Twisted conformations in complexes of N-(3-imidazol-1-yl-propyl)-1,8-naphthalimide and fluorescence properties†
Jayanta Kumar
Nath
and
Jubaraj B.
Baruah
*
Department of Chemistry, Indian Institute of Technology Guwahati, Guwahati 781 039, Assam, India. E-mail: juba@iitg.ernet.in; Web: http://www.iitg.ernet.in/juba Fax: +91-361-2690762; Tel: +91-361-2582311
First published on 26th March 2014
Abstract
A series of complexes of N-(3-imidazol-1-yl-propyl)-1,8-naphthalimide (L) with divalent ions of manganese, cobalt, zinc, cadmium and mercury are structurally characterized. The metal complexes [ML2Cl2] {M = Zn (1), Cd (2), Hg (3)} are isomorphous and have a distorted tetrahedral geometry with a bend conformation of L. The thiocyanate complex [ZnL2(SCN)2] has a distorted tetrahedral geometry with L in bent conformation but with a different geometry from that in the structure of complex 1. The manganese and cobalt thiocyanate complexes [ML4(SCN)2]·2CH3CN (M = Mn, Co) are isomorphous and have a distorted octahedral geometry with the thiocyanate ligands occupying the axial positions. The cadmium complex [CdL3(SCN)2DMF]·DMF has a distorted octahedral geometry with thiocyanate ligands in the axial positions. The tetrahedral complexes [ML2Cl2] {M = Zn (1), Cd (2)} in the solid state show emission at shorter wavelengths than the single emission peak observed from the ligand, whereas the fluorescence emission of [ML2(SCN)2] {M = Zn (1), Cd (2)} occurred at longer wavelengths than L. On the other hand, [HgL2Cl2] (3) showed a single emission peak with higher intensity but at 31 nm shorter wavelength than the emission peak of the parent ligand. Two types of bend orientations of the ligand L, namely parallel arrangement of imidazole with the 1,8-naphthalimide ring and non-parallel arrangement in the tetrahedral complexes are observed. The former case favors intra-molecular charge transfer to show shorter wavelength emission, whereas the non-parallel arrangement facilitates exciplex leading to emission at longer wavelengths.
Introduction
Naphthalimide derivatives are commonly used as fluorescence probes1 and optical materials.2 Structurally modified naphthalimide compounds show selectivity in binding ions.3 The nature of self-assemblies formed by naphthalimide derivatives is guided by functional groups.4 A study on chromogenic properties of naphthalimide derivatives helps us to understand biological interactions.5 Naphthalimides have vast photochemistry6 and they are used to make bio-models for anion transport.7 Solar cells are developed based on ruthenium complexes of naphthalimides.8 Stacking interactions between naphthalimides generate interesting structures.9 Naphthalimides also bind to DNA10 and act as anticancer agents.11 Naphthalimide based dyads are potent inhibitors against human N-acetyl-D-hexosaminidase.12 Imidazole based naphthalimides have been shown to have interesting DNA binding ability and anticancer activity.13 Beside these, the N-functionalised 1,8-naphthalimide compounds having a nitrogen-heterocyclic tether have gained special interest for their interesting dual fluorescence properties. Such compounds show dual fluorescence originating from charge transfer via an excited state with extended conjugation (ESEC).14 In such cases it was shown that the different orientation of an N-heterocyclic ring over a 1,8-naphthalimide ring can change the intensity as well as the position of the absorptions. It was earlier shown by us that the anion–π interactions guide the fluorescence emission of imidazole tethered 1,8-naphthalimide derivatives. Since variation of anions resulted in the change of fluorescence, it is essential to understand the role of cations in stabilization of different geometries of ligands such as L (Fig. 1) and understand their fluorescence properties. So far, such observations have not been related to the coordination effect of metal ions, as for example, an N-functionalised 1,8-naphthalimide ligand such as L having a flexible chain to connect the 1,8-naphthalimide with an imidazole ring would have a provision to adopt a linear or bent structure as illustrated in Fig. 1. Such a geometry may be guided by a metal ion coordinating the orientation of the ligand to affect the intramolecular charge transfer or photo-electron transfer mechanism operative in such a system to show characteristic fluorescence emission.3 While pursuing our study with metal complexes of L, we observe such structures in metal complexes of the ligand L and as a consequence, characteristic fluorescence emissions are observed. Thus, we present here the results of such a study to establish a correlation between the fluorescence and structures. | ||
| Fig. 1 (a) Linear and (b) bend form of the ligand L. Geometries of ligand L attached to a metal ion (c) linear form; and bend forms having (d) parallel or (e) non-parallel orientation of the imidazole ring over the 1,8-naphthalimide ring. | ||
Experimental
All reagents and solvents were obtained from commercial sources. The IR spectra were recorded on a Perkin-Elmer SpectrumOne FT-IR spectrometer in the range 4000–400 cm−1. The 1H-NMR spectra were recorded using a Varian Mercury plus 400 MHz and Bruker 600 MHz instrument. Powder X-ray Diffraction (PXRD) were carried out using a Bruker D2 phaser with the Cu-Kα source (λ = 154 Å) on glass surfaces of air dried samples. Thermogravimetric analyses (TGA) were performed on a Mettler Toledo TGA/SDTA 851e module. Samples were placed in open alumina pans in the temperature range 25–600 °C and were purged with a stream of dry N2 flowing at 100 mL min−1 with a heating rate of 5 °C. The UV-Vis and solid state fluorescence emission spectra were recorded on a Perkin-Elmer-Lambda 750 UV-Vis spectrometer and a Fluoromax-4 fluorimeter respectively at room temperature. The solid state fluorescence emission spectra of powdered samples of equal weight (30 mg) were recorded in each case.The quantum yield of fluorescence was determined by using quinine sulphate as a reference in water at room temperature.

The Q.S, DMF and R.I are quinine sulphate, dimethylformamide and refractive index, respectively. And area means the area covered by a fluorescence curve of the respective compound.
The N-(3-imidazol-1-yl-propyl)-1,8-naphthalimide (L) was synthesized by a reported procedure3f using DMF as a solvent and refluxing at 100 °C for 12 h. Then the reaction mixture was cooled and ice cold water was added to it. The silky white precipitate of L formed was filtered and dried.
[ZnL2Cl2] (1)
The ligand L (0.61 g, 2 mmol) was dissolved in warmed DMF and added to a methanolic solution of zinc chloride (0.136 g, 1 mmol). The reaction mixture was refluxed for 4 hours and cooled to room temperature, and the filtered solution was kept undisturbed for crystallization. After one week white crystals appeared. Yield, 51%. Elemental anal. calcd for C36H30Cl2N6O4Zn; C, 57.89; H, 4.05; N, 11.25; found C, 57.95; H, 4.07; N, 11.45. IR (KBr, cm−1): 3414 (br), 2963 (s), 1695 (s), 1653 (s), 1586 (s), 1441 (m), 1344 (m), 1261 (s), 1242 (m), 1170 (w), 1098 (s), 1057 (w), 1026 (w), 850 (w), 801 (m), 785 (m), 654 (w), 544 (w). 1H-NMR (600 MHz, DMSO-d6): 8.48 (d, 1H, 6.6 Hz), 8.42 (br, 1H), 7.84 (t, 1H, 7.8 Hz), 7.44 (s, 1H), 7.02 (s, 1H), 4.17 (t, 2H, 6.6 Hz), 4.10 (t, 2H, 5.4 Hz), 2.12 (m, 2H).[CdL2Cl2] (2)
Complex 2 was prepared by a procedure similar to that for complex 1. Cadmium chloride was used instead of zinc chloride. One week later colourless crystals of 2 were obtained. Yield, 39%. Elemental anal. calcd C36H30Cl2N6O4Cd; C, 54.46; H, 3.81; N, 10.58; found C, 54.51; H, 3.82; N, 10.61. IR (KBr, cm−1): 3140 (m), 1694 (m), 1653 (s), 1586 (m), 1440 (w), 1342 (m), 1242 (s), 1170 (w), 1097 (m), 849 (m), 783 (s), 750 (w), 544 (w). 1H-NMR (600 MHz, DMSO-d6): 8.51 (d, 1H, 24 Hz), 7.87 (bs, 1H), 7.71 (br, 1H), 7.22 (s, 1H), 6.88 (s, 1H), 4.08 (t, 2H, 6 Hz), 4.05 (t, 2H, 7.2 Hz), 2.08 (bs, 2H).[HgL2Cl2] (3)
Complex 3 was prepared by a procedure similar to that for complex 1 but mercuric chloride was used instead of cadmium chloride. On standing for 6–7 days, colorless crystals were observed. Isolated yield, 41%. Elemental anal. calcd C36H30Cl2N6O4Hg; C, 49.01; H, 3.43; N, 9.53; found C, 49.10; H, 3.44; N, 9.61. IR (KBr, cm−1): 3139 (s), 2123 (w), 1693 (m), 1654 (m), 1625 (s), 1518 (m), 1440 (m), 1342 (m), 1241 (s), 1096 (m), 1170 (w), 1096 (m), 1056 (w), 848 (s), 782 (s), 543 (m). 1H NMR (600 MHz, DMSO-d6): 8.47 (d, 1H, 7.2 Hz), 8.42 (d, 1H, 8.5 Hz), 7.83 (t, 1H, 7.8 Hz), 7.31 (s, 1H), 6.93 (s, 1H), 4.10 (t, 2H, 7.2 Hz), 4.03 (t, 2H, 7.2 Hz), 2.08 (m, 2H).[MnL4(SCN)2]·2CH3CN (4)
To a binary mixture solution of acetonitrile and methanol of L (0.61 g, 2 mmol), manganese chloride tetrahydrate (0.10 g, 0.5 mmol) was added. After stirring the mixture for five minutes, potassium thiocyanate (0.10 g, 1 mmol) was added. The reaction mixture was refluxed for 4 hours, and then cooled, filtered and allowed to evaporate at room temperature. Brown colored crystals were observed after a week. Isolated yield, 54%. Elemental anal. calcd C78H66N16O8S2Mn; C, 63.53; H, 4.51; N, 15.20; found C, 63.73; H, 4.61; N, 16.01. IR (KBr, cm−1): 3466 (bs), 3121 (m), 2958 (w), 2059 (s), 1698 (m), 1661 (s), 1591 (s), 1515 (s), 1443 (s), 1349 (s), 1238 (s), 1174 (w), 1088 (w), 931 (m), 844 (s), 775 (s), 661 (m), 537 (w). Thermogravimetry: loss of 5.11% weight in the temperature range 70–170 °C (calcd 5.56% for loss of acetonitrile molecules).[CoL4(SCN)2]·2CH3CN (5)
Complex 5 was synthesized by the same procedure as that of the complex 4, cobalt chloride hexahydrate (0.12 g, 0.5 mmol) was used instead of MnCl2·4H2O. Yield, 51%. Elemental anal. calcd C78H66N16O8S2Co; C, 63.36; H, 4.50; N, 15.16; found C, 63.38; H, 4.51; N, 15.34. IR (KBr, cm−1): 3444 (br), 3126 (w), 2071 (s), 1698 (s), 1660 (s), 1591 (s), 1515 (s), 1442 (s), 1349 (s), 1237 (s), 1174 (m), 1104 (w), 1089 (w), 935 (w), 844 (m), 775 (s), 661 (m), 537 (w). Thermogravimetry: loss of the 5.39% weight for acetonitrile molecules in the range 88–200 °C (calcd 5.55%).[ZnL2(SCN)2] (6)
It was synthesized by a similar procedure as that of the complex 4 where zinc nitrate hexahydrate was used. Colorless crystals were obtained after 7–8 days. Isolated yield, 39%. Elemental anal. calcd C37H30N8O4S2Zn; C, 56.96; H, 3.88; N, 14.36; found C, 56.49; H, 3.87; N, 14.23. IR (KBr, cm−1): 3130 (m), 2954 (w), 2075 (s), 1691 (w), 1657 (m), 1588 (m), 1535 (m), 1438 (s), 1346 (w), 1237 (s), 1173 (w), 844 (s), 755 (s), 653 (s), 630 (m), 539 (w), 409 (w). 1H NMR (600 MHz, DMSO-d6): 8.42–8.39 (m, 6H, naphthalene protons), 7.81–7.56 (m, 3H, imidazole protons), 4.19 (m, 2H, N-CH2), 4.04 (t, 2H, 4.8 Hz, N-CH2), 2.15 (m, 2H, –CH2–).[CdL3(SCN)2DMF]·DMF (7)
It was synthesized by a similar procedure as that of the complex 4 where cadmium nitrate tetrahydrate was used instead of zinc nitrate hexahydrate. Brown crystals were formed after 8 days. Isolated yield, 45%. IR (KBr, cm−1): 3112 (w), 2954 (w), 2062 (s), 1702 (m), 1661 (s), 1590 (m), 1513 (s), 1443 (s), 1236 (s), 1091 (m), 843 (m), 774 (s), 659 (s), 539 (m). 1H-NMR (600 MHz, DMSO-d6): 8.45–8.41 (m, 18H, naphthalene protons), 7.94 (s, 2H, OC-H of DMF), 7.83–7.43 (m, 9H, imidazole protons), 4.15 (t, 6H, 6.6 Hz, NCH2), 4.02 (t, 6H, 7.2 Hz, NCH2), 2.88 (s, 6H, CH3 of DMF), 2.72 (s, 6H, CH3 of DMF), 2.10 (m, 6H, –CH2–). Thermogravimetry: weight loss of 8.34% in the range of 120–182 °C corresponds to the loss of two dimethylformamide molecules (calcd 8.98%).Structure determination
The X-ray single crystal diffraction data for complex 1, complex 3, complex 4 and complex 7 were collected at 298 K with Mo Kα radiation (λ = 0.71073 Å) with the use of a Bruker Nonius SMART APEX CCD diffractometer equipped with a graphite monochromator and an Apex CCD camera; SMART software was used for data collection and also for indexing the reflections and determining the unit cell parameters;15a whereas the data of complexes 2, 5 and 6 were collected on an Oxford SuperNova diffractometer. For the data collected on the SuperNova diffractometer, data refinement and cell reductions were carried out by CrysAlisPro.15b Data reduction and cell refinement were performed using the SAINT software.15a The structures were solved by direct methods and refined by full-matrix least-squares calculations using the SHELXTL software. All the non-H atoms were refined in the anisotropic approximation against F2 of all reflections. The H-atoms were placed at their calculated positions and refined in the isotropic approximation; those attached to nitrogen and oxygen atoms were located in the difference Fourier maps and refined with isotropic displacement coefficients. Crystallographic data collection was done at room temperature, and the data are tabulated in Table 1.| Compound no. | Complex 1 | Complex 2 | Complex 3 | Complex 4 | Complex 5 | Complex 6 | Complex 7 |
|---|---|---|---|---|---|---|---|
| Formulae | C36H30Cl2N6O4Zn | C36H30Cl2N6O4Cd | C36H30Cl2N6O4Hg | C78H66N16O8S2Mn | C78H66N16O8S2Co | C38H30N8O4S2Zn | C62H59N13O8S2Cd |
| CCDC no. | 928185 | 928178 | 928182 | 928183 | 928180 | 928184 | 928179 |
| Mol. wt. | 746.93 | 793.96 | 882.15 | 1474.53 | 1478.52 | 792.19 | 1290.74 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Triclinic | Triclinic | Monoclinic | Triclinic |
| Space group | C2/c | C2/c | C2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/c |
P |
| a/Å | 14.6120(18) | 14.6314(6) | 14.7824(3) | 10.6242(7) | 10.6146(8) | 14.5214(4) | 10.3324(3) |
| b/Å | 11.9825(18) | 11.9993(4) | 12.0395(3) | 11.2787(6) | 11.2239(12) | 14.1478(5) | 16.9948(4) |
| c/Å | 19.488(3) | 19.7572(8) | 19.7994(5) | 15.7505(9) | 15.7625(14) | 22.8454(8) | 17.5964(4) |
| α/° | 90.00 | 90.00 | 90.00 | 95.575(4) | 95.493(8) | 90.00 | 91.4080(10) |
| β/° | 104.612(11) | 105.630(4) | 104.923(2) | 103.794(4) | 104.366(7) | 128.618(2) | 95.6650(10) |
| γ/° | 90.00 | 90.00 | 90.00 | 99.258(4) | 99.105(8) | 90.00 | 106.9570(10) |
| V/Å3 | 3276.9(8) | 3340.4(2) | 3404.91(14) | 1790.86(18) | 1778.3(3) | 3667.1(2) | 2936.48(13) |
| Z | 4 | 4 | 4 | 1 | 1 | 4 | 2 |
| Density/g cm−3 | 1. 514 | 1.579 | 1.721 | 1.367 | 1.381 | 1.435 | 1.460 |
| Abs. coeff./mm−1 | 0.964 | 0.864 | 4.727 | 0.312 | 0.370 | 0.836 | 0.511 |
| F(000) | 1536 | 1608 | 1736 | 767 | 769 | 1632 | 1332 |
| Total no. of reflections | 2919 | 3032 | 3066 | 6370 | 6422 | 6629 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 470 470 |
| Reflections, I > 2σ(I) | 2156 | 2638 | 2402 | 4095 | 4116 | 4765 | 7921 |
| Max. 2θ/° | 50.50 | 50.50 | 50.48 | 50.50 | 50.50 | 50.50 | 50.50 |
| Ranges (h, k, l) | −16 ≤ h ≤ 17 | −17 ≤ h ≤ 17 | −17 ≤ h ≤ 17 | −12 ≤ h ≤ 12 | −12 ≤ h ≤ 12 | −16 ≤ h ≤ 17 | −12 ≤ h ≤ 12 |
| −14 ≤ k ≤ 12 | −14 ≤ k ≤ 10 | −13 ≤ k ≤ 14 | −13 ≤ k ≤ 13 | −11 ≤ k ≤ 13 | −16 ≤ k ≤ 16 | −19 ≤ k ≤ 18 | |
| −22 ≤ l ≤ 22 | −23 ≤ l ≤ 23 | −23 ≤ l ≤ 23 | −18 ≤ l ≤ 18 | −18 ≤ l ≤ 18 | −26 ≤ l ≤ 27 | −21 ≤ l ≤ 20 | |
| Complete to 2θ (%) | 98.1 | 99.8 | 99.0 | 98.3 | 99.8 | 99.8 | 98.3 |
| Data/restraints/parameters | 2919/0/222 | 3032/0/255 | 3066/0/222 | 6370/0/476 | 6422/0/476 | 6629/0/478 | 10470/0/779 |
| Goof (F2) | 0.984 | 1.043 | 1.104 | 1.005 | 1.019 | 1.114 | 1.044 |
| R indices [I > 2σ(I)] | 0.0488 | 0.0351 | 0.0720 | 0.0433 | 0.0699 | 0.0473 | 0.0320 |
| R indices (all data) | 0.0627 | 0.0427 | 0.1093 | 0.0790 | 0.1076 | 0.0732 | 0.0450 |
| WR2 [I > 2σ(I)] | 0.1353 | 0.0793 | 0.1346 | 0.0676 | 0.1426 | 0.0774 | 0.0714 |
| WR2 (all data) | 0.1446 | 0.0835 | 0.1432 | 0.0781 | 0.1624 | 0.0863 | 0.0811 |
Results and discussion
A series of divalent metal (M = Mn, Co, Zn, Cd, Hg) complexes of the ligand L with various compositions as depicted in Scheme 1 were synthesized and characterized by various spectroscopic techniques and finally by single crystal X-ray diffraction. The powder XRD of all samples were recorded and compared with the simulated spectra, which showed the purity of the bulk samples in the solid state. For the sake of comparison the powder pattern of the complex 1 is shown in Fig. 2a and others are available as ESI.† The metal chlorides of zinc, cadmium and mercury independently reacted with L led to a series of isomorphous complexes [ML2Cl2] {M = Zn (1), Cd (2), Hg (3)}. Each of the three complexes adopts a distorted tetrahedral geometry. Since these three complexes are structurally similar, only the representative structure of the zinc complex (1) is shown in Fig. 2b. In the solid state structure the zinc ion lies on a two-fold axis. The ligand L in these cases adopts a bend structure and such a bend form of the ligand is stabilized by weak C–H⋯O and π-stacking interactions (ESI Fig. 2S†). Such interactions arise from intermolecular interactions between the carbonyl group of 1,8-naphthalimide with one C–H of the propylene attached to an imidazole unit. Another C–H bond of the propylene group at the α-position next to the imidazole also interacts with the carbonyl group of the 1,8-naphthalimide group. The formation of intra-molecular stacks of imidazole and naphthalene rings is favored by the flexible propylene group. The metal–ligand bond parameters of the complexes 1–3 are listed in Table 2. From Table 2 it is clear that metal–ligand bond distances are in the sequence Zn < Cd < Hg. The observed trend is due to the increase in the size of the metal ions (Table 2). | ||
| Scheme 1 Different metal complexes of L. | ||
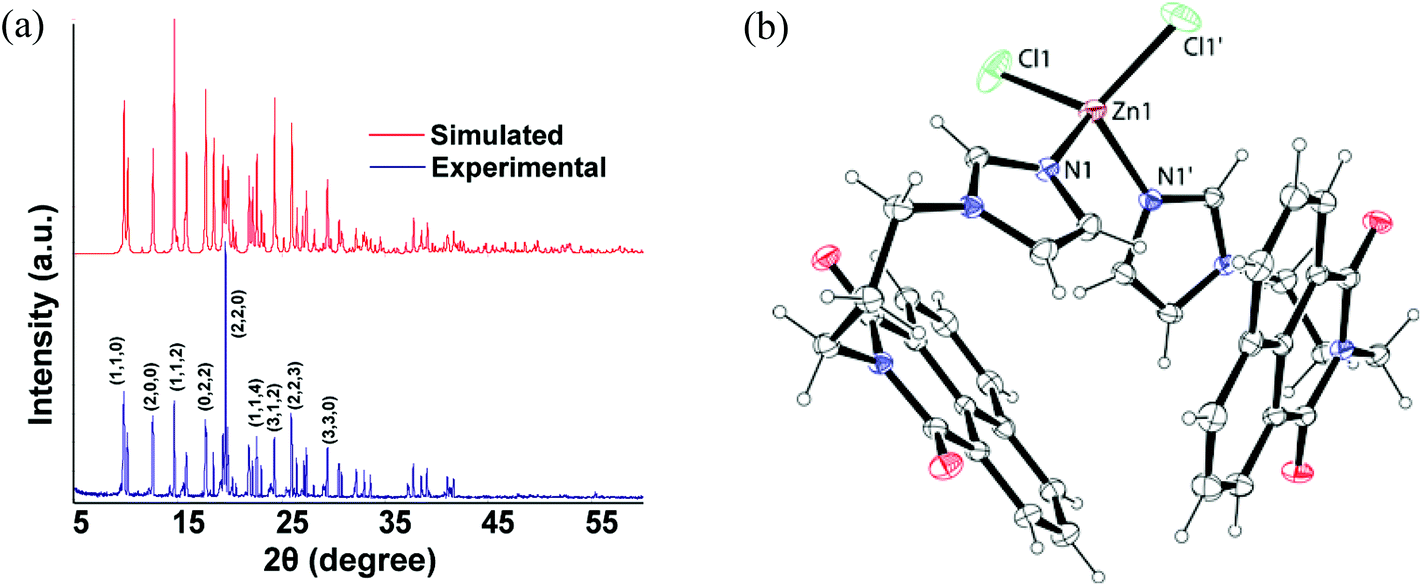 | ||
| Fig. 2 (a) Powder-XRD of the complex 1 (top: simulated, bottom: experimental). (b) Crystal structure of the complex 1 (ORTEP drawn with 30% probability; equivalent symmetry is generated through 2 − x, y, 1/2 − z). | ||
| Complex | M–L | Bond-length (Å) | <L–M–L | Angle (°) | <L–M–L | Angle (°) |
|---|---|---|---|---|---|---|
| Complex 1 | Zn1–N1 | 2.028(2) | N1–Zn1–N1 | 99.41(13) | N1–Zn1–Cl1 | 112.63(7) |
| Zn1–Cl1 | 2.2378(11) | N1–Zn1–Cl1 | 107.74(7) | Cl1–Zn1–Cl1 | 115.58(8) | |
| Complex 2 | Cd1–N1 | 2.231(2) | N1–Cd1–N1 | 94.99(12) | N1–Cd1–Cl1 | 112.84(7) |
| Cd1 Cl1 | 2.4131(10) | N1–Cd1–Cl1 | 107.93(7) | Cl1–Cd1–Cl1 | 118.00(7) | |
| Complex 3 | Hg1–N1 | 2.268(7) | N1–Hg1–N1 | 92.3(4) | N1–Hg1–Cl1 | 111.5(2) |
| Hg1–Cl1 | 2.451(5) | N1–Hg1–Cl1 | 106.3(2) | Cl1–Hg1–Cl1 | 124.2(3) |
In the thiocyanate containing metal complexes the ratio of the ligand to the metal ions varies. For example the 1:2 complex was formed in the case of zinc, 1:3 in cadmium and 1:4 in the case of manganese and cobalt. The metal complexes bearing the thiocyanate ligand have gained special interest due to their ease of degradability and their labile nature.16 The manganese complex 4 and cobalt complex 5 have a similar composition, namely [ML4(SCN)2]·2CH3CN and they are isomorphous (Fig. 3).
 | ||
| Fig. 3 The structure of the complex: (a) 4 (symmetry of equivalent atoms −x, 1 − y, −z); (b) 6 and (c) 7 (ORTEP drawn with 30% thermal ellipsoid). | ||
The manganese atom in the complex 4 and the cobalt atom in the complex 5 lie on an inversion centre. These hexa-coordinated complexes 4 and 5 have a distorted octahedral geometry with four nitrogen atoms of the imidazole in one plane forming a dative bond with a metal ion (Fig. 3a) and metal-ligand bond parameters of complex 4 and complex 5 are shown in Table 3. The mono-dentate thiocyanate ligands coordinating through nitrogen atoms occupy the axial positions. The structure of the complex 4 contains a center of inversion. The interesting feature of the structure is that the naphthalimide rings are π-stacked with another naphthalimide ring of neighboring molecules on each side so that layered structures are formed. The ligand L adopts a linear structure in the complex. While forming such structures the imidazole rings are not in one plane with respect to each other across the metal centers. The zinc thiocyanate complex [ZnL2(SCN)2] (6) is a tetra-coordinated complex (Fig. 3b) with a distorted tetrahedral geometry. The complex is stable in solution and the 1H-NMR show a significant shift from the parent ligand; the spectra are- shown in Fig. 21S of ESI.† On the other hand [CdL3(SCN)2DMF]·DMF (7) is a hexa-coordinated complex with three ligands (L) and one DMF occupying one plane and two thiocyanates occupying the axial positions of a distorted octahedron (Fig. 3c) and metal-ligand bond parameters of the complexes 6 and 7 are listed in Table 3. The complex 7 is also characterized by its 1H-NMR which is shown in the ESI Fig. 22S.† It shows methyl signals for two DMF molecules, one corresponding to coordinated and to the other free dimethylformamide which appears at 2.88 and 2.72 ppm respectively. This is supportive of the structure determined by X-ray crystallography. The integration compound shows the presence of two molecules of DMF per molecule. In thermogravimetry complex 7 shows a weight loss of 8.34 wt% (calcd 8.98 wt%) in the range of 120–182 °C corresponding to the loss of one lattice dimethylformamide molecule and one coordinated dimethylformamide molecule. The IR spectra of thiocyanate complexes showed characteristic stretching frequencies in the region of 2051–2075 cm−1. The values support the coordination of thiocyanate through a nitrogen atom in each case.17
| Complex no. | M–X | Bond-length (Å) | <L–M–L | Angle (°) | <L–M–L | Angle (°) |
|---|---|---|---|---|---|---|
| Complex 4 | Mn1–N1 | 2.2758(19) | N1–Mn1–N4 | 88.12 | N1′–Mn1–N4′ | 91.88 |
| Mn1–N4 | 2.2606(19) | N1–Mn1–N7 | 89.67 | N1′–Mn1–N7 | 90.33 | |
| Mn1–N7 | 2.236(2) | N4–Mn1–N7 | 92.67 | N4′–Mn1–N7 | 87.33 | |
| Complex 5 | Co1–N1 | 2.169(4) | N1–Co1–N4 | 88.92 | N1′–Mn1–N4 | 91.08 |
| Co1–N4 | 2.161(4) | N1–Co1–N7 | 90.37 | N1′–Mn1–N7 | 89.63 | |
| Co1–N7 | 2.157(4) | N4–Co1–N7 | 92.64 | N4′–Mn1–N7 | 87.46 | |
| Complex 6 | Zn1–N1 | 1.986(3) | N1–Zn1–N4 | 114.52(10) | N4–Zn1–N8 | 102.27(11) |
| Zn1–N4 | 1.997(3) | N1–Zn1–N7 | 103.77(11) | N7–Zn1–N8 | 111.58(12) | |
| Zn1–N7 | 1.972(3) | N1–Zn1–N8 | 115.03(12) | |||
| Zn1–N8 | 1.940(3) | N4–Zn1–N7 | 109.85(12) | |||
| Complex 7 | Cd1–N1 | 2.327(2) | N1–Cd1–N4 | 178.09(7) | N4–Cd1–N11 | 90.36(8) |
| Cd1–N4 | 2.335(2) | N1–Cd1–O7 | 88.72(8) | N4–Cd1–N7 | 87.37(7) | |
| Cd1–N7 | 2.339(2) | N1–Cd1–N11 | 91.18(9) | N4–Cd1–N10 | 90.92(8) | |
| Cd1–N10 | 2.340(3) | N1–Cd1–N7 | 93.77(7) | N7–Cd1–N10 | 92.63(9) | |
| Cd1–N11 | 2.343(3) | N1–Cd1–N10 | 87.50(9) | N11–Cd1–N10 | 177.69(9) | |
| Cd1–O7 | 2.337(2) | N4–Cd1–O7 | 90.20(8) | O7–Cd1–N7 | 176.79(8) | |
| O7–Cd1–N11 | 88.57(9) | O7–Cd1–N10 | 89.50(9) |
The complexes have UV-visible absorptions in the range of 315–360 nm originating from the ligand. By comparing with analogous compounds it may be suggested to originate from the n → π* transition.18 The naphthalimide derivatives are fluorescent19 and in the solid state the ligand L shows a strong emission at 461 nm. The solid samples of the complexes 1–5 showed emissions in the range of 425–481 nm upon excitation at 350 nm [Fig. 4(i) and (ii)]. The complexes 3, 6 or 7 show fluorescence enhancement with respect to the ligand (L). On the other hand the complexes 4 and 5 show quenching of fluorescence with a small shift towards longer wavelength. The central metal ions in complex 4 and complex 5 are manganese (d5, high-spin) and cobalt (d7) in +2 oxidation state, respectively, which are paramagnetic. Both complexes show lower fluorescence intensity than the parent ligand due to the paramagnetic nature of the ions.1f These complexes have stretched conformations of the ligand and hence follow conventional fluorescence emission resembling the emission of the parent ligand.
 | ||
| Fig. 4 (i) Solid state fluorescence emission of the ligand L and complexes: (a) complex 1; (b) complex 5; (c) complex 2; (d) complex 4, (e) complex 3. In each case, the intensity is normalized with respect to the lowest emission intensity shown by complex 1. (ii) The solid state fluorescence emission of (a) complex 6; (b) complex 7 at room temperature (λex = 350 nm) with intensities normalised with respect to the ligand. | ||
The fluorescence emission changes of solid samples of these tetrahedral complexes are interesting; they show a characteristic emission on variation of anions and cations. The increase and decrease in intensity of fluorescence emission shown by these complexes are schematically presented in Scheme 2. The complexes 1–3 have chloride ions, among them, the complexes 1 and 2 show fluorescence emissions at 428 nm and 425 nm, respectively, which are lower than the fluorescence emission of the ligand at 461 nm. The emission peak of mercury complex 3 has higher intensity than that of the free ligand that occurs at 430 nm, which is 31 nm less than the fluorescence emission of the ligand in the solid state. On the other hand, the complexes 6 and 7 have fluorescence emissions of longer wavelength than the parent ligand. A close examination of the structures 1, 2 and 6 shows that the orientation of 1,8-naphthalimide rings are similar in complexes 1 and 2 but different from complexes 6 and 7. Whereas, complex 6 has a non-parallel bent orientation, but complex 7 has a stretched orientation of the ligand L. Thus, twisting of the ligand to adopt near parallel orientation between the imidazole ring and 1,8-naphthalimide rings favors shorter-wavelength emission due to stabilization of the ground state by intramolecular charge transfer. A stretched orientation or an orientation not favoring stabilization of the ground state of L would help in stabilization of the inter-molecular charge transfer excited state, taking the emission to a longer wavelength. The mercury being large and having a more diffused charge shows intra-molecular interaction between imidazole and the naphthalimide ring, helps in the stabilization of the ground state, and accordingly emits at shorter wavelengths.
 | ||
| Scheme 2 The fluorescence selectivity in four coordinated complexes of L. | ||
The fluorescence emission of L in DMF solution by adding an aqueous solution of chloride salts of zinc, cadmium or mercury was independently studied with complexes 1–3 (Fig. 5). As anticipated, addition of mercury ions showed enhancement of fluorescence whereas the addition of zinc chloride or cadmium chloride quenched the emission causing emission at two wavelengths (Fig. 5a and 5b). We find an analogy between solution and solid state fluorescence emission study from the observed changes. The single emission peak of the ligand changed on titration with zinc chloride, and at the end of titration we observed emission at 385 nm and 475 nm; whereas similar titration with cadmium chloride yielded emissions at 385 nm and 552 nm, respectively. The role of anions in the fluorescence emission of naphthalimide fluorophores interacting with various anions is well established.2b Heagy and coworkers have shown that depending on the orientations of pyridine over a naphthalimide ring causes fluorescence to shift to either a shorter wavelength (SW) or to a longer wavelength (LW).14 Thus, in solution the addition of zinc ions facilitated the stretching of the ligand leading to emission at longer wavelength to occur from stabilization of exciplex through intermolecular π–π interactions.3f
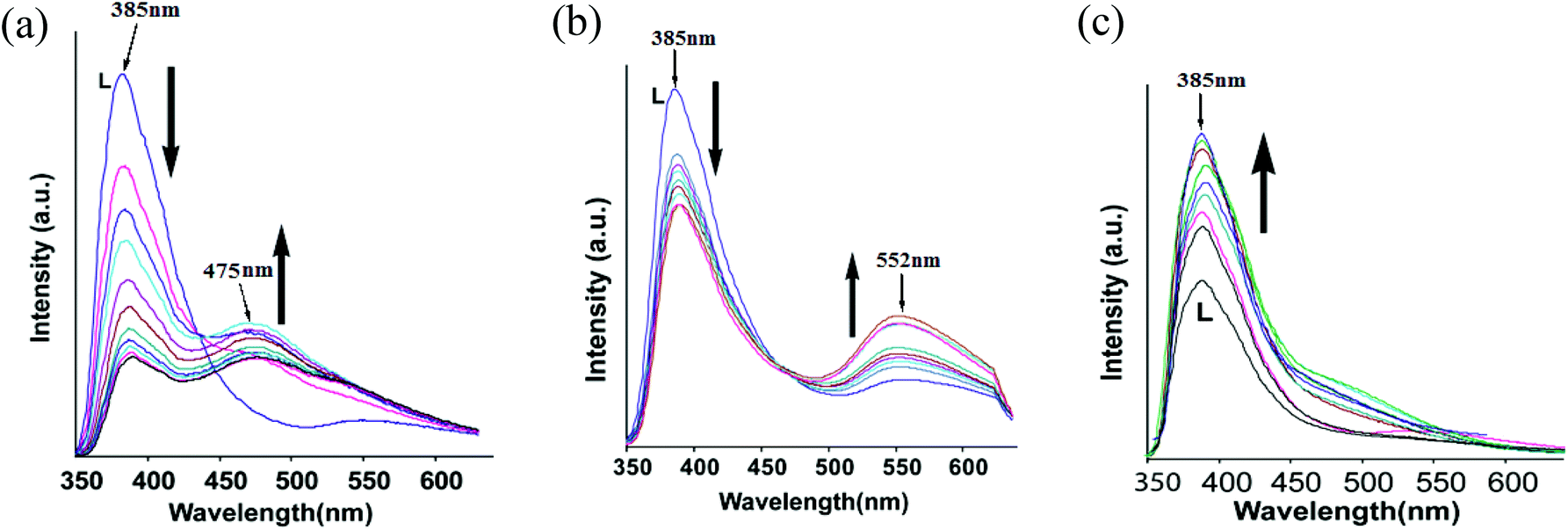 | ||
| Fig. 5 The changes in fluorescence emission intensity of (λex = 340 nm) the ligand L (2 mL of 10−4 M solution in DMF) on addition of (a) zinc chloride, (b) cadmium chloride and (c) mercuric chloride (5 μL aliquots of a 10−2 M solution in water). | ||
The fluorescence emission spectra of the complexes in solutions were determined and are shown in Fig. 6. It is clear from Fig. 6 that the fluorescence quenching with respect to the ligand occurred in solution in each metal complex. However, the exception was the mercury complex 3, which showed fluorescence enhancement. There are multiple components of fluorescence in each case, pointing out a number of orientations present in solutions. This makes a clear distinction between mercury and other ions. There are terpyridine ligands distinguishing mercury20 in aqueous medium; but our system can also distinguish mercury in aqueous solution containing cadmium and zinc ions. The quantum yields of the ligand as well as all the complexes were determined from their respective solution in dimethylformamide (Table 2S of ESI†). The quantum yield of the ligand L is 0.36 and the complexes lie in the range of 0.37–0.50.
 | ||
| Fig. 6 Fluorescence emission spectra (λex 330 nm) of (a) complex 3, (b) complex 2; (c) complex 6; (d) complex 1; (e) complex 7; (f) complex 5; (g) complex 4 and ligand (L) in dimethylformamide (concentration of 10−4 M each). | ||
To understand the effect of temperature on the fluorescence changes, we recorded the temperature dependent fluorescence emissions at different temperatures for the complexes 1–3 and compared these data with the ligand. This is carried out with the anticipation that as the thermal energy increases the ligand would open up from a bend geometry, which disfavors intramolecular charge transfer interaction. Three illustrative examples on the changes of fluorescence in solution with respect to temperatures are shown in Fig. 7. Quenching of fluorescence in the case of complexes 1 and 2 were observed as the temperature was increased, while mercury complex 3 showed enhancement of fluorescence emission as the temperature was increased. It may be mentioned that the nitrogen heterocycles attached to 1,8-naphthalimides are important as some of them show dual fluorescence and these are related to coplanar or orthogonal geometries of the 1,8-naphthalimide part with respect to the nitrogen containing heterocyclic units attached to them.2c,14b Thus, the complexes 1 and 2 show a decrease of fluorescence intensity on heating, suggesting the decrease in photo-electron energy transfer contribution to the emission process by change of orientation. Whereas the mercury complex having a folded structure, stretches out on heating to show an increase in the fluorescence emission intensity. When there is no metal ion to guide the orientation of the ligand L, the lone pair of electrons present on the nitrogen atom of imidazole contributes to photo-electron energy transfer (PET) mechanism, causing a partially quenched state at the start of heating; on heating, the orientation of L changes, and accordingly the intensity grows.
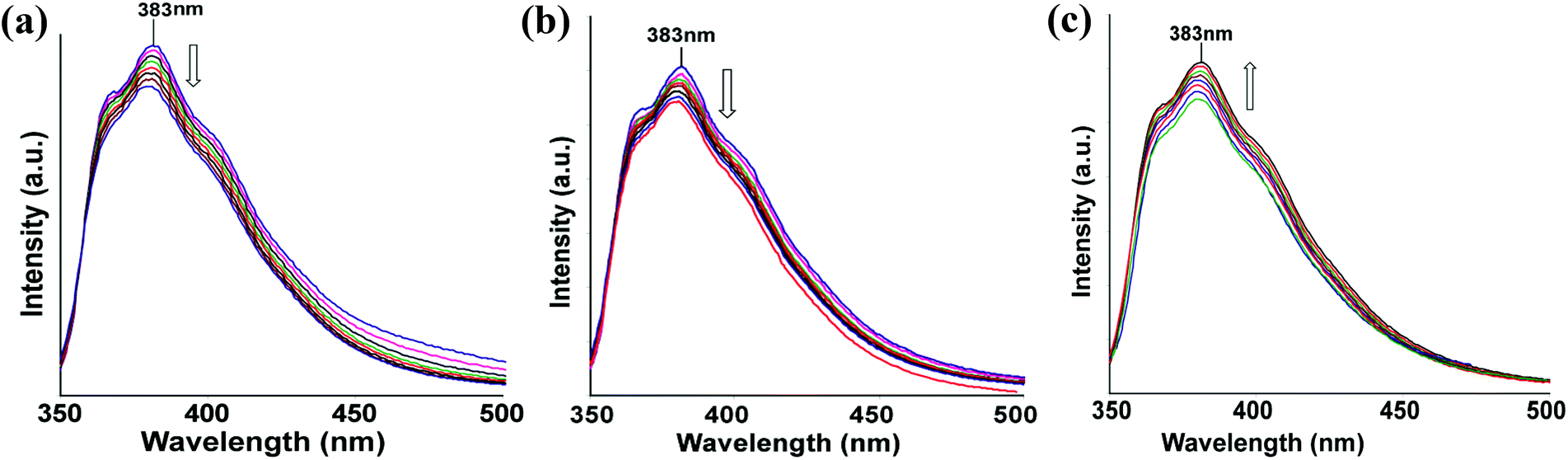 | ||
| Fig. 7 Temperature dependent fluorescence spectra of (a) complex 1; (b) complex 2 and (c) complex 3 (30 °C to 100 °C at 10 °C interval of temperature; 0.1 mM solution each in DMF; λex = 330 nm, downward arrow shows decease in intensity). | ||
The stability of the complexes in solution was ascertained from the respective 1H-NMR spectra of the complexes 1–3, 6 and 7. The 1H-NMR spectra of the complexes 1–3 and ligand are compared in Fig. 8, which clearly indicate that the complexes are stable in solution and on complexation of the ligand to zinc or cadmium, the chemical shift of the methylene group adjacent to imidazole is affected and so are the protons on the imidazole ring. Analogously, the complexes 6 and 7 also show clear shifts of the 1H-NMR peaks and some of the peaks were broadened in these complexes as shown in ESI (Fig. 21S and 22S†).
 | ||
| Fig. 8 1H-NMR spectra of the ligand and (400 MHz, CDCl3) and complexes 1–3 (600 MHz, DMSO-d6). | ||
In conclusion, the structures in the solid state have helped us to explain the fluorescence behavior of such complexes by invoking intramolecular charge-transfer (ICT) to stabilize the ground state in bent geometry (d of Fig. 1) to cause emissions at lower wavelength, whereas a stretched ligand helps exciplex through π-stacking of 1,8-naphthalimides to show emissions at long wavelengths (e of Fig. 1). The free ligand as well as the complexes in solution show fluorescence enhancement and on heating, show enhancement of fluorescence intensity through a PET mechanism21 arising from the lone pair of electrons in the nitrogen atom of imidazole. On heating, conformational changes disfavor participation of lone pairs in the nitrogen atom of imidazole in such a process, and hence enhancement of the intensity of fluorescence of the ligand was observed. Whereas, in the solid state the coordination of the ligand to d10-metal ions provides orientation to the ligand so as to favor or disfavor intra-molecular charge-transfer emission (d, e of Fig. 1). The mercury ions show fluorescence enhancement over the cadmium or zinc ions, hence it is distinguishable from the latter. From the series of study it is clear that the conformation shown as d of Fig. 1 favors the low wavelength emission, while the other stretched geometry helps in exciplex formation taking the fluorescence to longer wavelengths.
References
- (a) R. M. Duke, E. M. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC; (b) T. Gunnlaugsson, P. E. Kruger, P. Jensen, F. M. Pfeffer and G. M. Hussey, Tetrahedron Lett., 2003, 44, 8909–8913 CrossRef CAS PubMed; (c) X. Poteau, A. I. Brown, R. G. Brown, C. Holmes and D. Matthew, Dyes Pigm., 2000, 47, 91–105 CrossRef CAS; (d) A. P. De Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed; (e) S. Guha, F. S. Goodson, L. J. Corson and S. Saha, J. Am. Chem. Soc., 2012, 134, 13679–13691 CrossRef CAS PubMed; (f) Z. Xu, J. Yoon and D. R. Spring, Chem. Commun., 2010, 46, 2563–2565 RSC.
- (a) K.-Y. Lee, Chem. Rev., 1993, 93, 449–486 CrossRef; (b) H. D. P. Ali, P. E. Kruger and T. Gunnlaugsson, New J. Chem., 2008, 32, 1153–1161 RSC; (c) P. Nandhikonda, M. P. Begaye, Z. Cao and M. D. Heagy, Chem. Commun., 2009, 4941–4943 RSC; (d) J. Gan, H. Tian, Z. Wang, K. Chen, J. Hill, P. A. Lane, M. D. Rahn, A. M. Fox and D. D. C. Bradley, J. Organomet. Chem., 2002, 645, 168–175 CrossRef CAS.
- (a) Z. Liu, C. Zhang, X. Wang, W. He and Z. Guo, Org. Lett., 2012, 14, 4378–4381 CrossRef CAS PubMed; (b) M. Formica, V. Fusi, L. Giorgi and M. Micheloni, Coord. Chem. Rev., 2012, 256, 170–192 CrossRef CAS PubMed; (c) V. F. Pais, P. Remon, D. Collado, J. Anderssson, E. Perez-Inestros and U. Pischel, Org. Lett., 2011, 13, 5572–5575 CrossRef CAS PubMed; (d) L. Prodi, F. Bolletta, M. Montali and N. Zaccheroni, Coord. Chem. Rev., 2000, 205, 59–85 CrossRef CAS; (e) S. Guha and S. Saha, J. Am. Chem. Soc., 2010, 132, 17674–17677 CrossRef CAS PubMed; (f) J. K. Nath and J. B. Baruah, New J. Chem., 2013, 37, 1509–1519 RSC; (g) J. K. Nath and J. B. Baruah, J. Fluoresc. DOI:10.1007/s10895-014-1353-8.
- (a) C. Thalacker, A. Miura, S. De Feyter, F. De Schryver and F. Wurthner, Org. Biomol. Chem., 2005, 3, 414–422 RSC; (b) D. L. Reger, J. D. Elgin, R. F. Semeniuc, P. J. Pellechia and M. D. Smith, Chem. Commun., 2005, 4068–4070 RSC.
- (a) G. Loving and B. Imperiali, Bioconjugate Chem., 2009, 20, 2133–2141 CrossRef CAS PubMed; (b) M. E. Vazques, J. B. Blanco and B. Imperiali, J. Am. Chem. Soc., 2005, 127, 1300–1306 CrossRef PubMed; (c) G. Loving and B. Imperiali, J. Am. Chem. Soc., 2008, 130, 13630–13638 CrossRef CAS PubMed.
- (a) D. W. Cho, M. Fujisuka, K. H. Choi, M. J. Park, U. C. Yoon and T. Majima, J. Phys. Chem. B, 2006, 110, 4576–4582 CrossRef CAS PubMed; (b) Y. Xu, X. Qian, W. Yao, P. Mao and J. Cui, Bioorg. Med. Chem., 2003, 11, 5427–5433 CrossRef CAS PubMed.
- (a) V. Gorteau, G. Bollot, J. Mareda, A. Perez-valasco and S. Matile, J. Am. Chem. Soc., 2005, 128, 14788–14789 CrossRef PubMed; (b) J. T. Davis, Nat. Chem., 2010, 2, 516–517 CrossRef CAS PubMed; (c) R. E. Sawson, A. Hennig, D. P. Weimann, D. Emery, V. Ravikumar, J. Montenegro, T. Takeuchi, S. Gabutti, M. Mayor, J. Mareda, C. A. Schalley and S. Matile, Nat. Chem., 2010, 2, 533–538 CrossRef PubMed.
- D. V. Pogozhev, M. J. Bezdek, P. A. Schauer and C. P. Berlinguette, Inorg. Chem., 2013, 52, 3001–3006 CrossRef CAS PubMed.
- (a) D. L. Reger, A. Debreczeni, B. Reinecke, V. Rasslov and M. D. Smith, Inorg. Chem., 2009, 48, 8911–8924 CrossRef CAS PubMed; (b) D. L. Reger, A. Debreczeni and M. D. Smith, Inorg. Chim. Acta, 2010, 364, 10–15 CrossRef CAS PubMed; (c) D. L. Reger, J. J. Horger, A. Debreczeni and M. D. Smith, Inorg. Chem., 2011, 50, 4669–4670 CrossRef CAS; (d) R. J. Sarma, C. Tamuly, N. Barooah and J. B. Baruah, J. Mol. Struct., 2007, 829, 29–36 CrossRef CAS PubMed; (e) D. Singh and J. B. Baruah, Cryst. Growth Des., 2012, 12, 2109–2121 CrossRef CAS; (f) D. Singh and J. B. Baruah, Inorg. Chim. Acta, 2012, 390, 37–40 CrossRef CAS PubMed.
- (a) I. Saito, M. Takayama, H. Sugiyama and K. Nakatani, J. Am. Chem. Soc., 1995, 117, 6406–6407 CrossRef CAS; (b) M. F. Brafia, M. Cacho, A. Ramos, M. T. Dominguez, J. M. Pozuelo, C. Abradelo, M. F. Rey-Stolle, M. Yuste, C. Carrsco and C. Bailly, Org. Biomol. Chem., 2003, 1, 648–654 RSC; (c) E. B. Veale and T. Gunnlaugsson, J. Org. Chem., 2010, 75, 5513–5525 CrossRef CAS PubMed.
- (a) L. Ingrassia, F. Lefranc, R. Kiss and T. Mijatovic, Curr. Med. Chem., 2009, 161, 192–121 Search PubMed; (b) M. Ly and H. Xu, Curr. Med. Chem., 2009, 16, 797–813 Search PubMed; (c) J. L. Nitiss, J. Zhou, A. Rose, Y. Huiung, K. C. Gale and N. Osheroff, Biochemistry, 1998, 37, 3078–3085 CrossRef CAS PubMed; (d) S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2013, 42, 1601–1618 RSC; (e) K. Wang, Y. Wang, X. Yan, H. Chen, G. Ma, P. Zhang, J. Li, X. Li and J. Zhang, Bioorg. Med. Chem. Lett., 2012, 22, 937–941 CrossRef CAS PubMed; (f) P. Gasioski, K. S. Danel, M. Matusiewicz, T. Uchacz, W. Kuznik and A. V. Kityk, J. Fluoresc., 2012, 81–91 CrossRef PubMed.
- P. Guo, Q. Chen, T. Liu, L. Xu, Q. Yang and X. Qian, Med. Chem. Lett., 2013, 4, 527–531 CrossRef CAS.
- K. J. Kilpin, C. M. Clavel, F. Edafe and P. J. Dyson, Organometallics, 2012, 31, 7031–7039 CrossRef CAS.
- (a) S. Paudel, P. Nandhikonda and M. D. Heagy, J. Fluoresc., 2009, 19, 681–691 CrossRef CAS PubMed; (b) P. Nadhinkonda, M. P. Begaye, Z. Chao and M. D. Heagy, Org. Biomol. Chem., 2010, 8, 3195–3201 RSC; (c) L. Biczok, P. Valat and V. Wintgens, Phys. Chem. Chem. Phys., 1999, 1, 4759–4766 RSC; (d) H. Cao, V. Chang, R. Hernandez and M. D. Heagy, J. Org. Chem., 2005, 70, 4929–4934 CrossRef CAS PubMed; (e) J. V. Mello and N. S. Finney, Angew. Chem., Int. Ed., 2001, 40, 1536–1538 CrossRef CAS.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (b) CrysAlisPro, Oxford Diffraction Ltd, version 1,171. 33. 34d (release crysAlis 171.NET) 2009 Search PubMed.
- A. Abedi, V. Amani, R. Boca, L. Dlhan, H. K. Khavasi and N. Safari, Inorg. Chim. Acta, 2013, 395, 58–66 CrossRef CAS PubMed.
- P. C. H. Mitchell and R. J. P. Williams, J. Chem. Soc., 1960, 1912–1918 RSC.
- P. Kucheryavy, G. Li, S. Vyas, C. Hadad and K. D. Glusac, J. Phys. Chem. A, 2009, 113, 6453–6646 CrossRef CAS PubMed.
- (a) D. L. Reger, A. Debreczeni, J. J. Horger and M. D. Smith, Cryst. Growth Des., 2011, 11, 4068–4079 CrossRef CAS; (b) D. L. Reger, A. Debreczeni, A. E. Pascui and M. D. Smith, Polyhedron, 2013, 52, 1317–1322 CrossRef CAS PubMed; (c) S.-M. Fang, Q. Zhang, M. Hu, X.-G. Yang, L.-M. Zhou, M. Du and C.-S. Liu, Cryst. Growth Des., 2010, 10, 4773–4785 CrossRef CAS; (d) S.-L. Zheng, J.-H. Yang, X.-L. Yu, X.-M. Chen and W.-T. Wong, Inorg. Chem., 2004, 43, 830–838 CrossRef CAS PubMed.
- (a) R. Shunmugam, G. J. Gabriel, C. E. Smith, K. A. Aamer and G. N. Tew, Chem. – Eur. J., 2008, 14, 3904–3907 CrossRef CAS PubMed; (b) H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2012, 41, 3210–3244 RSC.
- D. W. Cho, M. Fujisuka, A. Sugimoto, U. C. Yoon, P. S. Mariano and T. Majima, J. Phys. Chem. B, 2006, 110, 11062–11068 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The packing diagrams, powder XRD-patterns and their comparison with simulated patterns, solid state and solution UV-Vis spectra and thermogravimetric analysis are available. CCDC 928178, 928179, 928180, 928182, 92813, 928164 and 9281885 for 1–7. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00028e |
| This journal is © the Partner Organisations 2014 |