Modulating the copper–sulfur interaction in type 1 blue copper azurin by replacing Cys112 with nonproteinogenic homocysteine†
Kevin M.
Clark
a,
Yang
Yu
b,
Wilfred A.
van der Donk
*acd,
Ninian J.
Blackburn
e and
Yi
Lu
*abc
aUniversity of Illinois at Urbana–Champaign, Department of Biochemistry, Urbana, IL 61801, USA. E-mail: yi-lu@illinois.edu
bUniversity of Illinois at Urbana–Champaign, Center for Biophysics and Computational Biology, Urbana, IL 61801, USA
cUniversity of Illinois at Urbana–Champaign, Department of Chemistry, Urbana, IL 61801, USA
dHoward Hughes Medical Institute, Urbana, IL 61801, USA
eOregon Health & Sciences University, Institute of Environmental Health, Portland, OR 97239, USA
First published on 13th January 2014
Abstract
The Cu–SCys interaction is known to play a dominant role in defining the type 1 (T1) blue copper center with respect to both its electronic structure and electron transfer function. Despite this importance, its role has yet to be probed by mutagenesis studies without a dramatic change in its T1 copper character. We report herein replacement of the conserved Cys112 in azurin with the nonproteinogenic amino acid homocysteine. Based on electronic absorption, electron paramagnetic resonance, and extended X-ray absorption fine structural spectroscopic studies, this variant displays typical type 1 copper site features. Surprisingly, instead of increasing the strength of the Cu–sulfur interaction by the introduction of the extra methylene group, the Cys112Hcy azurin showed a decrease in the covalent interaction between SHcy and Cu(II) when compared with the WT SCys–Cu(II) interaction. This is likely due to geometric adjustment of the center that resulted in the copper ion moving out of the trigonal plane defined by two histidines and one Hcy and closer to Met121. These structural changes resulted in an increase in reduction potential by 35 mV, consistent with lower Cu–S covalency. These results suggest that the Cu–SCys interaction is close to being optimal in native blue copper protein. It also demonstrates the power of using nonproteinogenic amino acids in addressing important issues in bioinorganic chemistry.
Introduction
The blue or type 1 (T1) copper centers in cupredoxins are a major class of redox centers commonly found in many biological systems. They are also among the most useful redox agents with high electron transfer (ET) efficiency.1–11 A T1 copper center consists of a unique His2Cys ligand set in a trigonal plane with long-range axial interactions from other residues such as Met (Fig. 1). Extensive spectroscopic1,3,12–14 and crystallographic studies15–17 have defined the roles of each ligand in contributing to the structure and function of the T1 Cu center.1,3,18–28 The equatorial Cys is shown to play a dominant role. This Cys residue defines the unique spectroscopic properties such as the sulfur-to-Cu(II) ligand to metal charge transfer band (LMCT). This LMCT causes the strong blue color of the protein and the small hyperfine splitting in the parallel region of Cu(II) electron paramagnetic resonance spectroscopy, as well as the strong Cu(II)–S covalency that contributes to the efficient ET.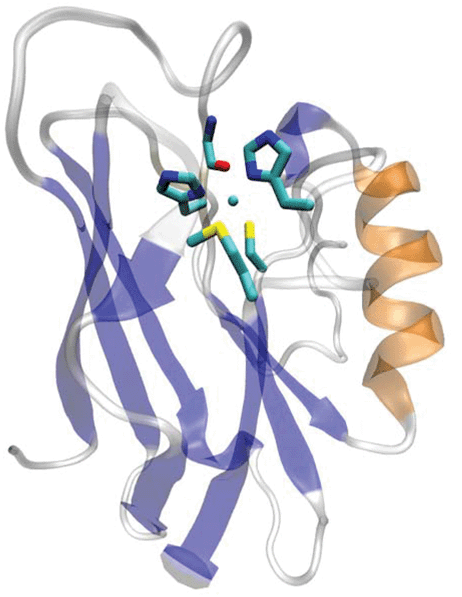 | ||
| Fig. 1 The overall and active site structure of type 1 blue copper azurin from Pseudomonas aeruginosa (PDB ID: 4AZU). | ||
In addition to defining the roles of a conserved amino acid by studying native proteins, biochemists or chemical biologists often provide additional experimental support and deeper insights by perturbation studies; an important test of how much we understand the protein is demonstration of how we can modulate the protein by replacing a certain amino acid with its analogs. Unfortunately, unlike successful replacement of other amino acids in the active site (e.g., Met121), substitution of the Cys with any of the 19 other proteinogenic amino acids by site-directed mutagenesis resulted in complete loss of T1 Cu character,29–32 including the strong blue color and small hyperfine coupling constant. Interestingly, one variant containing a Cys112Asp mutation in azurin displayed the small EPR hyperfine coupling characteristic of type 1 copper proteins.31,33,34 However, the missing sulphur-to-Cu coordination renders the C112D mutant without a LMCT band at 625 nm. Accordingly, this mutant has been designated as a type 0 copper protein.33 Therefore, even though the importance of the Cys has been implicated from previous studies, its role in T1 Cu proteins remains to be clearly defined by mutagenesis studies. Without maintaining the T1 copper character, it is difficult to demonstrate modulation of the Cu–Cys interaction.
One reason for the difficulty in using other amino acids to probe the role of Cys in T1 copper proteins is the restriction to the 20 proteinogenic amino acids. The limited functional group availability often complicates interpretation of the results for many reasons, including simultaneously changing multiple factors such as electronic and steric effects. Recent successes in incorporating nonproteinogenic amino acids into metalloproteins have firmly established that the use of amino acids beyond the 20 canonical amino acids can fine tune function, define metal ligand functionality, and act as an initial step in modulating protein function to engineer proteins with new functions.18,19,21,22,27
Pseudomonas aeruginosa azurin (Az) is an excellent metalloprotein model system for the incorporation of nonproteinogenic amino acids to gain insight into the role of each ligand in its T1 metal binding site.18,19,21,22,27 Previous reports from our laboratories using expressed protein ligation (EPL) to incorporate the nonproteinogenic amino acid selenocysteine (Sec) at position 112 thus far remain the only reported mutation of Cys112 with a nonproteinogenic amino acid. The Cys112Sec mutant was shown to retain both the strong blue color and other type 1 copper site characteristics.18,19,21,22,27 However, such an isostructural replacement does not address the key question of how to modulate the Cu–S interaction, particularly the Cu–S covalency. Likewise, replacing Cys112 with other natural amino acids perturbs multiple factors, such as thiolate ligation and steric interactions. To minimize these issues while still probing the role of Cys112 in a T1 Cu center, we report herein the replacement of Cys112 in Az with a homocysteine (Hcy). The extra methylene group brings the Hcy thiolate closer to the Cu core permitting better investigation of the interaction with the copper ion. Spectroscopic studies by UV-vis, EPR, and XAS methods indicate that azurin in which Cys112 is replaced by Hcy maintains all the type 1 blue Cu characteristics, with a slightly longer SHcy–Cu(II) distance and less covalency. The implication of these results for T1 copper centers in ET function is discussed.
Results and discussion
Computational modeling of the C112Hcy mutation
To evaluate the potential effects of the C112Hcy mutation on the structure of the blue copper center, molecular modeling using the NAMD software39,40 was undertaken. This program, like most other similar programs, has been shown to be effective in evaluating geometric changes relative to each other, rather than providing absolute distances.39,40 An overlay of the blue copper centers in WT Az and the C112Hcy Az modeled using the same procedure and parameters (Fig. 2) shows that the C112HCy mutation resulted in minimal changes of the T1 copper center, with His46 and His117 occupying similar positions as those of WT Az and Cu–His distances within the error of the modeling. The major effect of the extra methylene group in Hcy112 is to shorten the Cu–S distance by ∼0.09 Å in C112Hcy Az. At the same time, the copper ion is pushed away from the trigonal plane defined by His46, His117 and Hcy112, shortening the Cu–Met distance by almost 1 Å. These results suggest that the extra methylene group in Hcy112 could potentially shorten the Cu–S bond and increase its bond covalency.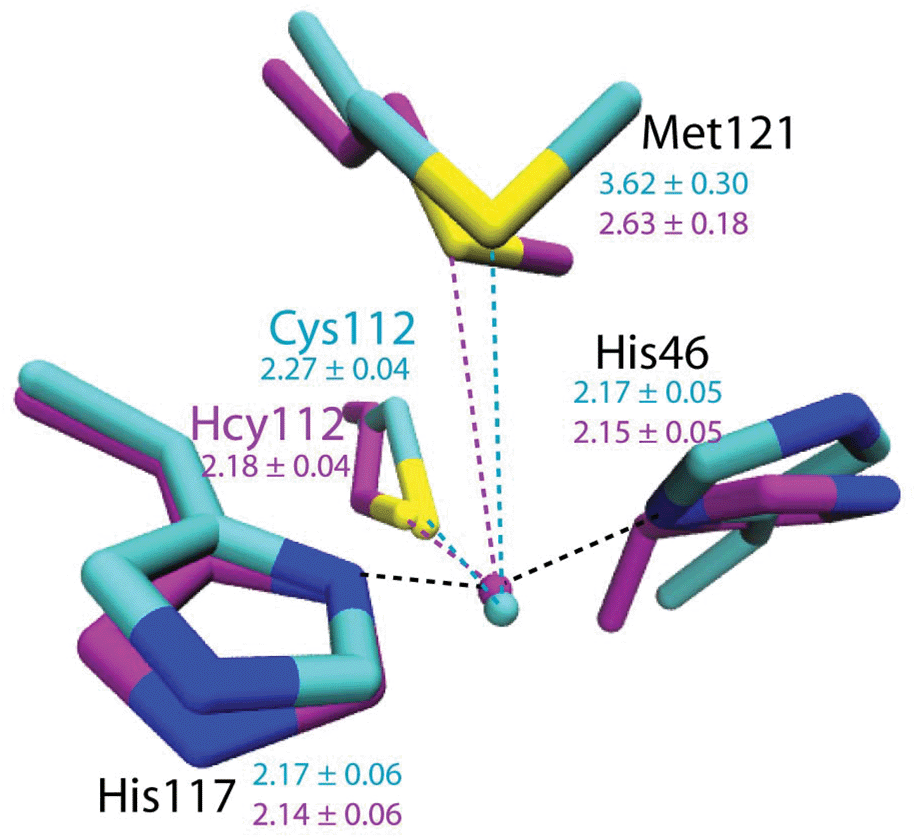 | ||
| Fig. 2 Computational modeling of WT Az (cyan) and C112Hcy Az (magenta) using NAMD.35,36 Average distances between Cu and ligands are shown in Å with standard deviations for WT Az (cyan) and C112Hcy Az (magenta). | ||
Preparation of C112Hcy by EPL
The use of Hcy for EPL has previously been reported both at the ligation point and within a synthetic peptide of interest.37,38 However, the use of Hcy as a means to probe the roles of Cys in metalloproteins is limited. Using a similar methodology to our other reported nonproteinogenic amino acid incorporation studies on the azurin type 1 copper site,18 a peptide consisting of the last 17 amino acids of azurin was prepared using standard Fmoc-based solid phase peptide synthesis. A truncated azurin-fusion protein containing azurinΔ,17 a modified Mxe-intein, and a chitin-binding domain (CBD) (New England Biolabs (NEB) – PtxB1 vector) was expressed and EPL was completed in the presence of mercaptophenyl acetic acid (MPAA) as the transthioesterification mediator.39 The molecular weight (MW) of C112Hcy azurin apo protein agreed within experimental error of the expected value as determined by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (apo-C112Hcy Az: calcd MW, 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 945; obsd MW, 13948: holo-C112Hcy Az: calcd MW, 14023; obsd MW, 14027).
945; obsd MW, 13948: holo-C112Hcy Az: calcd MW, 14023; obsd MW, 14027).
Characterization of Cu(II)-C112Hcy: implications to covalency and other physical properties
Addition of Cu(II) to the colorless apo-C112Hcy Az resulted in the appearance of a strong blue color and absorption at 618 nm (Fig. 3). A similar strong absorption at 628 nm in WT Az has previously been assigned to S(Cys112)-3pπ to Cu(II)-3dx2−y2 charge transfer.1,3,40–43 It is likely that the absorption at 618 nm by the Cys112Hcy Az variant is also a S(Hcy112)→Cu(II) charge transfer band, blue-shifted by 7 nm.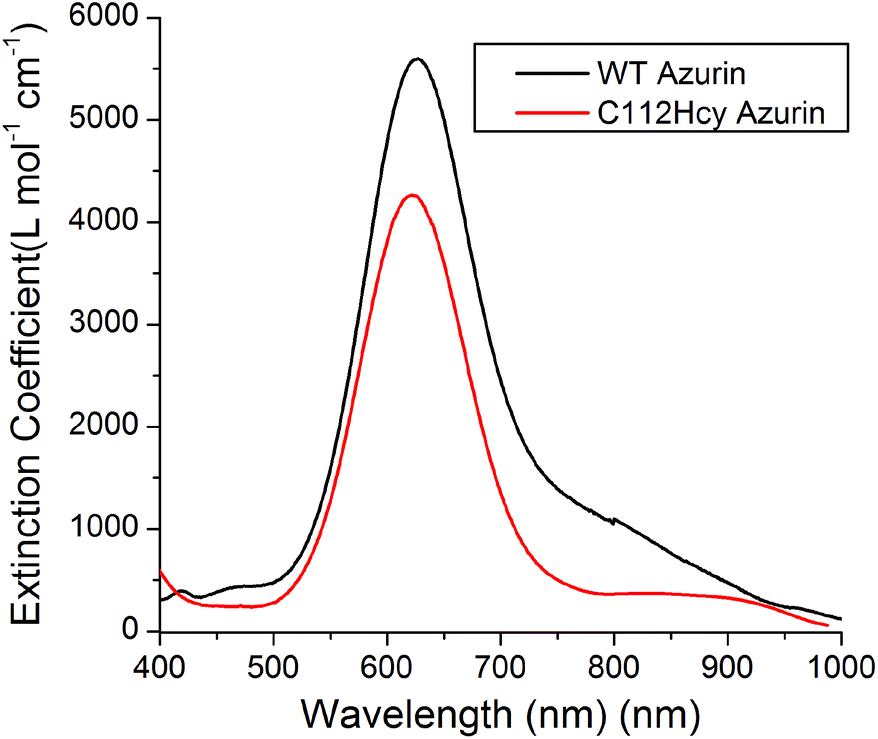 | ||
| Fig. 3 Visible absorption spectroscopy of C112Hcy azurin: The S(Hcy112)–Cu(II) charge transfer band is at 618 nm. The WT azurin charge transfer band is at 628 nm. | ||
The X-band EPR spectrum (Fig. 4) of the C112Hcy Az is also similar to that of WT azurin, with deviation from the axial symmetry observed in the WT Az. Simulation of the spectrum gave g-values of 2.009, 2.051, and 2.253. Compared with g-values from WT Az (2.039, 2.056, and 2.260),44 the large difference between gx and gy for C112Hcy Az suggests a more rhombic geometry for the type 1 Cu center. This result is consistent with the computational modeling shown in Fig. 2, which indicates the Cu(II) moving from the trigonal plane into a more tetrahedral environment, which has less axial geometry.
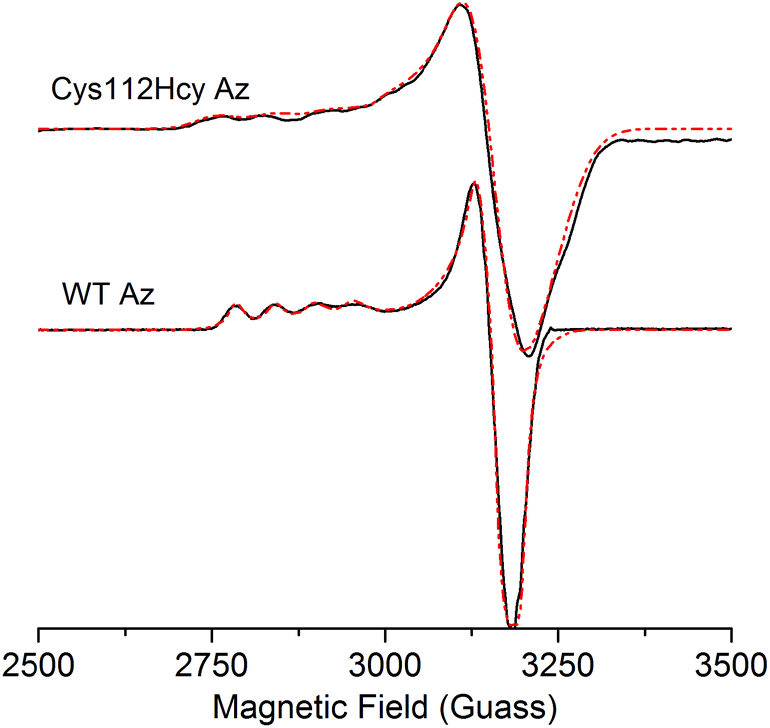 | ||
| Fig. 4 X-band EPR spectra of C112Hcy Az and WT Az (in black) and simulated EPR spectra (in red). EPR conditions: field center = 3000 G, field sweep = 2000 G, modulation amplification = 3 G, frequency = 9.049 GHz, power = 0.2 mW, temperature = 30 K. | ||
In addition, the C112Hcy Az displays a larger parallel hyperfine splitting (A∥= 84 × 10−4 cm−1) than that of WT azurin (63 × 10−4 cm−1).22,44 This value is still within the range observed for other T1 blue Cu proteins (<100 × 10−4 cm−1).45 The small A∥ is a primary characteristic of the T1 blue Cu center and has been attributed to the strong covalency of the Cu–Scys bond, which results in the unpaired electron on the Cu(II) being more delocalized onto the SCys ligand.3 Given the similar g values of the two proteins, especially in the gz (2.253 for C112Hcy Az vs. 2.260 for WT Az), the slightly larger A∥ suggests a reduced covalency between the SHcy ligand and the Cu(II).
The reduction potential of the C112Hcy Az was measured by cyclic voltammetry using a protocol reported previously.46,47 In comparison to that of WT Az (325 mV vs. NHE),45,46 the observed reduction potential for C112Hcy Az (360 mV vs. NHE) is 35 mV higher. This result is consistent with a decreased covalent character between the SHcy and Cu(II) as observed by EPR spectroscopy. The less covalent bond makes the SHcy less efficient in stabilizing the Cu(II) form, resulting in the raised reduction potential. The effect of the attenuated covalency of the SHcy–Cu bond on redox potential has been observed previously in mutants affecting a H-bond to Cys21,48 and has been estimated by Solomon and coworkers to be ∼−12 mV per % Sp.49
XAS data on the C112HCys variant were collected and compared with the WT protein (Fig. 5). Compared to crystallography, EXAFS is more accurate in determining ligand–metal distances and can minimize the effects of photoreduction.3,50,51 The spectra are similar to a typical cupredoxin site with scattering from two histidines and one S from homocysteine. However, notable differences between the WT and variant are observed. First, the variant has a Cu–S bond length of 2.19 Å, which is longer than reported WT azurin Cu–S bond lengths (2.14–2.18 Å) from EXAFS studies. This suggests a less covalent Cu–S interaction in the homocysteine variant, as also indicated by the increased Cu parallel hyperfine splitting in EPR spectra. Second, distinct changes exist on the absorption edge. Thirdly, the best fit (Fig. 5c, see Table 1) includes a contribution, albeit weak, from the methionine ligand, a feature generally not observed in WT Az EXAFS data. Again, this result suggests a decrease in the covalency of the Cu–S(thiolate) and increase in the interaction of the Cu–S(thioether) components of the spectra. Larger Debye–Waller terms for the Cu–N(His) shell are observed in the Hcy variant pointing to a more disordered Cu–N(His) environment. This result is consistent with the molecular dynamics simulation shown in Fig. 2. Collectively, all the data (molecular modeling, UV-vis, EPR and EXAFS) support the presence of a type 1 blue Cu center, in which the strongly covalent Cu–S(thiolate) interaction relaxes, leading to reorientation of the remaining copper ligands.
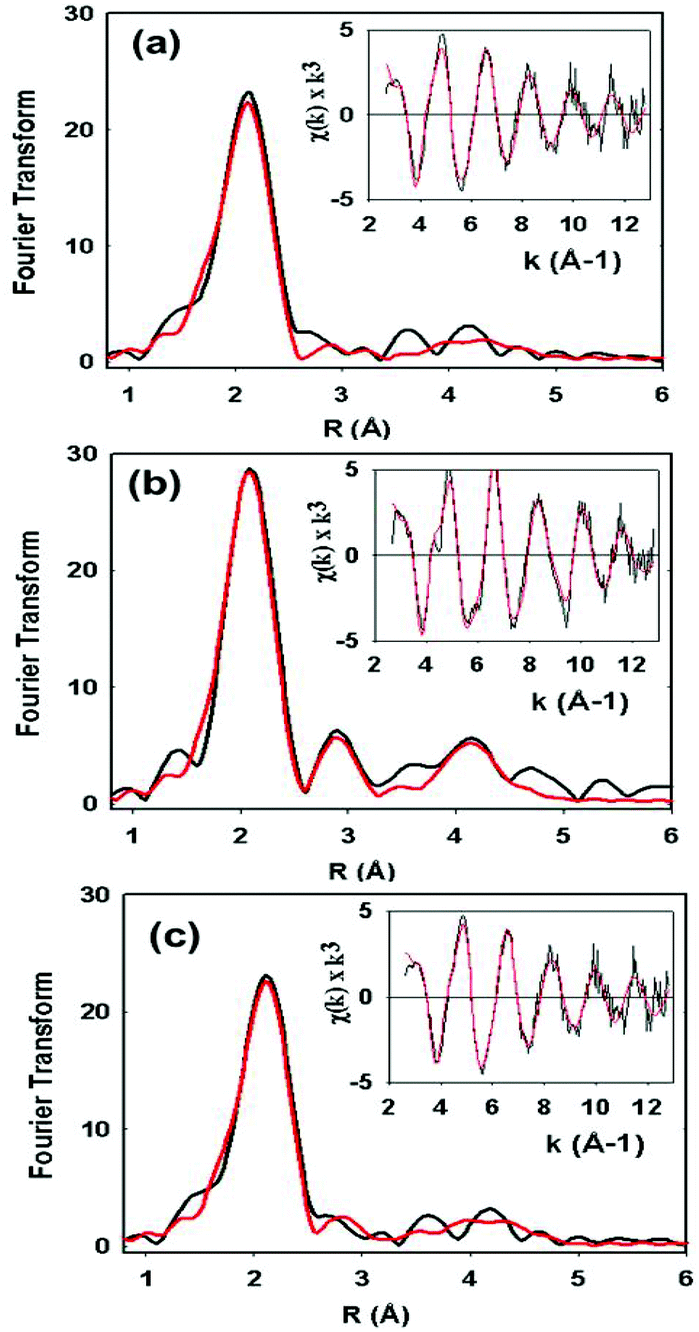 | ||
| Fig. 5 EXAFS data (inset) and their Fourier transformed spectra of C112Hcy (a and c) compared with the WT azurin spectrum (b). The experimental data are in black and the fitted results are in red. The fit in (a) does not include contributions from Met121 while the fit in (c) does. | ||
| Cu–N(His)a | Cu–S(Cys) | Cu–S(Met) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | F | No.c | R (Å)d | 2σe | No.c | R (Å)d | 2σe | No.c | R (Å)d | 2σe | E 0 |
a Fits modeled histidine coordination by an imidazole ring, which included single and multiple scattering contributions from the second shell (C2/C5) and third shell (C3/N4) atoms, respectively. The Cu–N–Cx angles were as follows: Cu–N–C2 126°, Cu–N–C3–126°, Cu–N–N4 163°, Cu–N–C5–163°.
b
F is a least-squares fitting parameter defined as 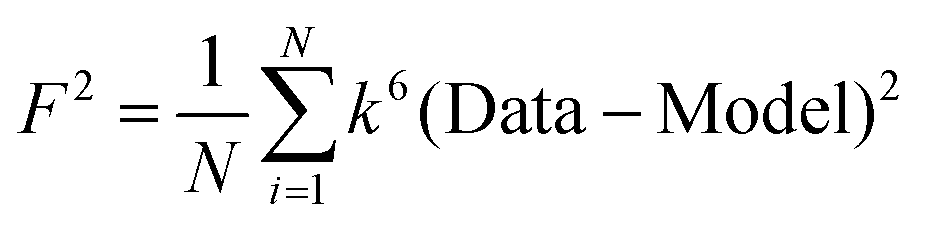 .
c Coordination numbers are generally considered accurate to ±25%.
d In any one fit, the statistical error in bond-lengths is ±0.005 Å. However, when errors due to imperfect background subtraction, phase-shift calculations, and noise in the data are compounded, the actual error is probably closer to ±0.02 Å.
e Debye–Waller factors are given as 2σ2 values.
f Both Fits A and B model histidine coordination using single and multiple scattering contributions. In Fit A the C2 and C5 carbon atoms require splitting by an unreasonably large value (Cu–C2 = 2.95 Å, Cu–C5 = 3.14 Å), whereas in Fit B the splitting is much smaller (Cu–C2 = 2.87 Å, Cu–C5 = 2.98 Å). Fit B, therefore, is more chemically reasonable with respect to the geometry of the histidine side chain. .
c Coordination numbers are generally considered accurate to ±25%.
d In any one fit, the statistical error in bond-lengths is ±0.005 Å. However, when errors due to imperfect background subtraction, phase-shift calculations, and noise in the data are compounded, the actual error is probably closer to ±0.02 Å.
e Debye–Waller factors are given as 2σ2 values.
f Both Fits A and B model histidine coordination using single and multiple scattering contributions. In Fit A the C2 and C5 carbon atoms require splitting by an unreasonably large value (Cu–C2 = 2.95 Å, Cu–C5 = 3.14 Å), whereas in Fit B the splitting is much smaller (Cu–C2 = 2.87 Å, Cu–C5 = 2.98 Å). Fit B, therefore, is more chemically reasonable with respect to the geometry of the histidine side chain.
|
|||||||||||
| WT | 0.38 | 2 | 1.96 | 0.009 | 1 | 2.18 | 0.003 | −1.35 | |||
| C112Hcys Fit Af | 0.59 | 2 | 1.95 | 0.016 | 1 | 2.19 | 0.006 | −0.87 | |||
| C112Hcys Fit Bf | 0.51 | 2 | 1.96 | 0.016 | 1 | 2.19 | 0.006 | 1 | 2.80 | 0.024 | −1.15 |
Conclusions
In summary, an azurin variant containing a Cys112Hcy mutation has been obtained through EPL to probe whether the extra methylene group can influence the Cu–S distance and thus its covalency. The variant displays typical type 1 copper site features, but with a decrease in the covalent interaction between SHcy–Cu(II) when compared with the WT SCys–Cu(II) interaction. This observation is surprising, given the fact that the inserted Hcy has a longer side chain than Cys. In principle, the Hcy thiolate should be able to approach Cu closer than Cys, thereby forming a stronger covalent interaction. Molecular modeling, EPR and EXAFS data suggest that this change is due to rearrangement of the blue copper site to accommodate the extra methylene group, by moving the copper ion out of the trigonal plane defined by the two histidines and one Cys (or Hcy) and closer to Met121. Further detailed studies, such as the use of X-ray crystallography and NMR spectroscopy,52 may shed more light on the structural features responsible for these observations. Nonetheless, this study provides a new insight into the Cu–SCys interaction. This study shows that the Cu–ligand covalency in native blue copper proteins is close to optimal and any further change of such interaction will result in a corresponding geometric adjustment of the T1 site. This work further shows that the incorporation of nonproteinogenic amino acids into metalloprotein scaffolds can provide an alternative approach to studying metal sites in metalloproteins without major perturbations of protein characteristics.Acknowledgements
We thank Dr Saumen Chakraborty and Ms Niloufar Hafezi-Mashhadi for help in editing the manuscript. This report is based on work supported by the National Science Foundation under award no. CHE 1058959 to Y. L. and the National Institute of Health award no. GM05480 to N. B.Notes and references
- H. B. Gray, B. G. Malmström and R. J. P. Williams, J. Biol. Inorg. Chem., 2000, 5, 551 CrossRef CAS.
- L. Banci, R. Pierattelli and A. J. Vila, in Adv. Protein Chem., ed. E. B. G. Joan Selverstone Valentine, Academic Press, 2002, vol. 60 Search PubMed.
- E. I. Solomon, R. K. Szilagyi, S. DeBeer George and L. Basumallick, Chem. Rev., 2004, 104, 419 CrossRef CAS PubMed.
- E. I. Solomon and R. G. Hadt, Coord. Chem. Rev., 2011, 255, 774 CrossRef CAS PubMed.
- O. Farver and I. Pecht, Coord. Chem. Rev., 2011, 255, 757 CrossRef CAS PubMed.
- C. Dennison, Coord. Chem. Rev., 2005, 249, 3025 CrossRef CAS PubMed.
- U. Kolczak, C. Dennison, A. Messerschmidt and G. W. Canters, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd, 2011 Search PubMed.
- A. J. Vila and C. O. Fernandez, in Handbook on Metalloproteins, ed. I. Bertini, A. Sigel and H. Sigel, Marcel Dekker, New York, NY, 2001 Search PubMed.
- J. J. Warren, K. M. Lancaster, J. H. Richards and H. B. Gray, J. Inorg. Biochem., 2012, 115, 119 CrossRef CAS PubMed.
- T. D. Wilson, Y. Yu and Y. Lu, Coord. Chem. Rev., 2013, 257, 260 CrossRef CAS PubMed.
- Y. Lu, in Comprehensive Coordinaion Chemistry II, ed. J. A. McCleverty, T. J. Meyer, Elsevier, Pergamon, Oxford, 2003, vol. 8 Search PubMed.
- B. G. Karlsson, R. Aasa, B. G. Malmström and L. G. Lundberg, FEBS Lett., 1989, 253, 99 CrossRef CAS.
- A. Messerschmidt, L. Prade, S. J. Kroes, J. Sanders-Loehr, R. Huber and G. W. Canters, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3443 CrossRef CAS.
- J. R. Winkler, P. Wittung-Stafshede, J. Leckner, B. G. Malmstrom and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4246 CrossRef CAS.
- B. G. Karlsson, L.-C. Tsai, H. Nar, J. Sanders-Loehr, N. Bonander, V. Langer and L. Sjolin, Biochemistry, 1997, 36, 4089 CrossRef CAS PubMed.
- H. Nar, A. Messerschmidt, R. Huber, M. van de Kamp and G. W. Canters, J. Mol. Biol., 1991, 221, 765 CrossRef CAS.
- A. Romero, C. W. G. Hoitink, H. Nar, R. Huber, A. Messerschmidt and G. W. Canters, J. Mol. Biol., 1993, 229, 1007 CrossRef CAS PubMed.
- S. M. Berry, M. D. Gieselman, M. J. Nilges, W. A. van der Donk and Y. Lu, J. Am. Chem. Soc., 2002, 124, 2084 CrossRef CAS PubMed.
- S. M. Berry, M. Ralle, D. W. Low, N. J. Blackburn and Y. Lu, J. Am. Chem. Soc., 2003, 125, 8760 CrossRef CAS PubMed.
- I. Bertini, C. O. Fernández, B. G. Karlsson, J. Leckner, C. Luchinat, B. G. Malmström, A. M. Nersissian, R. Pierattelli, E. Shipp, J. S. Valentine and A. J. Vila, J. Am. Chem. Soc., 2000, 122, 3701 CrossRef CAS.
- K. M. Clark, Y. Yu, N. M. Marshall, N. A. Sieracki, M. J. Nilges, N. J. Blackburn, W. A. van der Donk and Y. Lu, J. Am. Chem. Soc., 2010, 132, 10093 CrossRef CAS PubMed.
- D. K. Garner, M. D. Vaughan, H. J. Hwang, M. G. Savelieff, S. M. Berry, J. F. Honek and Y. Lu, J. Am. Chem. Soc., 2006, 128, 15608 CrossRef CAS PubMed.
- J. A. Guckert, M. D. Lowery and E. I. Solomon, J. Am. Chem. Soc., 1995, 117, 2817 CrossRef CAS.
- C. Immoos, M. G. Hill, D. Sanders, J. A. Fee, C. E. Slutter, J. H. Richards and H. B. Gray, J. Biol. Inorg. Chem., 1996, 1, 529 CrossRef CAS.
- V. Kofman, O. Farver, I. Pecht and D. Goldfarb, J. Am. Chem. Soc., 1996, 118, 1201 CrossRef CAS.
- M. H. Olsson and U. Ryde, J. Biol. Inorg. Chem., 1999, 4, 654 CrossRef CAS.
- M. Ralle, S. M. Berry, M. J. Nilges, M. D. Gieselman, W. A. van der Donk, Y. Lu and N. J. Blackburn, J. Am. Chem. Soc., 2004, 126, 7244 CrossRef CAS PubMed.
- S. Yanagisawa, K. Sato, M. Kikuchi, T. Kohzuma and C. Dennison, Biochemistry, 2003, 42, 6853 CrossRef CAS PubMed.
- S. DeBeer, C. N. Kiser, G. A. Mines, J. H. Richards, H. B. Gray, E. I. Solomon, B. Hedman and K. O. Hodgson, Inorg. Chem., 1999, 38, 433 CrossRef CAS PubMed.
- S. Faham, T. J. Mizoguchi, E. T. Adman, H. B. Gray, J. H. Richards and D. C. Rees, J. Biol. Inorg. Chem., 1997, 2, 464 CrossRef CAS.
- T. J. Mizoguchi, A. J. Di Bilio, H. B. Gray and J. H. Richards, J. Am. Chem. Soc., 1992, 114, 10076 CrossRef CAS.
- M. Piccioli, C. Luchinat, T. J. Mizoguchi, B. E. Ramirez, H. B. Gray and J. H. Richards, Inorg. Chem., 1995, 34, 737 CrossRef CAS.
- K. M. Lancaster, S. D. George, K. Yokoyama, J. H. Richards and H. B. Gray, Nat. Chem., 2009, 1, 711 CrossRef CAS PubMed.
- K. M. Lancaster, K. Yokoyama, J. H. Richards, J. R. Winkler and H. B. Gray, Inorg. Chem., 2009, 48, 1278 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781 CrossRef CAS PubMed.
- H. Yu and K. Schulten, PLoS Comput. Biol., 2013, 9, e1002892 CAS.
- K. Pachamuthu and R. R. Schmidt, Synlett, 2003, 0659 CAS.
- R. S. Roy, O. Allen and C. T. Walsh, Chem. Biol., 1999, 6, 789 CrossRef CAS.
- E. C. B. Johnson and S. B. H. Kent, J. Am. Chem. Soc., 2006, 128, 6640 CrossRef CAS PubMed.
- K. Pierloot, J. O. A. De Kerpel, U. Ryde and B. O. Roos, J. Am. Chem. Soc., 1997, 119, 218 CrossRef CAS.
- E. I. Solomon, M. J. Baldwin and M. D. Lowery, Chem. Rev., 1992, 92, 521 CrossRef CAS.
- B. L. Vallee and R. J. Williams, Proc. Natl. Acad. Sci. U. S. A., 1968, 59, 498 CrossRef CAS.
- R. J. Williams, Eur. J. Biochem., 1995, 234, 363 CAS.
- W. E. Antholine, P. M. Hanna and D. R. McMillin, Biophys. J., 1993, 64, 267 CrossRef CAS.
- P. J. Hart, A. M. Nersissian and S. D. George, in Encyclopedia of Inorganic Chemistry, John Wiley & Sons, Ltd, 2006 Search PubMed.
- J. Hirst and F. A. Armstrong, Anal. Chem., 1998, 70, 5062 CrossRef CAS.
- N. M. Marshall, D. K. Garner, T. D. Wilson, Y.-G. Gao, H. Robinson, M. J. Nilges and Y. Lu, Nature, 2009, 462, 113 CrossRef CAS PubMed.
- S. Yanagisawa, M. J. Banfield and C. Dennison, Biochemistry, 2006, 45, 8812 CrossRef CAS PubMed.
- R. G. Hadt, N. Sun, N. M. Marshall, K. O. Hodgson, B. Hedman, Y. Lu and E. I. Solomon, J. Am. Chem. Soc., 2012, 134, 16701 CrossRef CAS PubMed.
- J. E. Penner-Hahn, M. Murata, K. O. Hodgson and H. C. Freeman, Inorg. Chem., 1989, 28, 1826 CrossRef CAS.
- M. L. Barrett, I. Harvey, M. Sundararajan, R. Surendran, J. F. Hall, M. J. Ellis, M. A. Hough, R. W. Strange, I. H. Hillier and S. S. Hasnain, Biochemistry, 2006, 45, 2927 CrossRef CAS PubMed.
- M.-E. Zaballa, L. A. Abriata, A. Donaire and A. J. Vila, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9254 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3qi00096f |
| This journal is © the Partner Organisations 2014 |