Sandwich-like carbon-anchored ultrathin TiO2 nanosheets realizing ultrafast lithium storage†
Yongfu
Sun‡
a,
Jinbao
Zhu‡
a,
Liangfei
Bai
a,
Qiuyang
Li
a,
Xing
Zhang
a,
Wei
Tong
b and
Yi
Xie
*a
aHefei National Laboratory for Physical Sciences at Microscale, Collaborative Innovation Center of Chemistry for Energy Materials, University of Science & Technology of China, Hefei, Anhui 230026, P.R. China. E-mail: yxie@ustc.edu.cn
bHigh Magnetic Field Laboratory, Chinese Academy of Sciences, Hefei, Anhui 230031, P.R. China
First published on 16th December 2013
Abstract
Lithium-ion batteries have long been considered as the most promising energy storage technology for hybrid, plug-in hybrid and electric vehicle applications. However, their large-scale applications are still limited by the low electrical conductivity, easy agglomeration and inferior cycling stability of the active materials. Herein, sandwich-like carbon-anchored ultrathin nanosheets are put forward for the first time as an excellent platform to achieve ultrafast lithium storage kinetics and superior cycling stability. Taking the synthetic sandwich-like carbon-anchored ultrathin TiO2 nanosheets as an example, a capacity of 101.9 mA h g−1 is achieved at a current density as high as 40 C (6.8 A g−1), while a capacity of 150.4 mA h g−1 is obtained even after 1200 cycles at a discharge rate of 5 C. This work develops an in situ carbonization of organic octylamine for fabricating sandwich-like carbon-anchored ultrathin nanosheets, holding great promise for the future design and synthesis of high-performance active materials for lithium-ion batteries.
Introduction
Among the currently available energy storage technologies, lithium-ion batteries have received intense attention in the scientific and industrial fields, owing to their high electromotive force and large energy density.1–4 However, the large-scale applications of lithium-ion batteries have been severely limited by several major technological barriers, such as high cost, low electrical conductivity and poor cycling stability.5 To circumvent these challenges, use of ultrathin two-dimensional nanosheets and their composites with carbonaceous materials have recently emerged as the most promising strategies to improve their rate capability and cycling performance. On the one hand, our previous studies have demonstrated that the ultrathin thickness and the huge surface area could enable the atom-thick two-dimensional nanosheets to exhibit fast lithium storage and superior cycling life.6 For example, ultrathin Co3O4 nanosheets with a thickness of 1.5 nm achieved a specific capacity of 812.8 mA h g−1 with a negligible capacity loss per cycle (0.58%), which was superior to most reported anode materials.6a On the other hand, the addition of carbonaceous materials enabled the resulting composites to yield better performances than each individual component and the total sum of the individual effects.7–10 In this regard, Zhou et al. reported that the presence of graphene could dramatically improve the capacity, rate capability and cycling stability of NiO nanosheet–graphene composites through a synergistic effect between the two components.7 Also, our recent work has illustrated that the conducting graphene layers in ultrathin β-Ni(OH)2/graphene nanohybrids could act as electron “superhighways” for fast electron transfer and further improved their high-rate electrochemical performance.11 Despite these attractive advantages, the utilization of graphene-based composites has also encountered some limitations for applications in lithium-ion batteries. For instance, the fabrication of graphene usually needed a very long and complex process through exfoliation of graphite or required a very high carbonization temperature from carbonaceous materials.12 Also, most of the active materials were simply decorated on the surface of graphene rather than being confined between graphene sheets, and thus could not effectively avoid their severe aggregation and shedding due to the lack of oxygen bridges between graphene and the active materials.7 In this case, compared with the complicated synthetic process for graphene, the carbon-based composites would be a good choice for significantly improving the lithium-ion battery performance,13 thanks to the several advantages of carbon such as easy synthesis, high electronic conductivity, outstanding flexibility, large surface area and excellent chemical stability.Herein, sandwich-like carbon-anchored ultrathin two-dimensional nanosheets are put forward as an excellent platform to achieve ultrafast lithium storage kinetics and superior cycling stability. The ultrathin thickness of two-dimensional nanosheets could shorten the lithium ion diffusion length, while the huge surface area allows for a large contact area with the electrolyte, thus ensuring fast kinetics of lithium intercalation/deintercalation and high specific capacity.14–17 Also, the ultrathin nanosheets guarantee facile strain relaxation without the fracture and shedding that usually occur in bulk or micron-sized materials, thus achieving a more stable cycling life. Moreover, in this sandwich-like structure, the anchored carbon can be uniformly and tightly distributed on the surface of the ultrathin nanosheets through an oxygen bridge, which not only contributes to significantly enhancing their conductivity but also helps to hinder their agglomeration and shedding, thus enabling outstanding high-rate performance.14 Furthermore, the distance between the ultrathin nanosheets and flexible carbon not only allows easy electrolyte infiltration and extra storage space for lithium ions but also serves as an elastic zone to buffer volume changes during the lithium intercalation/deintercalation, which facilitates good contact with the electrolyte and hence achieves high cycling performance.18,19
Notably, titanium dioxides (TiO2) have been recently regarded as one of the most promising candidates for anode materials, thanks to their fascinating advantages including ease of synthesis, low density/molar mass, high abundance, non-toxicity, better thermal stability and structural integrity over many charge/discharge cycles.20 In spite of these advantages, the practical applications of the TiO2-based lithium-ion batteries are still hindered by the inferior chemical diffusion of lithium, resulting from the sluggish lithium ion diffusion and poor electronic conductivity. Inspired by the aforementioned concepts, sandwich-like carbon-anchored ultrathin TiO2 nanosheets are successfully synthesized by a facile and scalable strategy, taking advantage of an intermediate precursor of lamellar TiO2–octylamine hybrid nanosheets. As shown in Scheme 1, one can clearly see that the octylamine plays a crucial role in the formation of the lamellar inorganic–organic hybrid intermediate, since only TiO2 nanosheets are obtained under the same conditions except with a decreased concentration of octylamine (Fig. S1†). Note that after annealing the precursor lamellar TiO2–octylamine hybrid nanosheets at 450 °C for 2 h under argon, the sandwich-like carbon-anchored ultrathin TiO2 nanosheets are obtained as a result of an in situ carbonization of organic octylamine layers. As expected, the as-obtained carbon-anchored ultrathin TiO2 nanosheet-based electrode exhibits a capacity of 101.9 mA h g−1 at a large current density as high as 40 C (6.8 A g−1), while it still delivers a capacity of 150.4 mA h g−1 even after 1200 cycles at a discharge rate of 5 C. This lithium battery performance is superior to most previously reported values for TiO2-based electrodes, thus demonstrating their ultrafast lithium storage and long-term cycling stability.
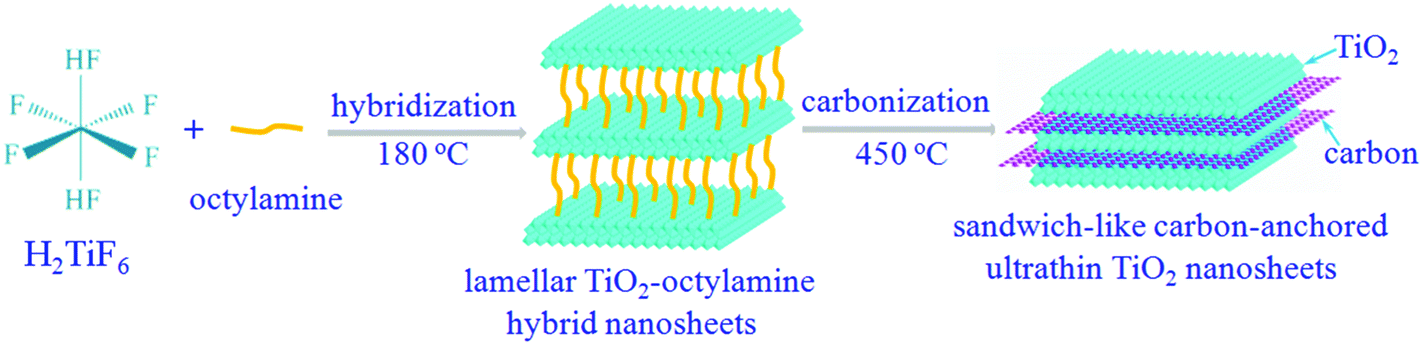 | ||
| Scheme 1 The formation process of sandwich-like carbon-anchored ultrathin TiO2 nanosheets, taking advantage of an intermediate precursor, lamellar TiO2–octylamine hybrid nanosheets. | ||
The above-obtained intermediate precursor lamellar TiO2–octylamine hybrid nanosheets and the sandwich-like carbon-anchored ultrathin TiO2 nanosheets were collected for the following characterization. As shown by the transmission electron microscopy (TEM) image in Fig. 1D, the as-synthesized precursor lamellar TiO2–octylamine hybrid nanosheets possessed a lateral size of about 100 nm. In addition, as depicted in Fig. 1A, the series of small-angle X-ray diffraction (XRD) peaks for the as-synthesized precursor could be assigned to “00L” (L = 1, 2, 3…), providing direct evidence for the presence of an ordered layered structure with a layer spacing of 2.18 nm. Taking the layer thickness of anatase TiO2 and the length of octylamine into account, the precursor could be identified as octylamine intercalated TiO2, which was further supported by the FT-IR, solid-state 13C and 1H NMR spectra in Fig. S2 and S5.† Note that the annealing of the lamellar TiO2–octylamine hybrid nanosheets at 450 °C for 2 h helped to give anatase TiO2 (Fig. 1C), while the corresponding small-angle XRD pattern in Fig. 1B also revealed their lamellar structure with a layer spacing of 1.82 nm, suggesting that the annealing process did not destroy their ordered structure. The TEM image in Fig. 1E showed that they also showed a sheet-like morphology with a thickness of 3–5 nm, further confirming that their structure did not change obviously compared with the lamellar TiO2–octylamine hybrid nanosheets. As shown by the inset HRTEM image in Fig. 1E, the interplanar spacing of 0.35 nm corresponded to the d spacing of the (011) plane and the corresponding dihedral angle of 90° was fairly consistent with the calculated angle between the (101) and (011) planes of anatase TiO2, indicating their [111] preferred orientation. It was noticeable that the heat treatment process at 450 °C resulted in the in situ carbonization of the intercalated organic components in the lamellar TiO2–octylamine hybrid nanosheets, which indicated the formation of sandwich-like carbon-anchored ultrathin TiO2 nanosheets (Scheme 1), further verified by the corresponding small-angle XRD patterns in Fig. 1A and B. This led to the uniform dispersion of the carbon component on the surface of the ultrathin TiO2 nanosheets, which was further evidenced by the corresponding elemental mapping analysis in Fig. 1F.
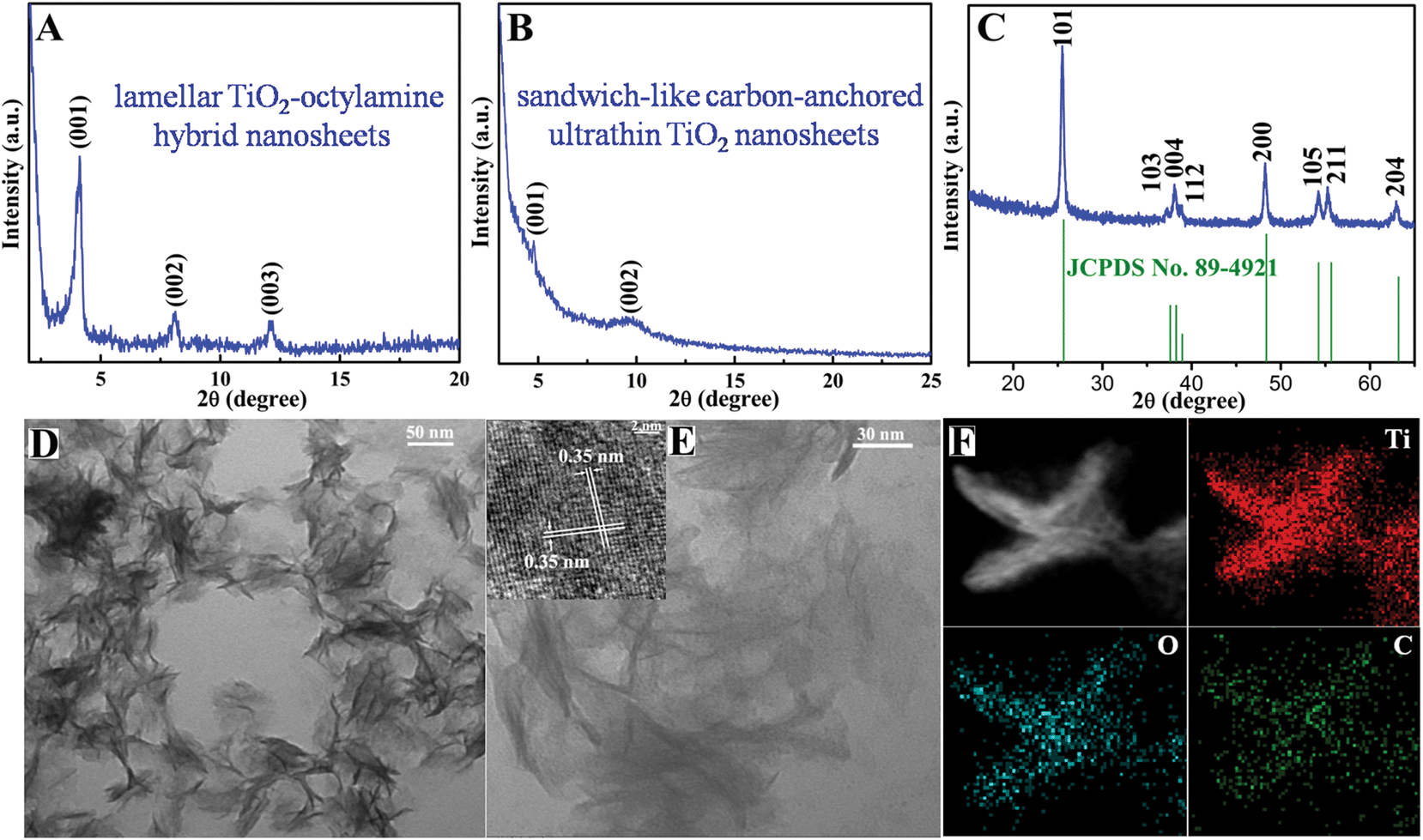 | ||
| Fig. 1 (A) Small-angle XRD pattern and (D) TEM image for the as-synthesized precursor lamellar TiO2–octylamine hybrid nanosheets. (B, C) Small-angle and wide-angle XRD patterns, (E) TEM and inset HRTEM images, (F) TEM image and the corresponding elemental mapping for the sandwich-like carbon-anchored ultrathin TiO2 nanosheets. | ||
The presence of carbon in the final products was further confirmed by their Raman spectrum in Fig. S3A.† Apart from the anatase TiO2 Raman feature,21 there were another two Raman peaks at 1339 and 1603 cm−1 for the sandwich-like carbon-anchored ultrathin TiO2 nanosheets, which originated from the disordered and ordered graphitic carbon,22 thus further evidencing the existence of the carbon coating layer. Also, the high ratio between the D and G bands indicated the formation of a reasonable degree of graphitization (Fig. S3A†).23 In addition, thermo-gravimetric analysis (TGA) for the sandwich-like carbon-anchored ultrathin TiO2 nanosheets further revealed that the weight fraction of carbon in the resulting final products was about 9.8% (Fig. S3B†); such a large amount of carbon could dramatically improve their electronic conductivity and hence achieve ultrafast lithium storage. Moreover, the open channels with large inter-spacing for the sandwich-like carbon-anchored ultrathin TiO2 nanosheets were also confirmed by the nitrogen physisorption measurements. As displayed in Fig. S4,† the adsorption–desorption curve for the sandwich-like carbon-anchored ultrathin TiO2 nanosheets showed a huge surface area of 221.2 m2 g−1 with a pore volume of 0.42 cm3 g−1, significantly higher than the previously reported values for porous TiO2 nanostructures (100–170 m2 g−1) and other ultrathin nanosheets.24 Intriguingly, the presence of open channels in this sandwich-like structure could not only facilitate the infiltration of electrolyte but also act as an elastic zone to buffer volume changes during the lithium uptake/release, thus promoting the capacity and cycling life. Furthermore, the solid-state 13C and 1H NMR spectra were further employed to support the proposed formation process of the sandwich-like carbon-anchored ultrathin TiO2 nanosheets. As depicted in Fig. S5A,† the organic carbon component in the final products could be identified using the cross polarization/magic angle spinning (CP/MAS) solid state 13C NMR spectrum: there was a distinct 13C signal around 120 ppm, which suggested the formation of orthocarbonate bands.25 In addition, the XPS analysis in Fig. S6† also revealed the presence of Ti–O–C carbonaceous bonds in the sandwich-like carbon-anchored ultrathin TiO2 nanosheets, indicating that the anchored carbon was bound to the ultrathin TiO2 nanosheets through a C–O–Ti linkage.26 In general, it is anticipated that the removal of octylamine layers will lead to the complete collapse of the layered titanate structure during the long annealing processes at 450 °C for 2 h. However, the MAS solid state 1H NMR spectrum in Fig. S5B† showed that these organic components were partially carbonized at 450 °C in situ, which could be ascribed to the strong bonding force between the titanate and organic layers. This provided solid evidence for the formation of nanocarbon in the final products. Accordingly, it could be reasonably concluded that the final structure was sandwich-like carbon-anchored ultrathin TiO2 nanosheets, in which the carbon was anchored on the ultrathin TiO2 nanosheets through a C–O–Ti linkage.
Benefiting from the unique sandwich-like structure with huge surface area, ultrathin thickness, and large inter-spacing features, as well as the homogeneous dispersion of a carbon layer in each TiO2 layer, the sandwich-like carbon-anchored ultrathin TiO2 nanosheet-based electrode should achieve ultrafast lithium storage performance. To evaluate the electrochemical performance, a cyclic voltammetry (CV) experiment was conducted at a scanning rate of 0.2 mV s−1. As shown in Fig. 2A, there were two well-defined current peaks observed at 1.7 V (cathodic sweep) and 2.2 V (anodic sweep), which were in good agreement with the previous studies.27 The peak at 1.7 V corresponded to the biphasic transition from tetragonal anatase to orthorhombic LiχTiO2 when the insertion coefficient χ reached ∼0.5.28 Interestingly, it could be observed that the intensities of both peaks gradually increased during the subsequent scans, suggesting a possible activating process in the electrode material.29 More strikingly, there was no apparent irreversible process observed in the first cathodic scan, indicating a high coulombic efficiency for the lithium extraction. This observation was quite unusual for anatase TiO2, suggesting its high lithium battery performance. The capacity and cycling stability for the carbon-anchored ultrathin TiO2 nanosheet-based electrodes were evaluated at a large current density of 5 C (1 C = 170 mA g−1) in comparison with the bare TiO2 nanosheet-based electrodes. As shown in Fig. 2B, one can clearly see that the carbon-anchored ultrathin TiO2 nanosheet-based electrode did not display obvious discharge capacity loss during the initial cycle, while the bare TiO2 nanosheet-based electrode possessed an initial rapid loss of capacity. This provided evidence that there was not an obvious irreversible process in the designed carbon-anchored ultrathin TiO2 nanosheets, fairly consistent with the corresponding CV result in Fig. 2A. Also, it was striking to note that the carbon-anchored ultrathin TiO2 nanosheet-based electrode achieved a very high and stable reversible capacity of about 155.1 mA h g−1 in the initial 10 cycles, while it still possessed a capacity of 150.4 mA h g−1 and the morphology was preserved quite well even after 1200 cycles (Fig. 2B and Fig. S7†). By contrast, for the bare TiO2 nanosheet-based electrode, the capacity dramatically decreased to ca. 7 mA h g−1 after 200 cycles, which was significantly lower than that of the carbon-anchored ultrathin TiO2 nanosheet-based electrode. As far as we know, the capacity of the carbon-anchored ultrathin TiO2 nanosheet-based electrode was still higher than those of most previously reported TiO2-based electrodes (100–150 mA h g−1),30 suggesting its superior lithium battery performance. Moreover, since the rate capability was also very important for practical applications, the effect of rate capacity on the carbon-anchored ultrathin TiO2 nanosheet-based electrode was also examined, as displayed in Fig. 2C. Compared to the bare TiO2 nanosheet-based electrode, the carbon-anchored ultrathin TiO2 nanosheet-based electrode exhibited remarkably improved specific capacity and cycling stability at high charging rates. Fig. 2C revealed that the bare TiO2 nanosheet-based electrode showed a capacity of 220 mA h g−1 at a low discharge rate of 1 C, which quickly decreased to ca. 40 mA h g−1 and 2.5 mA h g−1 with the discharge rate increasing to 2 C and 40 C, indicating their much poorer lithium kinetics. Contrastingly, at the relatively lower 1 C and 5 C rates, the carbon-anchored ultrathin TiO2 nanosheet-based electrode possessed discharge capacities of 201.1 and 162.8 mA h g−1, respectively. Upon increasing the discharging rate to 20 C and 30 C, it could also achieve capacities of 123.5 and 110.4 mA h g−1, respectively. Even at the highest rate of 40 C (6.8 A g−1), it still delivered a capacity as high as 101.9 mA h g−1, strikingly higher than that of the bare TiO2 nanosheet-based electrode and demonstrating its ultrafast kinetics for lithium insertion. Notably, a stable capacity of 200 mA h g−1 for the carbon-anchored ultrathin TiO2 nanosheet-based electrode could also be resumed when the current rate was reduced back to 1 C, while the bare TiO2 nanosheet-based electrode only retained an average capacity of ca. 50 mA h g−1. This suggested the good structural stability of the carbon-anchored ultrathin TiO2 nanosheet-based electrode, which was crucial for practical applications. As far as we know, the electrochemical performance for the carbon-anchored ultrathin TiO2 nanosheet-based electrode was superior to that of most previously reported TiO2 nanomaterial-based electrodes under similar testing conditions,30,31 indicating its fascinating lithium battery performance.
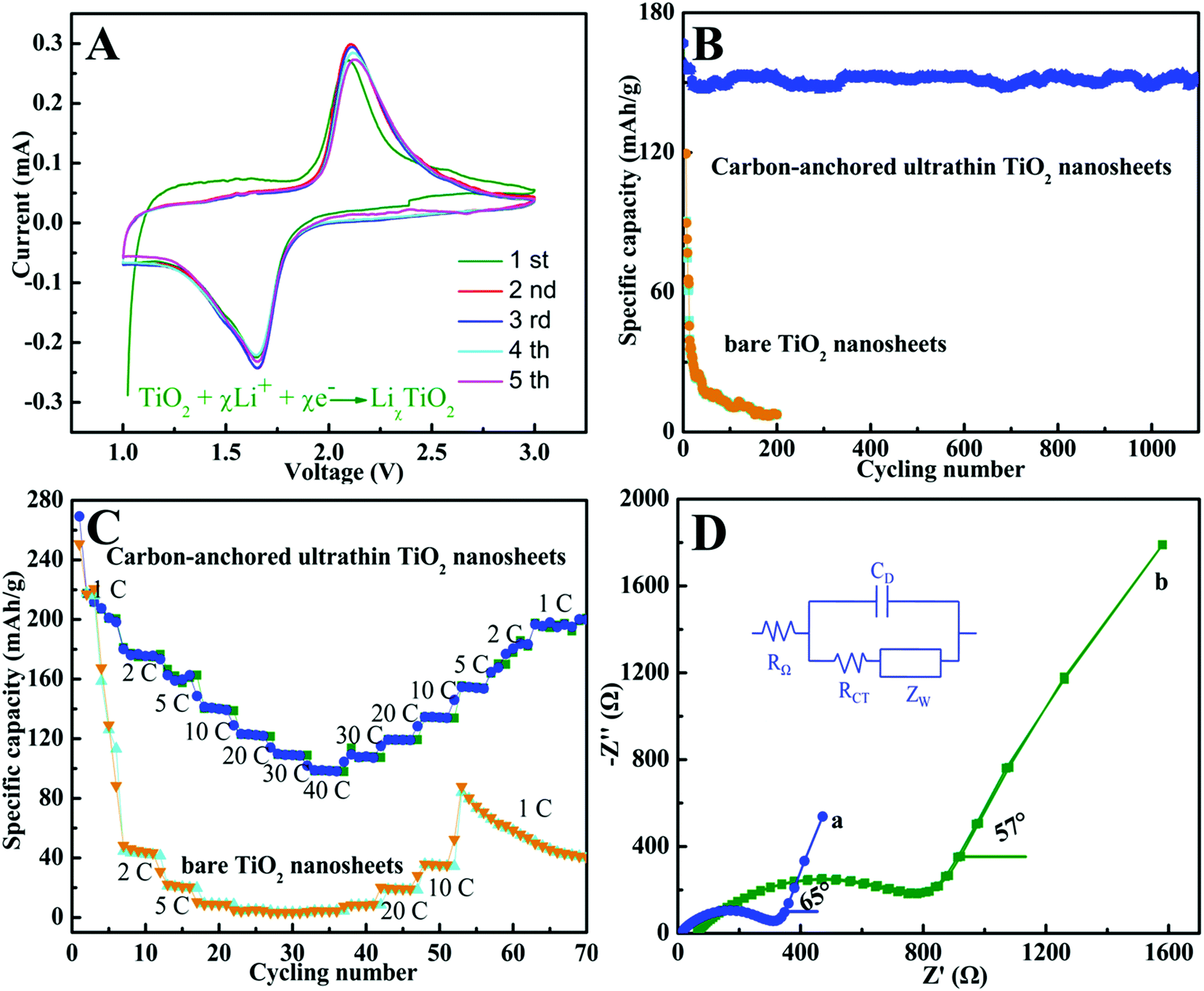 | ||
| Fig. 2 (A) Representative cyclic voltammograms of the sandwich-like carbon-anchored ultrathin TiO2 nanosheet-based electrode at a scanning rate of 0.2 mV s−1. Cycling performance for the carbon-anchored ultrathin TiO2 nanosheets and bare TiO2 nanosheet-based electrodes (B) at a constant current drain of 5 C and (C) at different charge–discharge rates (1 C = 170 mA g−1). (D) Nyquist plots for (a) the carbon-anchored ultrathin TiO2 nanosheets and (b) bare TiO2 nanosheet-based electrodes obtained by applying a sine wave with amplitude of 10.0 mV over the frequency range from 100 kHz to 0.01 Hz. | ||
Of note, the dramatically improved capacity, rate capability and cycling performance for the sandwich-like carbon-anchored ultrathin TiO2 nanosheet-based electrode could be ascribed to the synergistic effect between the ultrathin TiO2 nanosheets and the anchored carbon through a C–O–Ti bridge. As shown in Scheme 2, the ultrathin thickness of the synthetic TiO2 nanosheets helped to shorten the diffusion length of the lithium ions across the thickness dimension, thus enabling faster lithium insertion and extraction. This is due to the fact that the equilibrium time (τeq = L2/2DLi) for Li diffusion can be efficiently decreased by reducing the particle size (L).7 Importantly, the oxygen bridges between the ultrathin TiO2 nanosheets and the anchored carbon contributed a striking enhancement of their electrical conductivity, which directly correlated with the excellent electrochemical performance and could be verified by electrochemical impedance spectroscopy (EIS) measurements. As shown in Fig. 2D, the Nyquist plots showed that the semicircle diameter for the carbon-anchored ultrathin TiO2 nanosheet-based electrode in the high–medium frequency region was much smaller than that of the bare TiO2 nanosheet-based electrode, suggesting their lowered contact and charge-transfer resistance.32 The exact kinetic differences for these two electrodes were inspected by modeling AC impedance spectra based on the modified Randles equivalent circuit (inset Fig. 2D).33 The Ohmic resistance (RΩ) and charge-transfer resistance (RCT) for the carbon-anchored ultrathin TiO2 nanosheet-based electrode were 19.84 and 202.1 Ω, which were significantly smaller than those of the bare TiO2 nanosheet-based electrode (75.97 and 431.8 Ω). Note that the low-frequency slope angle for the carbon-anchored ultrathin TiO2 nanosheet-based electrode was 65°, higher than that of the bare TiO2 nanosheet-based electrode (57°), in which the steeper low-frequency tail indicated the higher lithium ion conductivity in the carbon-anchored ultrathin TiO2 nanosheets.7,34 Moreover, it was generally accepted that the intercalation of lithium ions into the active materials inevitably caused their volume expansion, pulverization and shedding, thus gradually leading to their capacity fading. As for the carbon-anchored ultrathin TiO2 nanosheet-based electrode, the flexible carbon could contribute to buffering the volume expansion of the ultrathin TiO2 nanosheets during lithium intercalation/deintercalation, thus improving their cycling behavior. In addition, the presence of carbon also helped to prevent the restacking of the ultrathin TiO2 nanosheets, which improved the utilization of active material and hence promoted their specific capacity. Furthermore, as revealed by the BET results in Fig. S4,† the carbon-anchored ultrathin TiO2 nanosheets reasonably possessed open channels in their sandwich-like structure, which not only offered easy electrolyte infiltration and extra lithium storage space but also served as an elastic zone to accommodate volume changes during lithium uptake–release, thus ensuring good electric contact and achieving excellent electrochemical performance.9
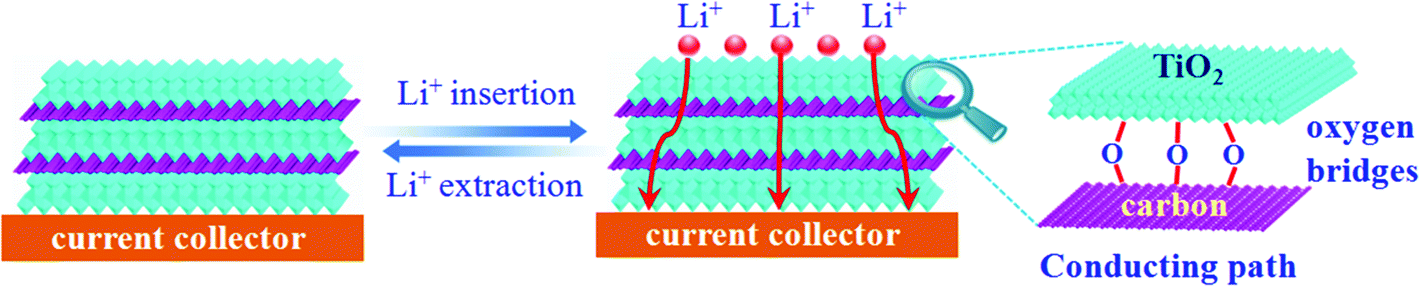 | ||
| Scheme 2 Advantages of using the sandwich-like carbon-anchored ultrathin TiO2 nanosheets as the lithium ion battery electrode; the ultrathin thickness facilitates fast lithium insertion/extraction, while the anchored carbon provides conducting paths through the Ti–O–C bridge. | ||
Conclusion
In summary, sandwich-like carbon-anchored ultrathin nanosheets are put forward for the first time as an excellent platform to achieve ultrafast lithium storage kinetics and superior cycling stability. As an example, sandwich-like carbon-anchored ultrathin TiO2 nanosheets were first successfully synthesized by a facile and scalable strategy, taking advantage of an intermediate precursor, lamellar TiO2–octylamine hybrid nanosheets. In this sandwich-like structure, oxygen bridges were formed between the ultrathin TiO2 nanosheets and the anchored carbon, producing a synergistic effect to greatly improve the lithium storage. The ultrathin thickness and huge surface area of the TiO2 nanosheets helped to shorten the lithium ion diffusion length and enhance the surface area contact with the electrolyte, thus achieving faster kinetics and higher capacity. The carbon, derived in situ from the carbonization of octylamine, contributed to providing increased conductivity and better strain accommodation, thus ensuring superior cycling stability and rate capability. As expected, the carbon-anchored ultrathin TiO2 nanosheet-based electrode delivered a capacity of 101.9 mA h g−1 at discharge rates as high as 40 C (6.8 A g−1), while it still possessed a capacity of 150.4 mA h g−1 even after 1200 cycles at a large current density of 5 C, which was superior to most previously reported TiO2-based electrodes, thus demonstrating the ultrafast lithium storage and long-term cycling stability. Briefly, this work not only provides a convenient in situ carbonization of organic octylamine for fabricating sandwich-like carbon-anchored ultrathin nanosheets but also proves that the peculiar structures are excellent platforms for optimizing lithium battery performance, holding great promise for triggering breakthroughs in the field of energy storage or energy conversion.Acknowledgements
This work was financially supported by the National Natural Science Foundation (21331005, 11079004, 90922016, 21201157, 11321503) and the Chinese Academy of Science (XDB01020300).References
- B. Kang and G. Ceder, Nature, 2009, 458, 190 CrossRef CAS PubMed.
- J. Maier, Nat. Mater., 2005, 4, 805 CrossRef CAS.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2390 CrossRef PubMed.
- (a) J. Y. Wang, N. L. Yang, H. J. Tang, Z. H. Dong, Q. Jin, M. Yang, D. Kisailus, H. J. Zhao, Z. Y. Tang and D. Wang, Angew. Chem., Int. Ed., 2013, 52, 6417 CrossRef CAS PubMed; (b) X. Y. Lai, J. E. Halpert and D. Wang, Energy Environ. Sci., 2012, 5, 5604 RSC; (c) L. Hu, H. Wu, Y. Cao, A. Cao, H. Li, J. McDough, X. Xie, M. Zhou and Y. Cui, Adv. Energy Mater., 2011, 1, 523 CrossRef CAS.
- Z. H. Chen, I. Belharouak, Y. K. Sun and K. Amine, Adv. Funct. Mater., 2013, 23, 959 CrossRef CAS.
- (a) J. Zhu, L. Bai, Y. Sun, X. Zhang, Q. Li, B. Cao, W. Yan and Y. Xie, Nanoscale, 2013, 5, 5241 RSC; (b) J. Zhu, Q. Li, W. Bi, L. Bai, X. Zhang, J. Zhou and Y. Xie, J. Mater. Chem. A, 2013, 1, 8154 RSC.
- G. M. Zhou, D. W. Wang, L. C. Yin, N. Li, F. Li and H. M. Cheng, ACS Nano, 2012, 6, 3214 CrossRef CAS PubMed.
- H. Wang, L. F. Cui, Y. Yang, H. Sanchez Casalongue, J. T. Robinson, Y. Liang, Y. Cui and H. Dai, J. Am. Chem. Soc., 2010, 132, 13978 CrossRef CAS PubMed.
- G. M. Zhou, D. W. Wang, F. Li, L. L. Zhang, N. Li, Z. S. Wu, L. Wen, G. A. Lu and H. M. Cheng, Chem. Mater., 2010, 22, 5306 CrossRef CAS.
- X. K. Guo, Q. Fan, L. Yu, J. J. Liang, W. X. Ji, L. M. Peng, X. F. Guo, W. P. Ding and Y. F. Chen, J. Mater. Chem. A, 2013, 1, 11534 CAS.
- J. F. Xie, X. Sun, N. Zhang, K. Xu, M. Zhou and Y. Xie, Nano Energy, 2013, 2, 65 CrossRef CAS PubMed.
- (a) Y. F. Sun, B. Y. Qu, Q. Liu, S. Gao, Z. X. Yan, W. S. Yan, B. C. Pan, S. Q. Wei and Y. Xie, Nanoscale, 2012, 4, 3761 RSC; (b) J. Y. Hong, K. Y. Shin, O. S. Kwon, H. Kang and J. Jang, Chem. Commun., 2011, 47, 7182 RSC.
- (a) L. Zhang, G. Q. Zhang, H. B. Wu, L. Yu and X. W. Lou, Adv. Mater., 2013, 25, 2589 CrossRef CAS PubMed; (b) S. J. Ding, D. Y. Zhang, H. B. Wu, Z. C. Zhang and X. W. Lou, Nanoscale, 2012, 4, 3651 RSC.
- Y. F. Sun, Z. H. Sun, S. Gao, H. Cheng, Q. H. Liu, J. Y. Piao, T. Yao, C. Z. Wu, S. L. Hu, S. Q. Wei and Y. Xie, Nat. Commun., 2012, 3, 1057 CrossRef PubMed.
- (a) Y. F. Sun, H. Cheng, S. Gao, Z. H. Sun, Q. H. Liu, Q. Liu, F. C. Lei, T. Yao, J. F. He, S. Q. Wei and Y. Xie, Angew. Chem., Int. Ed., 2012, 51, 8727 CrossRef CAS PubMed; (b) Y. F. Sun, F. C. Lei, S. Gao, B. C. Pan, J. F. Zhou and Y. Xie, Angew. Chem., Int. Ed., 2013, 52, 10569 CrossRef CAS PubMed.
- Y. F. Sun, H. Cheng, S. Gao, Q. H. Liu, Z. H. Sun, C. Xiao, C. Z. Wu, S. Q. Wei and Y. Xie, J. Am. Chem. Soc., 2012, 134, 20294 CrossRef CAS PubMed.
- Y. F. Sun, Z. H. Sun, S. Gao, H. Cheng, Q. H. Liu, F. C. Lei, S. Q. Wei and Y. Xie, Adv. Energy Mater., 2013 DOI:10.1002/aenm.201300611.
- Y. F. Sun, S. S. Jiang, W. T. Bi, C. Z. Wu and Y. Xie, J. Power Sources, 2011, 196, 8644 CrossRef CAS PubMed.
- A. Q. Pan, H. B. Wu, L. Yu and X. W. Lou, Angew. Chem., Int. Ed., 2013, 52, 2226 CrossRef CAS PubMed.
- (a) X. X. Li, Y. J. Xiong, L. F. Zou, M. T. Wang and Y. Xie, Microporous Mesoporous Mater., 2008, 112, 641 CrossRef CAS PubMed; (b) J. S. Chen and X. W. Lou, Mater. Today, 2012, 15, 246 CrossRef CAS; (c) J. Ye, W. Liu, J. Cai, S. Chen, X. Zhao, H. Zhou and L. Qi, J. Am. Chem. Soc., 2011, 133, 933 CrossRef CAS PubMed; (d) J. Y. Shin, J. H. Joo, D. Samuelis and J. Maier, Chem. Mater., 2012, 24, 543 CrossRef CAS; (e) N. L. Yang, Y. Zhang, J. E. Halpert, J. Zhai, D. Wang and L. Jiang, Small, 2012, 8, 1762 CrossRef CAS PubMed.
- X. B. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746 CrossRef CAS PubMed.
- X. Zhang, Z. Xing, L. L. Wang, Y. C. Zhu, Q. W. Li, J. W. Liang, Y. Yu, T. Huang, K. B. Tang, Y. T. Qian and X. Y. Shen, J. Mater. Chem., 2012, 22, 17864 RSC.
- L. C. Liu, Q. Fan, C. Z. Sun, X. R. Gu, H. Li, F. Gao, Y. F. Chen and L. Dong, J. Power Sources, 2013, 221, 141 CrossRef CAS PubMed.
- (a) I. Moriguchi, R. Hidaka, H. Yamada, T. Kudo, H. Murakami and N. Nakashima, Adv. Mater., 2006, 18, 69 CrossRef CAS; (b) J. T. Jang, S. Jeong, J. W. Seo, M. C. Kim, E. Sim, Y. Oh, S. Nam, B. Park and J. Cheon, J. Am. Chem. Soc., 2011, 133, 7636 CrossRef CAS PubMed; (c) C. Wang, Y. Zhou, M. Y. Ge, X. B. Xu, Z. L. Zhang and J. Z. Jiang, J. Am. Chem. Soc., 2010, 132, 46 CrossRef CAS PubMed; (d) J. Zhu, L. Bai, Y. Sun, X. Zhang, Q. Li, B. Cao, W. Yan and Y. Xie, Nanoscale, 2013, 5, 5241 RSC.
- E. M. Rockafellow, X. Fang, B. G. Trewyn, K. Schmidt-Rohr and W. S. Jenks, Chem. Mater., 2009, 21, 1187 CrossRef CAS.
- (a) O. Akhavan, R. Azimirad, S. Safa and M. M. Larijani, J. Mater. Chem., 2010, 20, 7386 RSC; (b) L. C. Chen, Y. C. Ho, W. S. Guo, C. M. Huang and T. C. Pan, Electrochim. Acta, 2009, 54, 3884 CrossRef CAS PubMed.
- (a) J. S. Chen, Y. L. Tan, C. M. Li, Y. L. Cheah, D. Y. Luan, S. Madhavi, F. Y. C. Boey, L. A. Archer and X. W. Lou, J. Am. Chem. Soc., 2010, 132, 6124 CrossRef CAS PubMed; (b) C. H. Sun, X. H. Yang, J. S. Chen, Z. Li, X. W. Lou, C. Z. Li, S. C. Smith, G. Q. Lu and H. G. Yang, Chem. Commun., 2010, 46, 6129 RSC; (c) S. J. Ding, J. S. Chen, D. Y. Luan, F. Y. C. Boey, S. Madhavi and X. W. Lou, Chem. Commun., 2011, 47, 5780 RSC.
- M. Wagemaker, A. P. M. Kentgens and F. M. Mulder, Nature, 2002, 418, 397 CrossRef CAS PubMed.
- J. S. Chen and X. W. Lou, Electrochem. Commun., 2009, 11, 2332 CrossRef CAS PubMed.
- (a) Y. S. Hu, L. Kienle, Y. G. Guo and J. Maier, Adv. Mater., 2006, 18, 1421 CrossRef CAS; (b) J. S. Chen, H. Liu, S. Z. Qiao and X. W. Lou, J. Mater. Chem., 2011, 21, 5687 RSC; (c) K. Saravanan, K. Ananthanarayanan and P. Balaya, Energy Environ. Sci., 2010, 3, 939 RSC; (d) J. Y. Shin, D. Samuelis and J. Maier, Adv. Funct. Mater., 2011, 21, 3464 CrossRef CAS.
- (a) Z. S. Hong, M. D. Wei, T. B. Lan, L. L. Jiang and G. Z. Cao, Energy Environ. Sci., 2012, 5, 5408 RSC; (b) J. Wang, Y. K. Zhou, Y. Y. Hu, R. O'Hayre and Z. P. Shao, J. Phys. Chem. C, 2011, 115, 2529 CrossRef CAS; (c) D. Dambournet, I. Belharouak and K. Amine, Chem. Mater., 2010, 22, 1173 CrossRef CAS; (d) J. S. Chen and X. W. Lou, Chem. Sci., 2011, 2, 2219 RSC; (e) J. F. Ye, W. Liu, J. G. Cai, S. A. Chen, X. W. Zhao, H. H. Zhou and L. M. Qi, J. Am. Chem. Soc., 2011, 133, 933 CrossRef CAS PubMed; (f) Z. Q. Sun, J. H. Kim, Y. Zhao, F. Bijarbooneh, V. Malgras, Y. Lee, Y. M. Kang and S. X. Dou, J. Am. Chem. Soc., 2011, 133, 19314 CrossRef CAS PubMed.
- (a) D. H. Wang, D. W. Choi, J. Li, Z. G. Yang, Z. M. Nie, R. Kou, D. H. Hu, C. M. Wang, L. V. Saraf, J. G. Zhang, I. A. Aksay and J. Liu, ACS Nano, 2009, 3, 907 CrossRef CAS PubMed; (b) S. B. Yang, X. L. Feng and K. Mullen, Adv. Mater., 2011, 23, 3575 CrossRef CAS PubMed.
- Y. M. Lin, P. R. Abel, A. Heller and C. B. Mullins, J. Phys. Chem. Lett., 2011, 2, 2885 CrossRef CAS.
- Y. Ma, C. Zhang, G. Ji and J. Y. Lee, J. Mater. Chem., 2012, 22, 7845 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3qi00050h |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2014 |