 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of an anti-metastatic ruthenium(III) prodrug on extracellular protein–protein interactions: studies by bio-layer interferometry†
Aviva
Levina
and
Peter A.
Lay
*
Room 307. School of Chemistry, F11, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: peter.lay@sydney.edu.au; Fax: +61-2-93513329; Tel: +61-2-93514269
First published on 16th December 2013
Abstract
In the first application of bio-layer interferometry in medicinal inorganic chemistry, Fe(III)–transferrin (Tf) binds strongly to Tf receptor 1 (TfR1), but an apo–Tf adduct of the anti-metastatic prodrug, NAMI-A does not bind specifically to TfR1. Binding of NAMI-A to albumin affects its interactions with other blood proteins, such as immunoglobulins.
Development of Ru-based anti-cancer prodrugs, particularly those with specific anti-metastatic (rather than cytotoxic) action, is currently one of the most active fields in medicinal inorganic chemistry, which has been promoted by the widespread clinical success of cisplatin.1,2 Typically, these metal complexes contain relatively labile ligands (such as chlorido) that are exchanged with donor groups of both intra- and extracellular biological macromolecules, which leads to kinetically inert metal–biomolecule adducts.2 The resulting structural alterations, which affect the interactions between biological macromolecules, are among the most general mechanisms of biological activity of metal complexes.1,2
Extracellular protein–protein interactions (PPIs), including high-affinity receptor-substrate binding and transient interactions of the cell surface with the extracellular matrix, provide an essential means of communication in any multi-cellular organism.3 In particular, a crucial role of extracellular interactions in the anti-metastatic activity of Ru complexes has been proposed.2,4 Bio-layer interferometry (BLI), which is based on measuring the changes in light reflectance from an optical surface that are caused by macromolecule binding,5 is an emerging tool for the studies of PPIs in cell-free systems.3 These studies have been greatly assisted by the availability of soluble recombinant proteins that represent the substrate-binding (extracellular) portions of common receptors.3 In addition to a multi-channel instrument (Octet®) that can be used for high-throughput screening of PPIs,3 a portable single-channel instrument (BLItz™), which is suitable for directed small-scale studies, has now become available.5 To our knowledge, no applications of BLI to studies of the biochemistry of protein adducts of metal-based prodrugs have been reported as yet.
One of the outstanding questions in medicinal inorganic chemistry is the role of transferrin (Tf, the main Fe transport protein in blood plasma) in the cellular uptake of “alternative” metal ions, including Ru that is derived from Ru anti-cancer prodrugs.6–10 Many biological and abiological metal ions bind to Tf with affinities that are comparable to those for Fe(III),6–8 but the resulting adducts are not necessarily recognized and taken up by the cell.9,10 Transport of essential Fe into cells involves the binding of Fe(III) ions by the two specific sites of Tf, which changes the conformation of the Tf molecule to allow its high-affinity binding to transferrin receptor 1 (TfR1) on the cell surface.11 The resulting Fe(III)–Tf–TfR1 complex is taken into the cell inside an endosome, where Fe(III) is released by a combination of changes in acidity (pH < 6) and yet unknown chelators, but the Tf–TfR1 complex remains intact.11 The complex is then returned to the cell surface (pH = 7.4), where it dissociates and releases Tf into extracellular space for more Fe(III) binding.11 Various stages of Fe(III)–Tf–TfR1 interactions have been studied in detail,11 using radiolabelling techniques,12 immunostaining assays,13 rapid kinetic measurements by electronic or fluorescent spectroscopy,8,14 and surface plasmon resonance (SPR) measurements.15 The latter technique is related to, but not identical with, BLI.5,15
Here we present the first application of BLI to studies of Tf–TfR1 interactions, using a BLItz instrument5 (see ESI† for experimental details). A (His)6-tagged recombinant human protein that represents the extracellular portion of TfR1 (Cys89-Phe760; Sino Biological, China) was loaded on a Ni(II)–NTA-coated optical probe (Fig. S1 in ESI;† NTA = nitrilotriacetate(3−)). The reaction of Fe(III)-loaded human Tf (1.0 μM Tf, 2.0 μM Fe(III)) with this probe at pH = 7.40 (20 mM HEPES,‡ 25 mM NaHCO3, 140 mM NaCl, 295 K) led to rapid Tf binding to TfR1, followed by slow dissociation when the probe was incubated with the buffer alone (red line in Fig. 1, segments B and C). Application of simulated endosomal conditions (100 mM MES,‡ 4.0 mM Na2edta,‡ 300 mM KCl, pH = 5.60)14 caused little change in the probe response within 5 min (segment D in Fig. 1), which is consistent with the Tf–TfR1 complex remaining intact while Fe(III) is released.11,14 A subsequent change to pH = 7.40 caused a rapid dissociation of the Tf–TfR1 complex (segment E in Fig. 1), in agreement with the literature data.11,14 Functional TfR1 remained attached to the probe, and could be re-loaded with Fe(III)–Tf several times without significant loss of performance (Fig. S2 in ESI†). The use of apo–Tf (1.0 μM) instead of Fe(III)–Tf resulted in a much lower response of the probe (black line in Fig. 1), although some binding was still observed due to the traces of Fe(III) in the buffer.12 To our knowledge, this is the first observation of the full Fe(III)–Tf–TfR1 receptor binding and dissociation cycle in a single experiment in cell-free systems. A comparison of normalised association and dissociation kinetic curves for Fe(III)–Tf and apo–Tf (segments B and C in Fig. 1) is shown in Fig. 2.
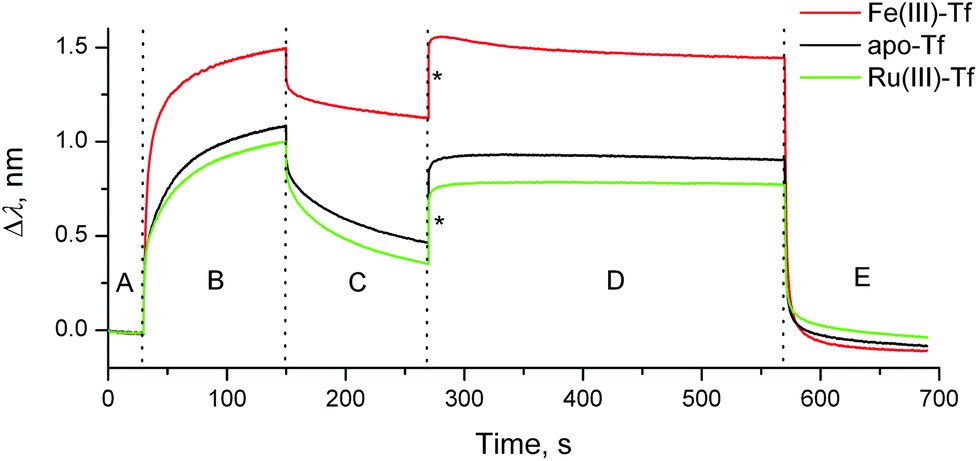 | ||
| Fig. 1 Typical BLI response curves (295 K) for the binding and dissociation of human Tf (1.0 μM) in the presence or absence of metal ions (2.0 μM) to recombinant human TfR1 immobilized on a Ni(II)–NTA probe (see ESI† for experimental details). Designations of the segments: (A) initial buffer background; (B) association of Tf to TfR1; (C) dissociation of Tf from TfR1 (buffer alone); (D) removal of metal ion under endosomal–mimetic conditions; and (E) dissociation of metal-free Tf from TfR1 under cell surface conditions.11,14 Buffer for segments (A–C) and (E): 20 mM HEPES, 25 mM NaHCO3, 140 mM NaCl, pH = 7.40. Buffer for segment (D): 100 mM MES, 4.0 mM Na2edta, 300 mM KCl, pH = 5.60.14 Jumps due to the changing ionic strengths of solutions are designated with asterisks. | ||
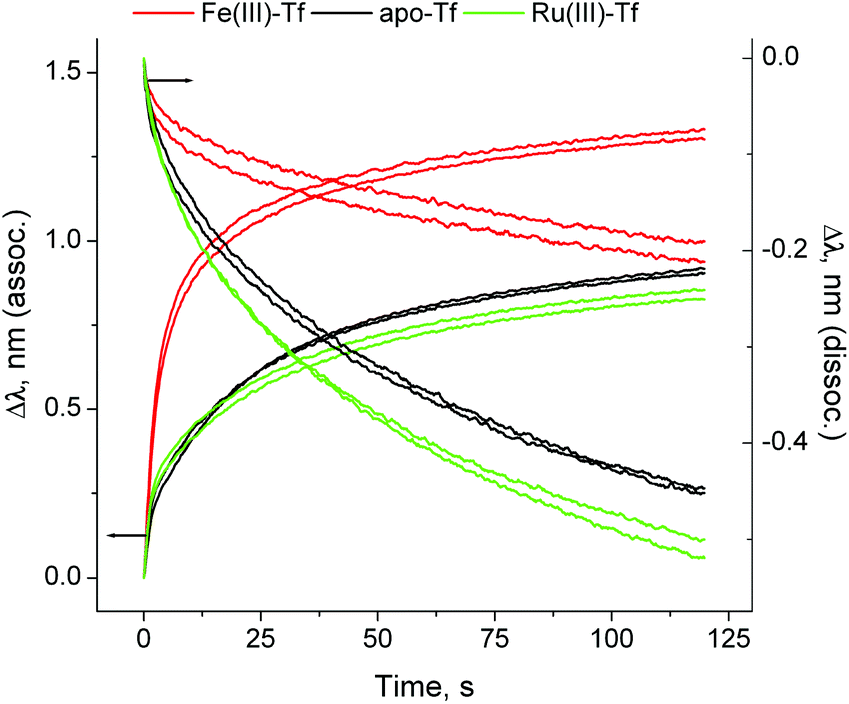 | ||
| Fig. 2 Normalised association and dissociation curves for the Tf–TfR1 interactions (corresponding to segments (B) and (C) in Fig. 1). Each condition is represented by two measurements, performed on different probes. Kinetic parameters are given in Table S1 and Fig. S3, ESI.† | ||
The dissociation constant (KD; a standard measure of the protein–protein binding affinity)11–15 for the Fe(III)–Tf–TfR1 interaction was calculated from segments B and C of the binding curve (red line in Fig. 1), using the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model (the only one currently available in the BLItz software),5 which led to KD = 12 ± 2 nM (mean and SD of four measurements using two different probes). The literature values of KD depend on the experimental technique, but are generally in the low nM or sub-nM range.8,11–15 Our data were most consistent with the SPR results,15 which used the 2:2 binding model (dimeric TfR1 sequentially binds two Fe(III)–Tf molecules) and led to KD1 = 1.9 nM and KD2 = 29 nM (KD = 12 nM approximates the average of these two constants). However, our preliminary kinetic studies suggested that the Fe(III)–Tf–TfR1 binding mechanism may be more complicated than this, since both association and dissociation kinetic curves were best fitted with sequences of three first-order reactions (Table S1 and Fig. S3 in ESI†). Detailed kinetic studies of these reactions are in progress since they may have unravelled previously unrecognised steps in the binding and release that were not obvious in data obtained from other methodologies.
:1 binding model (the only one currently available in the BLItz software),5 which led to KD = 12 ± 2 nM (mean and SD of four measurements using two different probes). The literature values of KD depend on the experimental technique, but are generally in the low nM or sub-nM range.8,11–15 Our data were most consistent with the SPR results,15 which used the 2:2 binding model (dimeric TfR1 sequentially binds two Fe(III)–Tf molecules) and led to KD1 = 1.9 nM and KD2 = 29 nM (KD = 12 nM approximates the average of these two constants). However, our preliminary kinetic studies suggested that the Fe(III)–Tf–TfR1 binding mechanism may be more complicated than this, since both association and dissociation kinetic curves were best fitted with sequences of three first-order reactions (Table S1 and Fig. S3 in ESI†). Detailed kinetic studies of these reactions are in progress since they may have unravelled previously unrecognised steps in the binding and release that were not obvious in data obtained from other methodologies.
An anti-metastatic Ru(III) complex, NAMI-A (Chart 1), is currently in Phase II human clinical trials.1 In agreement with literature data,16 a reaction of two molar equivalents of NAMI-A with human apo–Tf ([Ru(III)] = 200 μM, [Tf] = 100 μM, 20 mM HEPES, 25 mM NaHCO3, 140 mM NaCl, pH = 7.40, 4 h at 310 K) caused the formation of an absorbance band at ∼500 nm in electronic spectra (Fig. S4 in ESI†), similar to that for Fe(III)–Tf.17 As shown in Fig. S4,† this absorbance was only observed in the presence of HCO3− (a synergistic ion for Fe(III)–Tf binding).17 These features are indicative of specific Ru(III) binding to the Fe(III) binding sites of Tf,16 although this has not yet been unambiguously demonstrated (non-specific Ru(III) binding to surface His residues of Tf is also likely to occur).2,10 As shown in Fig. 1 and 2 (green lines), the binding of Ru(III)–Tf (1.0 μM apo–Tf, 2.0 μM Ru(III)) to TfR1 was weaker (slower association and faster dissociation) compared not only with that of Fe(III)–Tf, but also with that of apo–Tf. Like Fe(III)–Tf and apo–Tf, both the association and dissociation kinetic curves for Ru(III)–Tf were best fitted with three sequential first-order reactions (Table S1 and Fig. S3†). These results indicated that the binding of Ru(III) to the Fe(III) binding sites of Tf inhibited its interaction with TfR1, so that Tf is unlikely to serve as a specific carrier of NAMI-A into cancer cells.2 Similar results were recently reported for the binding of various Ru anti-cancer prodrugs to Tf, and for the interactions of the resultant Ru-Tf adducts with TfR1, although no experimental details for NAMI-A were provided.10 Compared with the previous work that used immunosorption assays to study the Tf–TfR1 binding,10 the use of BLI provides a simpler and faster option for assessing the TfR1 binding of various metal–Tf complexes.
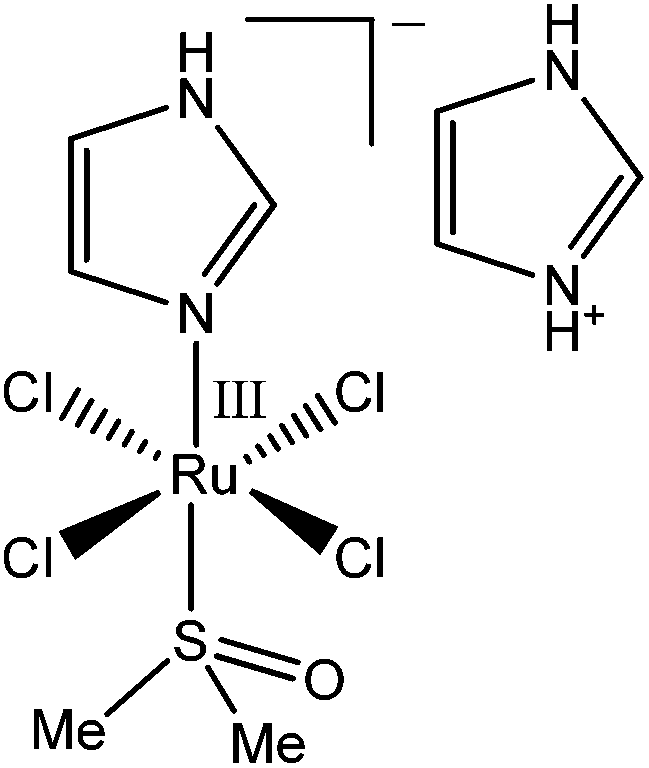 | ||
| Chart 1 Structure of NAMI-A ((ImH+)trans-[RuIIICl4(Im)(S-dmso)], where Im = imidazole and dmso = dimethylsulfoxide).1 | ||
In contrast to the binding of Fe(III)–Tf to TfR1 that has been studied by a variety of experimental techniques,11 the removal of Fe(III) from the Tf–TfR1 complex under simulated endosomal conditions has only been studied by stopped-flow fluorescence spectroscopy in solutions.8,14 Although the changes in BLI response during the metal ion removal were small (probably due to changes in protein conformation; see segment D in Fig. 1 and 3), they were sufficient for the kinetic analyses (Table S1 and Fig. S5 in ESI†), which offers a valuable alternative technique for the studies of this process. The data displayed in Fig. 3 indicated that the strong binding of apo–Tf to TfR at pH < 611,12 was further enhanced in the presence of Ru(III), probably due to the cross-linking of the two proteins by this kinetically inert metal ion.2 The presence of Ru(III) was also manifested by a slight decrease in the dissociation rate of apo–Tf–TfR1 complex at pH = 7.40, and by partial retention of Tf on the probe after this process (segment E in Fig. 1; see Table S1 and Fig. S5 and S6 in ESI† for details). These data suggested that blocking of Fe(III) delivery to fast-metabolising cancer cells through Ru(III)–Tf binding may be one of the mechanisms of anti-cancer activity of Ru(III),2,9 but the possible extent of this process in vivo is not yet clear.
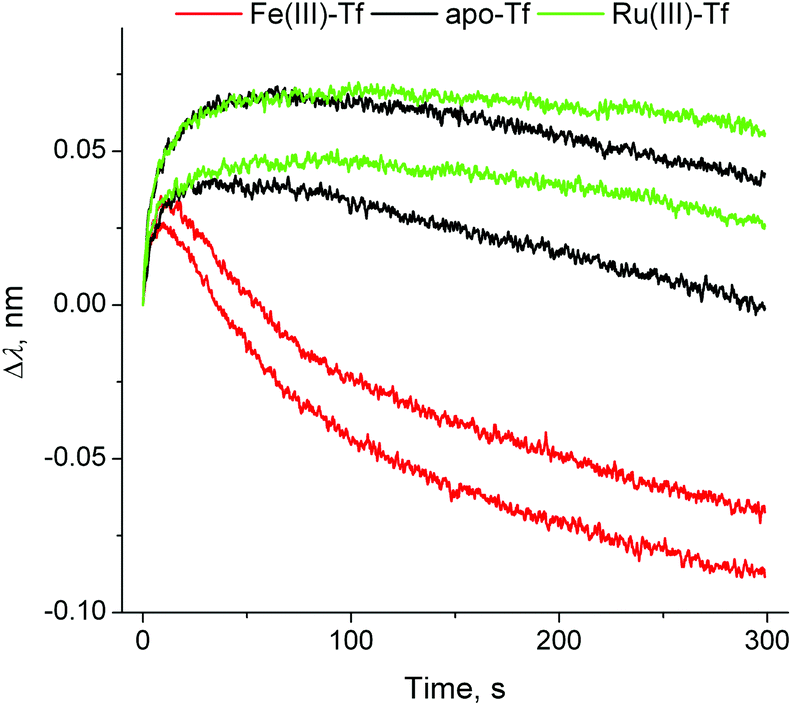 | ||
| Fig. 3 Normalised BLI response curves for the metal ion removal from the Tf–TfR1 complex under endosomal–mimetic conditions14 (segment (D) in Fig. 1). Each condition is represented by two measurements, performed on different probes. Kinetic parameters are given in Table S1 and Fig. S5, ESI.† | ||
Notably, the formation of a Ru(III)–Tf complex (measured from its absorbance at ∼500 nm) could be partially suppressed by the addition of an equimolar amount of human serum albumin (HSA), as shown in Fig. S4.† This observation is consistent with those of human clinical trials, which showed that NAMI-A and other Ru-based anti-cancer prodrugs were bound predominantly (>90%) to albumin in the blood of patients (the HSA:Tf molar ratio in human blood is ∼15:1).18 Previously, we have shown that Ru(III)–albumin adducts are likely to be responsible for the anti-metastatic action of NAMI-A,4 despite the fact that the coordination environment of Ru(III) in such adducts is completely different from that in the original prodrug.4,19 In the current study, we tested the influence of Ru(III)–HSA binding on non-specific interactions20 of HSA with another major group of blood proteins, immunoglobulins G (IgG; typical total blood plasma concentrations in humans, 7.5–22 mg mL−1, compared with ∼40 mg mL−1 for HSA).21
Loading of IgG was performed by incubation of a probe coated with protein G (a bacterial protein that strongly binds to human IgG)5 with a dilute solution of human IgG (50 μg mL−1) for 2 min at 295 K in phosphate-buffered saline (PBS; 20 mM H2PO4−/HPO42−, 150 mM NaCl, pH = 7.40), followed by a brief wash with PBS (segments B and C in Fig. 4). This was followed by the association step with concentrated solutions of HSA (0.50 mM in PBS), or of a Ru(III)–HSA adduct (pre-formed by the incubation of 0.50 mM HSA with 0.50 mM NAMI-A in PBS for 4 h at 310 K),4,21 and by the dissociation step in PBS alone (segments D and E in Fig. 4). A comparison of normalised kinetic curves for the association and dissociation steps (Fig. 5) showed that Ru(III) binding significantly inhibited both processes compared with those for native HSA. The kinetics of both processes (either in the absence or presence of Ru(III)) were best described with two sequential first-order reactions (Table S1 and Fig. S7 in ESI†), which means that the HSA–IgG interactions could not be adequately described by simple KD values.20 Inhibition of protein–protein dissociation in the presence of Ru(III) (Fig. 5 and S6†) can be particularly important for the enhancement of Ru(III)–HSA interactions with collagen, which is a likely crucial step in the anti-metastatic action of NAMI-A.2,4,19
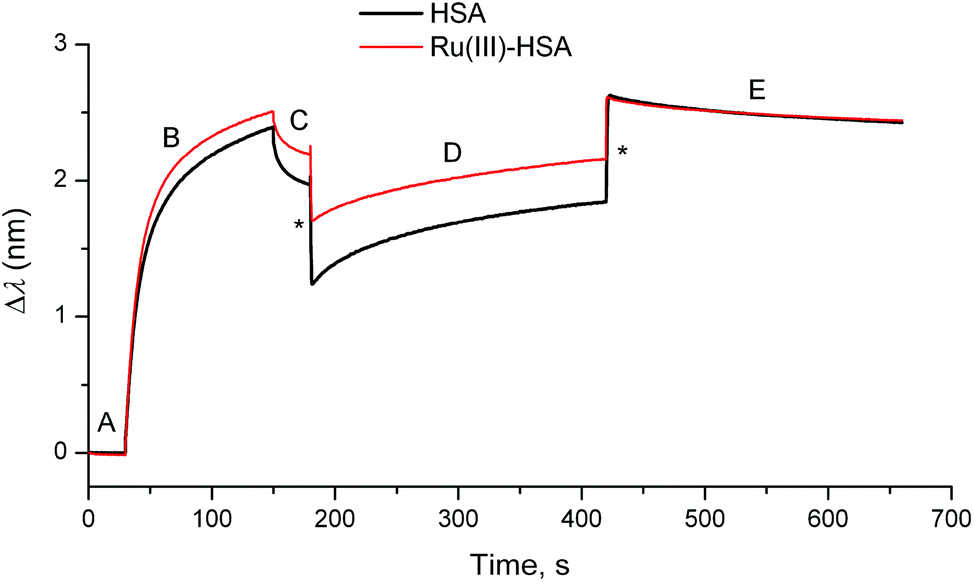 | ||
| Fig. 4 Typical BLI response curves (295 K) for the binding of HSA or the Ru(III)–HSA adduct (0.50 mM) to human IgG loaded on a protein G probe (see ESI† for experimental details). Designations of the segments: (A) initial buffer background; (B) loading of IgG (50 μg mL−1); (C) washing of the probe with the buffer; (D) association of HSA; and (E) dissociation of HSA (buffer alone). Buffer used in all the segments: 20 mM H2PO4−/HPO42−, 150 mM NaCl, pH = 7.40. Jumps due the changing solution viscosity are designated with asterisks. | ||
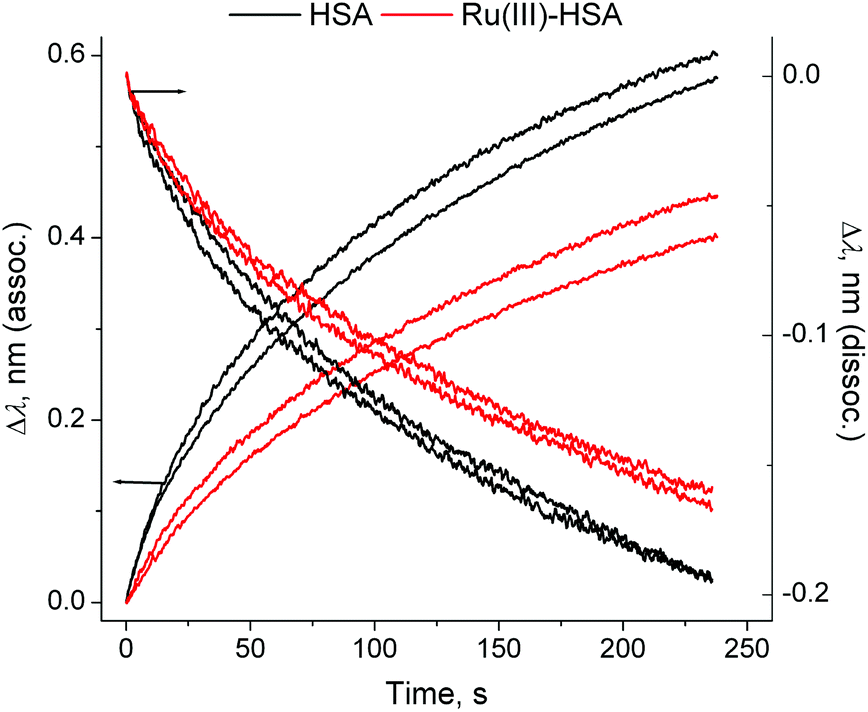 | ||
| Fig. 5 Normalised association and dissociation curves for the HSA–IgG interactions (corresponding to the segments (D) and (E) in Fig. 4). Each condition is represented by two measurements, performed on different probes. Kinetic parameters are given in Table S1 and Fig. S7, ESI.† | ||
Conclusions
This proof-of-concept work has shown the considerable potential of BLI in the studies of the influences of medicinal metal ions on both strong (Tf–TfR1) and weak (IgG–HSA) protein–protein interactions, using an anti-metastatic Ru(III) prodrug, NAMI-A, as an example. Prior to these studies, the method has been validated using the well-understood binding of Fe(III)–Tf to TfR1. Unlike for other experimental techniques, BLI can be used to study the full cycle of Tf–TfR1 binding and dissociation in a single experiment. Specific binding of NAMI-A to the Fe(III) binding sites of Tf can occur in the absence of excess Fe(III) and albumin, but it is unlikely to have major effects on the cellular uptake of NAMI-A in vivo. Results of IgG–HSA binding studies support the hypothesis that cross-linking of extracellular proteins by Ru(III) is involved in its anti-metastatic activity.2,4 The BLI technique can be expanded to the studies of the roles of metal ions in protein–lipid and protein–nucleic acid interactions.22,23Acknowledgements
Financial support of this work has been provided by the Australian Research Council (ARC) Discovery grant, including an ARC Professorial Fellowship, to P. A. L. (DP0984722) and funding for an ARC Senior Research Associate for A. L. (DP130103566). We thank Dr Donna Lai (Bosch Institute, The University of Sydney) for assistance in using the BLItz instrument, and Ms. Hannah O'Riley (School of Chemistry, The University of Sydney) for the synthesis of NAMI-A.Notes and references
- A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90 CrossRef CAS PubMed.
- A. Levina, A. Mitra and P. A. Lay, Metallomics, 2009, 1, 458 RSC.
- L. C. Gonzalez, Methods, 2012, 57, 448 CrossRef CAS PubMed.
- M. Liu, Z. J. Lim, Y. Y. Gwee, A. Levina and P. A. Lay, Angew. Chem., Int. Ed., 2010, 49, 1661 CrossRef CAS PubMed.
- ForteBio®, Menlo Park, CA, USA, http://www.fortebio.com, 2013.
- H. Li, P. J. Sadler and H. Sun, Eur. J. Biochem., 1996, 242, 387 CAS.
- J. B. Vincent and S. Love, Biochim. Biophys. Acta, 2012, 1820, 362 CrossRef CAS PubMed.
- J.-M. El Hage Chanine, M. Hémadi and N.-T. Ha-Duong, Biochim. Biophys. Acta, 2012, 1820, 334 CrossRef PubMed.
- M. Pongratz, P. Schluga, M. A. Jakupec, V. B. Arion, C. G. Hartinger, G. Allmaier and B. K. Keppler, J. Anal. At. Spectrom., 2004, 19, 46 RSC.
- W. Guo, W. Zheng, Q. Luo, X. Li, Y. Zhao, S. Xiong and F. Wang, Inorg. Chem., 2013, 52, 5328 CrossRef CAS PubMed.
- K. M. Mayle, A. M. Le and D. T. Kamei, Biochim. Biophys. Acta, 2012, 1820, 264 CrossRef CAS PubMed.
- A. Dautry-Varsat, A. Ciechanover and H. F. Lodish, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 2258 CrossRef CAS.
- H. Fuchs and R. Geßner, Biochim. Biophys. Acta, 2002, 1570, 19 CrossRef CAS.
- A. N. Steere, S. L. Byrne, N. D. Chasteen and A. B. Mason, Biochim. Biophys. Acta, 2012, 1820, 326 CrossRef CAS PubMed.
- J. A. Lebrón, M. J. Bennett, D. E. Vaughn, A. J. Chirino, P. M. Snow, G. A. Mintier, J. N. Feder and P. J. Bjorkman, Cell, 1998, 93, 111 CrossRef.
- L. Messori, F. Kratz and E. Alessio, Met.-Based Drugs, 1996, 3, 1 CrossRef CAS PubMed.
- P. Aisen, R. Aasa, B. G. Malmström and T. Vänngård, J. Biol. Chem., 1967, 242, 2484 CAS.
- J. M. Rademaker-Lakhai, D. van den Bongard, D. Pluim, J. H. Beijnen and J. H. M. Schellens, Clin. Cancer Res., 2004, 10, 3717 CrossRef CAS PubMed; M. M. Henke, H. Richly, A. Drescher, M. Grubert, D. Alex, D. Thyssen, U. Jaehde, M. E. Scheulen and R. A. Hilger, Int. J. Clin. Pharmacol. Ther., 2009, 47, 58 CrossRef.
- A. Levina, J. B. Aitken, Y. Y. Gwee, Z. J. Lim, M. Liu, A. Mitra Singharay, P. F. Wong and P. A. Lay, Chem.–Eur. J., 2013, 19, 3609 CrossRef CAS PubMed.
- ForteBio® Application Note 14. Biomolecular Binding Kinetics Assays on the Octet Platform, http://www.fortebio.com, 2013 Search PubMed.
- Sigma-Aldrich®. Normal Serum, Ascites and Cell Supernatant. Typical Immunoglobulin Concentration Ranges. http://www.scribd.com/doc/12344361, 2013 Search PubMed.
- J. Wallner, G. Lhota, D. Jeschek, A. Mader and K. Vorauer-Uhl, J. Pharm. Biomed. Anal., 2013, 72, 150 CrossRef CAS PubMed.
- C. C. Thompson, S. S. Nicod, D. S. Malcolm, S. S. Grieshaber and R. A. Carabeo, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 10546 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures; loading curves of TfR1 on Ni(II)-NTA probes; results of repeated stripping and re-binding of Fe(III)-Tf to a TfR1-loaded probe; typical electronic spectra of the reaction products of NAMI-A with aqueous buffers in the presence or absence of Tf; and typical results of kinetic analyses of the association and dissociation curves for the Tf-TfR1 and IgG-HSA bindings. See DOI: 10.1039/c3qi00054k |
| ‡ HEPES = 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid; MES = 2-(N-morpholino)ethanesulfonic acid; edta = N,N,N′,N′-ethane-1,2-diaminetetraacetate(4−). |
| This journal is © the Partner Organisations 2014 |