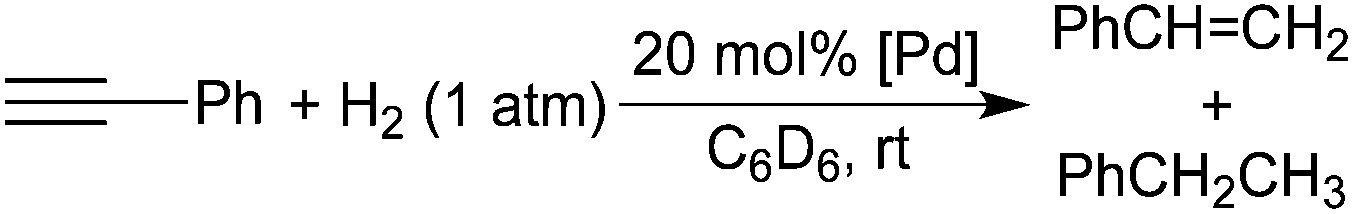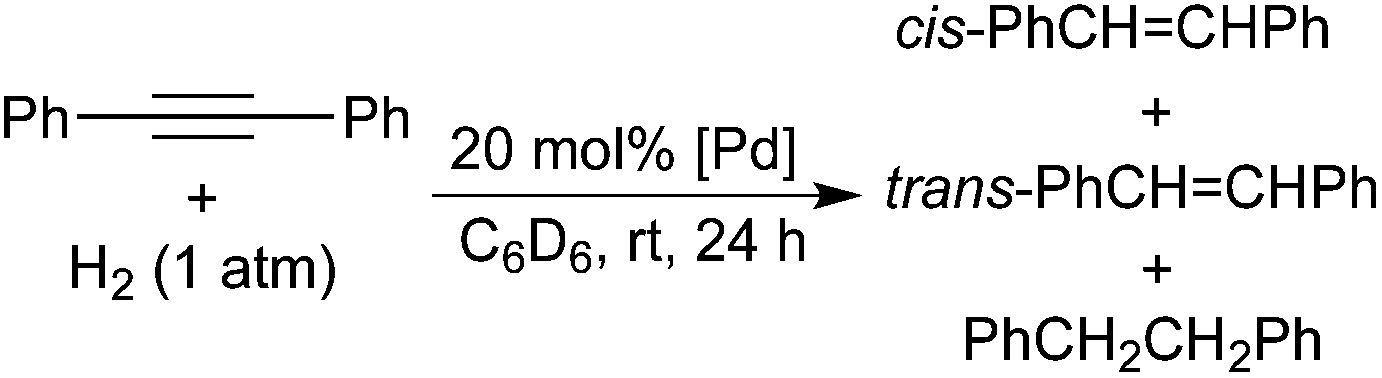Interaction of alkynes with palladium POCOP-pincer hydride complexes and its unexpected relation to palladium-catalyzed hydrogenation of alkynes†
Received
16th October 2013
, Accepted 27th November 2013
First published on 16th December 2013
Abstract
Palladium POCOP-pincer hydride complexes [2,6-(R2PO)2C6H3]PdH (R = tBu, 2a; R = iPr, 2b; R = cPe, 2c, cPe = cyclopentyl) have been synthesized from [2,6-(R2PO)2C6H3]PdCl (1a–c) and LiAlH4 or LiBEt3H. These hydride complexes react with phenylacetylene to afford H2, [2,6-(R2PO)2C6H3]PdC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh (3a–c) and a small amount of styrene. When the R groups are isopropyl groups, a second palladium species is generated, and it has been identified as an alkenyl complex (E)-[2,6-(iPr2PO)2C6H3]PdCH
CPh (3a–c) and a small amount of styrene. When the R groups are isopropyl groups, a second palladium species is generated, and it has been identified as an alkenyl complex (E)-[2,6-(iPr2PO)2C6H3]PdCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh (4b). Mechanistic studies have shown that decomposition of these palladium pincer complexes and related palladium methyl complexes [2,6-(R2PO)2C6H3]PdCH3 (5a–c) occurs at room temperature in the presence of H2 (1 atm or lower), resulting in the leaching of palladium particles. These particles have been shown to catalyze the hydrogenation of phenylacetylene and diphenylacetylene to their alkene and alkane products. A mechanism for the formation of palladium particles has been proposed. The structures of 1a, 1c, 2a, 2c, 3a, 4b and 5b have been studied by X-ray crystallography.
CHPh (4b). Mechanistic studies have shown that decomposition of these palladium pincer complexes and related palladium methyl complexes [2,6-(R2PO)2C6H3]PdCH3 (5a–c) occurs at room temperature in the presence of H2 (1 atm or lower), resulting in the leaching of palladium particles. These particles have been shown to catalyze the hydrogenation of phenylacetylene and diphenylacetylene to their alkene and alkane products. A mechanism for the formation of palladium particles has been proposed. The structures of 1a, 1c, 2a, 2c, 3a, 4b and 5b have been studied by X-ray crystallography.
Introduction
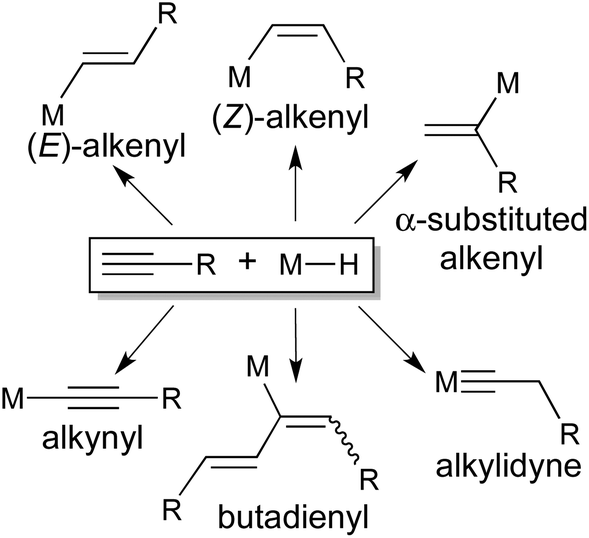
The reaction of an alkyne with a transition metal hydride is fundamentally important due to its involvement in the catalytic cycles for reduction (e.g., hydrogenation, hydroboration and hydrosilylation) and oligomerization of alkynes.1 The most common outcome of the reaction is cis addition of M–H across the CC bond, although in some cases the trans-addition product has been obtained through either direct insertion or isomerization of the kinetically formed cis-addition product. The interaction between specifically a terminal alkyne and a transition metal hydride is often more complicated and less predictable (Scheme 1). In addition to an (E)-, (Z)-, or α-substituted-alkenyl complex stemmed from cis-1,2-,2trans-1,2-,3 or 2,1-insertion,2d,f,3,4 an alkylidyne complex may be produced via the rearrangement of an η2-alkenyl intermediate.5 Moreover, facile alkyne-to-vinylidene isomerization6 makes it possible to incorporate 2 equiv. of alkyne for the synthesis of a butadienyl complex.7 The formation of a σ-alkynyl complex has also been reported.8
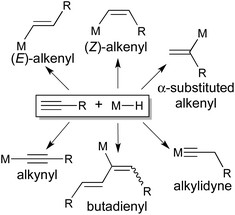 |
| | Scheme 1 | |
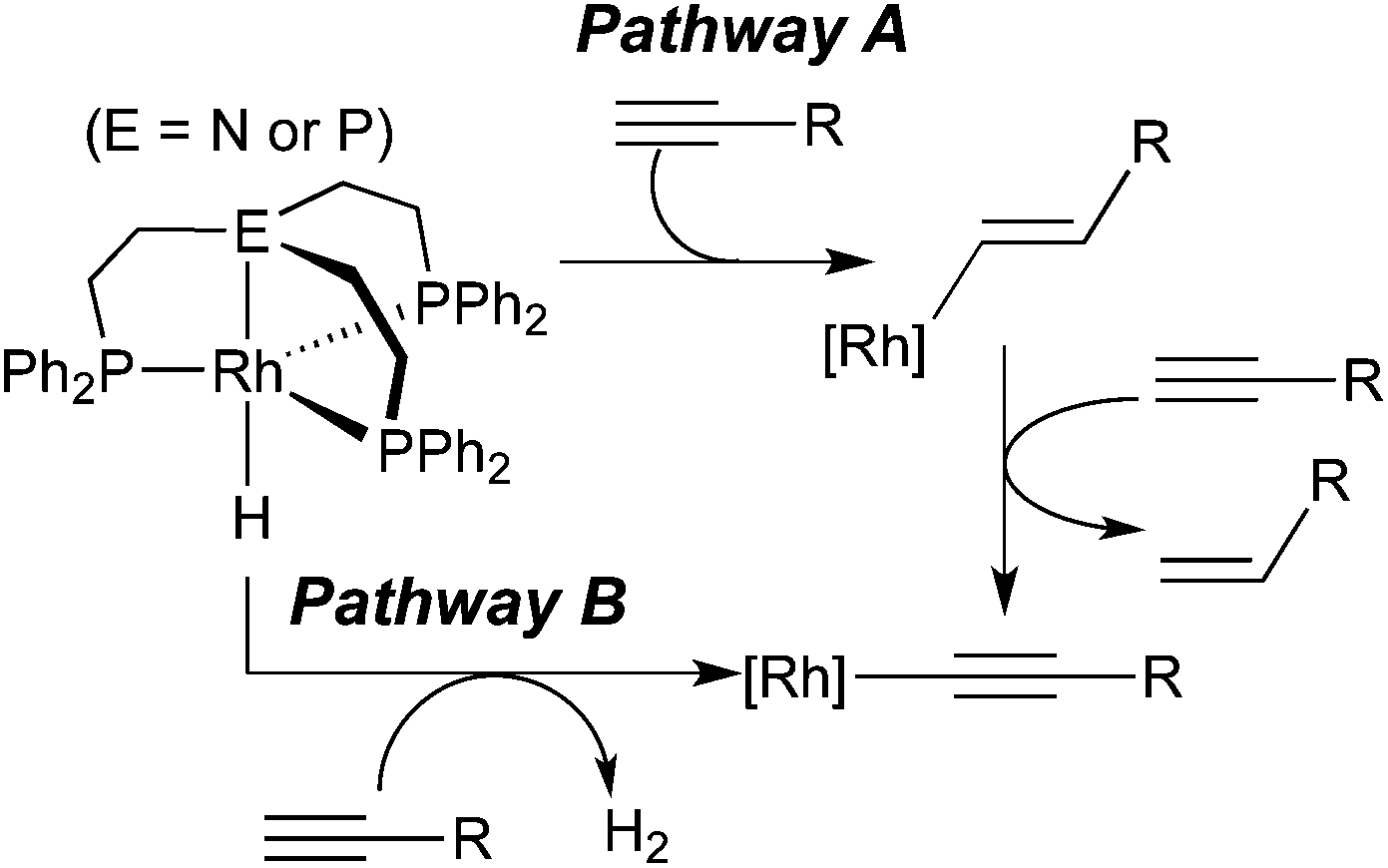
From the hydrogenation point of view, generating a σ-alkynyl complex from a terminal alkyne and a hydride appears to be the least productive pathway as far as formal oxidation states of the alkyne carbons are concerned. It is, however, not completely irrelevant within the broader context of alkyne reduction. Bianchini and co-workers have shown that σ-alkynyl complexes are the major organometallic species during a series of reactions between tripodal-ligated Rh(I) hydride complexes and 1-alkynes.8a The organic products of these reactions mainly consisted of oligomers of alkynes along with some 1-alkenes. Mechanistic analysis suggested that alkenes originated from alkyne insertion followed by the cleavage of the Rh–C(sp2) bond with another alkyne molecule (Scheme 2, pathway A). The net reaction for rhodium hydride complexes is the conversion to σ-alkynyl complexes. A second path to such species involves the loss of H2 without using alkyne as a sacrificial hydrogen acceptor (pathway B). It should be mentioned that in this study H2 has been found from the headspace of the reaction mixtures. Several other reports on metal hydrides have proposed pathway A as the single route to σ-alkynyl complexes, all based on a stoichiometric amount of alkene produced as the by-product.4a,8b,d Kirchner et al. have suggested a similar process (pathway A) from HCCR and MH to σ-alkynyl complexes, which are catalytically active species for dimerization of alkynes.9
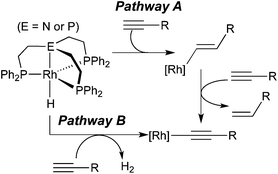 |
| | Scheme 2 | |
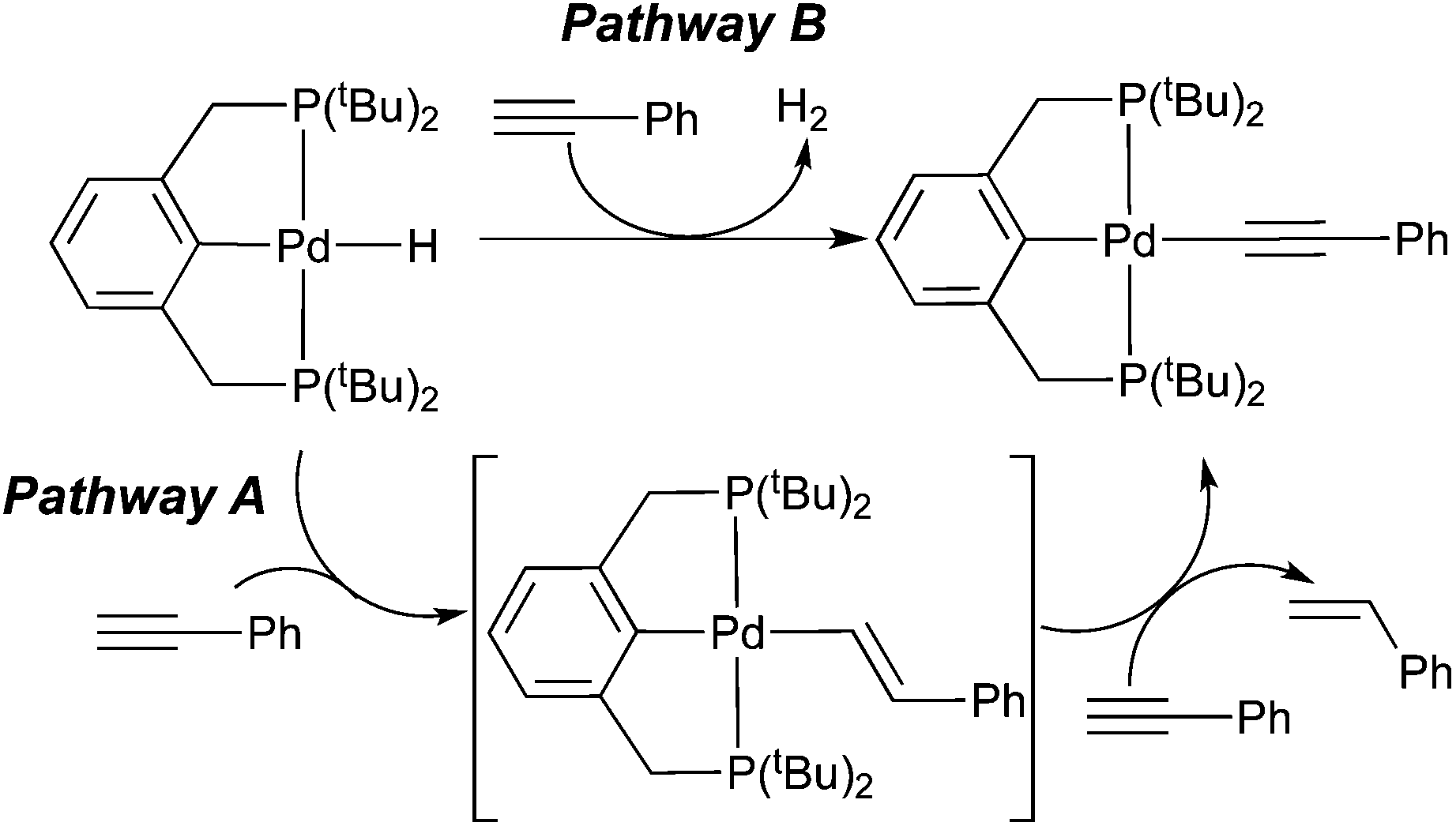
A study of particular interest to us is that by Johansson and Wendt,8e which has proposed that both pathway A and pathway B are operative for the reaction between a palladium PCP-pincer hydride complex and phenylacetylene (Scheme 3). Although H2 was not detected by 1H NMR spectroscopy, likely due to its minute amount in solution, the substoichiometric amount of styrene (0.7 equiv.) made during the reaction is consistent with the dual process. Unlike the rhodium system described above, the alkenyl complex is not an observable intermediate, suggesting that C–H bond exchange between the alkenyl complex and phenylacetylene is much faster than the insertion step.
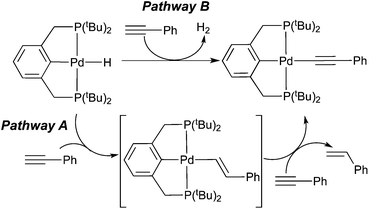 |
| | Scheme 3 | |
We have been interested in the chemistry of Group 10 metal hydride complexes bearing bis(phosphinite)-based POCOP-pincer ligands.10 One of our recent studies has demonstrated that steric and electronic differences between PCP- and POCOP-pincer systems have a profound influence on the reactivity of the hydride moiety.10d This has made us wonder how different it could be for palladium POCOP-pincer hydride complexes to react with terminal alkynes, and if a well-defined catalytic system could be developed for the hydrogenation of alkynes. In this paper, we will show that indeed palladium POCOP-pincer hydride complexes behave differently from their PCP analogues when mixed with terminal alkynes. Our study suggests that alkene formation is not a consequence of pathway A, but rather a result of direct hydrogenation of alkynes by palladium particles that are released from the pincer complexes. We will also report on the hydrogenation of alkynes catalyzed by related palladium POCOP-pincer complexes.
Results and discussion
Preparation of [2,6-(R2PO)2C6H3]PdH (2a–c)
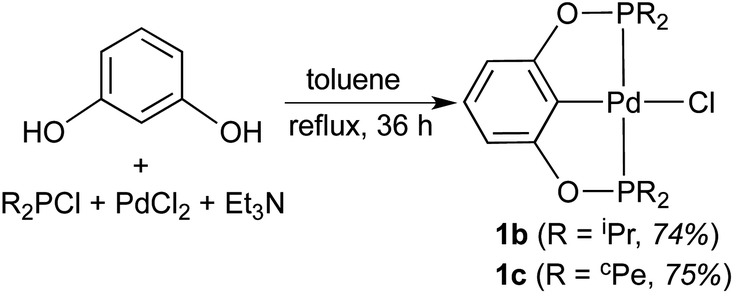
Although palladium POCOP-pincer complexes have been known for more than a decade now,11 their hydride derivatives have never been reported prior to this work. One of the most widely used methods to synthesize compounds of this type is to treat palladium chloride complexes with an appropriate hydride donor such as NaBH4,12 LiAlH4,13 LiEt3BH,14 or NaH.15 Our initial efforts were thus focused on preparing a series of palladium POCOP-pincer chloride complexes as the precursors to the targeted hydride complexes. The P-substituents of the POCOP-pincer ligand were varied in order to investigate how ligand modification can tune the reactivity at the palladium center.
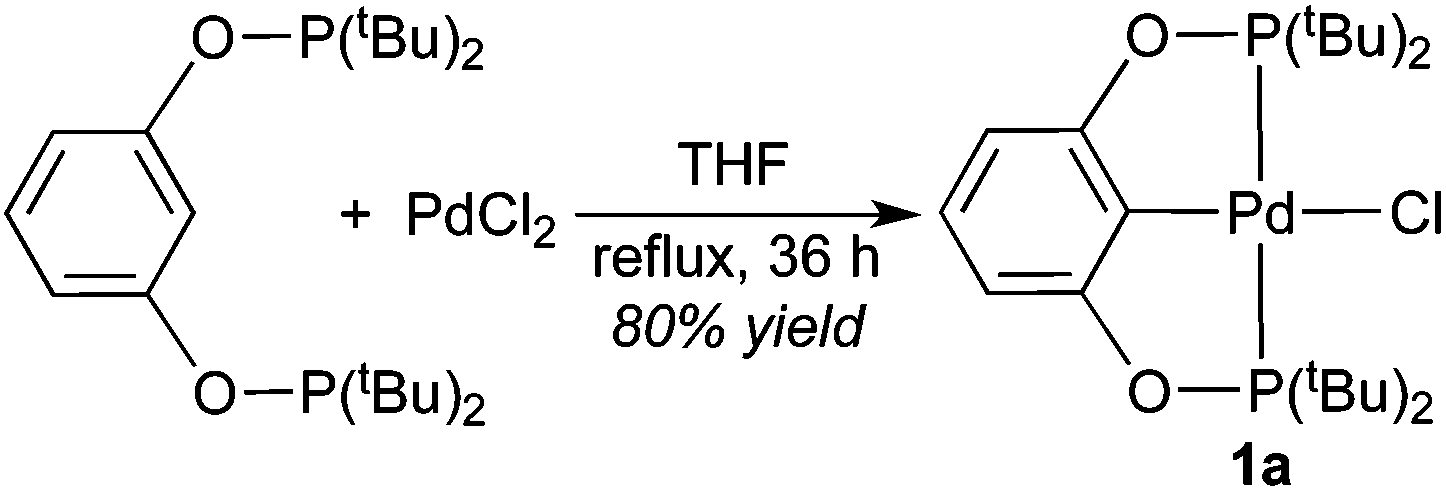
Complex 1a, which contains a relatively bulky POCOP-pincer ligand, has been previously prepared in 31% isolated yield via cyclometalation of 1,3-bis(di-tert-butylphosphinito)benzene16 with Pd(PhCN)2Cl2 in refluxing 2-methoxyethanol.17 In our hands a much higher yield was obtained when PdCl2 was used as the source of palladium and THF was employed as the solvent (eqn (1)). NMR data of 1a are consistent with literature values,17 and the structure of the molecule was further confirmed by X-ray crystallography (Fig. 1).
| | 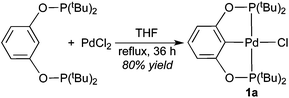 | (1) |
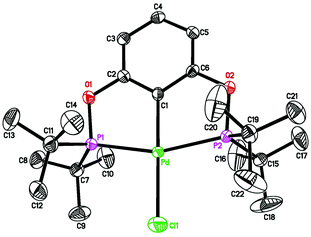 |
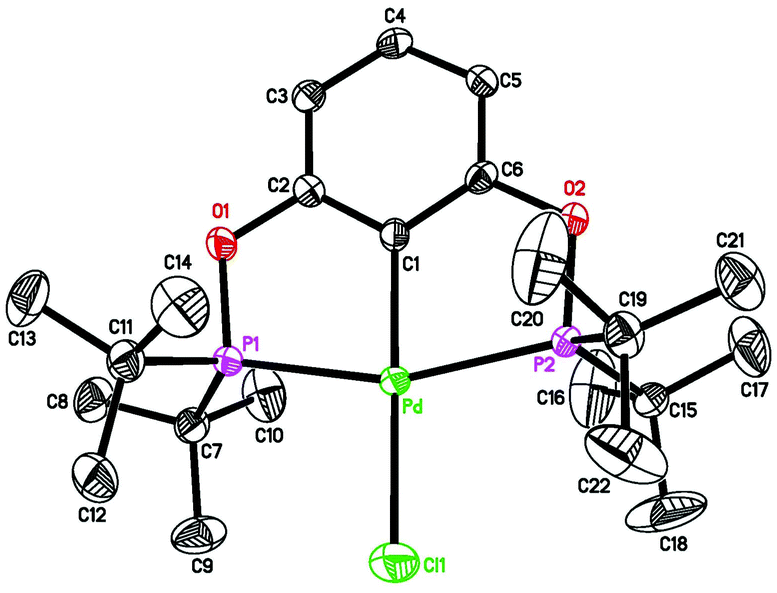
| | Fig. 1 ORTEP drawing of [2,6-(tBu2PO)2C6H3]PdCl (1a) at the 50% probability level. Selected bond lengths (Å) and angles (°): Pd–Cl(1) 2.3813(8), Pd–C(1) 2.011(3), Pd–P(1) 2.2926(7), Pd–P(2) 2.2954(7), P(1)–Pd–P(2) 160.12(3), C(1)–Pd–Cl(1) 179.55(8). | |
With a less crowded POCOP-pincer ligand, palladium chloride complexes were more conveniently prepared in a one-pot synthesis without isolating the bis(phosphinite) ligand first (eqn (2)). Such a strategy has been previously utilized by Song and co-workers18 for the synthesis of [2,6-(Ph2PO)2C6H3]PdCl (1d) and [2,6-(Cy2PO)2C6H3]PdCl. The new complex 1c (cPe = cyclopentyl) was characterized by NMR spectroscopy, elemental analysis and X-ray crystallography (Fig. 2). The Pd–P and Pd–Cipso bonds of 1c were found to be similar to those of 1b,11b but slightly shorter (by 0.02 Å) than those of 1a possibly due to reduced steric congestion around the palladium center.
| | 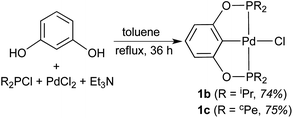 | (2) |
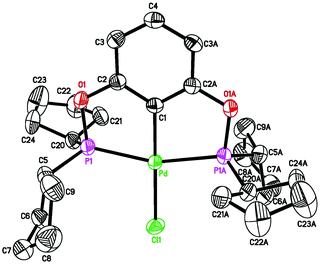 |
| | Fig. 2 ORTEP drawing of [2,6-(cPe2PO)2C6H3]PdCl (1c) at the 50% probability level. Selected bond lengths (Å) and angles (°): Pd–Cl(1) 2.3763(9), Pd–C(1) 1.988(4), Pd–P(1) and Pd–P(1A) 2.2748(8), P(1)–Pd–P(1A) 160.38(4), C(1)–Pd–Cl(1) 180.0. | |
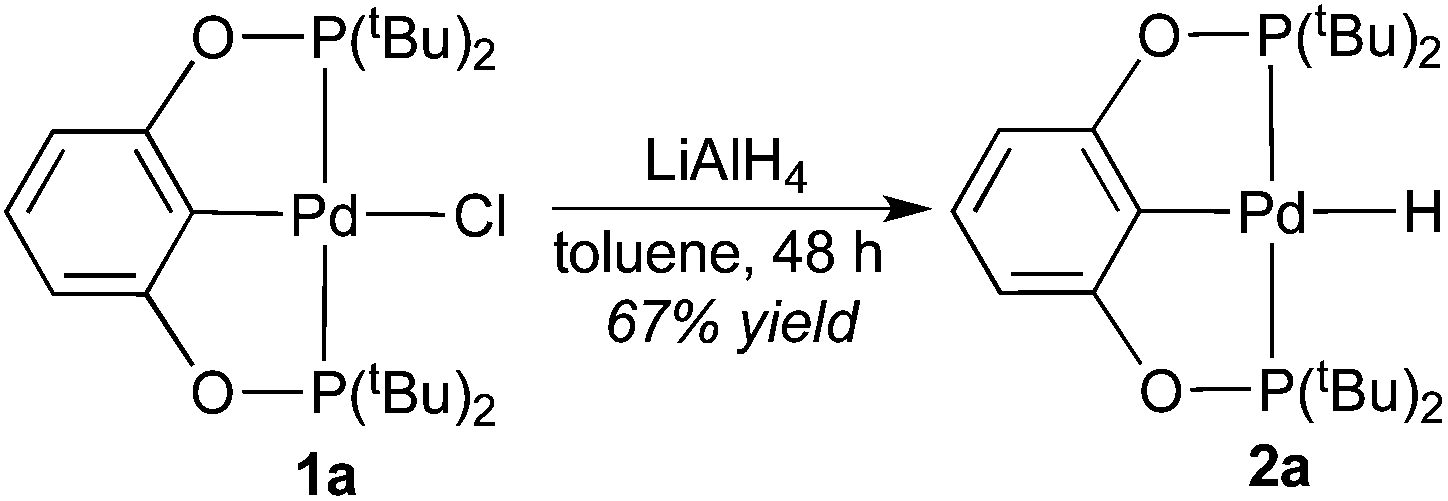
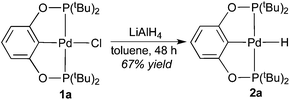
The synthesis of palladium hydride complexes turned out to be nontrivial. Our previously developed procedures10a,c using LiAlH4 to convert nickel POCOP-pincer chloride complexes to the corresponding hydrides could be extended to the palladium system, but only for the tBu-substituted POCOP-pincer complex. Thus, palladium hydride 2a was readily isolated as a white solid in good yield after stirring the mixture of 1a and LiAlH4 in toluene for 48 h (eqn (3)). The 1H NMR spectrum of 2a in C6D6 showed a characteristic hydride resonance as a triplet at −2.48 ppm (JP−H = 16.0 Hz). The IR spectrum of 2a revealed a strong band at 1756 cm−1, which is expected for the Pd–H stretch.12a,19 The hydride ligand in 2a was also located by X-ray diffraction of its single crystals (Fig. 3). The Pd–Cipso bond distance is elongated by 0.02 Å upon conversion from 1a to 2a, which reflects a stronger trans-influence from the hydride. Another noticeable difference between the two complexes is that the Pd–P bonds in 2a are shorter by 0.03 Å, possibly due to reduced steric congestion near palladium or perhaps due to increased palladium-to-phosphorus back-donation with a more electron-donating hydride ligand.
| |  | (3) |
 |
| | Fig. 3 ORTEP drawing of [2,6-(tBu2PO)2C6H3]PdH (2a) at the 50% probability level. Selected bond lengths (Å) and angles (°): Pd–H 1.75(3), Pd–C(1) 2.036(2), Pd–P(1) 2.2634(6), Pd–P(2) 2.2627(6), P(1)–Pd–P(2) 159.93(2). | |
Replacing the tBu groups on the phosphorus donors with smaller groups created some synthetic challenges. The mixture of 1c and LiAlH4 in toluene turned black within a few hours, a phenomenon that was absent for the reaction of 1a. Standard work-up procedures (filtration followed by solvent evaporation) led to the isolation of a colorless oil. Surprisingly, the 1H NMR spectrum of this material (in C6D6) showed no resonance that could be assigned to the hydride species. The most notable resonance was a doublet of triplet at 3.30 ppm with coupling constants of 192.0 and 8.0 Hz. 31P{1H} NMR spectrum of the same material displayed only one resonance at −35.7 ppm. These data are consistent with the formation of (cPe)2PH, which is likely to be released from degradation of the pincer complex. A similar decomposition pathway involving the cleavage of the pincer P–O bonds has been observed by the Milstein group and us in ruthenium20 and nickel10d,21 systems, respectively. The success of synthesizing 2a can thus be explained by well-shielded P–O bonds in 1a that prevent the breakdown of the pincer framework. Reactions of 1b and 1d with LiAlH4 exhibited a similar decomposition process as seen in the case of 1c.
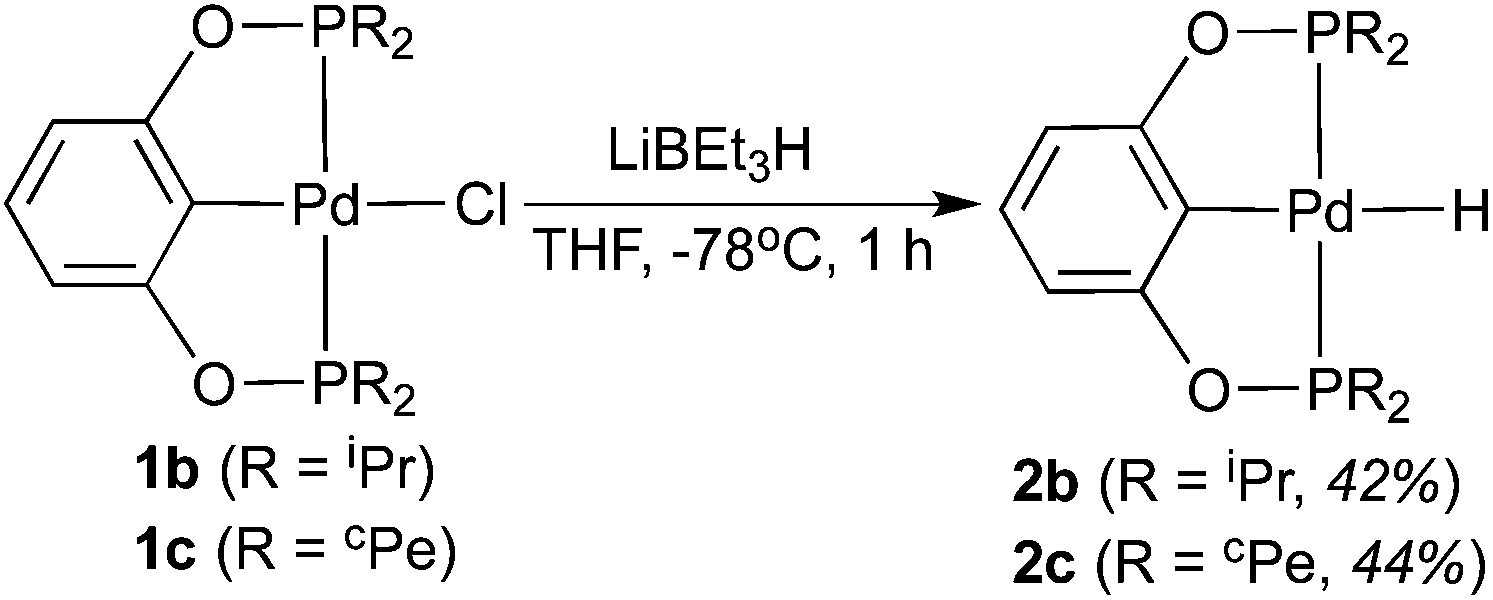
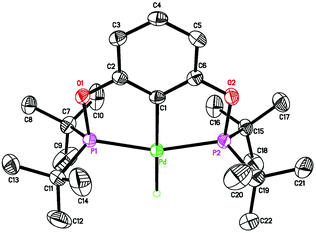
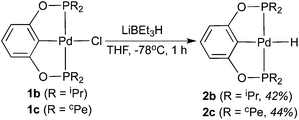
Attempts to synthesize hydrides from 1b–d using other hydride donors such as NaBH4 and NaH also failed. In each case, a complicated mixture of multiple palladium species was obtained, as judged by 31P{1H} NMR spectroscopy. Somewhat promising results came from the reactions of 1b and 1c with LiBEt3H. Hydride species along with some impurities were identified from the isolated products. After extensive optimization, hydrides 2b and 2c could be isolated in an analytically pure form as long as the reaction and the work-up procedures were performed at low temperatures. The reaction with LiBEt3H must be carried out at −78 °C for not more than 1 h (eqn (4)), and solvent evaporation as well as recrystallization processes should be kept below 0 °C. However, once isolated as pure compounds, 2b and 2c are thermally stable at room temperature. Boron-containing byproduct (BEt3) or a small amount of unreacted LiBEt3H may facilitate the decomposition of the hydrides at ambient temperature. In 1H NMR spectra, the hydride resonances of 2b and 2c appeared as triplets at −2.40 ppm (JP–H = 20.8 Hz) and −2.37 ppm (JP–H = 20.0 Hz), respectively. A strong IR band in the 1700–1800 cm−1 region (2b: 1765 cm−1; 2c: 1755 cm−1) further supported the presence of a Pd–H bond.12a,19 The structure of 2c was also established by X-ray crystallography (Fig. 4).22 Compared to 1c, the Pd–Cipso bond distance of 2c is 0.04 Å longer while the Pd–P bond distances are 0.02 Å shorter. Unfortunately, the reaction of 1d with LiBEt3H gave intractable products even at low temperatures. We10a and others23 have experienced a similar difficulty in preparing other Group 10 metal hydride complexes with PPh2-containing pincer ligands.
| |  | (4) |
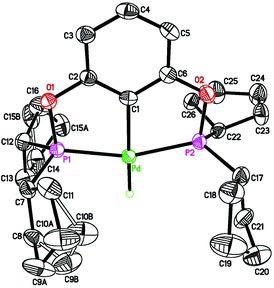 |
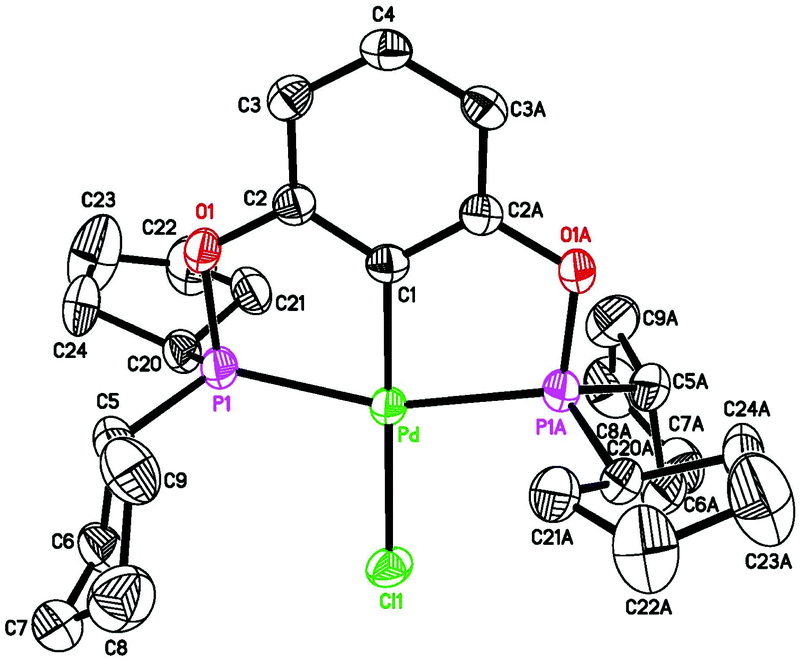
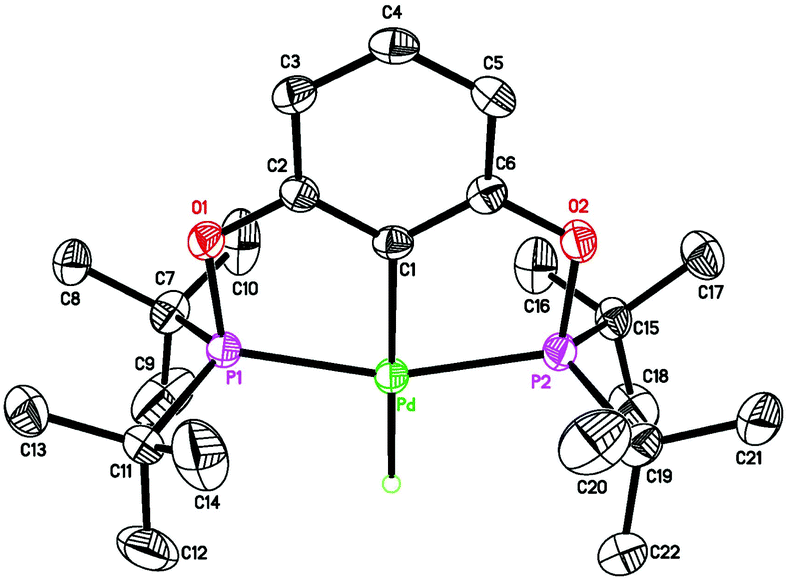
| | Fig. 4 ORTEP drawing of [2,6-(cPe2PO)2C6H3]PdH (2c) at the 50% probability level. Selected bond lengths (Å) and angles (°): Pd–H 1.53(4), Pd–C(1) 2.031(3), Pd–P(1) 2.2518(8), Pd–P(2) 2.2525(9), P(1)–Pd–P(2) 160.25(3). | |
Reactions of 2a–c with phenylacetylene
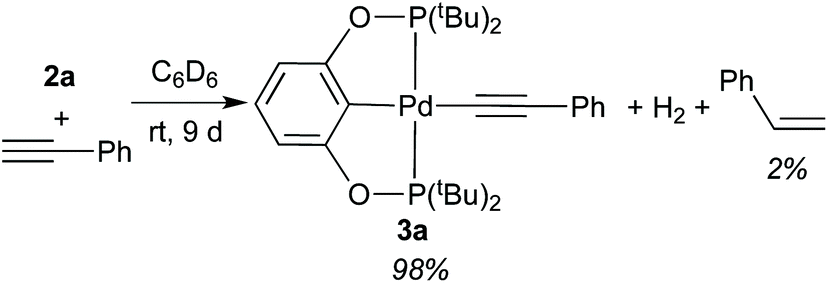
A solution of 2a in C6D6 was treated with an equimolecular amount of freshly distilled phenylacetylene, and the reaction was monitored by 1H and 31P{1H} NMR spectroscopy. At room temperature, 2a disappeared very slowly, requiring as long as 9 days to be fully converted to phenylacetylide complex 3a (eqn (5)). Also observed by 1H NMR were H2 (singlet at 4.47 ppm) and a small amount of styrene. For reasons unknown to us, this reaction was complete within 24 h when the alkyne was not distilled.24 However, the purity of phenylacetylene appeared to have little effect on the composition of the products. At 50 °C, the reaction of 2a with HCCPh (distilled) afforded 3a in 65% yield after 24 h, while styrene was already formed in 10% yield. At that point, H2 remained present in the solution, and some of the starting materials 2a and HCCPh were still left unreacted.| | 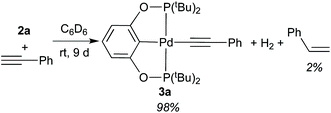 | (5) |
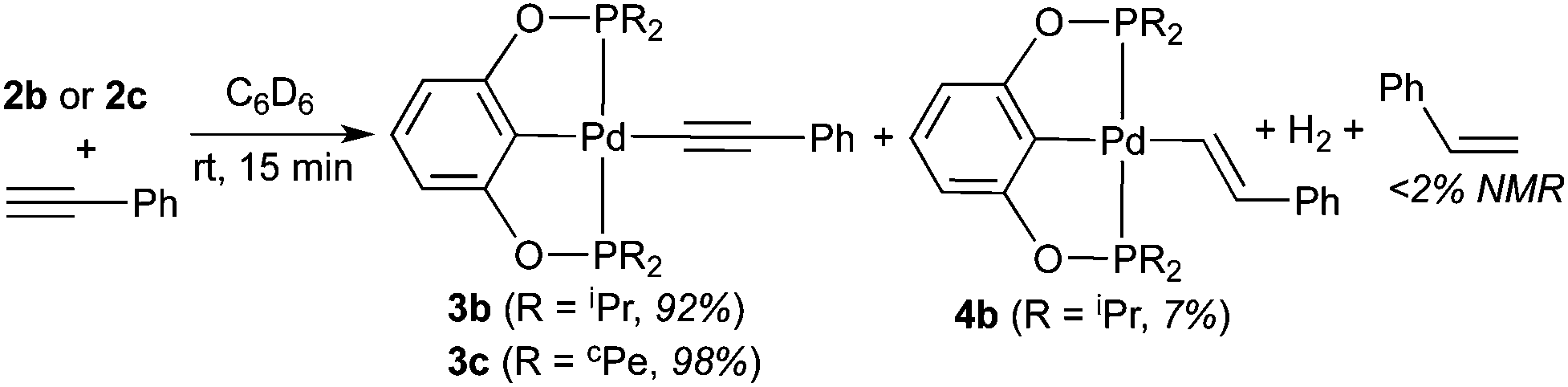
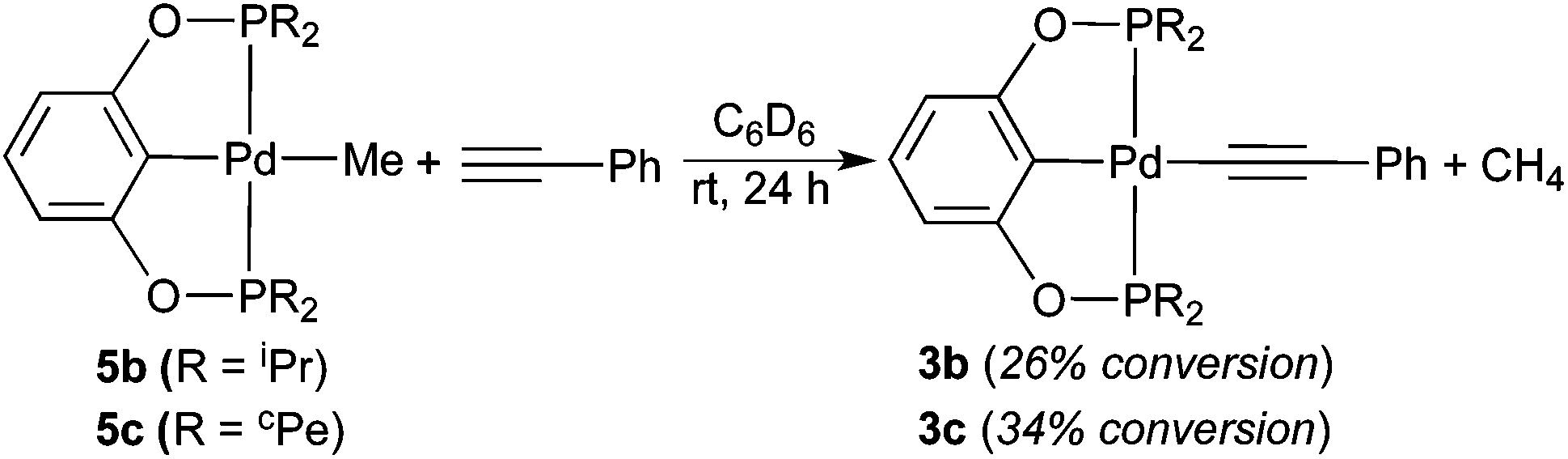
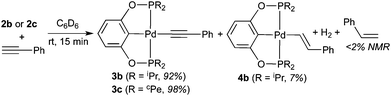
The less bulky palladium hydrides 2b and 2c are significantly more reactive. Their reactions with phenylacetylene finished within 15 min, whether or not the alkyne was distilled. The main palladium-containing products were identified as phenylacetylide complexes 3b and 3c, and similar to the observation for 2a, only a negligible amount of styrene was found (eqn (6)). The anomaly was the reaction of 2b, which gave rise to a second palladium pincer complex 4b with a phosphorus resonance at 187.2 ppm. 1H NMR spectrum revealed a broad doublet at 8.20 ppm, and the relatively large coupling constant of 20.0 Hz suggested that 4b could be an (E)-alkenyl complex. The reaction of 2c also generated a second palladium species as judged by 31P{1H} NMR spectroscopy, although its quantity was too insignificant (∼1%) to provide any useful structural information.
| |  | (6) |
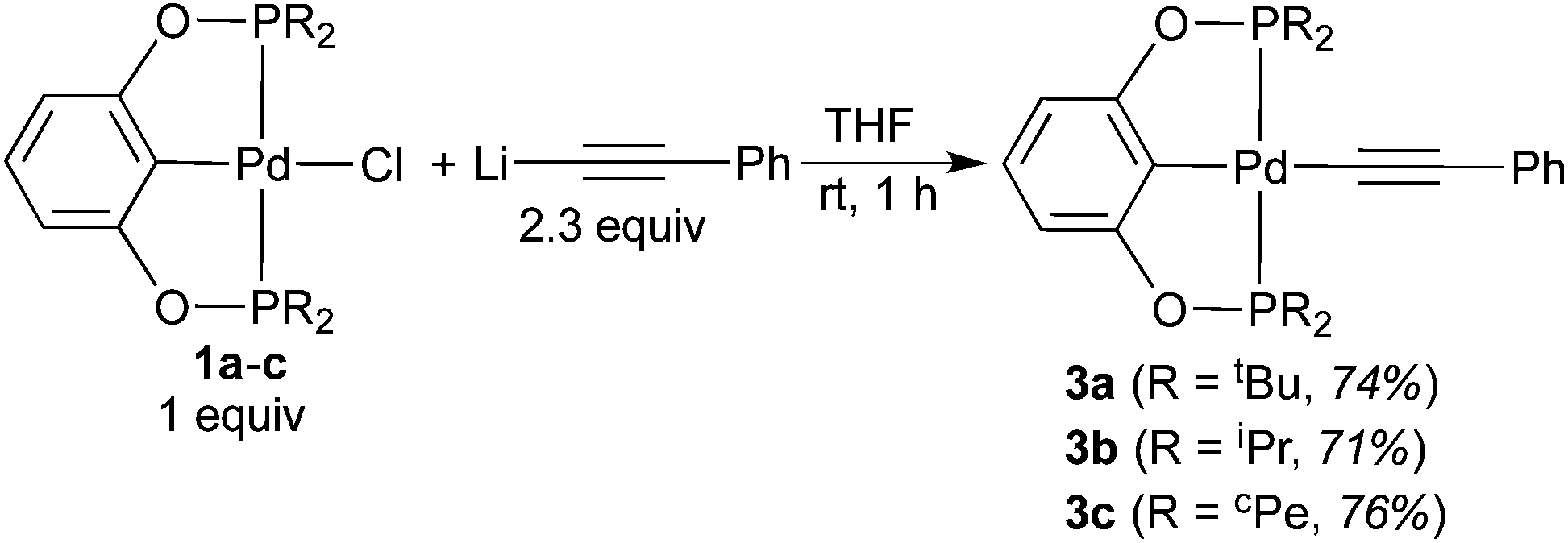
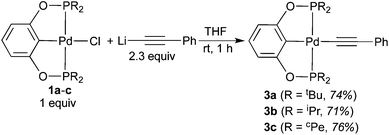
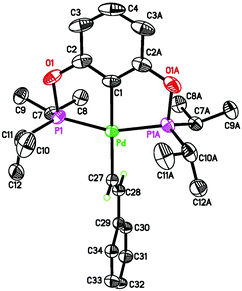
As further confirmation of the structures proposed in eqn (5) and (6), independent syntheses of 3a–c and 4b were pursued. The phenylacetylide complexes were readily prepared in good yield by mixing 1a–c with a large excess of lithium phenylacetylide (eqn (7)). Lowering the equivalents of LiCCPh would result in partial conversion of the palladium chloride complexes, suggesting that this reaction might be reversible. Consistent with this hypothesis, mixing pure 3a–c with 1 equiv. of LiCl in THF at room temperature for 48 h led to the formation of 1a–c in about 3% yield. Compounds 3a–c were characterized by NMR and IR spectroscopy as well as elemental analysis. Complex 3a was also characterized by X-ray crystallography (Fig. 5).25 The CC stretching frequencies of these molecules (3a: 2100 cm−1; 3b: 2085 cm−1; 3c: 2095 cm−1) are comparable to those reported in the literature for other phenylacetylide complexes.8c,26
| |  | (7) |
 |
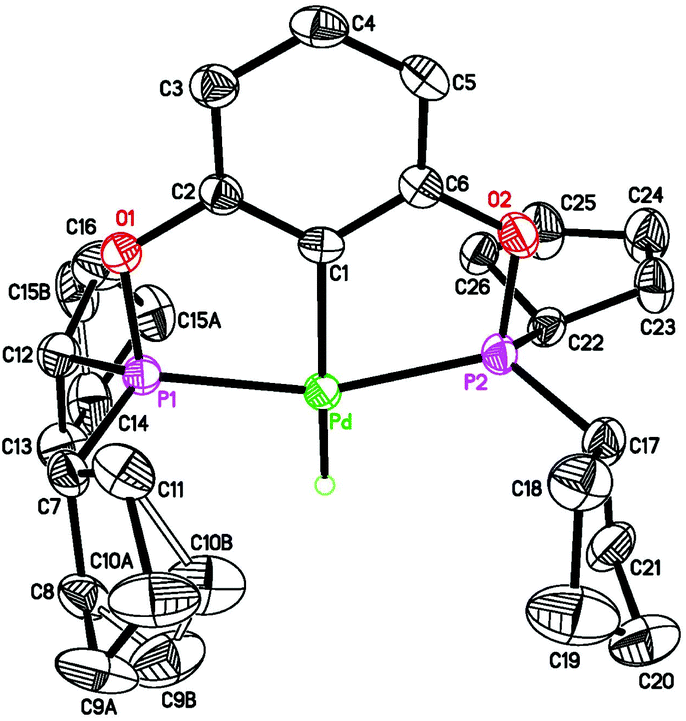
| | Fig. 5 ORTEP drawing of [2,6-(tBu2PO)2C6H3]PdCCPh (3a) at the 50% probability level. Selected bond lengths (Å) and angles (°): Pd(2)–C(1B) 2.020(4), Pd(2)–C(31B) 2.027(4), Pd(2)–P(1B) 2.2802(10), Pd(2)–P(2B) 2.2753(10), C(31B)–C(32B) 1.211(5), P(1B)–Pd(2)–P(2B) 159.79(4), C(1B)–Pd(2)–C(31B) 178.85(17). | |
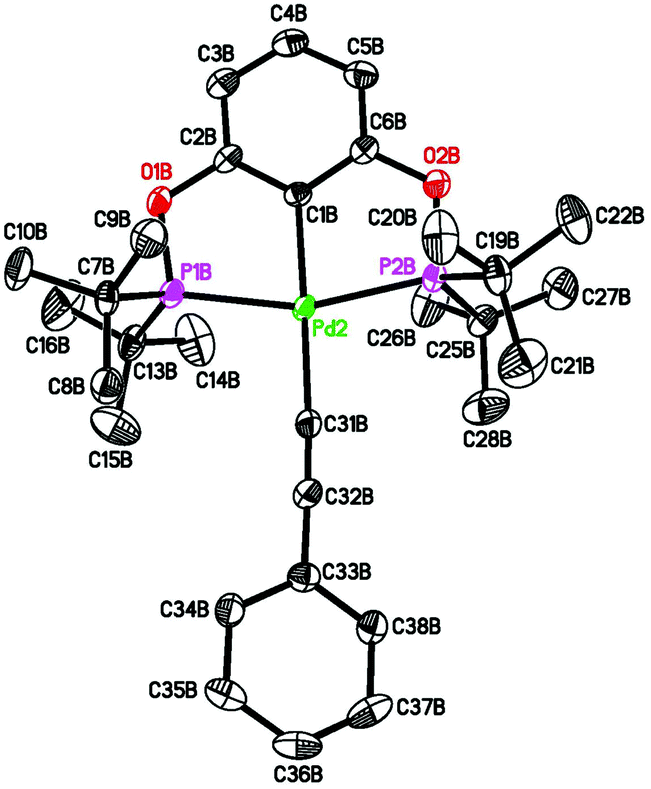
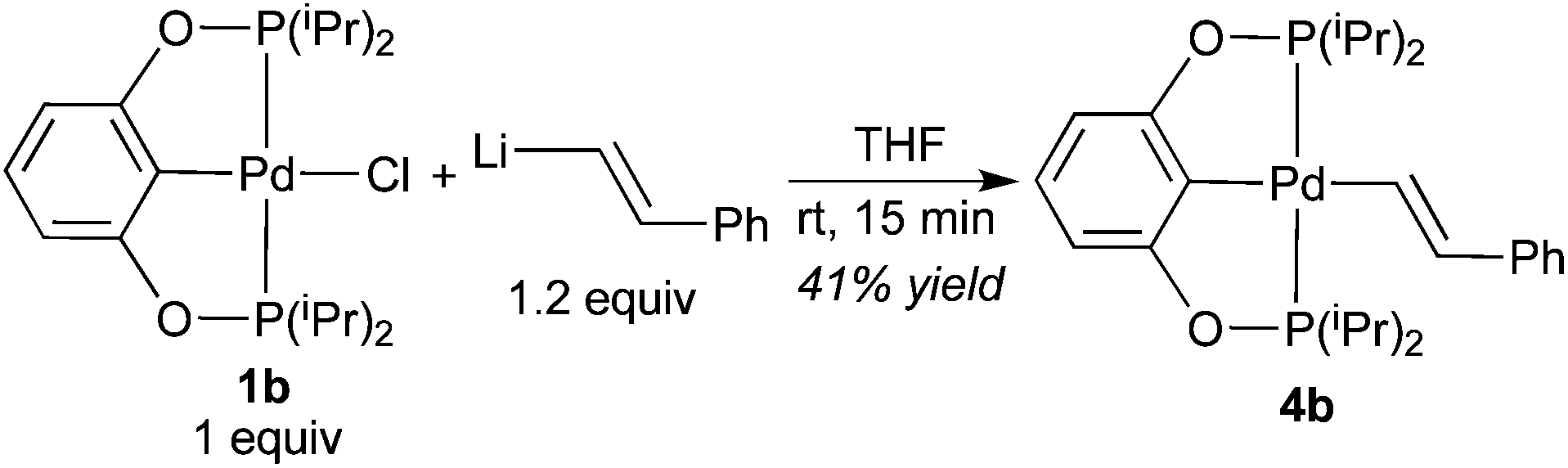
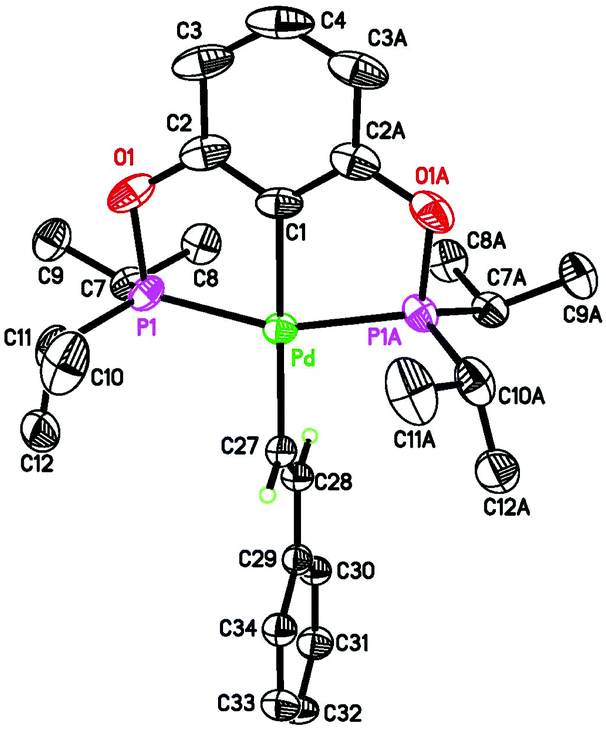
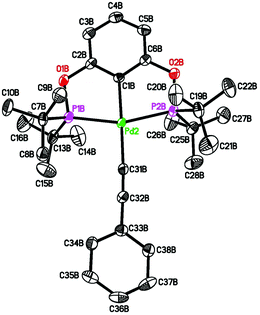
Pure 4b was obtained from salt metathesis reaction of 1b with (E)-2-phenylethenyllithium (prepared in situ from trans-β-iodostyrene and nBuLi) as illustrated in eqn (8). The 1H and 31P{1H} NMR spectra of 4b in C6D6 match well with what has been described for the second observable palladium species during the reaction between 2b and HCCPh (eqn (6)). The characteristic vinylic resonance at 8.20 ppm, however, appeared as a doublet of triplets (J = 20.0 and 4.0 Hz) instead of a broad doublet. The smaller coupling constant of 4.0 Hz, which is presumably due to phosphorus–hydrogen coupling, was somehow better resolved for the pure sample. The configuration of the CC bond of this compound was unambiguously established by X-ray crystallographic study (Fig. 6). Interestingly, the CHCHPh moiety adopts a conformation that is perpendicular to the coordination plane with a dihedral angle of 89.9(2)° between the two phenyl rings. This is probably due to sterics rather than being driven by back-donation from palladium dxy orbital to the π* orbital of the alkenyl group; the C(27)–C(28) distance of 1.329(5) Å indicates no appreciable elongation from a normal CC bond.
| | 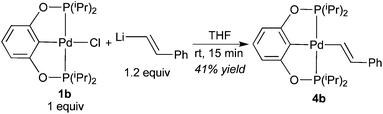 | (8) |
 |
| | Fig. 6 ORTEP drawing of (E)-[2,6-(iPr2PO)2C6H3]PdCHCHPh (4b) at the 50% probability level. Selected bond lengths (Å) and angles (°): Pd–C(1) 2.031(3), Pd–C(27) 2.066(3), Pd–P(1) and Pd–P(1A) 2.2549(6), C(27)–C(28) 1.329(5), P(1)–Pd–P(1A) 159.45(3), C(1)–Pd–C(27) 179.84(12). | |
Mechanism for the conversion of phenylacetylene to styrene
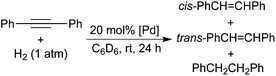
The presence of both 3b and 4b in the reaction of 2b with phenylacetylene (eqn (6)) seems to support the dual mechanism described in Schemes 2 and 3. The fact that styrene was generated in small quantities could be explained by pathway A being the minor process. Since 4b could be synthesized independently, investigating its reaction with HCCPh would give us a better picture of the overall process. It should be mentioned that well-defined reactions of this type are well known in the literature. The Bianchini group has demonstrated in rhodium8a and cobalt4a systems that alkenyl complexes undergo facile M–C(sp2) bond cleavage by HCCCO2Et to generate alkynyl complexes while releasing alkenes. They have suggested a mechanism involving oxidative addition of the alkyne C–H bond followed by reductive elimination of the alkene products. Eisen and co-workers have reported similar C–H exchange reactions between alkenyl actinide complexes and HCCiPr,27 and have proposed a σ-bond metathesis mechanism as anticipated for f-element complexes. In our case, mixing 4b with 1 equiv. of HCCPh in C6D6 at room temperature did not yield any styrene within 15 min. However, after 24 h, styrene was obtained in 10% yield along with the same amount of 3b. This result stands in strong contrast to the rapid C–H exchange process postulated for the PCP-pincer system (Scheme 3).8e The reason for the smaller amount of styrene produced in our POCOP-pincer system is therefore twofold: less favorable formation of the alkenyl species and more sluggish C–H exchange between the alkenyl species and HCCPh. As expected, increasing the ratio of HCCPh to 2b from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 2:1 while keeping [2b] the same as in eqn (6) gave almost the same product ratios for 3b, 4b and styrene after 15 min. Surprisingly, extending the reaction time to 24 h resulted in more styrene (31% yield with respect to 2b) without much expense of 4b. In other words, even if all 4b present in the solution were converted to 3b, it would not account for the amount of styrene generated. This also implies that styrene must be predominantly formed from a different pathway. Perhaps, 4b acts as a catalyst for the hydrogenation of the excess HCCPh using H2 produced during the reaction, and the hydride 2b is being regenerated. To test this hypothesis, a solution of 4b in C6D6 was treated with 1 atm of H2 and the reaction was monitored by 1H NMR spectroscopy. At room temperature after 12 h, only 7% of 4b was converted to 2b. If the palladium pincer hydride were a true active species, regenerating it from the hydrogenolysis of 4b would be too slow to be catalytically viable.
:1 to 2:1 while keeping [2b] the same as in eqn (6) gave almost the same product ratios for 3b, 4b and styrene after 15 min. Surprisingly, extending the reaction time to 24 h resulted in more styrene (31% yield with respect to 2b) without much expense of 4b. In other words, even if all 4b present in the solution were converted to 3b, it would not account for the amount of styrene generated. This also implies that styrene must be predominantly formed from a different pathway. Perhaps, 4b acts as a catalyst for the hydrogenation of the excess HCCPh using H2 produced during the reaction, and the hydride 2b is being regenerated. To test this hypothesis, a solution of 4b in C6D6 was treated with 1 atm of H2 and the reaction was monitored by 1H NMR spectroscopy. At room temperature after 12 h, only 7% of 4b was converted to 2b. If the palladium pincer hydride were a true active species, regenerating it from the hydrogenolysis of 4b would be too slow to be catalytically viable.
Since H2 was observed during the reactions between the hydride complexes and phenylacetylene, hydrogenolysis of the palladium alkynyl complexes 3a–c could be another mechanism for styrene formation. A solution of 3b in C6D6 was exposed to 1 atm of H2 and mixed well at room temperature. After 24 h, no styrene was found, thereby ruling out such a mechanistic pathway. An alternative but remote possibility of converting 3b to styrene is somehow for the alkynyl moiety to abstract hydrogen atoms from phenylacetylene. However, mixing complex 3b with 1 equiv. of HCCPh at room temperature for 24 h did not yield any appreciable product. In contrast, carrying out a similar reaction under 1 atm of H2 did generate styrene in 5% yield after 24 h. This result is more consistent with a mechanism in which styrene is produced by reduction of the alkyne with molecular hydrogen and this process is catalyzed by 3b. This hypothesis was further substantiated by the fact that 31P NMR did not show any new resonance throughout the reaction.
Catalytic hydrogenation of alkynes
Palladium alkynyl complexes 3a–c were then subjected to catalytic studies using phenylacetylene as the substrate and the reactions were conducted at room temperature under 1 atm of H2 pressure. As shown in eqn (9) and Table 1, in the presence of 20 mol% 3a, after 24 h, only 6% of phenylacetylene was converted to styrene (entry 1). Under the same conditions using 3b as a catalyst (entry 2), a higher conversion of phenylacetylene was observed. The amount of styrene produced in this case (30% yield) is greater than 20%, confirming that this process is catalytic in palladium. Catalyst 3c is slightly less reactive than 3b, providing styrene in 23% yield after 24 h (entry 3).| | 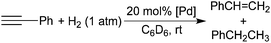 | (9) |
Table 1 Palladium-catalyzed hydrogenation of phenylacetylenea
| Entry |
[Pd] |
Time (h) |
PhCHCH2b (%) |
PhCH2CH3b (%) |
|
Reaction conditions: phenylacetylene (62.5 μmol), palladium catalyst (12.5 μmol), and 1,4-dioxane (25 μmol) in 0.40 mL of C6D6 at room temperature under 1 atm of H2.
NMR yield.
|
| 1 |
3a
|
24 |
6 |
0 |
| 2 |
3b
|
24 |
30 |
0 |
| 3 |
3c
|
24 |
23 |
0 |
| 4 |
5a
|
12 |
92 |
8 |
| 5 |
5b
|
2 |
53 |
31 |
| 6 |
5b
|
12 |
0 |
100 |
| 7 |
5c
|
2 |
11 |
4 |
| 8 |
5c
|
12 |
4 |
79 |
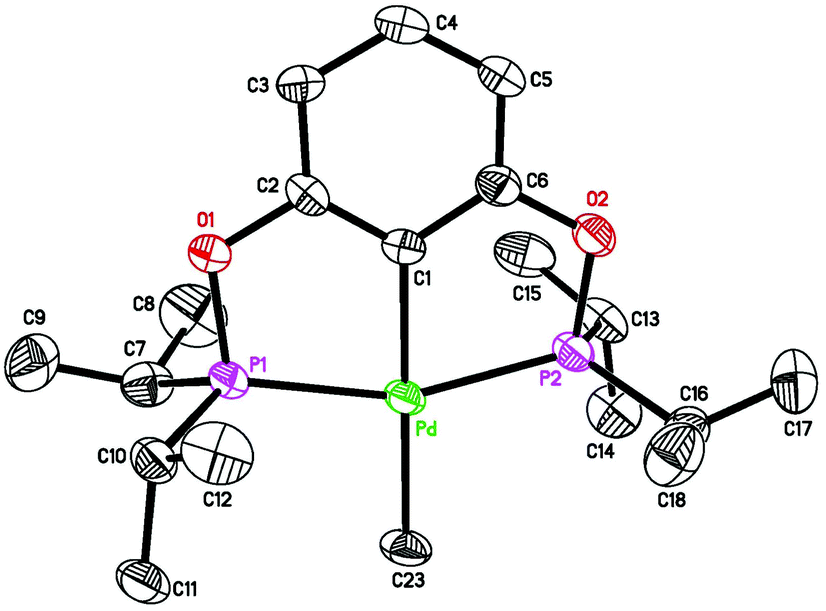
To further facilitate the hydrogenation process, the catalyst structure was modified through the replacement of the alkynyl group with a methyl group. It was hypothesized that the more σ-donating methyl group would promote dihydrogen activation at the palladium center. Palladium POCOP-pincer methyl complexes 5a–c were readily synthesized from the reaction of 1a–c with CH3Li (see the Experimental section for details), and compound 5b was also crystallographically characterized (Fig. 7). These methyl complexes indeed are catalytically more reactive than the alkynyl complexes. As shown in Table 1, when 5a was used as a catalyst (entry 4), after 12 h, 92% of phenylacetylene was converted to styrene while the rest of the alkyne substrate was fully reduced to ethylbenzene. By comparison, reactions catalyzed by 5b and 5c under otherwise the same conditions yielded ethylbenzene as the major product (entries 6 and 8). However, when the hydrogenation reactions were stopped at 2 h, styrene became the main hydrogenation product (entries 5 and 7). Another noticeable feature of the reactions catalyzed by 5b and 5c was that after 12 h, about 10% and 20% of palladium species turned into the alkynyl complexes 3b and 3c, respectively. This is due to a C–H exchange reaction between the methyl complexes and HCCPh. In separate experiments, 5b and 5c were shown to react with HCCPh slowly to give 3b and 3c as well as CH4 (eqn (10)). Similar reaction with the more bulky complex 5a did not proceed even at 50 °C.
| | 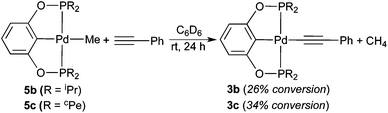 | (10) |
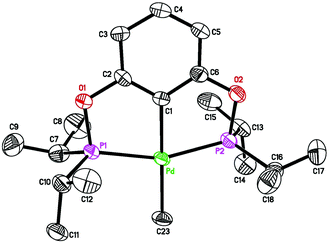 |
| | Fig. 7 ORTEP drawing of [2,6-(iPr2PO)2C6H3]PdCH3 (5b) at the 50% probability level. Selected bond lengths (Å) and angles (°): Pd–C(1) 2.026(3), Pd–C(23) 2.125(3), Pd–P(1) 2.2628(7), Pd–P(2) 2.2591(7), P(1)–Pd–P(2) 158.87(3), C(1)–Pd–C(23) 179.45(12). | |
The hydrogenation protocol could also be applied to internal alkynes such as diphenylacetylene. At room temperature, alkynyl complexes 3a–c did not show any catalytic activity. On the other hand, methyl complexes 5a–c proved to be active catalysts for the hydrogenation of diphenylacetylene (eqn (11) and Table 2). Catalytic activity of these palladium complexes followed the decreasing order of 5b > 5c > 5a, which is the same trend observed for the hydrogenation of phenylacetylene. Although cis-stilbene was the major hydrogenation product in each catalytic run, a noticeable amount of bibenzyl was detected, indicating that the hydrogenation process did not stop at the olefin stage.
| |  | (11) |
Table 2 Palladium-catalyzed hydrogenation of diphenylacetylenea
| Entry |
[Pd] |
cis-Stilbeneb (%) |
trans-Stilbenec (%) |
Bibenzylb (%) |
|
Reaction conditions: diphenylacetylene (62.5 μmol), palladium catalyst (12.5 μmol), and 1,4-dioxane (25 μmol) in 0.40 mL of C6D6 at room temperature under 1 atm of H2.
NMR yield.
GC yield.
|
| 1 |
5a
|
57 |
0 |
3 |
| 2 |
5b
|
82 |
1 |
6 |
| 3 |
5c
|
74 |
5 |
3 |
Mechanism for the hydrogenation of alkynes
Palladium-catalyzed hydrogenation of alkynes is a well-established process, particularly in the field of heterogeneous catalysis.28 In recent years, there have also been a number of reports focusing on the development of palladium-based homogeneous catalysts for selective hydrogenation of alkynes.29 One common mechanistic feature of these studies involves the intermediacy of a palladium hydride, which is thought to interact with the alkyne substrate to form a palladium alkenyl species. Given these precedents and the observation of 4b from the reaction between 2b and HCCPh (eqn (6)), one might have anticipated that the hydrogenation of alkynes to alkenes described in this paper would also proceed via alkyne insertion into a palladium–hydrogen bond. However, several pieces of mechanistic information argue against such a mechanism. First, cleavage of the Pd–C bond in 4b by either H2 or HCCPh is much slower than the formation of styrene. Furthermore, it is difficult to rationalize why the palladium methyl complexes 5a–c are catalytically more reactive than the palladium alkynyl complexes 3a–c. Neither class of complexes reacts with H2 to give palladium hydrides, the presumed active species.
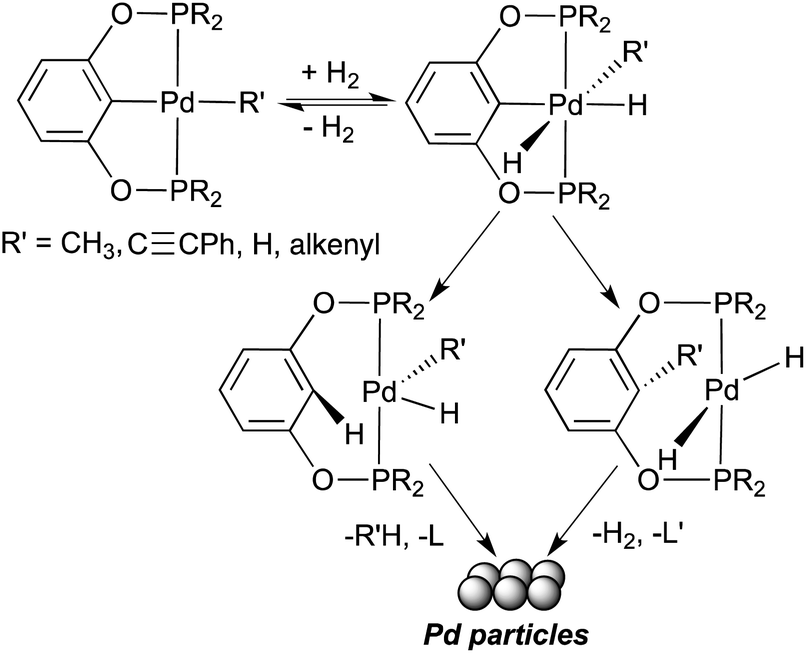
The substantial amounts of over-reduction products shown in Tables 1 and 2 indicated to us that the hydrogenation reactions might be catalyzed by nano-sized palladium particles released from the metal complexes. Thus, two catalytic reactions (entry 2 in both Tables 1 and 2) were repeated with added elemental mercury (200 equiv. relative to the palladium complexes). After 24 h, neither reaction showed hydrogenation products. The positive mercury test30 is in agreement with palladium particles being the true catalytically active species. Further evidence supporting the formation of palladium particles came from the observation that the solution of 5b in toluene31 changed from colorless to faint yellow upon exposure to 1 atm of H2 for 12 h. As a control experiment, the same solution without H2 remained colorless after 48 h. The former solution was then centrifuged, resulting in some black particles. Analyzing the particle size using dynamic light scattering (DLS) techniques revealed particles distributed in the range of 0.18–0.30 μm.
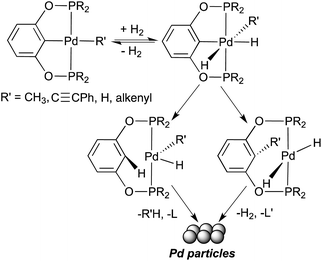
Generating palladium particles from palladium complexes with well-defined structures is not an unusual phenomenon, but the conditions to form these particles in our system are quite rare. A more frequent scenario for palladium particles leached from a pincer complex happens in cross-coupling reactions, where a base and a relatively high temperature are typically used.32 A report by the groups of Sherrill, Jones and Weck has suggested that decomposition of pincer complexes is initiated by displacing one of the pincer arms by a base such as triethylamine.32c A different study by Williams and co-workers, although focused on platinum pincer complexes, has shown that CO can initiate a similar decomposition pathway to platinum particles.33 By comparison, our reactions of palladium pincer complexes with alkynes as well as catalytic hydrogenation reactions were performed at room temperature, under neutral conditions and without an obvious nucleophile to displace the pincer arms. The control experiment described earlier suggests that the release of palladium particles is caused by H2. A plausible mechanism is thus outlined in Scheme 4, in which oxidative addition of H2 to a pincer complex generates a Pd(IV) intermediate. Subsequent reductive elimination to change the coordination mode of the pincer ligand from meridional to bidentate becomes possible, which leads to the eventual decomplexation of all ligands from palladium. Efforts to detect the newly formed diphosphinites by 31P{1H} spectroscopy were, however, unsuccessful. It is likely that the amount of diphosphinites is too small to be observed by NMR.
 |
| | Scheme 4 | |
The mechanism depicted in Scheme 4 can be used to rationalize the reactivity differences between palladium methyl complexes 5a–c and alkynyl complexes 3a–c in catalyzing the hydrogenation of alkynes. With a more σ-donating methyl group, the former complexes have more electron-rich palladium centers that favor oxidative addition of H2. As a result, palladium particles are more quickly released from the metal complexes, leading to higher catalytic activities. The fact that palladium complexes bearing a tBu-substituted pincer ligand are inferior catalysts can be explained by their lower tendency to react with H2 to generate sterically crowded 6-coordinate Pd(IV) intermediates. Finally, styrene produced in eqn (5) and (6) is a consequence of hydrogenation of HCCPh catalyzed by palladium particles that are formed from decomposition of 3a–c as well as 4b.
Conclusions
We have investigated stoichiometric reactions between palladium POCOP-pincer hydride complexes and phenylacetylene. Analogous to other reactions involving transition metal hydrides and terminal alkynes, both palladium alkynyl and alkenyl complexes along with styrene have been identified as products. Through isolation of these metal species and examination of their reactivity, we have discounted the commonly proposed mechanism for alkene formation (in this case styrene), which is via a C–H bond exchange process between the alkenyl complexes and phenylacetylene. Instead, we have provided evidence supporting that leached palladium particles are responsible for the reduction of phenylacetylene to styrene. We have also shown that in catalytic hydrogenation of alkynes, palladium methyl or alkynyl complexes are merely the precursors to the active palladium particles. Unlike cross-coupling reactions or related catalytic processes where colloidal metal particles are often produced under harsh conditions (high temperature and in the presence of a base), palladium particles are released from the POCOP-pincer complexes at room temperature under atmospheric or lower H2 pressure. We expect these results have important implications in designing homogeneous palladium catalysts for the reduction of alkynes.
Experimental section
General procedures
Unless otherwise mentioned, all the organometallic compounds were prepared and handled under an argon atmosphere using standard glovebox and Schlenk techniques. Dry and oxygen-free solvents for carrying out syntheses (THF, pentane, and toluene) were collected from an Innovative Technology solvent purification system. Solvents for column chromatography (CH2Cl2 and hexanes) were purchased from commercial sources and used without purification or degassing. Benzene-d6 was distilled from Na and benzophenone under an argon atmosphere. Unless otherwise mentioned, HCCPh was freshly distilled prior to use. [2,6-(Ph2PO)2C6H3]PdCl (1d)18 and trans-β-iodostyrene34 were prepared as described in the literature.
Synthesis of [2,6-(tBu2PO)2C6H3]PdCl (1a).
This compound was prepared according to a slightly modified procedure from the one described by Koridze and co-workers.17 A suspension of NaH (500 mg, 21 mmol) in THF (20 mL) was added dropwise to a Schlenk flask containing a solution of resorcinol (1.10 g, 10 mmol) in THF (40 mL). The resulting mixture was refluxed for 3 h. After cooling to room temperature, a solution of tBu2PCl (3.98 mL, 21 mmol) in THF (20 mL) was added and the reaction mixture was refluxed again for 3 h. The volatiles were removed under vacuum and the residue was extracted with pentane (3 × 35 mL). The combined pentane solution was pumped to dryness to give a white solid, which was mixed with PdCl2 (1.77 g, 10 mmol) in 60 mL of THF and then heated to reflux for 36 h. Upon cooling, the insolubles were filtered off, resulting in a faint yellow solution, which was concentrated under vacuum to yield the crude product. Further purification was performed by column chromatography (eluted with 1:1 CH2Cl2–hexanes) to provide 1a as a white solid (4.30 g, 80% yield). 1H NMR (400 MHz, CDCl3, δ): 1.45 (t, JP–H = 8.0 Hz, CH3, 36H), 6.56 (d, JH–H = 8.0 Hz, ArH, 2H), 6.97 (t, JH–H = 8.0 Hz, ArH, 1H). 13C{1H} NMR (101 MHz, CDCl3, δ): 27.7 (t, JP–C = 4.0 Hz, CH3), 39.6 (t, JP–C = 7.1 Hz, C(CH3)3), 105.7 (t, JP–C = 7.1 Hz, ArC), 127.6 (s, ArC), 129.9 (s, ArC), 167.1 (t, JP–C = 6.1 Hz, ArC). 31P{1H} NMR (162 MHz, CDCl3, δ): 192.2 (s).
Synthesis of [2,6-(cPe2PO)2C6H3]PdCl (1c).
A mixture of resorcinol (275 mg, 2.5 mmol) and triethylamine (1.40 mL, 10 mmol) in 40 mL of toluene was stirred at room temperature for 15 min, after which cPe2PCl (1.0 mL, 5.2 mmol) was added, and the mixture was stirred for another 15 min. Following the addition of PdCl2 (443 mg, 2.5 mmol), the reaction mixture was refluxed for 36 h. Upon cooling, the insolubles were filtered off, and the resulting solution was concentrated under vacuum to give the crude product. Washing the solid with pentane (10 mL) yielded analytically pure 1c as a white solid (1.10 g, 75% yield). 1H NMR (400 MHz, CDCl3, δ): 1.58–1.64 (m, CH2, 8H), 1.76–1.80 (m, CH2, 8H), 1.88–2.02 (m, CH2, 16H), 2.53–2.61 (m, PCH, 4H), 6.51 (d, JH–H = 8.0 Hz, ArH, 2H), 6.95 (t, JH–H = 8.0 Hz, ArH, 1H). 13C{1H} NMR (101 MHz, CDCl3, δ): 26.5 (t, JP–C = 3.5 Hz, CH2), 26.6 (t, JP–C = 4.5 Hz, CH2), 27.5 (t, JP–C = 4.2 Hz, CH2), 28.4 (s, CH2), 39.8 (t, JP–C = 12.9 Hz, CH), 106.0 (t, JP–C = 7.2 Hz, ArC), 128.1 (s, ArC), 129.4 (t, JP–C = 2.7 Hz, ArC), 166.1 (t, JP–C = 6.7 Hz, ArC). 31P{1H} NMR (162 MHz, CDCl3, δ): 177.9 (s). Anal. Calcd for C26H39P2O2PdCl: C, 53.16; H, 6.69; Cl, 6.04. Found: C, 53.26; H, 6.86; Cl, 5.93.
Synthesis of [2,6-(iPr2PO)2C6H3]PdCl (1b).
This compound was prepared in 74% yield by a procedure similar to that used for 1c. 1H and 31P NMR data are consistent with the reported values.11b
Synthesis of [2,6-(tBu2PO)2C6H3]PdH (2a).
The mixture of 1a (300 mg, 0.56 mmol) and LiAlH4 (317 mg, 8.3 mmol) in 20 mL of toluene was stirred at room temperature for 48 h. The resulting mixture was filtered through a short plug of Celite to give a colorless solution. After the solvent was evaporated under vacuum, the desired hydride 2a was isolated as a white solid (188 mg, 67% yield). 1H NMR (400 MHz, C6D6, δ): −2.48 (t, JP–H = 16.0 Hz, PdH, 1H), 1.28 (t, JP–H = 8.0 Hz, CH3, 36H), 6.88 (d, JH–H = 8.0 Hz, ArH, 2H), 7.01 (t, JH–H = 8.0 Hz, ArH, 1H). 13C{1H} NMR (101 MHz, C6D6, δ): 28.3 (t, JP–C = 4.0 Hz, CH3), 38.2 (t, JP–C = 8.1 Hz, C(CH3)3), 105.3 (t, JP–C = 7.1 Hz, ArC), 128.2 (s, ArC), 145.1 (t, JP–C = 6.1 Hz, ArC), 166.8 (t, JP–C = 6.1 Hz, ArC). 31P{1H} NMR (162 MHz, C6D6, δ): 213.8 (s). ATR-IR (solid): ν(Pd–H) = 1756 cm−1. Anal. Calcd for C22H40P2O2Pd: C, 52.33; H, 7.99. Found: C, 52.35; H, 8.00.
Synthesis of [2,6-(iPr2PO)2C6H3]PdH (2b).
A 1.0 M solution of LiBEt3H in THF (2.1 mL, 2.1 mmol) was added dropwise to a chilled (−78 °C) Schlenk flask containing a THF (5 mL) solution of 1b (1.0 g, 2.1 mmol). The reaction mixture was stirred at −78 °C for 1 h, followed by evaporation of the solvent while keeping the flask cold (0 °C). The residue was extracted with pentane (2 × 30 mL), and the combined pentane solution was concentrated to 2 mL. Faint yellow crystals were obtained within 24 h when the solution was kept at −35 °C (400 mg, 42% yield). 1H NMR (400 MHz, C6D6, δ): −2.40 (t, JP–H = 20.8 Hz, PdH, 1H), 1.08–1.18 (m, CH3, 24H), 2.05–2.08 (m, CH, 4H), 6.90 (d, JH–H = 7.6 Hz, ArH, 2H), 7.04 (t, JH–H = 7.6 Hz, ArH, 1H). 13C{1H} NMR (101 MHz, C6D6, δ): 17.3 (s, CH3), 18.6 (t, JP–C = 5.1 Hz, CH3), 29.4 (t, JP–C = 12.1 Hz, CH), 105.3 (t, JP–C = 7.1 Hz, ArC), 128.6 (s, ArC), 144.2 (t, JP–C = 6.1 Hz, ArC), 165.9 (t, JP–C = 6.1 Hz, ArC). 31P{1H} NMR (162 MHz, C6D6, δ): 202.1 (s). ATR-IR (solid): ν(Pd–H) = 1765 cm−1. Anal. Calcd for C18H32P2O2Pd: C, 48.17; H, 7.19. Found: C, 48.35; H, 7.34.
Synthesis of [2,6-(cPe2PO)2C6H3]PdH (2c).
This compound was prepared in 44% yield by a procedure similar to that used for 2b. 1H NMR (400 MHz, C6D6, δ): −2.37 (t, JP–H = 20.0 Hz, PdH, 1H), 1.33–1.38 (m, CH2, 8H), 1.53–1.64 (m, CH2, 8H), 1.71–1.79 (m, CH2, 8H), 1.88–1.95 (m, CH2, 8H), 2.26–2.34 (m, PCH, 4H), 6.93 (d, JH–H = 8.0 Hz, ArH, 2H), 7.05 (t, JH–H = 8.0 Hz, ArH, 1H). 13C{1H} NMR (101 MHz, C6D6, δ): 26.6 (t, JP–C = 3.6 Hz, CH2), 26.8 (t, JP–C = 4.2 Hz, CH2), 29.2 (s, CH2), 29.3 (t, JP–C = 5.8 Hz, CH2), 40.6 (t, JP–C = 13.7 Hz, CH), 105.3 (t, JP–C = 6.8 Hz, ArC), 128.7 (s, ArC), 144.5 (t, JP–C = 6.2 Hz, ArC), 165.9 (t, JP–C = 6.8 Hz, ArC). 31P{1H} NMR (162 MHz, C6D6, δ): 195.7 (s). ATR-IR (solid): ν(Pd–H) = 1755 cm−1. Anal. Calcd for C26H40P2O2Pd: C, 56.47; H, 7.29. Found: C, 56.17; H, 7.09.
Procedures for a stoichiometric reaction between a palladium hydride complex and phenylacetylene
In a J. Young NMR tube, palladium hydride 2a (12.5 μmol) was mixed with phenylacetylene (12.5 μmol) and 1,4-dioxane (NMR internal standard, 25 μmol) in 0.4 mL of C6D6. The progress of the reaction at an appropriate temperature was monitored by 1H and 31P{1H} NMR spectroscopy. NMR yields for phenylacetylide complex 3a and styrene were calculated based on the integrations of their proton resonances versus the integration of CH2 resonance (3.35 ppm) of the internal standard. Selected 1H NMR data (400 MHz, C6D6, δ) for styrene: 5.07 (d, JH–H = 10.8 Hz), 5.60 (d, JH–H = 17.6 Hz), 6.58 (dd, JH–H = 17.6 and 10.8 Hz). The reactions of other palladium hydride complexes with phenylacetylene were carried out under the same conditions (temperatures, concentrations, etc.).
Synthesis of [2,6-(tBu2PO)2C6H3]PdC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) CPh (3a).
A 1.6 M solution of nBuLi in hexanes (0.80 mL, 1.28 mmol) was added dropwise to a chilled (−78 °C) Schlenk flask containing a pentane (4 mL) solution of phenylacetylene (154 μL, 1.4 mmol). The flask was gradually warmed to room temperature within 15 min. The resulting suspension was added slowly via cannula to a THF (16 mL) solution of 1a (300 mg, 0.56 mmol) at −78 °C. After stirring the mixture at room temperature for 1 h, the solvent was removed under vacuum. Extraction of the residue with pentane (2 × 30 mL) followed by evaporation of the solvent under vacuum gave the product as a white solid (250 mg, 74% yield). 1H NMR (400 MHz, CDCl3, δ): 1.46 (t, JP–H = 8.0 Hz, CH3, 36H), 6.59 (d, JH–H = 8.0 Hz, ArH, 2H), 6.96 (t, JH–H = 8.0 Hz, ArH, 1H), 7.09 (t, JH–H = 8.0 Hz, ArH, 1H), 7.20 (t, JH–H = 8.0 Hz, ArH, 2H), 7.28 (d, JH–H = 8.0 Hz, ArH, 2H). 13C{1H} NMR (101 MHz, CDCl3, δ): 28.0 (t, JP–C = 3.0 Hz, CH3), 39.5 (t, JP–C = 8.1 Hz, C(CH3)3), 105.0 (t, JP–C = 7.1 Hz, ArC), 113.2 (t, JP–C = 16.2 Hz, CCPh), 117.8 (s, CCPh), 124.9 (s, ArC), 127.7 (s, ArC), 127.9 (s, ArC), 129.1 (s, ArC), 130.9 (s, ArC), 138.5 (t, JP–C = 3.0 Hz, ArC), 167.3 (t, JP–C = 6.1 Hz, ArC). 31P{1H} NMR (162 MHz, CDCl3, δ): 200.2 (s). ATR-IR (solid): ν(CC) = 2100 cm−1. Anal. Calcd for C30H44P2O2Pd: C, 59.55; H, 7.33. Found: C, 59.26; H, 7.26.
CPh (3a).
A 1.6 M solution of nBuLi in hexanes (0.80 mL, 1.28 mmol) was added dropwise to a chilled (−78 °C) Schlenk flask containing a pentane (4 mL) solution of phenylacetylene (154 μL, 1.4 mmol). The flask was gradually warmed to room temperature within 15 min. The resulting suspension was added slowly via cannula to a THF (16 mL) solution of 1a (300 mg, 0.56 mmol) at −78 °C. After stirring the mixture at room temperature for 1 h, the solvent was removed under vacuum. Extraction of the residue with pentane (2 × 30 mL) followed by evaporation of the solvent under vacuum gave the product as a white solid (250 mg, 74% yield). 1H NMR (400 MHz, CDCl3, δ): 1.46 (t, JP–H = 8.0 Hz, CH3, 36H), 6.59 (d, JH–H = 8.0 Hz, ArH, 2H), 6.96 (t, JH–H = 8.0 Hz, ArH, 1H), 7.09 (t, JH–H = 8.0 Hz, ArH, 1H), 7.20 (t, JH–H = 8.0 Hz, ArH, 2H), 7.28 (d, JH–H = 8.0 Hz, ArH, 2H). 13C{1H} NMR (101 MHz, CDCl3, δ): 28.0 (t, JP–C = 3.0 Hz, CH3), 39.5 (t, JP–C = 8.1 Hz, C(CH3)3), 105.0 (t, JP–C = 7.1 Hz, ArC), 113.2 (t, JP–C = 16.2 Hz, CCPh), 117.8 (s, CCPh), 124.9 (s, ArC), 127.7 (s, ArC), 127.9 (s, ArC), 129.1 (s, ArC), 130.9 (s, ArC), 138.5 (t, JP–C = 3.0 Hz, ArC), 167.3 (t, JP–C = 6.1 Hz, ArC). 31P{1H} NMR (162 MHz, CDCl3, δ): 200.2 (s). ATR-IR (solid): ν(CC) = 2100 cm−1. Anal. Calcd for C30H44P2O2Pd: C, 59.55; H, 7.33. Found: C, 59.26; H, 7.26.
Synthesis of [2,6-(iPr2PO)2C6H3]PdCCPh (3b).
This compound was prepared in 71% yield by a procedure similar to that used for 3a. 1H NMR (400 MHz, CDCl3, δ): 1.28–1.34 (m, CH3, 12H), 1.39–1.45 (m, CH3, 12H), 2.47–2.54 (m, CH, 4H), 6.60 (d, JH–H = 8.0 Hz, ArH, 2H), 7.00 (t, JH–H = 8.0 Hz, ArH, 1H), 7.10 (t, JH–H = 7.2 Hz, ArH, 1H), 7.20 (t, JH–H = 7.2 Hz, ArH, 2H), 7.31 (d, JH–H = 7.6 Hz, ArH, 2H). 13C{1H} NMR (101 MHz, CDCl3, δ): 17.1 (s, CH3), 17.8 (t, JP–C = 3.2 Hz, CH3), 29.3 (t, JP–C = 12.2 Hz, CH), 105.2 (t, JP–C = 7.0 Hz, ArC), 109.7 (t, JP–C = 17.5 Hz, CCPh), 118.4 (s, CCPh), 125.1 (s, ArC), 127.9 (s, ArC), 128.2 (s, ArC), 128.6 (s, ArC), 131.2 (s, ArC), 137.7 (t, JP–C = 3.8 Hz, ArC), 166.4 (t, JP–C = 6.5 Hz, ArC). 31P{1H} NMR (162 MHz, CDCl3, δ): 192.7 (s). ATR-IR (solid): ν(CC) = 2085 cm−1. Anal. Calcd for C26H36P2O2Pd: C, 56.89; H, 6.61. Found: C, 56.74; H, 6.47.
Synthesis of [2,6-(cPe2PO)2C6H3]PdCCPh (3c).
This compound was prepared in 76% yield by a procedure similar to that used for 3a except that the extraction of the residue was performed using toluene instead of pentane. The use of pentane for extraction resulted in a much lower isolated yield. 1H NMR (400 MHz, CDCl3, δ): 1.52–1.72 (m, CH2, 8H), 1.76–1.85 (m, CH2, 12H), 1.87–2.08 (m, CH2, 8H), 2.12–2.27 (m, CH2, 4H), 2.56–2.65 (m, PCH, 4H), 6.56 (d, JH–H = 8.0 Hz, ArH, 2H), 6.97 (t, JH–H = 8.0 Hz, ArH, 1H), 7.09 (t, JH–H = 8.0 Hz, ArH, 1H), 7.20 (t, JH–H = 8.0 Hz, ArH, 2H), 7.27 (d, JH–H = 8.0 Hz, ArH, 2H). 13C{1H} NMR (101 MHz, CDCl3, δ): 26.5 (t, JP–C = 3.2 Hz, CH2), 26.7 (t, JP–C = 4.2 Hz, CH2), 27.7 (t, JP–C = 4.1 Hz, CH2), 28.8 (s, CH2), 40.3 (t, JP–C = 13.4 Hz, CH), 105.1 (t, JP–C = 7.0 Hz, ArC), 111.3 (t, JP–C = 17.2 Hz, CCPh), 116.9 (s, CCPh), 124.9 (s, ArC), 127.9 (s, ArC), 128.2 (s, ArC), 128.9 (s, ArC), 131.0 (s, ArC), 137.8 (t, JP–C = 4.3 Hz, ArC), 166.2 (t, JP–C = 6.6 Hz, ArC). 31P{1H} NMR (162 MHz, CDCl3, δ): 183.8 (s). ATR-IR (solid): ν(CC) = 2095 cm−1. Anal. Calcd for C34H44P2O2Pd: C, 62.53; H, 6.79. Found: C, 62.25; H, 6.83.
Synthesis of (E)-[2,6-(iPr2PO)2C6H3]PdCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CHPh (4b).
A 1.6 M solution of nBuLi in hexanes (0.32 mL, 0.51 mmol) was added dropwise to a chilled (−78 °C) Schlenk flask containing a pentane (5 mL) solution of trans-β-iodostyrene (143 mg, 0.62 mmol). The reaction mixture was warmed to room temperature and stirred for 15 min. The resulting suspension was transferred via cannula to a THF (15 mL) solution of 1b (200 mg, 0.42 mmol) at −78 °C. After stirring the reaction mixture at room temperature for 15 min, the volatiles were removed under vacuum. Extraction of the residue with pentane (2 × 30 mL) followed by evaporation of the solvent produced 4b as a white solid. The product was further purified by recrystallization from pentane at −35 °C (95 mg, 41% yield). 1H NMR (400 MHz, C6D6, δ): 1.06–1.14 (m, CH3, 24H), 2.02–2.09 (m, CH(CH3)2, 4H), 6.85 (d, JH–H = 8.0 Hz, ArH, 2H), 6.97–7.08 (m, ArH + CHCHPh, 3H), 7.30 (t, JH–H = 8.0 Hz, ArH, 2H), 7.57 (d, JH–H = 8.0 Hz, ArH, 2H), 8.20 (dt, JH–H = 20.0 Hz, JP–H = 4.0 Hz, CHCHPh, 1H). 13C{1H} NMR (101 MHz, C6D6, δ): 16.9 (s, CH3), 17.6 (t, JP–C = 4.0 Hz, CH3), 28.7 (t, JP–C = 12.1 Hz, CH), 105.5 (t, JP–C = 7.1 Hz, ArC), 124.9 (s, ArC), 125.1 (s, ArC), 128.6 (s, ArC), 128.8 (s, ArC), 139.0 (t, JP–C = 4.0 Hz, PdCHCH), 140.9 (t, JP–C = 5.1 Hz, ArC), 142.7 (s, ArC), 150.9 (t, JP–C = 13.1 Hz, PdCHCH), 166.2 (t, JP–C = 6.1 Hz, ArC). 31P{1H} NMR (162 MHz, C6D6, δ): 187.2 (s). Anal. Calcd for C26H38P2O2Pd: C, 56.68; H, 6.95. Found: C, 56.54; H, 6.80.
CHPh (4b).
A 1.6 M solution of nBuLi in hexanes (0.32 mL, 0.51 mmol) was added dropwise to a chilled (−78 °C) Schlenk flask containing a pentane (5 mL) solution of trans-β-iodostyrene (143 mg, 0.62 mmol). The reaction mixture was warmed to room temperature and stirred for 15 min. The resulting suspension was transferred via cannula to a THF (15 mL) solution of 1b (200 mg, 0.42 mmol) at −78 °C. After stirring the reaction mixture at room temperature for 15 min, the volatiles were removed under vacuum. Extraction of the residue with pentane (2 × 30 mL) followed by evaporation of the solvent produced 4b as a white solid. The product was further purified by recrystallization from pentane at −35 °C (95 mg, 41% yield). 1H NMR (400 MHz, C6D6, δ): 1.06–1.14 (m, CH3, 24H), 2.02–2.09 (m, CH(CH3)2, 4H), 6.85 (d, JH–H = 8.0 Hz, ArH, 2H), 6.97–7.08 (m, ArH + CHCHPh, 3H), 7.30 (t, JH–H = 8.0 Hz, ArH, 2H), 7.57 (d, JH–H = 8.0 Hz, ArH, 2H), 8.20 (dt, JH–H = 20.0 Hz, JP–H = 4.0 Hz, CHCHPh, 1H). 13C{1H} NMR (101 MHz, C6D6, δ): 16.9 (s, CH3), 17.6 (t, JP–C = 4.0 Hz, CH3), 28.7 (t, JP–C = 12.1 Hz, CH), 105.5 (t, JP–C = 7.1 Hz, ArC), 124.9 (s, ArC), 125.1 (s, ArC), 128.6 (s, ArC), 128.8 (s, ArC), 139.0 (t, JP–C = 4.0 Hz, PdCHCH), 140.9 (t, JP–C = 5.1 Hz, ArC), 142.7 (s, ArC), 150.9 (t, JP–C = 13.1 Hz, PdCHCH), 166.2 (t, JP–C = 6.1 Hz, ArC). 31P{1H} NMR (162 MHz, C6D6, δ): 187.2 (s). Anal. Calcd for C26H38P2O2Pd: C, 56.68; H, 6.95. Found: C, 56.54; H, 6.80.
Synthesis of [2,6-(tBu2PO)2C6H3]PdCH3 (5a).
A 1.6 M solution of CH3Li in diethyl ether (375 μL, 0.60 mmol) was added slowly to a chilled (−78 °C) Schlenk flask containing a THF (20 mL) solution of 1a (270 mg, 0.50 mmol). The flask was warmed to room temperature and the reaction mixture was stirred at this temperature for 2 h. The volatiles were removed under vacuum and the residue was extracted with pentane (2 × 30 mL). Evaporating the solvent from the combined extracts yielded 5a as a white solid (240 mg, 92% yield). 1H NMR (400 MHz, CDCl3, δ): −0.01 (t, JP–H = 4.0 Hz, PdCH3, 3H), 1.33 (t, JP–H = 8.0 Hz, C(CH3)3, 36H), 6.57 (d, JH–H = 8.0 Hz, ArH, 2H), 6.91 (t, JH–H = 8.0 Hz, ArH, 1H). 13C{1H} NMR (101 MHz, CDCl3, δ): −17.8 (t, JP–C = 10.1 Hz, PdCH3), 28.0 (t, JP–C = 4.4 Hz, C(CH3)3), 39.4 (t, JP–C = 7.1 Hz, C(CH3)3), 104.4 (t, JP–C = 6.1 Hz, ArC), 126.7 (s, ArC), 143.1 (t, JP–C = 7.1 Hz, ArC), 165.9 (t, JP–C = 6.1 Hz, ArC). 31P{1H} NMR (162 MHz, CDCl3, δ): 194.1 (s). Anal. Calcd for C23H42P2O2Pd: C, 53.23; H, 8.16. Found: C, 52.98; H, 8.01.
Synthesis of [2,6-(iPr2PO)2C6H3]PdCH3 (5b).
This compound was prepared in 67% yield by a procedure similar to that used for 5a. 1H NMR (400 MHz, C6D6, δ): 0.32 (t, JP–H = 5.2 Hz, PdCH3, 3H), 1.08–1.15 (m, CH(CH3)2, 24H), 2.05–2.12 (m, PCH, 4H), 6.86 (d, JH–H = 8.0 Hz, ArH, 2H), 6.99 (t, JH–H = 8.0 Hz, ArH, 1H). 13C{1H} NMR (101 MHz, C6D6, δ): −20.0 (t, JP–C = 10.1 Hz, PdCH3), 17.1 (s, CH(CH3)2), 17.7 (t, JP–C = 3.0 Hz, CH(CH3)2), 29.1 (t, JP–C = 11.1 Hz, CH(CH3)2), 105.3 (t, JP–C = 6.1 Hz, ArC), 127.9 (s, ArC), 143.2 (t, JP–C = 7.1 Hz, ArC), 165.7 (t, JP–C = 7.1 Hz, ArC). 31P{1H} NMR (162 MHz, C6D6, δ): 188.1 (s). Anal. Calcd for C19H34P2O2Pd: C, 49.31; H, 7.40. Found: C, 49.20; H, 7.38.
Synthesis of [2,6-(cPe2PO)2C6H3]PdCH3 (5c).
This compound was prepared in a quantitative yield by a procedure similar to that used for 5a. 1H NMR (400 MHz, C6D6, δ): 0.37 (t, JP–H = 5.2 Hz, PdCH3, 3H), 1.31–1.44 (m, CH2, 8H), 1.58–1.66 (m, CH2, 8H), 1.71–1.80 (m, CH2, 8H), 1.84–2.02 (m, CH2, 8H), 2.31–2.40 (m, PCH, 4H), 6.89 (d, JH–H = 8.0 Hz, ArH, 2H), 7.01 (t, JH–H = 8.0 Hz, ArH, 1H). 13C{1H} NMR (101 MHz, C6D6, δ): −18.6 (t, JP–C = 10.1 Hz, PdCH3), 26.7 (t, JP–C = 4.0 Hz, CH2), 26.8 (t, JP–C = 4.0 Hz, CH2), 28.1 (t, JP–C = 4.0 Hz, CH2), 28.9 (s, CH2), 40.5 (t, JP–C = 12.7 Hz, PCH), 105.4 (t, JP–C = 6.9 Hz, ArC), 128.0 (s, ArC), 143.4 (t, JP–C = 6.8 Hz, ArC), 165.6 (t, JP–C = 7.0 Hz, ArH). 31P{1H} NMR (162 MHz, C6D6, δ): 177.9 (s). Anal. Calcd for C27H42P2O2Pd: C, 57.20; H, 7.47. Found: C, 57.46; H, 7.55.
Procedures for catalytic hydrogenation of alkynes
In a J. Young NMR tube, an alkyne substrate (62.5 μmol), a palladium catalyst (12.5 μmol, 20 mol%), and 1,4-dioxane (25 μmol) were mixed in 0.4 mL of C6D6. The mixture was degassed by a freeze–pump–thaw cycle and then placed under 1 atm of H2 at room temperature. The reaction was monitored by 1H NMR spectroscopy and the NMR yields for the hydrogenation products were calculated based on integrations of individual peaks. The resonances for trans-stilbene (from the hydrogenation of diphenylacetylene) were obscured by other resonances, and therefore its yield was obtained from GC using hexamethylbenzene as an internal standard.
X-ray structure determinations
Single crystals of 1a and 1c were obtained from recrystallization in toluene–pentane and CH2Cl2–pentane, respectively. Single crystals of 2a were obtained from cold (−30 °C) toluene solution. Single crystals of 2c, 3a, 4b and 5b were obtained from cold (−30 °C for 2c and 4b, −5 °C for 3a and 5b) pentane solutions. Crystal data collection and refinement parameters can be found in ESI.† Intensity data for 1a, 2a, 2c, 4b and 5b were collected at 150 K on a Bruker SMART6000 CCD diffractometer using graphite-monochromated Cu Kα radiation, λ = 1.54178 Å. Data for 1c and 3a were collected at Beamline 11.3.1 at Advanced Light Source at Lawrence Berkeley National Laboratory, λ = 0.8856 Å and 0.7749 Å, respectively. The data frames were processed using the program SAINT. The data were corrected for decay, Lorentz, and polarization effects as well as absorption and beam corrections based on the multi-scan technique. The structures were solved by a combination of direct methods in SHELXTL and the difference Fourier technique and refined by full-matrix least-squares procedures. Non-hydrogen atoms were refined with anisotropic displacement parameters. The hydride in 2a and 2c was located directly from the difference map and the coordinates refined. The remaining H-atoms were calculated and treated with a riding model. No solvent of crystallization was present in the lattice for any of the structures. Typical disorder was observed in two of the cyclopentyl rings of 2c; a suitable multi-component disorder model was applied. Three independent molecules were found in the lattice of 3a, and two of these molecules showed some disorder for the tBu carbons. The crystal structures for 1a, 1c, 2a, 2c, 3a, 4b and 5b have been deposited at the Cambridge Crystallographic Data Centre (CCDC) and allocated the deposition numbers CCDC 965919–965925.
Acknowledgements
We thank the National Science Foundation (CHE-0952083), Alfred P. Sloan Foundation (research fellowship to H.G.), and the University of Cincinnati Honors Program for supporting this research. We also thank Padmanabh Joshi and Prof. Peng Zhang at the University of Cincinnati for their assistance with the DLS experiments. Crystallographic data were collected on a Bruker SMART6000 diffractometer (funded by an NSF-MRI grant; CHE-0215950), or through the SCrALS (Service Crystallography at Advanced Light Source) program at Beamline 11.3.1 at the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory (supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under contract no. DE-AC02-05CH11231).
Notes and references
-
J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, CA, 2010, pp. 368–370 Search PubMed.
-
(a) H. C. Clark and C. R. Milne, J. Organomet. Chem., 1978, 161, 51–59 CrossRef CAS;
(b) H. Werner, M. A. Esteruelas and H. Otto, Organometallics, 1986, 5, 2295–2299 CrossRef CAS;
(c) M. A. Esteruelas, L. A. Oro and C. Valero, Organometallics, 1995, 14, 3596–3599 CrossRef CAS;
(d) M. Bassetti, P. Casellato, M. P. Gamasa, J. Gimeno, C. González-Bernardo and B. Martín-Vaca, Organometallics, 1997, 16, 5470–5477 CrossRef CAS;
(e) A. V. Marchenko, H. Gérard, O. Eisenstein and K. G. Caulton, New J. Chem., 2001, 25, 1244–1255 RSC;
(f) J. Navarro, E. Sola, M. Martín, I. T. Dobrinovitch, F. J. Lahoz and L. A. Oro, Organometallics, 2004, 23, 1908–1917 CrossRef CAS;
(g) H. Xia, T. B. Wen, Q. Y. Hu, X. Wang, X. Chen, L. Y. Shek, I. D. Williams, K. S. Wong, G. K. L. Wong and G. Jia, Organometallics, 2005, 24, 562–569 CrossRef CAS.
- M. A. Esteruelas, F. J. Lahoz, J. A. Lopez, L. A. Oro, C. Schlünken, C. Valero and H. Werner, Organometallics, 1992, 11, 2034–2043 CrossRef CAS.
-
(a) C. Bianchini, P. Innocenti, A. Meli, M. Peruzzini, F. Zanobini and P. Zanello, Organometallics, 1990, 9, 2514–2522 CrossRef CAS;
(b) W. Weng, S. Parkin and O. V. Ozerov, Organometallics, 2006, 25, 5345–5354 CrossRef CAS;
(c) L. She, X. Li, H. Sun, J. Ding, M. Frey and H.-F. Klein, Organometallics, 2007, 26, 566–570 CrossRef CAS.
- D. S. Frohnapfel, P. S. White and J. L. Templeton, Organometallics, 2000, 19, 1497–1506 CrossRef CAS.
-
(a) M. I. Bruce, Chem. Rev., 1991, 91, 197–257 CrossRef CAS;
(b) C. Bruneau and P. H. Dixneuf, Acc. Chem. Res., 1999, 32, 311–323 CrossRef CAS;
(c) H. Werner, Coord. Chem. Rev., 2004, 248, 1693–1702 CrossRef CAS PubMed;
(d) H. Katayama and F. Ozawa, Coord. Chem. Rev., 2004, 248, 1703–1715 CrossRef CAS PubMed;
(e) J. A. Varela and C. Saá, Chem.–Eur. J., 2006, 12, 6450–6456 CrossRef CAS PubMed;
(f) C. Bruneau and P. H. Dixneuf, Angew. Chem., Int. Ed., 2006, 45, 2176–2203 CrossRef CAS PubMed.
-
(a) H. E. Selnau and J. S. Merola, J. Am. Chem. Soc., 1991, 113, 4008–4009 CrossRef CAS;
(b) X. Li, C. D. Incarvito and R. H. Crabtree, J. Am. Chem. Soc., 2003, 125, 3698–3699 CrossRef CAS PubMed;
(c) X. Li, T. Vogel, C. D. Incarvito and R. H. Crabtree, Organometallics, 2005, 24, 62–76 CrossRef.
-
(a) C. Bianchini, A. Meli, M. Peruzzini, F. Vizza and P. Frediani, Organometallics, 1990, 9, 1146–1155 CrossRef CAS;
(b) C. Bianchini, C. Bohanna, M. A. Esteruelas, P. Frediani, A. Meli, L. A. Oro and M. Peruzzini, Organometallics, 1992, 11, 3837–3844 CrossRef CAS;
(c) O. V. Ozerov, L. A. Watson, M. Pink, M.-H. Baik and K. G. Caulton, Organometallics, 2004, 23, 4934–4943 CrossRef CAS;
(d) G. Albertin, S. Antoniutti, A. Bacchi, G. Pelizzi and M. Tollon, J. Organomet. Chem., 2005, 690, 4573–4582 CrossRef CAS PubMed;
(e) R. Johansson and O. F. Wendt, Organometallics, 2007, 26, 2426–2430 CrossRef CAS.
- C. Slugovc, K. Mereiter, E. Zobetz, R. Schmid and K. Kirchner, Organometallics, 1996, 15, 5275–5277 CrossRef CAS.
-
(a) S. Chakraborty, J. A. Krause and H. Guan, Organometallics, 2009, 28, 582–586 CrossRef CAS;
(b) S. Chakraborty, J. Zhang, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2010, 132, 8872–8873 CrossRef CAS PubMed;
(c) S. Chakraborty, Y. J. Patel, J. A. Krause and H. Guan, Polyhedron, 2012, 32, 30–34 CrossRef CAS PubMed;
(d) S. Chakraborty, J. Zhang, Y. J. Patel, J. A. Krause and H. Guan, Inorg. Chem., 2013, 52, 37–47 CrossRef CAS PubMed.
-
(a) D. Morales-Morales, R. Redón, C. Yung and C. M. Jensen, Chem. Commun., 2000, 1619–1620 RSC;
(b) D. Morales-Morales, C. Grause, K. Kasaoka, R. Redón, R. E. Cramer and C. M. Jensen, Inorg. Chim. Acta, 2000, 300–302, 958–963 CrossRef CAS.
-
(a) C. J. Moulton and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1976, 1020–1024 RSC;
(b) L. Fan, B. M. Foxman and O. V. Ozerov, Organometallics, 2004, 23, 326–328 CrossRef CAS.
- M. C. Denney, N. A. Smythe, K. L. Cetto, R. A. Kemp and K. I. Goldberg, J. Am. Chem. Soc., 2006, 128, 2508–2509 CrossRef CAS PubMed.
-
(a) R. B. Lansing Jr., K. I. Goldberg and R. A. Kemp, Dalton Trans., 2011, 40, 8950–8958 RSC;
(b) H.-W. Suh, T. J. Schmeier, N. Hazari, R. A. Kemp and M. K. Takase, Organometallics, 2012, 31, 8225–8236 CrossRef CAS.
- R. Gerber, T. Fox and C. M. Frech, Chem.–Eur. J., 2010, 16, 6771–6775 CrossRef CAS PubMed.
- I. Göttker-Schnetmann, P. White and M. Brookhart, J. Am. Chem. Soc., 2004, 126, 1804–1811 CrossRef PubMed.
- A. V. Polukeev, S. A. Kuklin, P. V. Petrovskii, S. M. Peregudova, A. F. Smol'yakov, F. M. Dolgushin and A. A. Koridze, Dalton Trans., 2011, 40, 7201–7209 RSC.
- J.-F. Gong, Y.-H. Zhang, M.-P. Song and C. Xu, Organometallics, 2007, 26, 6487–6492 CrossRef CAS.
- L. M. Martínez-Prieto, C. Melero, D. del Río, P. Palma, J. Cámpora and E. Álvarez, Organometallics, 2012, 31, 1425–1438 CrossRef.
- H. Salem, L. J. W. Shimon, Y. Diskin-Posner, G. Leitus, Y. Ben-David and D. Milstein, Organometallics, 2009, 28, 4791–4806 CrossRef CAS.
- J. Zhang, C. M. Medley, J. A. Krause and H. Guan, Organometallics, 2010, 29, 6393–6401 CrossRef CAS.
- Two of the cyclopentyl rings show some disorder. A multi-component disorder model is presented.
-
(a) H. Rimml and L. M. Venanzi, J. Organomet. Chem., 1984, 260, C52–C54 CrossRef CAS;
(b) B. J. Boro, E. N. Duesler, K. I. Goldberg and R. A. Kemp, Inorg. Chem., 2009, 48, 5081–5087 CrossRef CAS PubMed;
(c) N. Grüger, H. Wadepohl and L. H. Gade, Dalton Trans., 2012, 41, 14028–14030 RSC.
- Phenylacetylene from commercial sources sometimes shows light yellow color, but freshly distilled one should be colorless.
- Three independent molecules crystallize in the lattice. Only one molecule is shown here.
-
(a) G. Erker, W. Frömberg, R. Benn, R. Mynott, K. Angermund and C. Krüger, Organometallics, 1989, 8, 911–920 CrossRef CAS;
(b) A. B. Salah and D. Zargarian, Dalton Trans., 2011, 40, 8977–8985 RSC.
- A. K. Dash, J. Q. Wang and M. S. Eisen, Organometallics, 1999, 18, 4724–4741 CrossRef CAS.
- For recent reviews on heterogeneous palladium systems for catalytic hydrogenation of alkynes, see:
(a)
H. Arnold, F. Döbert and J. Gaube, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH, Weinheim, 2008, vol. 7, pp. 3266–3284 Search PubMed;
(b) M. Crespo-Quesada, F. Cárdenas-Lizana, A.-L. Dessimoz and L. Kiwi-Minsker, ACS Catal., 2012, 2, 1773–1786 CrossRef CAS;
(c) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114 DOI:10.1021/cr400133p , ASAP.
-
(a) A. M. Kluwer, T. S. Koblenz, T. Jonischkeit, K. Woelk and C. J. Elsevier, J. Am. Chem. Soc., 2005, 127, 15470–15480 CrossRef CAS PubMed;
(b) J. López-Serrano, S. B. Duckett, J. P. Dunne, C. Godard and A. C. Whitwood, Dalton Trans., 2008, 4270–4281 RSC;
(c) J. Broggi, V. Jurčík, O. Songis, A. Poater, L. Cavallo, A. M. Z. Slawin and C. S. J. Cazin, J. Am. Chem. Soc., 2013, 135, 4588–4591 CrossRef CAS PubMed.
- For recent examples on using a mercury test to discern the nature of the active species, see:
(a) M. C. Lipke, R. A. Woloszynek, L. Ma and J. D. Protasiewicz, Organometallics, 2009, 28, 188–196 CrossRef CAS;
(b) P. Singh, D. Das, O. Prakash and A. K. Singh, Inorg. Chim. Acta, 2013, 394, 77–84 CrossRef CAS PubMed.
- Catalytic hydrogenation of alkynes in toluene gave similar results as those obtained in Tables 1 and 2.
-
(a) M. R. Eberhard, Org. Lett., 2004, 6, 2125–2128 CrossRef CAS PubMed;
(b) D. Olsson, P. Nilsson, M. El Masnaouy and O. F. Wendt, Dalton Trans., 2005, 1924–1929 RSC;
(c) W. J. Sommer, K. Yu, J. S. Sears, Y. Ji, X. Zheng, R. J. Davis, C. D. Sherrill, C. W. Jones and M. Weck, Organometallics, 2005, 24, 4351–4361 CrossRef CAS;
(d) J. G. de Vries, Dalton Trans., 2006, 421–429 RSC;
(e) J. L. Bolliger, O. Blacque and C. M. Frech, Chem.–Eur. J., 2008, 14, 7969–7977 CrossRef CAS PubMed.
- M. L. Scheuermann, A. L. Rheingold and B. S. Williams, Organometallics, 2009, 28, 1613–1615 CrossRef CAS.
- J. J. Mousseau, J. A. Bull and A. B. Charette, Angew. Chem., Int. Ed., 2010, 49, 1115–1118 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of DLS experiments. CCDC 965919–965925 for 1a, 1c, 2a, 2c, 3a, 4b and 5b. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3qi00073g |
|
| This journal is © the Partner Organisations 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.