
A biomimetic photoelectrochemical device from a molecular heterometallic sodium–manganese water splitting catalyst†
C. R. Raymond
Gan
ab,
Zhaolin
Liu
b,
Shi-Qiang
Bai
b,
Siok Wei
Tay
b,
Xiaoming
Ge
b,
Ji-En
Wu
c and
T. S. Andy
Hor
*bc
aNUS Graduate School for Integrative Sciences and Engineering, Center for Life Sciences, #05-01, 28 Medical Drive, Singapore 117456, Singapore
bInstitute of Materials Research and Engineering, Agency for Science, Technology and Research, 3 Research Link, S117602, Singapore
cDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, S117543, Singapore. E-mail: andyhor@nus.edu.sg
First published on 10th September 2014
Abstract
A photoelectrochemical device for water splitting has been fabricated from a new water stable molecular [Na2Mn4] complex that acts as an in situ precursor towards NaMn2Ox nanoparticles under catalytic conditions when immobilized within a conductive PEDOT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PSS/Nafion composite film for optimal electron transport. The optimized photoanode architecture of ITO/TiO2/Ru/60% PEDOT:PSS/Na2Mn4/Nafion resulted in a water oxidation onset potential of 1.35 V (vs. RHE) and achieved a maximum current density of 1.31 mA cm−2 (initial) and 0.80 mA cm−2 (stabilized) at 1.8 V (vs. RHE) at pH 6.5.
:PSS/Nafion composite film for optimal electron transport. The optimized photoanode architecture of ITO/TiO2/Ru/60% PEDOT:PSS/Na2Mn4/Nafion resulted in a water oxidation onset potential of 1.35 V (vs. RHE) and achieved a maximum current density of 1.31 mA cm−2 (initial) and 0.80 mA cm−2 (stabilized) at 1.8 V (vs. RHE) at pH 6.5.
Introduction
Water splitting has long been dominated by bulk inorganic heterogeneous materials since TiO2 was discovered as the photoanode in a photoelectrochemical (PEC) device by Honda and Fujishima.1 Since then, numerous inorganic materials have been identified as possible photocatalyst candidates.2,3 While highly efficient photoelectrochemical cells (PEC) using GaAs p–n junction and p-type GaInP2 had been demonstrated by Turner (12.4%) and Licht (18.3%),4,5 these materials, typically used in satellite technology, are prohibitively expensive for mass market consumption. On the other hand, photosynthetic organisms provide an ideal natural model for harvesting of solar to chemical energy conversion. Starting with the [Mn4O4Ca] cluster in the heart of photosystem II (PSII),6 the mode of water oxidation to O2 operates in an electron cascade mode upon photon absorption with near unity quantum efficiency. Inspired by the photosynthetic [Mn4O4Ca] cluster, synthetic inorganic complexes consisting of Mn,7 Ru,8 Ir,9 Fe,10 and Co11 are developed as excellent models towards the structural and energetics studies of the water splitting process. Recently manganese oxides such as MnOOH, Mn3O4, Mn2O3, and MnO2 have also been unveiled as promising water oxidation catalysts (WOCs) and can be fabricated by inexpensive methods such as electrodeposition, atomic layer deposition, hydrothermal methods or sol–gel processes.12,13 While thin films of MnOx fabricated under such conditions demonstrate good photoelectrochemical activity in water oxidation, performance often gradually decreases due to the surface contamination and degradation due to direct exposure to the external environment over time.14 On the other hand, molecular manganese complexes, in particular,15 were found to exhibit water oxidation activities when doped into the Nafion matrix as a result of conversion into very small nano-particulate manganese oxide (MnOx) as the active species during the water splitting process and represent an interesting alternative to direct MnOx films or nanoparticles formed from external preparation techniques.16,17 Expanding from our previous work on the Co-Pi-Fe2O3 heterogeneous catalyst,18 a natural progression is towards bio-mimicking of the approach of PSII by related molecular manganese catalysts. It should also be noted that there have been no reports on heterometallic Mn clusters/aggregates which could give rise to new intermetallic oxide catalysts in situ within a membrane matrix environment. One of the most important strategies in bulk heterogeneous photocatalysts is the introduction of dopants within bulk semiconducting materials for creating intermediate electronic states towards reducing the thermodynamic and kinetic barriers in electronic transition for improving the catalytic activity.19 Thus, nanostructured heteroatom doped MnOx arising from heterometallic metal clusters/aggregates precursors in a suitable matrix environment could prove to be a masterstroke in developing the next generation of highly robust and efficient water oxidation catalysts.The membrane of choice is also critical in providing functionalities. It provides nurturing support for catalytic species formation, a medium for the immobilization of active catalytic species while maintaining solvent/byproduct transportation,20 and an electron conducting antenna into the anode. To date, Nafion has been commonly utilized, due to its excellent ionic conductivity and robustness. There are however limitations in its high electrical impedance which causes intermittent electron–hole recombination. The latter gives rise to low current densities in pure Nafion matrices which have so far restricted PECs from making the next quantum leap forward as a serious contender in alternative energy dominated by nuclear, geothermal, solar and wind energy.21,22
We herein report a new design by doping a conductive mixture of poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) with Nafion, in union with highly dispersed and very small nanoparticulate NaMn2Ox derived in situ from a new water stable [Na2Mn4] precursor complex formulated as [Mn2Na(H2O)3L]2·C2O4·Na4 {L = 2,2′,2′′,2′′′-(3,3′-methylenebis(2-hydroxy-5-nitro-3,1-phenylene))bis(methylene)bis(azanetriyl)tetraacetic acid, [C26H14N4O14]6−}. The ligand L features two highly electronegative nitro groups, two phenol groups and four carboxylate chelating arms, which enable it to support heterometallic coordination to manganese and sodium. The single crystal structure of the complex reveals a 1D polymeric chain in which the subunits are linked by Na+ through coordination (Fig. 1). Each subunit is derived from [Mn2Na(H2O)3L]2·C2O4·Na4 and the core is composed of a [Mn4O4Na2] moiety which has biological resemblance to the [Mn4O4Ca] cluster in PSII but with Ca2+ replaced by 2Na+. The hybrid PEDOT:PSS/Nafion membrane primarily serves as a protein matrix equivalent in PSII in which selective transportation of H2O, O2 and H+ can diffuse freely while remaining highly conductive for electron transport (Fig. 2).
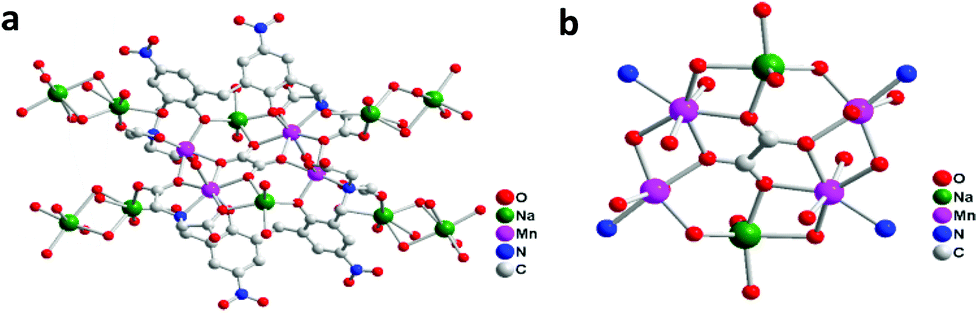 | ||
| Fig. 1 X-ray single-crystal structure of the [Na2Mn4] complex [Mn2Na(H2O)3L]2·C2O4·Na4 represented as (a) a polymeric chain and (b) its local coordination environment of Mn and Na within the chelating ligand framework. | ||
 | ||
| Fig. 2 Schematic representation of the device architecture (a) that is conceptually similar to that of PSII (b). | ||
Results and discussion
The PEDOT:PSS/Nafion composite is designed as the top layer of the PEC device due to the fact that the solution largely absorbs light in the UV region between 200 and 250 nm in conjunction with the 305 to 405 nm associated with the absorption peak of the [Na2Mn4] complex (Fig. S1 & S2†). This allows ample exposure of visible light towards the Ru dye layer to facilitate photosensitization without interference from the adjacent top membrane layer. The [Na2Mn4] complex is intercalated within the membrane, as suggested in time-of-flight secondary-ion-mass-spectroscopy (TOF-SIMS) analysis (Fig. S3†), which revealed the presence of Mn ions just beneath the hybrid PEDOT:PSS/Nafion membrane. The uniform thickness of the PEDOT:PSS/Na2Mn4/Nafion membrane and the TiO2 layer obtained from field emission scanning electron microscopy (FESEM) was determined to be ∼13.4 μm for the TiO2 layer and ∼11.6 μm for the membrane depicted in Fig. S4.† A comparison of the surface of the membrane and a natural specimen of the Rhodomyrtus tomentosa leaf (Fig. S5†) revealed the pores of the former to be several tens to hundreds of nanometers (Fig. S6†) and 6 μm in length scattered throughout its surface for the latter to facilitate the diffusion of CO2 and O2. The difference in morphology was expected, which shows that the nano-sized porosity of the PEDOT:PSS/Nafion membrane surface could also provide a mechanism for controlled diffusion of H2O, O2 and H+.
Linear sweep voltammetric (LSV) analysis was carried out to analyze the initial water oxidation activities in devices freshly assembled from different concentrations of PEDOT:PSS/Nafion membrane illuminated under 100 mW cm−2 over the range of 1.2 to 1.8 V (vs. RHE) in a 0.1 M Na2SO4 solution of pH 6.5 (Fig. 3a). Upon irradiation with a light intensity of 1 sun (100 mW cm−2), the photocurrent density of ITO/TiO2/Ru/12% PEDOT:PSS/Na2Mn4/Nafion increases steadily from an onset water oxidation potential of 1.35 V vs. RHE (overpotential of only 0.12 V) to reach a maximum of 0.84 mA cm−2 at 1.8 V (vs. RHE). Increasing the percentage of PEDOT:PSS in the Nafion matrix from 12% PEDOT:PSS to 60% PEDOT:PSS in the device did not affect the onset water oxidation potential significantly but has a profound effect on boosting the photocurrent density to a maximum of 0.93, 1.02, 1.14 and 1.31 mA cm−2 at 1.8 V (vs. RHE) for devices composed of 24, 36, 48 and 60% PEDOT:PSS composition respectively. As the concentration of the [Na2Mn4] complex within the PEDOT:PSS/Nafion membrane is kept relatively constant, the results indicate that the presence and concentration of PEDOT:PSS within the membrane not only has a notable influence on the onset potential of water oxidation but also on the photocurrent density.
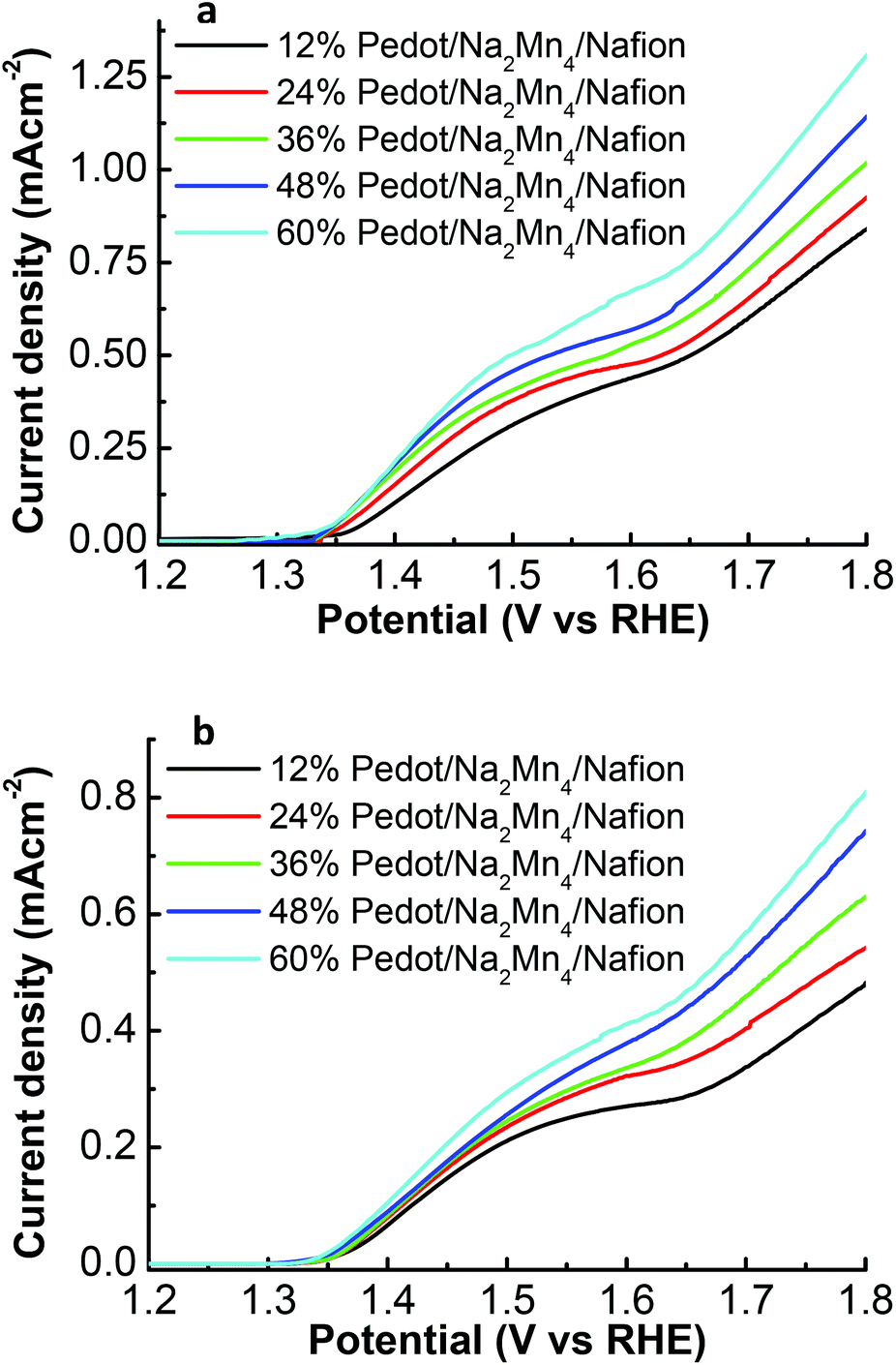 | ||
| Fig. 3 (a) Initial photocurrent density, (b) stabilized photocurrent density after 5 LSV cycles, vs. potential of the ITO/TiO2/Ru/x% PEDOT:PSS/Na2Mn4/Nafion series under illumination of 100 mW cm−2. | ||
Subsequent LSV reiteration on the same devices did not affect the onset potential for water oxidation of 1.35 V (vs. RHE) but resulted in a decrease in the initial photocurrent density prior to stabilization after 5 repeated LSVs cycles. Beyond 5 LSVs reiteration, the PEC devices maintained maximum photocurrent densities of 0.48, 0.55, 0.63, 0.74 and 0.80 mA cm−2 at 1.8 V (vs. RHE) for devices composed of 12, 24, 36, 48 and 60% PEDOT:PSS composition respectively (Fig. 3b). The decrease of the initial high photocurrent density to a stabilized photocurrent density after several cycles of LSV is possibly attributed to poorly embedded active catalytic species leaching out from the freshly prepared PEDOT:PSS/Nafion membrane into the electrolyte solution. The remaining catalytic active species that is sufficiently trapped within the membrane matrix thus gave rise to a stabilized photocurrent density response. This phenomenon will be elaborated further in the text.
A possible reason for the concentration dependent PEDOT:PSS/Nafion sheets’ influence on photocurrent density performance could be due to the correlation of electrical resistivity of the membrane as a function of PEDOT:PSS concentration in Nafion. A study was hence carried out to compare the sheet resistivity of the PEDOT:PSS/Na2Mn4/Nafion films using a 4 pin probe (Table 1). The results indeed show a decreasing sheet resistivity (249, 148, 124, 91 and 74 Ω cm−2) as the concentration of PEDOT:PSS increases (12, 24, 36, 48 and 60%) respectively, independent of the LSV reiteration, and confirm that an effective interfacial charge transfer within the robust membrane contributed to the high current density of the device. As faster electron transport will drive the electrons liberated from the 4e−/4H+ water cleavage process to the anode, it is expected to suppress electron and hole recombination, thereby capturing a greater percentage of the theoretical electron yields.23,24 Higher conductivity with low sheet resistivity has also been established as one of the main factors governing high performance in organic light emitting devices (OLED),25 organic photovoltaic devices (OPV),26 and dye sensitized solar cells.27 Higher concentration of PEDOT:PSS in the Nafion matrix tends to reduce the film's tensile and fracture strength. Films containing more than 60% PEDOT:PSS become increasingly brittle and are not suitable for use in the photoelectrochemical device. A compromise between the low sheet resistance and mechanical strength of about 60% PEDOT:PSS/Na2Mn4/Nafion hence produced the optimal photocurrent as a result of water oxidation catalytic activity.
:PSS/Na2Mn4/Nafion film on TiO2
| Percentage of PEDOT (%) | 12 | 24 | 36 | 48 | 60 |
| Sheet resistance (Ω cm−2) | 249 | 148 | 124 | 91 | 74 |
Stabilized photocurrent density values achieved through controlled potential electrolysis polarized at 1.8 V (vs. RHE) on the stabilized devices for 1000 s (Fig. 4) are in good agreement with LSV results discussed above. Upon illumination of one sun (100 mW cm−2), a sharp spike of the current density was observed for all [Na2Mn4] complex containing devices before gradually decaying to a more stable photocurrent density. The sharp spike is assigned to a sudden injection of electrons into the anode as witnessed in another molecular based PEC.28 Similarly to what was observed in the LSV, the 60% PEDOT:PSS/Na2Mn4/Nafion device achieved the highest steady state photocurrent (0.109 mA cm−2), followed by the 48%, 36%, 24% and 12% PEDOT:PSS devices (0.096, 0.084, 0.075 and 0.065 mA cm−2, respectively).
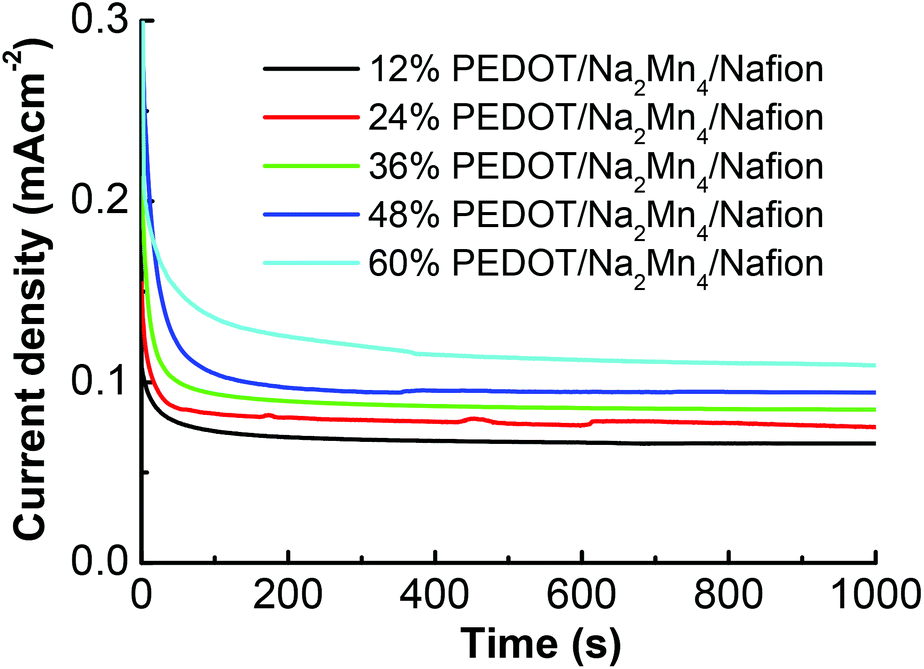 | ||
| Fig. 4 Amperograms for photocurrent of the stabilized ITO/TiO2/Ru/x% PEDOT:PSS/Na2Mn4/Nafion devices at an applied potential of 1.8 V vs. RHE under 100 mW cm−2 intensity. | ||
Electron paramagnetic resonance (EPR) measurements of the [Na2Mn4] complex were conducted during the course of the water oxidation reaction to provide information on the catalytic behavior under different oxidation environments (Fig. S7–S10†). In aqueous solution, the EPR signal is consistent with the long range order associated with weak antiferromagnetic interaction between distanced MnII atoms, the EPR signal is less complicated than the reported literature on mononuclear Mn2+ species with a standard hyperfine 6 line splitting due to the μ-oxalato heterometallic Mn2+–Na–Mn2+ bridged structure as part of the [Na2Mn4] complex that gave rise to large Mn–Mn distances of 5.8 Å.29 This consolidated additional support for the [Na2Mn4] structure as deduced from ESI-MS and single crystal XRD. Upon doping within 60% PEDOT:PSS/Nafion, the EPR signal of the [Na2Mn4] complex is preserved and suggests the integrity of the [Na2Mn4] complex structure even when doped within 60% PEDOT:PSS/Nafion. The fate of the [Na2Mn4] catalyst under catalytic conditions was further investigated by the application of a voltage polarization at 1.8 V (vs. RHE); the initial EPR signal broadens, indicating structural and chemical changes to the initial [Na2Mn4] catalyst. However when subjecting the photoelectrochemical cell to light illumination and potential bias simultaneously, a characteristic hyperfine six line Mn2+ signal could be observed in conjunction with the broad signals. This suggests liberation of Mn2+ under the mutual effect of voltage bias and light stimulus. As EPR could not satisfactorily identify the exact species formed during catalytic conditions within the 60% PEDOT:PSS/Nafion matrix, the 60% PEDOT:PSS/Na2Mn4/Nafion films after voltage polarization were carefully peeled off using a sharp razor blade, shredded into smaller pieces and immersed in deionized H2O to undergo intensive ultrasonication to dislodge the active species for analysis. Subsequent transmission electron microscopy (TEM) and energy dispersive X-ray (EDX) analyses of the dislodged electro-oxidized precipitates after centrifugation and loading onto a carbon coated copper grid revealed irregular ultra-fine nanoparticles of composition NaMn2Ox, x = ∼4.3, of approximate diameter size ∼5–20 nm (Fig. 5a and S11†). The EDX derived composition ratio of Na:Mn ≃ 1:2 also agrees well with the mole ratio of Na:Mn within the [Na2Mn4] complex [Mn2Na(H2O)3L] for the sample. This transformation to nanoparticulate NaMn2Ox rather than MnOx appears to be aided by a specifically designed chelating ligand that brings the Na and two Mn centers to closer proximity (3.67 Å) as compared to the large Mn–Mn distance (5.8 Å) in the initial [Na2Mn4] complex precursor. It should be noted that in the presence of PEDOT:PSS as part of the membrane matrix, in situ formation of NaMn2Ox resulted in nanoparticles with a smaller diameter size of 5–20 nm which expectedly differ in morphology and size distribution as compared to the catalytically active MnOx nanoparticles (30–100 nm in diameter) that utilized Mn(ClO4)2 as the precursor species in Nafion in a recent report;30 this disparity was expected, as the [Na2Mn4] complex precursor in this case is not [Mn(H2O)6]2+. It is hence evident that nanoparticle size and morphology formation in the membrane matrix are dependent on the composition of the membrane, oxidation state of the metal, nuclearity and the ligand coordination mode. These results together with the sheet resistivity measurements (Table 1) provide valuable clues to the low onset potential experienced by the PEDOT:PSS/Na2Mn4/Nafion devices as observed in the LSVs (Fig. 3). The implications of the LSV, EPR, EDX and TEM results therefore suggest cycling between the matrix constitution size dependent NaMn2Ox phase during voltage bias and a reduced Mn2+ product during light illumination within the PEDOT:PSS/Nafion matrix throughout the water oxidation activity. The mode of water oxidation thus agrees well with the geochemical-like cycle analysis of PEC devices.
 | ||
| Fig. 5 (a) TEM image of the electro-oxidized product resulting from the [Na2Mn4] complex from ITO/TiO2/Ru/60% PEDOT:PSS/Na2Mn4/Nafion. (b) Leached NaMn2Ox nanoparticles in the 0.1 M Na2SO4 electrolyte solution after the 1st LSV study. | ||
The NaMn2Ox nanoparticles extracted as the electro-oxidized products from the 60% PEDOT:PSS/Nafion membrane were subsequently subjected to X-ray photospectroscopy (XPS) in order to elucidate the oxidation state of Mn (Fig. S12 and S13†). The spin energy difference between Mn2p1/2 and Mn3p1/2 is 11.8 and 10.5 eV while that of Δ3s is 4.4 and 5.3 eV. This suggests that the surface oxidation state of the Mn under the catalytic conditions is likely a mixed valence Mn3/4+ oxide consistent with literature reports.31 Peaks attributed to Na1s were established by the high resolution scan at 1072.8 eV (Fig. S14†). The surface of an electrodeposited Na birnessite-type MnO2 film that is active in water oxidation was similarly shown to be composed of Mn3/4+ oxide and pertinent to the oxygen evolution reaction but has a large water oxidation onset overpotential of 0.4 V.32 Examination of related MnOx PECs in the literature revealed that while a current density in excess of 1 mA cm−2 could be obtained, the water oxidation onset potential typically requires overpotential in excess of 0.15 V.33–35 The uniqueness of our device architecture of ITO/TiO2/Ru/x% PEDOT:PSS/Na2Mn4/Nafion requires a significantly lower overpotential of 0.12 V at pH 6.5 for water oxidation regardless of device stability and indicates that the integrative TiO2/Ru/x% PEDOT:PSS/Nafion support is paramount for reducing the onset potential. Although direct comparisons with PECs systems based on other metals are difficult due to the variations in experimental conditions such as film thickness, surface area and morphology, oxidation state, catalyst loading, electrolyte conditions and scan rate, the onset potential of the PEDOT:PSS containing devices nevertheless approaches the range of modified hematite,36 cobalt,37,38 ruthenium,39 and IrO2 based PECs with water oxidation onset ranging from 0.5–1.3 V (vs. RHE).40
The decrease of the initial high photocurrent density to a stabilized photocurrent density after several cycles of repetitive LSV on the same devices is likely indicative of insecurely entrapped NaMn2Ox nanoparticles (diameter ∼5–20 nm) leaching out from the PEDOT:PSS/Nafion membrane (pore size ∼ several tens to hundreds of nanometers) into the electrolyte solution. The hypothesis was confirmed by TEM analysis of the electrolyte solution after LSV studies that revealed traces of aggregated NaMn2Ox nanoparticles of similar elemental composition and size distribution as compared to the ultrasonicated electro-oxidized specimens arising from the PEDOT:PSS/Nafion membrane (Fig. 5b).
The oxygen evolution on the stabilized ITO/TiO2/Ru/60% PEDOT:PSS/Na2Mn4/Nafion-light device was determined with the aid of a Clark electrode (Fig. 6). Under these conditions, an amount resulting in 0.30 μmol of O2 evolved at standard temperature and pressure can be obtained after 1000 s under light illumination of intensity 100 mW cm−2 biased at 1.8 V vs. RHE. The calculated O2 evolution based on the integration of the current density–time curve of ITO/TiO2/Ru/60% PEDOT:PSS/Na2Mn4/Nafion indicates good compliance with the measured Clark cell O2 evolution for 1000 s. Therefore, the cumulative charge as collected at 1000 s from the current density–time curve of ITO/TiO2/Ru/60% PEDOT:PSS/Mn/Nafion is calculated to be 0.12 C and corresponds to 1.24 μmol of electrons accordingly to Faraday's equation. Theoretically, based on a 4:1 stoichiometry of electrons to one O2 molecule, 0.31 μmol of O2 should be generated. The calculated electrons to actual measured O2 ratio stands at 4.1:1 and the faradic efficiency of the catalyst can thus be calculated as ∼100%.
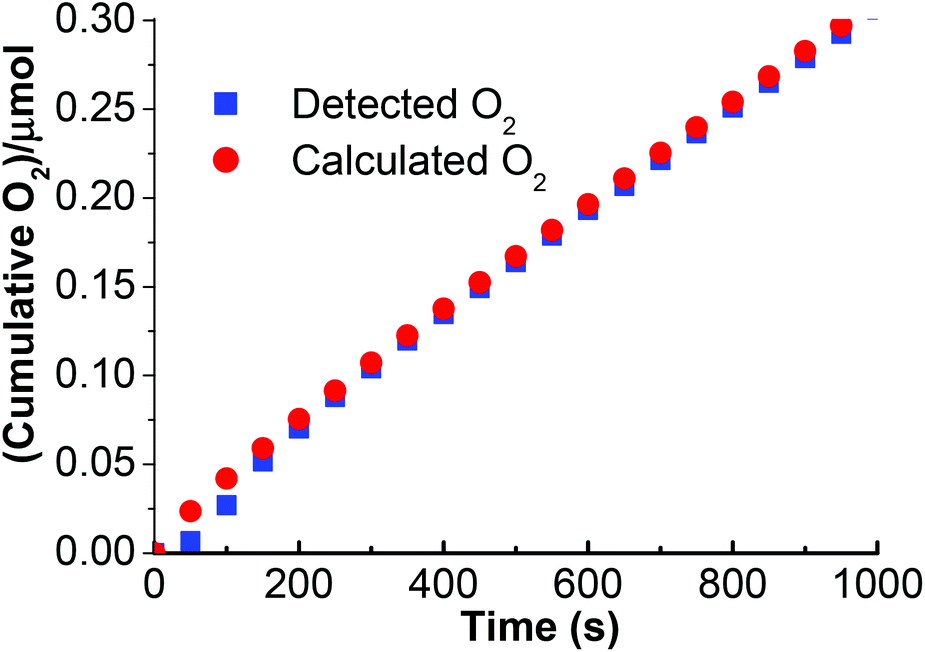 | ||
| Fig. 6 Detected vs. calculated O2 evolution vs. time based on the stabilized ITO/TiO2/Ru/60% PEDOT:PSS/Na2Mn4/Nafion device under illumination of light intensity of 100 mW cm−2 at a voltage polarization of 1.8 V (vs. RHE). | ||
The [Na2Mn4] complex was evaluated independently of its catalytic activity by adding only the [Na2Mn4] complex and [Ru(bpy)3]3+ into a 0.1 M Na2SO4 solution (pH 6.5) and illuminated with light (Fig. S15†). As there was no detectable O2 evolved, the result implied that the original [Na2Mn4] complex is non-catalytic. Instead, it serves as a precursor to the catalytically active NaMn2Ox nanoparticles uniquely within the PEDOT:PSS/Nafion environment. Cyclic voltammetry (vs. Ag/AgCl, 3 M KCl) of the [Na2Mn4] complex in a 1:1 EtOH, 0.1 M Na2SO4 solution revealed a redox wave at E1/2 (MnII/III) = 0.9 V as deducted from the well-defined oxidation peak (Epa) at 1.01 V and the reduction process (Epc) at 0.79 V. An irreversible reduction peak was surveyed at 0.19 V while the significant oxidation wave observed from 1.1 V onwards is attributed to water oxidation by the [Na2Mn4] complex under electrochemical conditions harsher than within the PEDOT:PSS/Nafion environment (Fig. 7 & Table 2). This further illustrates the importance of the encompassing PEDOT:PSS/Nafion matrix in its ability to transform the initial non-catalytic species into catalytic ones, controlled diffusion of the required species and electron conducting capability to the anode.
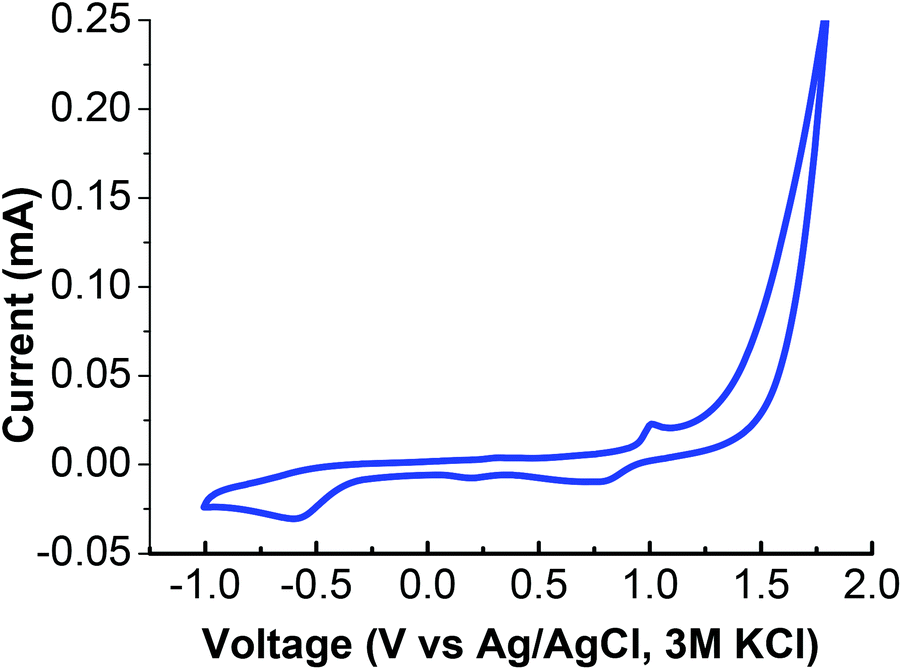 | ||
| Fig. 7 Cyclic voltammetry of the [Na2Mn4] complex in 0.1 M Na2SO4–EtOH (1:1). | ||
| [Epa, Epc]/V | ΔEp/V | E 1/2/V |
|---|---|---|
| [1.01, 0.79], [NA, 0.19] | 0.22 | 0.9 |
Conclusion
Overall water splitting has been achieved using the ITO/TiO2/Ru/x% PEDOT:PSS/Na2Mn4/Nafion architecture with the 60% PEDOT:PSS/Nafion membrane adjacent to a TiO2/Ru layer electrooxidized at a low overpotential (1.8 V vs. RHE and 100 mW cm−2 light intensity) to give a maximum photocurrent density of 1.31 mA cm−2 (initial) and 0.80 mA cm−2 (stabilized) as the optimal design. The active species was identified to be NaMn2Ox and uniquely achieved in situ within the environment of PEDOT:PSS/Nafion under voltage stimulus. Interestingly, solid CaMnIII/IV oxide has been extracted from the cell walls of photosynthetic organisms and is a possible indication that the [Mn4O4Ca] cluster in PSII could spontaneously form fragments of catalytically active CaMnIII/IV oxides which act as the catalytic active species before being reduced by the light-driven antenna process as part of the water oxidation catalytic cycle.41 The catalytic cycling process between [Mn4O4Ca] and CaMnIII/IV oxide is most likely aided by the amino acid ligation within PSII, highlighting the importance of a supportive local coordination environment. There is hence impetus on using small molecular heterometallic aggregates/clusters precursors towards achieving intermetallic oxide for water oxidation in membrane matrices. While our device is still short of the natural PSII in terms of performance,42 this biomimetic device shows promise in achieving improved photocurrent density through enhanced charge separation and transportation. An optimized photoelectrochemical cell depends on multiple factors, such as an underlying consistent and uniform TiO2 layer for uninterrupted electron transport, a Ru dye that enables absorption of photons in the visible region of the electromagnetic spectrum and subsequently promotion of electrons into the TiO2 layer, functional nanoparticulate metallic oxides capable of driving catalytic oxidation of water to O2 and release of H+, which then can be reduced at the platinum cathode. Equally important is a conductive yet robust membrane matrix whose composition can influence the in situ configuration and entrapment of NaMn2Ox nanoparticles that can minimize leaching loss of the active catalyst while allowing water, O2 and H+ to diffuse freely. All of these factors need to be optimized for the full potential of a hybrid device like PEDOT:PSS/Na2Mn4/Nafion PEC to be realized and put to practical use for driving water oxidation. We shall investigate other small molecular heterometallic precursors in new hybrid composite membranes for water oxidation activities.
Experimental section
Device fabrication
The devices were built by calcinating a thin coat of TiO2 as the underlying support on the indium tin oxide (ITO) coated glass substrate before soaking in an EtOH solution of a RuL′2(NCS)2·TBA (L′ = 2,2′-bipyridyl-4,4′-dicarboxylic acid, TBA = tetrabutylammonium) dye (0.3 mM) for a day to ensure uptake of the Ru dye onto the TiO2 surface. The primary function for the Ru dye that was absorbed on TiO2 is for rapid electron injection upon photosensitization that does not carry water oxidation function. The [Na2Mn4] complex and Nafion solution were subsequently mixed with different ratios of PEDOT:PSS (12%, 24%, 36%, 48% and 60%) and a 100 μl solution was drop-casted on the ITO/TiO2/Ru surface. Heat treatment at 40 °C for a day resulted in the fabrication of a device that produced a robust x% PEDOT:PSS/Na2Mn4/Nafion film on the TiO2/Ru layer (x = 12, 24, 36, 48 or 60) (Fig. S1†). The [Na2Mn4] complex is presumably situated within the cavity of the x% PEDOT:PSS/Nafion membrane (Fig. 2).
Structural characterization
Transmission electron microscopy (TEM) characterization was performed using a Philips CM300-FEG instrument with an operating voltage at 300 kV while energy-dispersive X-ray (EDX) spectra were collected with an Oxford INCA Energy Dispersive X-ray (EDX) spectrometer system. Field emission secondary electron microscopy (FESEM) images were obtained using a JEOL 6700F. ToF SIMS analysis was performed using the CAMECA IONTOF-SIMS IV system with a Bi 25 kV analysis gun at a scan area of 150 μm by 150 μm. XPS measurements were performed on the samples using a Thermo Scientific Theta Probe and the binding energies were calibrated with respect to the residual C1s peak at 285.0 eV.Photoelectrochemical measurements
All PEC measurements were performed in a three-electrode electrochemical cell utilizing a platinum foil as a counter electrode and a Ag/AgCl electrode (3 M KCl) as a reference electrode using an Autolab potentiostat (model PGSTAT30) by Echochimie and the device as the working electrode. A fiber-lite halogen lamp (MI-150, Dolan-Jemmer) was used as a white light source (with 100 mW cm−2 intensity) together with a 400 nm band cutoff filter. The devices were masked with black tape except for a working area of 0.8 cm × 1.25 cm. The evolved oxygen amount was measured with a Clark oxygen electrode (Rank Brothers Ltd). The electrolyte solution was prepared using 0.1 M of Na2SO4 at pH 6.5 and measured using a Metrohm pH meter. The potential vs. Ag/AgCl (3 M KCl) was then converted to a reversible hydrogen electrode (RHE) at pH 6.5 to facilitate discussion in the text; all voltage potential references in the main text are discussed with respect to RHE unless otherwise stated.Electron paramagnetic resonance measurements
The EPR measurements were carried out using a JEOL FA200 ESR Spectrometer at a microwave power of 1 mW, a frequency of 9200 MHz and a liquid nitrogen cooled temperature of −150 °C.Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35 CrossRef CAS.
- K. Maeda, D. Lu and K. Domen, Angew. Chem., Int. Ed., 2013, 52, 6488 CrossRef CAS PubMed.
- O. Khaselev and J. A. Turner, Science, 1998, 280, 425 CrossRef CAS.
- S. Licht, J. Phys. Chem. B, 2001, 105, 6281 CrossRef CAS.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831 CrossRef CAS PubMed.
- E. Y. Tsui, J. S. Kanady and T. Agapie, Inorg. Chem., 2013, 52, 13833 CrossRef CAS PubMed.
- C. Besson, Z. Huang, Y. V. Geletii, S. Lense, K. I. Hardcastle, D. G. Musaev, T. Lian, A. Proust and C. L. Hill, Chem. Commun., 2010, 46, 2784 RSC.
- N. D. McDaniel, F. J. Coughlin, L. L. Tinker and S. Bernhard, J. Am. Chem. Soc., 2008, 130, 210 CrossRef CAS PubMed.
- W. C. Ellis, N. D. McDaniel, S. Bernhard and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 10990 CrossRef CAS PubMed.
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342 CrossRef CAS PubMed.
- F. Zhou, A. Izgorodin, R. K. Hocking, L. Spiccia and D. R. MacFarlane, Adv. Energy Mater., 2012, 2, 1013 CrossRef CAS.
- K. L. Pickrahn, S. W. Park, Y. Gorlin, H.-B.-R. Lee, T. F. Jaramillo and S. F. Bent, Adv. Energy Mater., 2012, 2, 1269 CrossRef CAS.
- I. Zaharieva, P. Chernev, M. Risch, K. Klingan, M. Kohlhoff, A. Fischer and H. Dau, Energy Environ. Sci., 2012, 5, 7081 CAS.
- R. Brimblecombe, G. C. Dismukes, G. F. Swiegers and L. Spiccia, Dalton Trans., 2009, 9374 RSC.
- R. K. Hocking, R. Brimblecombe, L.-Y. Chang, A. Singh, M. H. Cheah, C. Glover, W. H. Casey and L. Spiccia, Nat. Chem., 2011, 3, 461 CAS.
- K. J. Young, Y. Gao and G. W. Brudvig, Aust. J. Chem., 2011, 64, 1221 CrossRef PubMed.
- Y. R. Hong, Z. Liu, S. F. B. S. A. Al-Bukhari, C. J. J. Lee, D. L. Yung, D. Chi and T. S. A. Hor, Chem. Commun., 2011, 47, 10653 RSC.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294 RSC.
- A. J. Seen, J. Mol. Catal. A: Chem., 2001, 177, 105 CrossRef CAS.
- G. F. Moore, J. D. Blakemore, R. L. Milot, J. F. Hull, H. Song, L. Cai, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Energy Environ. Sci., 2011, 4, 2389 CAS.
- L. Li, L. Duan, Y. Xu, M. Gorlov, A. Hagfeldt and L. Sun, Chem. Commun., 2010, 46, 7307 RSC.
- M. Grätzel, Nature, 2001, 414, 338 CrossRef PubMed.
- J. N. Clifford, E. Palomares, M. K. Nazeeruddin, M. Gratzel, J. Nelson, X. Li, N. J. Long and J. R. Durrant, J. Am. Chem. Soc., 2004, 126, 5225 CrossRef CAS PubMed.
- E. C. W. Ou, L. Hu, C. R. R. Gan, K. S. Ong, J. Pan, Z. Zheng, Y. Park, D. Hecht, G. Irvin, P. Drzaic and G. Gruner, ACS Nano, 2009, 3, 2258 CrossRef CAS PubMed.
- S.-I. Na, S.-S. Kim, J. Jo and D.-Y. Kim, Adv. Mater., 2008, 20, 4061 CrossRef CAS.
- K. Park, Q. Zhang, D. Myers and G. Cao, ACS Appl. Mater. Interfaces, 2013, 5, 1044 CAS.
- Y. Gao, X. Ding, J. Liu, L. Wang, Z. Lu, L. Li and L. Sun, J. Am. Chem. Soc., 2013, 135, 4219 CrossRef CAS PubMed.
- V. Tangoulis, G. Psomas, C. D. Samara, C. P. Raptopoulou, A. Terzis and D. P. Kessissoglou, Inorg. Chem., 1996, 35, 7655 CrossRef CAS.
- A. Singh, R. K. Hocking, S. L.-Y. Chang, B. M. George, M. Fehr, K. Lips, A. Schnegg and L. Spiccia, Chem. Mater., 2013, 25, 1098 CrossRef CAS.
- Y. Matsumoto and E. Sato, Mater. Chem. Phys., 1986, 14, 397 CrossRef CAS.
- Y. Gorlin, B. Lassalle-Kaiser, J. D. Benck, S. Gul, S. M. Webb, V. K. Yachandra, J. Yano and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 8525 CrossRef CAS PubMed.
- A. M. Mohammad, M. I. Awad, M. S. El-Deab, T. Okajima and T. Ohsaka, Electrochim. Acta, 2008, 53, 4351 CrossRef CAS PubMed.
- I. M. Sadiek, A. M. Mohammad, M. E. El-Shakre and M. S. El-Deab, Int. J. Hydrogen Energy, 2012, 37, 68 CrossRef CAS PubMed.
- B. A. Pinaud, Z. Chen, D. N. Abram and T. F. Jaramillo, J. Phys. Chem. C, 2011, 115, 11830 CAS.
- D. K. Zhong and D. R. Gamelin, J. Am. Chem. Soc., 2010, 132, 4202 CrossRef CAS PubMed.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238 CrossRef CAS PubMed.
- Y. Surendranath, D. A. Lutterman, Y. Liu and D. G. Nocera, J. Am. Chem. Soc., 2012, 134, 6326 CrossRef CAS PubMed.
- J. Kiyota, J. Yokoyama, M. Yoshida, S. Masaoka and K. Sakai, Chem. Lett., 2010, 39, 1146 CrossRef CAS.
- Y. Zhao, J. R. Swierk, J. D. Megiatto, B. Sherman, W. J. Youngblood, D. Qin, D. M. Lentz, A. L. Moore, T. A. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15612 CrossRef CAS PubMed.
- A. Schöler, I. Zaharieva, S. Zimmermann, M. Wiechen, A.-M. Manke, P. Kurz, C. Plieth and H. Dau, Eur. J. Inorg. Chem., 2014, 4, 780 CrossRef.
- G. Ananyev and G. C. Dismukes, Photosynth. Res., 2005, 84, 355 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The detailed synthesis procedure for the [Na2Mn4] complex is provided. The single crystal X-ray refinement data, PEDOT:PSS/Na2Mn4/Nafion solution preparation and UV-Vis spectrum, time-of-flight secondary-ion-mass-spectroscopy data, field emission scanning electron microscope images of the 60% PEDOT:PSS/Na2Mn4/Nafion device, electron paramagnetic resonance measurements, and control experiment are also included. CCDC 949196. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00081a |
| This journal is © the Partner Organisations 2014 |