 1 Crystal structure of (a) α-Sr3Sn2As4 and (b) β-Sr3Sn2As4. Sr atoms are plotted in purple spheres, and the polyanionic framework are represented by stick mode with Sn and As atoms shown in light blue and red colors, respectively. | ||
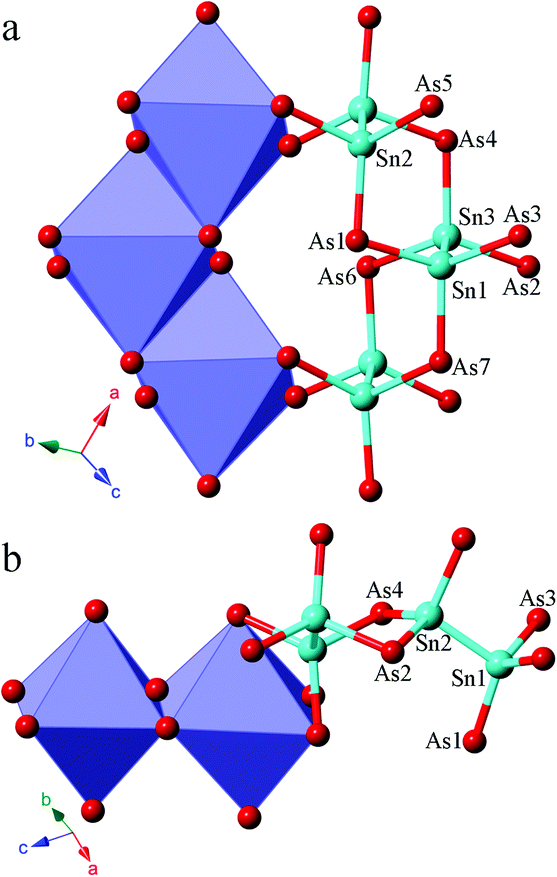 | ||
| Fig. 2 Polyhedral and ball-and-stick view of the structure of polyanion units of (a) α-Sr3Sn2As4 and (b) β-Sr3Sn2As4. Sn and As atoms are plotted as light blue and red spheres, respectively. [Sn2As6] octahedra are emphasized in polyhedral view. | ||
The Sr cations are six-coordinated in both compounds, which all reside in the centers of the distorted As6 octahedra. The Sn–Sn and Sn–As bonding distances are very similar in these two polymorphs. The corresponding Sn–Sn distances are 2.7659(9) to 2.7955(12) Å in α-Sr3Sn2As4, and 2.7173(9) Å in β-Sr3Sn2As4. The Sn–As interatomic distances are in the range of 2.5523(10) to 2.6563(9) Å in α-Sr3Sn2As4, and 2.5327(11) to 2.6362(10) Å in β-Sr3Sn2As4. These results are comparable to those Zintl compounds with similar bonding patterns,21–23 yet much shorter than the typical multicenter bonding distances.24,25 This is consistent with the Zintl concept that the interactions of anionic structures should be localized in two-center two-electron (2c–2e) bonding. The As–Sn–Sn bonding angles are in the range of 103.59(4)° to 111.73(3)° in α-Sr3Sn2As4 and 97.24(3)° to 117.87(3) ° in β-Sr3Sn2As4, which suggests an obviously more distorted tetrahedral coordination geometry for the Sn atoms in β-Sr3Sn2As4 due to the formation of the five-membered rings.
Thermal stability
TG-DSC measurements were performed in order to evaluate the thermal stability of both compounds, and the results are shown in Fig. S1 (ESI†). There is no obvious phase transition behavior observed between these two phases. For α-Sr3Sn2As4, two sequential cycles were performed to ensure its thermal behavior and the results of the 2nd cycle are shown in Fig. S2.† There is no significant mass loss observed in the whole measured process. Both DSC curves exhibit similar sharp endothermic peaks and exothermic peaks at 1185 K and 1160 K, respectively. Due to the good reproducibility, these two peaks should be attributed to melting and crystallization processes of this compound. Although based on the powder diffraction patterns (Fig. S3†) there are no significant impurities identified, the slight fluctuations of the curves suggest the possible existence of some uncertain amorphous impurities. A similar situation exists for β-Sr3Sn2As4; however, a significant mass loss is observed at above 800 K, which evidently indicates the decomposition process of β-Sr3Sn2As4. It is not surprising that both compounds have shown a distinct thermal behavior due to their different structures. The thermal behavior of the β-phase is very similar to its isostructural analogue Eu3Ge2As4,26 which also has a mass loss of about 7% in the range from 830 K to 1170 K.Optical absorption spectrum
UV–vis/near-IR diffuse reflectance spectra were recorded at room temperature for both α-Sr3Sn2As4 and β-Sr3Sn2As4 and the calculated optical absorption spectra are presented in Fig. S4.† The optical absorption spectra of compounds are very similar and both suggest semiconducting characters. The room temperature band gaps evaluated for α-Sr3Sn2As4 and β-Sr3Sn2As4 are 0.86(2) and 0.87(3) eV, respectively, which are consistent with their black colour. However, as mentioned above, the existence of some uncertain amorphous impurities may also affect the absorption behavior, which exhibit a small peak around 0.9 eV as well as the fluctuant baseline. Tauc plots were also tried to evaluate the band gaps (Fig. S5†) and the results are consistent with the previous findings.Electronic band structure
To better understand their optical properties, electronic band structure calculations based on the density functional method were carried out for both compounds and the results are shown in Fig. 3. As shown in Fig. 3, both structures clearly indicate the existence of a small band gap, 0.82 and 0.83 eV for α-Sr3Sn2As4 and β-Sr3Sn2As4, respectively, which suggest a semiconducting character as confirmed by the optical absorption spectra above. In addition, the valence band maximum (VBM) and the conduction band minimum (CBM) of the compounds are both located at the same k-vector, a clue that these compounds should be direct band gap semiconductors. Despite the similarities, the band structures of α-Sr3Sn2As4 and β-Sr3Sn2As4 are significantly different in the band width. The bands in α-Sr3Sn2As4 are obviously much narrower than those of β-Sr3Sn2As4, which means the electrons are more localized in α-Sr3Sn2As4 than in β-Sr3Sn2As4. By taking their crystal structures into account, such discrepancies are reasonable since in α-Sr3Sn2As4, the polyanion structure is basically composed of the isolated [Sn12As24]36− clusters, whereas for β-Sr3Sn2As4 the Sn and As atoms form infinite chains in favor of electron transport.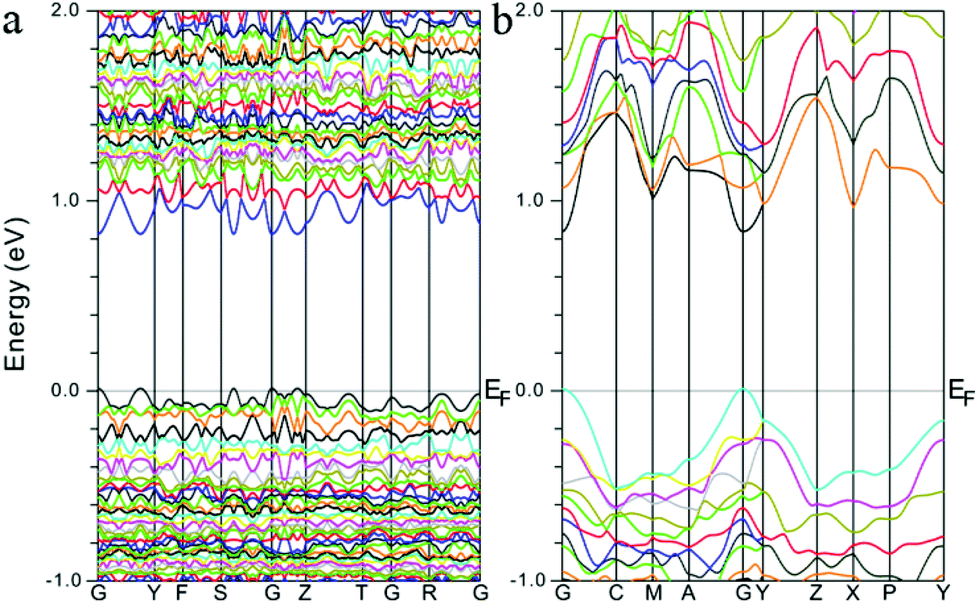 | ||
| Fig. 3 Calculated electronic band structures for (a) α-Sr3Sn2As4 and (b) β-Sr3Sn2As4, respectively. The Fermi level is chosen as the energy reference at 0 eV. | ||
Projections of the corresponding total and partial density of states (DOS) for both α-Sr3Sn2As4 and β-Sr3Sn2As4 are presented in Fig. 4. A qualitative comparison of the DOS between these two polymorphs reveals high similarity in the DOS curves: the conduction bands are generally composed of 4d orbitals of Sr and the antibonding states originated from the Sn–As interactions; the valence bands can be divided into two different regions—the lower region ranging from −8 to −4 eV are primarily contributed by the localized s states from As atoms and another region, which has relatively higher energy below the Fermi level, is composed mainly of Sr-4d, Sn-5s and 5p, and As-4p orbitals. The high hybridization between Sn and As orbitals in the valence band suggests strong covalent interactions within the anionic framework and the significant p–d mixing of Sr and As indicates non-negligible covalent bonding interactions between cations and anions. Although cations in Zintl phases are usually considered merely as charge suppliers and space fillers, many recent researches27–29 indicate that they can play more important roles, such as stabilizing the structure or affecting the transport properties. Despite the similarites above, there exists a sharp shoulder peak right below the Fermi level for α-Sr3Sn2As4, which predominantly consists of the 4p orbitals of As atoms and is not observed in β-Sr3Sn2As4. This peak should be designated as the non-bonding electrons or a lone pair on As atoms of α-Sr3Sn2As4 with the knowledge that the polyanion structure of α-Sr3Sn2As4 consists of the isolated [Sn12As24]36− clusters, which should more favor the electron localization than the chain structure of β-Sr3Sn2As4. These results are in accord with the electronic band structure calculations presented above and also are similar to our recent report on arsenide Zintl analogues.30,31
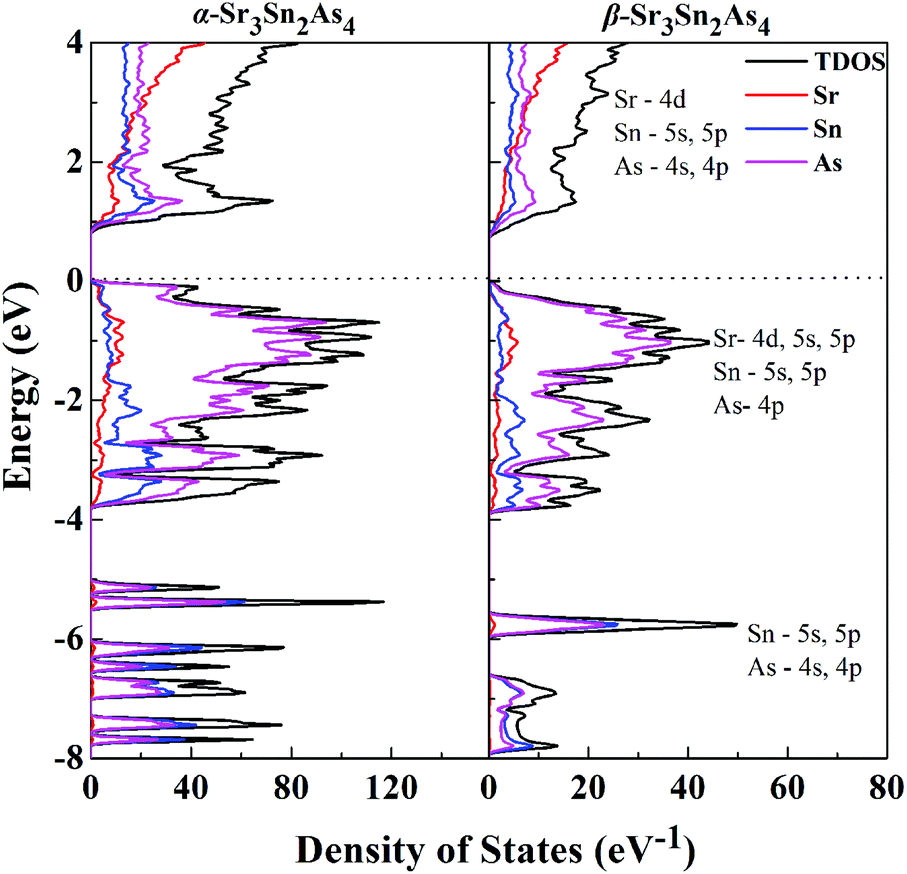 | ||
| Fig. 4 Calculated total DOS and projected DOS are plotted on the same energy scale for α-Sr3Sn2As4 and β-Sr3Sn2As4. The Fermi level is selected as the energy reference. | ||
Conclusions
In conclusion, two polymorphs of Sr3Sn2As4 have been synthesized and structurally characterized. Although they feature identical [Sn2As6] octahedral building units, the different connection patterns lead to the significantly different polyanionic structures. For α-Sr3Sn2As4, the structure can be described as made up of zero-dimensional [Sn12As24]36− clusters that are separated by the Sr cations and in the β-phase there exist one-dimensional infinite chains consisting of the edge-sharing [Sn2As6] octahedra. The measured optical absorption spectra for both compounds suggest a semiconducting character with band gaps of 0.86 and 0.87 eV for α-Sr3Sn2As4 and β-Sr3Sn2As4, respectively. These results are in good agreement with the theoretical calculations.Experimental section
Synthesis
All syntheses were performed in an argon-filled glovebox with the oxygen level below 0.1 ppm or under vacuum. The elements were commercially purchased and used as received: Sr (Alfa, granules, 99%), Sn (Alfa, shot, 99.9%), and As (Alfa, lump, 99.999%).:Sn:As = 3:10:4 were loaded into an alumina crucible and the reaction mixture was first heated to 1000 °C at a rate of 200 °C h−1, then homogenized at this temperature for 20 h, and slowly cooled down to 600 °C at a rate of 3 °C h−1. At this temperature, the ampule was quickly taken out of the furnace and the excess of Sn was removed by centrifugation. The utilization of low-fold metal flux (5-fold excess of Sn) is necessary in order to synthesize α-Sr3Sn2As4 and a high-fold metal flux, such as 20-fold excess or higher, will lead to another product Sr5Sn2As6.21 Stoichiometric reactions were also performed, but the main products of such reactions were mixtures of small irregular blocks of α-Sr3Sn2As4 as well as some uncertain amorphous phases.
:Sn:As = 1:7:2. The synthesis process is similar to that of α-Sr3Sn2As4 but a higher homogenization temperature (1050 °C) and lower flux separation temperature (500 °C) were applied. The products of this reaction consist of block-shaped α-Sr3Sn2As4 and some plate-shaped crystals of β-Sr3Sn2As4. After the crystal structure of β-Sr3Sn2As4 was identified, several stoichiometric reactions attempting to synthesize β-Sr3Sn2As4 were tried but the results were negative.
Single crystal structure determination
In order to avoid possible decomposition of the title compounds during the data collection, all crystals were picked in an argon-filled glovebox and cut in Paratone N oil to the desired dimensions. The X-ray diffraction data were collected on a Bruker CCD-based diffractometer at 296 K utilizing a Mo Kα radiation (0.71703 Å). Data reduction and integration, together with global unit cell refinements were done by the INTEGRATE program incorporated in the APEX2 software.32 Semi-empirical absorption corrections were applied using the SCALE program for the area detector.32 The structures were solved by direct methods and refined by full matrix least-squares methods on F2 using SHELX.33 In the last refinement cycles, the atomic positions of the two compounds were standardized using the program structure TIDY.34,35 All structures were refined to convergence with anisotropic displacement parameters. Crystallographic data and structural refinements are summarized in Table 1. Atomic positions and anisotropic displacement parameters and selected bond lengths are given in Tables S1 and S2, respectively. Further information in the form of CIF has been deposited with Fachinformationszentrum Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany (fax: (49) 7247-808-666; e-mail: crysdata@fiz-karlsruhe.de) – depository CSD 428135 and 428136 for α-Sr3Sn2As4 and β-Sr3Sn2As4, respectively.Powder X-ray diffraction
For phase identification, powder X-ray diffraction (PXRD) patterns were taken at room temperature by a Bruker AXS X-ray powder diffractometer using Cu-Kα radiation. The data were recorded in a 2θ mode with a step size of 0.02° and a counting time of 10 s. In Fig. S3 (in ESI†), the comparison between experimental and simulated data confirms the purity of the samples.Differential thermal analysis and thermogravimetry measurements (DTA/TG)
The thermal stabilities of α-Sr3Sn2As4 and β-Sr3Sn2As4 were tested using a Mettler–Toledo TGA/DSC/1600HT instrument. Thermogravimetric Analysis (TGA) and Differential Scanning Calorimetry (DSC) experiments were performed on polycrystalline material samples for the two phases under the protection of high-purity argon gas with a heating rate of 10 K min−1. In order to verify the thermal behavior of the α-phase, the TGA–DSC experiments were performed twice.UV–Vis–NIR diffuse reflectance spectrum
The optical diffuse reflectance spectrum for two polymorphs of Sr3Sn2As4 was measured on polycrystalline samples at room temperature using a Shimadzu UV-3101 PC spectrometer equipped with an integrating sphere attachment and BaSO4 as a reference. The absorption spectrum was calculated from the reflection spectrum via the Kubelka–Munk function: α/S = (1 − R)2/2R, in which α was the absorption coefficient, S was the scattering coefficient, and R was the reflectance.Computational details
The electronic band structure calculations of Sr3Sn2As4 were performed with the full potential linearized augmented plane wave method (FP-LAPW)36,37 as implemented in the Wien2k code.38 In this method, the unit cell is divided into non-overlapping muffin-tin (MT) spheres and an interstitial region. The wavefunctions in the interstitial regions are expanded in plane waves up to RMT × Kmax = 7, where RMT is the smallest radius of all MT spheres and Kmax is the plane wave cut-off. The valence wavefunctions inside the MT spheres are expanded up to lmax = 10 while the charge density was Fourier expanded up to Gmax = 12 au−1. The MT radii were chosen to be 2.4 Bohr for Sr and Sn atoms and 2.3 Bohr for As atoms. Calculations were performed using 1000 k-points and the exchange correlation potential was calculated using the Perdew–Burke–Ernzerhof generalized gradient approximation (PBE-GGA).39 The Brillouin Zone (BZ) integration was performed using the tetrahedron method and the self-consistent calculations were considered to have converged if the total energy and the charge of the system are stable within 10−4 Ryd and 10−4 e−, respectively.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (grant no. 51271098, 51021062, 51272129), the Independent Innovation Foundation of Shandong University (IIFSDU), the Program of Introducing Talents of Disciplines to Universities in China (111 program no. b06017).Notes and references
- E. Zintl, Angew. Chem., 1939, 52, 1–6 CrossRef CAS.
- S. M. Kauzlarich, Chemistry, Structure and Bonding of Zintl Phases and Ions, VCH Publishers, New York, 1996 Search PubMed.
- M. Rotter, M. Tegel and D. Johrendt, Phys. Rev. Lett., 2008, 101, 107006 CrossRef.
- X. C. Wang, Q. Q. Liu, Y. X. Lv, W. B. Gao, L. X. Yang, R. C. Yu, F. Y. Li and C. Q. Jin, Solid State Commun., 2008, 148, 538–540 CrossRef CAS PubMed.
- T. Nomura, S. W. Kim, Y. Kamihara, M. Hirano, P. V. Sushko, K. Kato, M. Takata, A. L. Shluger and H. Hosono, Supercond. Sci. Technol., 2008, 21, 125028 CrossRef.
- A. M. Goforth, H. Hope, C. L. Condron, S. M. Kauzlarich, N. Jensen, P. Klavins, S. MaQuilon and Z. Fisk, Chem. Mater., 2009, 21, 4480–4489 CrossRef CAS.
- D. J. Webb, R. Cohen, P. Klavins, R. N. Shelton, J. Y. Chan and S. M. Kauzlarich, J. Appl. Phys., 1998, 83, 7192 CrossRef CAS PubMed.
- X. Ma, J. Lu, J. B. Whalen and S. E. Latturner, Inorg. Chem., 2013, 52, 3342–3348 CrossRef CAS PubMed.
- J. R. Salvador, D. Bilc, S. D. Mahanti, T. Hogan, F. Guo and M. G. Kanatzidis, J. Am. Chem. Soc., 2004, 126, 4474–4475 CrossRef CAS PubMed.
- P. H. Tobash and S. Bobev, J. Am. Chem. Soc., 2006, 128, 3532–3533 CrossRef CAS PubMed.
- F. Gascoin, S. Ottensmann, D. Stark, S. M. Haïle and G. J. Snyder, Adv. Funct. Mater., 2005, 15, 1860–1864 CrossRef CAS.
- X. J. Wang, M. B. Tang, H. H. Chen, X. X. Yang, J. T. Zhao, U. Burkhardt and Y. Grin, Appl. Phys. Lett., 2009, 94, 092106 CrossRef PubMed.
- A. Zevalkink, W. G. Zeier, G. Pomrehn, E. Schechtel, W. Tremel and G. J. Snyder, Energy Environ. Sci., 2012, 5, 9121–9128 CAS.
- E. S. Toberer, A. F. May and G. J. Snyder, Chem. Mater., 2010, 22, 624–634 CrossRef CAS.
- B. Saparov, S.-Q. Xia and S. Bobev, Inorg. Chem., 2008, 47, 11237–11244 CrossRef CAS PubMed.
- J. Jiang, M. M. Olmstead, S. M. Kauzlarich, H.-O. Lee, P. Klavins and Z. Fisk, Inorg. Chem., 2005, 44, 5322–5327 CrossRef CAS PubMed.
- B. Eisenmann and U. Röβler, Z. Anorg. Allg. Chem., 1998, 624, 406–410 CrossRef CAS.
- B. Eisenmann and H. Schäfer, Angew. Chem., Int. Ed. Engl., 1980, 19, 480–481 CrossRef.
- B. Eisenman and H. Schäfer, Z. Anorg. Allg. Chem., 1982, 484, 142–152 CrossRef.
- M. Khatun, S. S. Stoyko and A. Mar, Inorg. Chem., 2014, 53, 7756–7762 CrossRef CAS PubMed.
- J. Wang, S.-Q. Xia and X.-T. Tao, Inorg. Chem., 2012, 51, 5771–5778 CrossRef CAS PubMed.
- B. Eisenmann, H. Jordan and H. Schäfer, Z. Naturforsch., Teil B: Anorg. Chem., Org. Chem., 1984, 39, 1151–1153 Search PubMed.
- R. Lam and A. Mar, Solid State Sci., 2001, 3, 503–512 CrossRef CAS.
- G. Haegg and A. G. Hybinette, Philos. Mag., 1935, 20, 913–929 Search PubMed.
- W. Willott and E. Evans, Philos. Mag., 1934, 18, 114–128 CAS.
- X. Liu, S. Xia, X. Lei, M. Pan and X. Tao, Eur. J. Inorg. Chem., 2014, 13, 2248–2253 CrossRef.
- S. Xia and S. Bobev, J. Am. Chem. Soc., 2007, 129, 4049–4057 CrossRef CAS PubMed.
- S.-J. Kim, S. Ponou and T. F. Fässler, Inorg. Chem., 2008, 47, 3594–3602 CrossRef CAS PubMed.
- P. Alemany, M. Llunell and E. Canadell, Inorg. Chem., 2006, 45, 7235–7241 CrossRef CAS PubMed.
- X. Liu, N. Lin, J. Wang, M. Pan, X. Zhao, X. Tao and S. Xia, Inorg. Chem., 2013, 52, 11836–11842 CrossRef CAS PubMed.
- S. S. Stoyko and A. Mar, Inorg. Chem., 2011, 50, 11152–11161 CrossRef CAS PubMed.
- Bruker APEX2, Bruker AXS Inc., Madison, WI, 2005 Search PubMed.
- G. M. Sheldrick, SHELXTL, University of Göttingen, Göttingen, Germany, 2001 Search PubMed.
- E. Parthé and L. M. Gelato, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1984, 40, 169 CrossRef.
- L. M. Gelato and E. Parthé, J. Appl. Crystallogr., 1987, 20, 139 CrossRef.
- G. K. H. Madsen, P. Blaha, K. Schwarz, E. Sjostedt and L. Nordstrom, Phys. Rev. B: Condens. Matter, 2001, 64, 195134 CrossRef.
- K. Schwarz, P. Blaha and G. K. H. Madsen, Comput. Phys. Commun., 2002, 147, 71–76 CrossRef.
- P. Blaha, K. Schwarz, G. K. H. Madsen, D. Kvasnicka and J. Luitz, WIEN2k, An Augmented Plane Wave+Local Orbitals Program for Calculating Crystal Properties, Technische Universität Vienna, Austria, 2001 Search PubMed.
- J. P. Perdew, S. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The X-ray crystallographic file in CIF format of the title compounds; positional and equivalent isotropic displacement parameters and selected bond distances of two compounds; TG-DSC measurements on both compounds; the TG-DSC curves of 2nd cycle for α-Sr3Sn2As4; powder X-ray diffraction patterns of the polycrystalline samples of two polymorphs of Sr3Sn2As4; the optical absorption spectrum measured on polycrystalline samples. See DOI: 10.1039/c4qi00106k |
| This journal is © the Partner Organisations 2014 |