Sm and Eu(III) lanthanide triple helicate cages based on N,N′-methylene-bis(pyridin-4-one)†
Yu-Bo
Shu
,
Xiao-Liang
Tang
and
Wei-Sheng
Liu
*
Key Laboratory of Nonferrous Metals Chemistry and Resources Utilization of Gansu Province and State Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, P. R. China. E-mail: liuws@lzu.edu.cn
First published on 4th February 2014
Abstract
Presented here are Sm and Eu(III) lanthanide triple helicate cages with two distinct metal environments based on the N,N′-methylene-bis(pyridin-4-one) (MBP) ligand. Spontaneous resolution occurs in the crystallization of the Sm(III) complex. Luminescence is investigated by excitation of the metal ions with near-UV light. For the Sm(III) complex, close π–π stacking experienced by the pyridinone ring increases the sensitization of the Sm(III) ion, revealed by the appearance of 4G5/2→6H11/2 emission, the long luminescence lifetimes (35 and 18 μs) and the relatively high quantum yield (1.7%).
Lanthanide complexes with discrete di- and polynuclear structures have received considerable interest amongst synthetic inorganic chemists because of their diverse architectures and fascinating optical and magnetic properties.1 Many efforts have succeeded in obtaining a variety of geometrically intriguing lanthanide supramolecules containing molecular helicates,2 polygons,3 wheels,4 polyhedrons5 and cages.6 Although the dinuclear complex is the simplest amongst lanthanide structures and is not so rare, we still thought it an important model system in order to understand the hard-to-predict self-organization behaviour and answer some fundamental questions regarding photo- and magnetophysical properties.
The most notable type of dinuclear lanthanide complex is the triple-stranded helicate based on a methylene-bridged bis(terdentate) ligand,2 because the methylene spacer allows for efficient binding of the tridentate units to the metal ions while limiting the number of possible conformations. However, spontaneous resolution,7 usually resulting in optically inactive conglomerates, has never occurred in the crystallization process—not to mention chiral symmetry breaking,8 giving rise to a non-zero enantiomeric excess. Except for chiral d–f heterodimetallic helicates,9 to the best of our knowledge, there has been no chiral lanthanide homometal di- and polynuclear complex synthesized from an achiral ligand without adding any chiral reagent. Moreover, it is of significance to prepare chiral solids due to their potential use in nonlinear or circularly polarized luminescent materials.10
The strategy for the synthesis of most coordination cages usually involves the use of ancillary terminal ligands to cover partial coordination sites of the metal ion. Two elegant examples are the use of a cis-chelating di-imine/amine as a capping group for square-planar coordinated Pd and Pt ions11 and an organo-phosphine group for the Pt ion.12 It is harder to choose capping ligands for lanthanide ions because of their well-known high coordination numbers and weak stereochemical preferences. Hence, lanthanide coordination cages are extremely rare. The only successful example of the construction of cage-shaped lanthanide complexes by the end-capping strategy is the assembly of Pr, Nd, Sm and Er(III) lanthanide triple helicate cages from two methylene-bridged N,N′-xylene-1,4-diylbis(pyridin-2-one) (p-XBP) ligands.13 In this work, we report Sm and Eu(III) lanthanide triple helicate cages based on a new, single methylene-bridged pyridinone-type ligand, N,N′-methylene-bis(pyridin-4-one) (MBP, Scheme 1).
 | ||
| Scheme 1 The ligand MBP. | ||
The MBP ligand can be generated by the hydroxy–ketone isomerization of N,N′-methylene-bis(4,4′-dihydroxy-bispyridinium)bromide [MBP(HBr)2]. Solvothermal reaction of MBP(HBr)2 with an excess of samarium(III) nitrate hexahydrate in MeCN and EtOH solution at 75 °C results in the formation of polyhedral crystals of [Sm2(MBP)3(NO3)6] (1). The europium complex [Eu2(MBP)3(NO3)6] (2) can be obtained by reaction of MBP(HBr)2 with europium(III) nitrate hexahydrate in ethanol solution. The single crystal X-ray analyses reveal that complexes 1 and 2 are isostructural. They both crystallize in the trigonal R3 space group. Their asymmetric units contain two crystallographically independent lanthanide ions (Ln1 and Ln2) (Fig. 1a). For complex 1, the environment of Sm1 is nine-coordinate while that of Sm2 is ten-coordinate. The Sm1 atom is bound to three bidentate nitrates [Sm–O in the range of 2.542(1)–2.552(1) Å] and three oxygens from three different MBP ligands [Sm–O 2.286(1) Å] with a distorted 4,4,4-tricapped trigonal prism14 coordination geometry (Fig. 1c). Sm2 is also bound to three bidentate nitrates [Sm–O in the range 2.561(1)–2.582(1) Å] and three other oxygens of three MBP ligands [Sm–O 2.376(1) Å], and lastly the irregular 3,4,4,4-tetracapped trigonal prism15 coordination geometry is completed by one water molecule [Sm–O, 2.512(1) Å] (Fig. 1b).
 | ||
| Fig. 1 (a) The triple helicate cage of complex 1. (b) The coordination geometry of the Sm2 atom. (c) The coordination geometry of the Sm1 atom. | ||
The MBP ligand serves as a ‘V’ shaped linker with a curved angle of about 114°. Three MBP ligands, in a P fashion, in turn bridge the two Ln centers forming a triple helicate cage structure with a small (123.2 Å3) cavity volume. The Ln⋯Ln separation is ca. 12.3 Å, and the distance between adjacent methylene groups is ca. 7.9 Å. The nitrate acts as a capping group in a cis-chelating mode. Three nitrates cover the rest of the Ln1 sites in a ligated fashion, while the rest of the Ln2 sites are occupied by three other nitrates and one water in a capped fashion, putting an end to the connectivity of the cage-like molecule. Both of the two complexes have a strictly molecular C3 axis passing through the two Ln centers along the crystallographic c axis (Fig. 2).
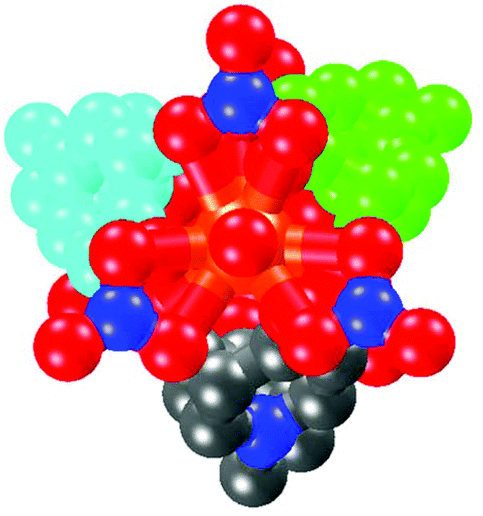 | ||
| Fig. 2 View down the molecular C3 axis (Sm2—Sm1) of complex 1. | ||
Complexes 1 and 2 exhibit the same supramolecular framework. For example, for 1, the three-dimensional framework was formed and stabilized by a richness of supramolecular interactions, classified as π–π stacking, nitrate–nitrate electrostatic repulsion and hydrogen bonding. The dominant intermolecular interaction gives rise to π–π stacking of paired pyridinone rings between one cage and the neighboring six units (the centroid–centroid separations being 3.393 Å, Fig. S6†), serving as both a supramolecular synthon and a supramolecular mediator of chirality.7 The Flack parameter of complex 1 is 0.014 (10), which demonstrates the homo-chirality of the single crystal. The result indicates that the crystallization undergoes spontaneous resolution, which was further evaluated by the determination of the solid-state circular dichroism (CD) spectrum of bulk crystals in a KCl matrix. As seen in Fig. 3, the bulk sample exhibits strong CD signals with a negative Cotton effect at 292 nm and a positive Cotton effect at 275 nm. The source of chirality is assigned to the inherent helicity of this complex.16 These two opposite Cotton effects are the result of “intranuclear” exciton coupling between long-axis π–π* transition dipole moments of pyridinone units located at the same metal center.9a,17 As a result of the “internuclear” exciton coupling between pyridinone units located on different metal centers, the absolute configurations of the metal centers are inversely correlated to the observed phases of Cotton effects.9b,18 Therefore, we suggest that a P-helix is favored for complex 1, with the local configurations at both the two metal centers being Λ.16
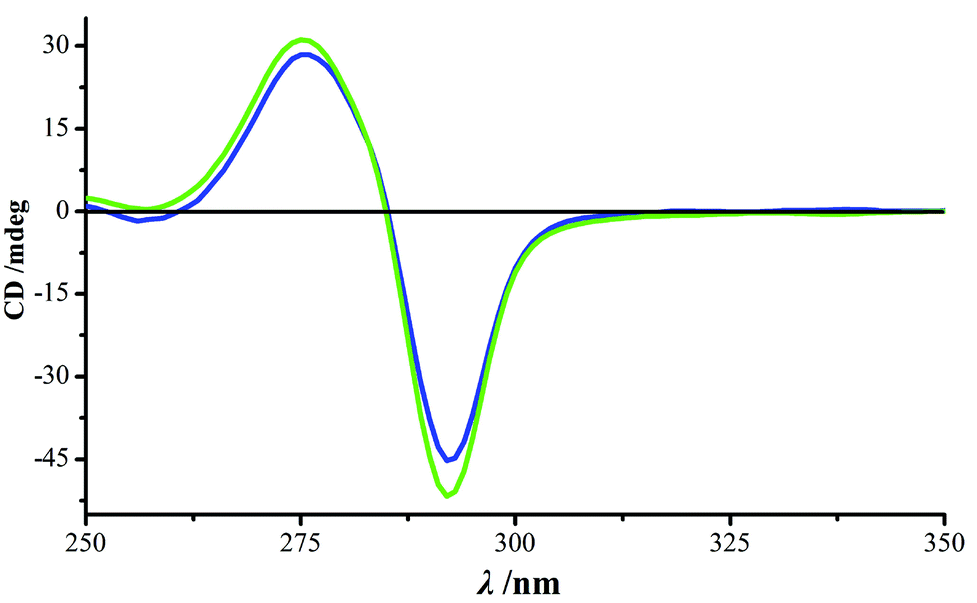 | ||
| Fig. 3 CD spectra of complex 1 determined from two batches of samples. | ||
Unfortunately, this kind of complex can easily dissociate in water and is poorly soluble in ordinary organic solvents, which limits its application in analytical sensors. However, we much anticipate its use in lighting and display devices,19 because the MBP ligand is easily accessible and, more importantly, there is continuing importance in the development of phosphors emitting desired colors of light upon near-UV (e.g., 380–420 nm) excitation.20 Thus, the solid-state luminescence was examined at room temperature. Although the ligand sensitization is not very effective, the maximum excitation wavelengths of complexes 2 and 1 are, respectively, located at 395 nm and 406 nm, which fall in the near-UV region (Fig. S5, ESI†). The wavelength λmax = 395 is from the 7F0→5L6 transition of the Eu(III) ion, and λmax = 406 corresponds to the 6H5/2→4F7/2 transition of the Sm(III) ion. Upon irradiation, they showed red and pink luminescence respectively (Fig. 4).
 | ||
| Fig. 4 Emission spectra of (a) complex 1 and (b) complex 2. | ||
The sharp lines in the emission spectrum of complex 2 are assigned to transitions between the first excited state, 5D0, and the ground septet, 7FJ (J = 0–4) of Eu(III). The symmetry-forbidden emission 5D0→7F0 is very weak and is situated at 580 nm; its degenerate single line indicates that both Eu1 and Eu2 are located at a C3 symmetrical site. The intensity of the 5D0→7F2 transition (electric dipole), which is hypersensitive to the site symmetry of the Eu(III) ion, is far greater than that of the 5D0→7F1 line (magnetic dipole), indicating the absence of inversion centers in the coordination environments of Eu1 and Eu2. The 5D0 emissive state exhibits a double-exponential decay, with lifetimes of 589 ± 23 and 384 ± 4 μs upon monitoring the most intense lines of the 5D0→7F2 emission, after excitation at 395 nm. The double exponential can be attributed to the two distinct environments experienced by Eu1 and Eu2. As shown in other luminescent Ln(III) complexes,21 in Eu2, the f–f luminescence can be vibrationally quenched to some extent by the O–H oscillators of coordinated water. These two metal environments may result from the strong nitrate–nitrate electrostatic repulsion (Fig. S7, ESI†). Upon excitation at 395 nm, the emission quantum yield was estimated to be 18.2% by the integrating sphere method.
The emission spectrum of complex 1 exhibits the typical emission lines of the Sm(III) ion, situated at 564, 597, 644, and 706 nm, which are assigned to the transitions of 4G5/2→6HJ (J = 5/2, 7/2, 9/2, 11/2); the most intense line is the hypersensitive transition 4G5/2→6H9/2 at 647 nm. After excitation at 406 nm, the 4G5/2 emissive state also exhibits a double-exponential decay with lifetimes of 35.4 ± 0.3 and 17.6 ± 0.5 μs upon monitoring the 4G5/2→6H9/2 emission. The emission quantum yield upon excitation at 406 nm was measured to be 1.74% using an integrating sphere. Comparing with the reported Sm(III) triple helicate cage based on the p-XBP ligand, complex 1 is found to be an example of the activation of the 4G5/2→6H11/2 transition. This fact, along with the long luminescence lifetimes and relatively high quantum yield,22 demonstrate that a pyridinone ring experiencing closer π–π stacking is more effective for increasing the sensitization of the Sm(III) ion.23 This provides a clear direction for the design of highly luminescent Sm(III) complexes.
Conclusions
In conclusion, the assembly of Sm and Eu(III) lanthanide triple helicate cages by using a single methylene-bridged pyridinone-type ligand is achieved. The single crystals of the Sm(III) complex are homochiral, and the bulk sample exhibits enantiomeric excess. The luminescence of Sm and Eu(III) complexes were investigated upon near-UV excitation. The increase in sensitization of the Sm(III) ion through reinforcing the π–π stacking of the pyridinone ring is confirmed by analysis of the emission spectrum and in combination with lifetime and quantum yield measurements.Experimental section
Synthesis of 1: Sm(NO3)3·6H2O (0.055 g, 0.120 mmol) and MBP(HBr)2 (0.020 g, 0.054 mmol) in mixed MeCN–EtOH solvent (2/2 mL) was placed in a 20 mL vial. After stirring for 5 min, the sample was heated at 75 °C for 6 days, and then cooled to room temperature. The resulting colorless crystals were filtered off, washed with ethanol and dried in air [92% yield, based on MBP(HBr)2]. A larger single crystal usable for single-crystal X-ray diffraction was obtained by reaction of 0.023 g Sm(NO3)3·6H2O with 0.010 g MBP(HBr)2 in 1ml MeCN and 1 ml EtOH for 3 days. IR (KBr, cm−1): 3442 (w), 3028 (w), 3056 (w), 1643 (vs), 1559 (vs), 1501 (vs), 1386 (s), 1354 (m), 1312 (vs), 1279 (s), 1197 (m), 1173(s), 1024(m), 863(m), 814 (w), 737 (w), 593 (m), 519 (w), 470 (w). Elemental analysis calcd (%) for C33H32N12O25Sm2 (1297.41): C 30.55, H 2.49, N 12.96; found: C 30.31, H 2.36, N 12.66.Synthesis of 2: Eu(NO3)3·6H2O (0.055 g, 0.0120 mmol) and MBP(HBr)2 (0.020 g, 0.054 mmol) in EtOH solvent (4 mL) was placed in a 20 mL vial. After stirring for 5 min, the sample was heated at 75 °C for 6 days, and then cooled to room temperature. Colorless crystals were filtered off, washed with ethanol and dried in air [87% yield, based on MBP(HBr)2]. A larger single crystal usable for single-crystal X-ray diffraction was obtained by reaction of 0.023 g Eu(NO3)3·6H2O with 0.010 g MBP(HBr)2 in 2 ml EtOH for 3 days. IR (KBr, cm−1): 3436 (w), 3028 (w), 3056 (w), 1644 (vs), 1560 (vs), 1502 (vs), 1385 (s), 1354 (m), 1314 (vs), 1281 (s), 1197 (m), 1174 (vs), 1024 (m), 863 (m), 814 (w), 739 (w), 593 (m), 520 (m), 471 (w). Elemental analysis calcd (%) for C33H32N12O25Eu2 (1300.63): C 30.47, H 2.48, N 12.93; found: C 30.54, H 2.36, N 12.73.
The single-crystal X-ray diffraction data were collected on a Bruker APEX-II CCD diffractometer operating at 50 kV and 30 mA using a graphite-monochromated Mo-Kα radiation source (λ = 0.71073 Å). An empirical absorption correction based on a comparison of redundant and equivalent reflections was applied using SADABS. Both structures were solved by the Patterson method and refined by full-matrix least-squares cycles on F2. Crystal data for 1: C33H32N12O25Sm2, M = 1297.41, trigonal, space group R3, a = 14.3524(7), b = 14.3524(7), c = 19.1378(8) Å, α = 90.00°, β = 90.00°, γ = 120.00°, V = 3414.1(4) Å3, Z = 3, Dc = 1.893 g cm−3. The final refinement converges to final R = 0.0209 and wR = 0.0389 with 218 parameters from 2668 independent reflections (I > 2σ(I)). The goodness of fit on F2 was 0.983. Flack parameter = 0.014(10). Crystal data for 2: C33H32N12O25Eu2, M = 1300.63, trigonal, space group R3, a = 14.359(2), b = 14.359(2), c = 19.115(4) Å, α = 90.00°, β = 90.00°, γ = 120.00°, V = 3413.1(14) Å3, Z = 3, Dc = 1.898 g cm−3. The final refinement converges to final R = 0.0661 and wR = 0.1430 with 218 parameters from 2690 independent reflections (I > 2σ(I)). The goodness of fit on F2 was 1.096. Flack parameter = 0.10(5). CCDC reference numbers 942075 and 965141.
Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (NSFC) (grant numbers 20931003, and 91122007) and the Specialized Research Fund for the Doctoral Program of Higher Education (grant number 20110211130002).Notes and references
- (a) J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 RSC; (b) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110 CrossRef CAS PubMed; (c) F. Habib and F. Murugesu, Chem. Soc. Rev., 2013, 42, 3278 RSC.
- (a) G. Bernardinelli, C. Piguet and A. F. Williams, Angew. Chem., Int. Ed., 1992, 31, 12 Search PubMed; (b) C. Piguet, J.-C. G. Bünzli, G. Bernardinelli, G. Hopfgartner and A. F. Williams, J. Am. Chem. Soc., 1993, 115, 8197 CrossRef CAS; (c) M. Elhabiri, R. Scopelliti, J.-C. G. Bünzli and C. Piguet, J. Am. Chem. Soc., 1999, 121, 10747 CrossRef CAS.
- (a) S. Zebret, N. Dupont, G. Bernardinelli and J. Hamacek, Chem.–Eur. J., 2009, 15, 3355 CrossRef PubMed; (b) G. Bozoklu, C. Marchal, C. Gateau, J. Pecaut, D. Imbert and M. Mazzanti, Chem.–Eur. J., 2010, 16, 6159 CrossRef PubMed; (c) M. U. Anwar, L. K. Thompson, L. N. Dawe, F. Habib and M. Murugesu, Chem. Commun., 2012, 48, 4576 RSC; (d) L. Ungur, S. K. Langley, T. N. Hooper, B. Moubaraki, E. K. Brechin, K. S. Murray and L. F. Chibotaru, J. Am. Chem. Soc., 2012, 134, 18554 CrossRef PubMed.
- (a) Y. Bretonnière, M. Mazzanti, J. Pécaut and M. M. Olmstead, J. Am. Chem. Soc., 2012, 134, 8372 CrossRef PubMed; (b) G. Bozoklu, C. Gateau, D. Imbert, J. Pécaut, K. Robeyns, Y. Filinchuk, F. Memon, G. Muller and M. Mazzanti, J. Am. Chem. Soc., 2002, 124, 9012 CrossRef PubMed.
- (a) R. J. Blagg, C. A. Muryn, E. J. L. McInnes, F. Tuna and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2011, 50, 6530 CrossRef CAS PubMed; (b) J. B. Peng, X. J. Kong, Y. P. Ren, L. S. Long, R. B. Huang and L. S. Zheng, Inorg. Chem., 2012, 51, 2186 CrossRef CAS PubMed; (c) H. Tian, M. Wang, L. Zhao, Y. N. Guo, Y. Guo, J. Tang and Z. Liu, Chem.–Eur. J., 2012, 18, 442 CrossRef CAS PubMed.
- (a) Y.-B. Dong, P. Wang, J.-P. Ma, X.-X. Zhao, H.-Y. Wang, B. Tang and R.-Q. Huang, J. Am. Chem. Soc., 2007, 129, 4872 CrossRef CAS PubMed; (b) B. El Aroussi, L. Guénée, P. Pal and J. Hamacek, Inorg. Chem., 2011, 50, 8588 CrossRef CAS PubMed.
- L. Pérez-García and D. B. Amabilino, Chem. Soc. Rev., 2002, 31, 342 RSC.
- (a) S.-Z. Hou, D.-K. Cao, Y.-Z. Li and L.-M. Zheng, Inorg. Chem., 2008, 47, 10211 CrossRef CAS PubMed; (b) D. K. Kondepudi, Acc. Chem. Res., 2001, 34, 946 CrossRef CAS PubMed.
- (a) M. Cantuel, G. Bernardinelli, G. Muller, J. P. Riehl and C. Piguet, Inorg. Chem., 2004, 43, 1840 CrossRef CAS PubMed; (b) S. G. Telfer, N. Tajima, R. Kuroda, M. Cantuel and C. Piguet, Inorg. Chem., 2004, 43, 5302 CrossRef CAS PubMed.
- (a) S. Dang, J.-H. Zhang, Z.-M. Sun and H.-J. Zhang, Chem. Commun., 2012, 48, 11139 RSC; (b) F. Stomeo, C. Lincheneau, J. P. Leonard, J. E. O'Brien, R. D. Peacock, C. P. McCoy and T. Gunnlaugsson, J. Am. Chem. Soc., 2009, 131, 9636 CrossRef CAS PubMed; (c) E. Peeters, M. P. T. Christiaans, R. A. J. Janssen, H. F. M. Schoo, H. P. J. M. Dekkers and E. W. Meijer, J. Am. Chem. Soc., 1997, 119, 9909 CrossRef CAS.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS PubMed.
- M. Wang, Y.-R. Zheng, K. Ghosh and P. J. Stang, J. Am. Chem. Soc., 2010, 132, 6282 CrossRef CAS PubMed.
- D. M. L. Goodgame, S. P. W. Hill and D. J. Williams, J. Chem. Soc., Chem. Commun., 1993, 1019 RSC.
- (a) R. B. King, J. Am. Chem. Soc., 1969, 91, 7211 CrossRef CAS; (b) R. B. King, J. Am. Chem. Soc., 1970, 92, 6455 CrossRef CAS.
- R. B. King, J. Am. Chem. Soc., 1970, 92, 6460 CrossRef CAS.
- M. Ziegler and A. von Zelewsky, Coord. Chem. Rev., 1998, 177, 257 CrossRef CAS.
- D. Shirotani, T. Suzuki and S. Kaizaki, Inorg. Chem., 2006, 45, 6111 CrossRef CAS PubMed.
- (a) M. Albrecht, S. Schmid, S. Dehn, C. Wickleder, S. Zhang, A. P. Bassett, Z. Pikramenou and R. Fröhlich, New J. Chem., 2007, 31, 1755 RSC; (b) S. G. Telfer, R. Kuroda and T. Sato, Chem. Commun., 2003, 1064 RSC; (c) S. G. Telfer, N. Tajima and R. Kuroda, J. Am. Chem. Soc., 2004, 126, 1408 CrossRef CAS PubMed.
- (a) L. D. Carlos, R. A. S. Ferreira, V. de Zea Bermudez, B. Julián-López and P. Escribano, Chem. Soc. Rev., 2011, 40, 536 RSC; (b) Y. Kuramochi, T. Nakagawa, T. Yokoo, J. Yuasa, T. Kawai and Y. Hasegawa, Dalton Trans., 2012, 41, 6634 RSC.
- C. C. Lin and R.-S. Liu, J. Phys. Chem. Lett., 2011, 2, 1268 CrossRef CAS.
- (a) K. L. Wong, G.-L. Law, Y.-Y. Yang and W.-T. Wong, Adv. Mater., 2006, 18, 1051 CrossRef CAS; (b) B.-L. Chen, L.-B. Wang, F. Zapata, G.-D. Qian and E. B. Lobkovsky, J. Am. Chem. Soc., 2008, 130, 6718 CrossRef CAS PubMed.
- (a) A. P. Bassett, S. W. Magennis, P. B. Glover, D. J. Lewis, N. Spencer, S. Parsons, R. M. Williams, L. De Cola and Z. Pikramenou, J. Am. Chem. Soc., 2004, 126, 9413 CrossRef CAS PubMed; (b) K. Miyata, T. Nakagawa, R. Kawakami, Y. Kita, K. Sugimoto, T. Nakashima, T. Harada, T. Kawai and Y. Hasegawa, Chem.–Eur. J., 2011, 17, 521 CrossRef CAS PubMed.
- J. P. Leonard, P. Jensen, T. McCabe, J. E. O'Brien, R. D. Peacock, P. E. Kruger and T. Gunnlaugsson, J. Am. Chem. Soc., 2007, 129, 10986 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectra, powder XRD patterns, excitation spectra and packing diagrams. CCDC reference numbers 942075 and 965141. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3qi00089c |
| This journal is © the Partner Organisations 2014 |