
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cellulose grafting by photoinduced controlled radical polymerisation†
Emma
Larsson
a,
Samuel A.
Pendergraph
a,
Tahani
Kaldéus
b,
Eva
Malmström
a and
Anna
Carlmark
*a
aKTH Royal Institute of Technology, School of Chemical Science and Engineering, Department of Fibre and Polymer Technology, Teknikringen 56, SE-100 44 Stockholm, Sweden. E-mail: annac@kth.se; Fax: +468 790 8283; Tel: +468 790 8027
bKTH Royal Institute of Technology, School of Chemical Science and Engineering, Wallenberg Wood Science Center, Teknikringen 56, SE-100 44 Stockholm, Sweden
First published on 24th December 2014
Abstract
The photoinduced controlled radical polymerisation (CRP) technique has been utilised to graft methyl acrylate (MA) and di(ethylene glycol) ethyl ether acrylate (DEGA) from filter paper. Grafting of MA was performed from α-bromoisobutyryl bromide functionalised papers. The amount of polymer grafted on the surface could be regulated by modifying the target DP of the reaction. SEC of cleaved linear polymer grafts showed that the grafting from filter papers proceeded with different kinetics compared to polymerisation from a free initiator added to the reaction mixture, resulting in higher dispersity. Furthermore, filter papers were polymerised with α-chloro-ε-caprolactone by surface-initiated ring opening polymerisation, yielding linear grafts containing initiating functions through-out the main chain. This functionality was subsequently utilised for the photoinduced CRP grafting of DEGA, yielding a graft-on-graft structure, which resulted in a thermoresponsive cellulose surface.
Introduction
Cellulose has emerged as a prominent material for composite applications due to its robust mechanical properties, inherent functional groups, and its natural abundance in plants rendering it available in many different climates. Numerous examples of the utilisation of cellulose have been demonstrated in several applications ranging from robust composites to advanced electronic displays.1–4 While the use of cellulose in more advanced applications than paper and cardboard has gained momentum, some challenges remain which hinder the utility of these bio-based polymers. One major challenge with cellulose is the incompatibility as a filler material in composites with hydrophobic polymer matrices. One strategy to circumvent this problem is to graft polymers on the fibre surface, either through a “grafting-to” or a “grafting-from” approach.1,3–13 Both methods have been effectively utilised to modify several cellulose substrates with polymers ranging from polyesters to poly(meth)acrylates.1,3–14Atom transfer radical polymerisation (ATRP) is one of the most utilised controlled polymerisation methods as it can be employed to polymerise a wide variety of monomers. End group fidelity is maintained throughout polymerisation, making it an attractive method for the synthesis of block copolymers and polymers with advanced architectures.1,6–9 Guan and Smart discovered that visible light had an effect on the ATRP of methyl methacrylate (MMA).15 They observed that the rate of polymerisation was increased by light compared to when the reaction was performed in the dark. They also discovered that there was an increase in the control of the polymerisation when it was exposed to light during the reaction. More recently, several studies using ultra-violet (UV) and visible light have been demonstrated to trigger metal complex mediated radical polymerisations. Examples of this include work by Hawker and co-workers, in which a bipyridine/iridium complex was used to absorb visible light and control the reaction.16,17 Matyjaszewski and co-workers utilised pyridine-based ligands with copper bromide to polymerise methacrylates with sunlight/visible light.18,19 While these methods were effective, their synthetic procedures required unconventional ligands. Yagci and co-workers reported the first photoinduced controlled radical polymerisation (CRP) utilising common ATRP reactants, polymerising MMA using copper(II) bromide (Cu(II)Br2) and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) to form the catalyst complex.20,21 Haddleton and co-workers have recently reported the use of a system based on Cu(II)Br2 and tris(2-(dimethylamino)ethyl)amine (Me6TREN) to form a catalyst complex, used for the CRP of methyl acrylate (MA) in DMSO by exposure to UV light.22 The system is similar to a SET–LRP system previously utilised by the Haddleton group.23–25 To further investigate potential applications of this technique, MA polymerisation was also performed in a continuous flow reactor.26 To broaden the scope of this polymerisation technique, Haddleton and co-workers investigated additional solvents and monomers that can be utilised.27 While metal complex-mediated photoinduced CRP has produced well-defined polymers in solution, there are no reports on the effect of grafting polymers on cellulose surfaces utilising this technique.
ATRP and ARGET-ATRP have previously been employed for the grafting of cellulose surfaces in several studies.7–9,28–31 Barner-Kowollik and co-workers have reported on the utilisation of light to graft polymeric chains onto cellulosic surfaces but, as has been shown, “grafting-from” cellulose typically results in higher grafting densities than “grafting-to” cellulose.11,12 The method developed by Haddleton and co-workers, utilising UV light to induce polymerisation, is an attractive alternative for the “grafting-from” of a surface, as it facilitates the possibility to control the chain growth by turning the light source off and on. Furthermore, it requires smaller amounts of copper than traditional ATRP, while still resulting in high conversions and low dispersities (Đ) of the final polymer. In this work, we present the first use of this technique from a cellulose substrate. In addition, we characterise and compare free polymers formed from the sacrificial initiator in solution with the grafted polymer by initiating polymerisation on the surface from a cleavable initiator. The grafted surfaces were characterised by field emission scanning electron microscopy (FE-SEM), Fourier transform infrared spectroscopy (FT-IR), contact angle measurements (CAM) and thermogravimetric analysis (TGA).
In order to increase the functionality on the cellulose substrate, surface initiated ring-opening polymerisation (SI-ROP) was employed to graft ε-caprolactone (ε-CL) and α-chloro-ε-caprolactone (αClεCL) from the surface of filter paper. The naturally inherent OH-groups found in cellulose were utilised as initiators for this polymerisation. There are several publications where a cyclic monomer/lactone, containing a halogen functionality in the α-position, has been polymerised by ROP, which allows for further derivatisation by ATRP.32–36 Pan and co-workers copolymerised αClεCL and ε-caprolactone (ε-CL) in different proportions followed by ARGET-ATRP of N-isopropylacrylamide (NIPAAm), producing graft copolymers with well-defined structures.32 A similar approach was utilised herein to create a polymer brush from the cellulose surface, containing numerous ATRP-initiating sites. The sites were subsequently utilised for the photoinduced CRP of di(ethylene glycol) ethyl ether acrylate (DEGA), in a graft-on-graft approach, resulting in a thermoresponsive cellulose paper.
Experimental section
Materials
α-Bromoisobutyryl bromide (BiB, 98%), ethyl α-bromoisobutyrate (EBiB, 98%), 4-(dimethylamino)pyridine (DMAP, 99%), copper(II) bromide (Cu(II)Br2, 99%), tris(2-carboxyethyl)phosphine hydrochloride (TCEP, 98%), tin(II) 2-ethylhexanoate (SnOct2, 92.5%), and Whatman 1 filter paper were purchased from Aldrich. Triethylamine (TEA, 99%), dimethyl sulfoxide (DMSO, 99.0%) and benzyl alcohol (99.5%) were purchased from Merck. Dithiothreitol (DTT, 99%) was purchased from Apollo Scientific. Tris(2-(dimethylamino)ethyl)amine (Me6TREN),37 2-((2-((2-bromo-2-methylpropanoyl)oxy)ethyl)disulfanyl)ethyl 4-chloro-4-oxobutanoate (S–S BiB),38 and α-chloro-ε-caprolactone (αClεCL)39 were synthesised as reported elsewhere. Methyl acrylate (MA, 99%) and di(ethylene glycol) ethyl ether acrylate (DEGA, 90%) were purchased from Aldrich and passed through a column of basic, activated, aluminium oxide prior to use. ε-Caprolactone (εCL, 99%) was purchased from Alfa Aesar and dried by distillation over calcium hydride under reduced pressure before use. All materials were used as received unless stated otherwise.Instrumentation
UV photoinduced CRP was conducted in a UV nail gel curing box, equipped with four 9 W light bulbs, λmax ≈ 360 nm, as previously developed by Haddleton et al.22 The polymers were obtained after 90 min exposure with an intensity of approximately 40 mW cm−2.1H NMR spectra were recorded at room temperature with a Bruker Avance 400 MHz spectrometer, using CDCl3 solvent. Tetramethylsilane (TMS) and the solvent residual peak were used as internal standards.
Fourier transform infrared spectroscopy (FT-IR) was performed using a Perkin-Elmer Spectrum 2000 FT-IR equipped with a MKII Golden Gate, single reflection ATR System from Specac Ltd, (London, UK). The ATR-crystal used was a MKII heated Diamond 45° ATR Top Plate. For each spectrum 16 scans were recorded.
Size exclusion chromatography (SEC) was performed using two separate systems. System 1: A TOSOH EcoSEC HLC-8320GPC system equipped with an EcoSEC RI detector and three columns (PSS PFG 5 μm; Microguard, 100 Å, and 300 Å) (MW resolving range: 300–100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) from PSS GmbH was used for the analysis. Dimethylformamide (DMF) (0.2 mL min−1, 50 °C) was used as the mobile phase. A conventional calibration method was employed using narrow linear poly(methyl methacrylate) (PMMA) standards (800–1600000 Da). Corrections for flow rate fluctuations were made using toluene as an internal standard. PSS WinGPC Unity software version 7.2 was used to process data. System 2: A Verotech PL-GPC 50 Plus system equipped with a PL-RI detector and two PLgel 5 μm MIXED-D (300 × 7.5 mm) columns from Varian was used for the analysis. Chloroform (CHCl3) (1 mL min−1, 30 °C) was used as the mobile phase. A conventional calibration method was employed using narrow polystyrene standards (PS) (162–371100 Da). Corrections for flow rate fluctuations were made using toluene as an internal standard. Cirrus GPC Software was used to process data.
000 Da) from PSS GmbH was used for the analysis. Dimethylformamide (DMF) (0.2 mL min−1, 50 °C) was used as the mobile phase. A conventional calibration method was employed using narrow linear poly(methyl methacrylate) (PMMA) standards (800–1600000 Da). Corrections for flow rate fluctuations were made using toluene as an internal standard. PSS WinGPC Unity software version 7.2 was used to process data. System 2: A Verotech PL-GPC 50 Plus system equipped with a PL-RI detector and two PLgel 5 μm MIXED-D (300 × 7.5 mm) columns from Varian was used for the analysis. Chloroform (CHCl3) (1 mL min−1, 30 °C) was used as the mobile phase. A conventional calibration method was employed using narrow polystyrene standards (PS) (162–371100 Da). Corrections for flow rate fluctuations were made using toluene as an internal standard. Cirrus GPC Software was used to process data.
Field emission scanning electron microscopy (FE-SEM) images were recorded on a Hitachi S-4800 FE-SEM. The samples were mounted on a substrate with carbon tape and coated with 5 nm of palladium.
Contact angles were measured at 50% RH and 23 °C on a KSV instrument CAM 200 equipped with a Basler A602f camera, using 5 μL droplets of Milli-Q water.
Immobilisation of an ATRP initiator (BiB) on filter paper
The procedure for the modification of filter paper with BiB was adopted from Carlmark and Malmström.28 Pieces of filter paper (2 × 3 cm2) were washed in ethanol (EtOH), acetone, and tetrahydrofuran (THF) by ultrasonication in each solvent for 2 min. Each filter paper was then immersed into a solution of BiB (305 mg, 1.33 mmol), TEA (148 mg, 1.46 mmol), and catalytic amounts of DMAP (approx. 10 mg) in THF (20 mL). The reaction proceeded for 1 h at room temperature on a shaking device. The initiator immobilised filter papers were thoroughly washed in THF and EtOH to remove any remaining reactants or byproducts formed. Finally, the filter papers were dried in a vacuum oven (50 °C) overnight.Immobilisation of a cleavable ATRP initiator (S–S BiB) on filter paper
The modification of the filter paper was performed in a similar manner as previously described by Malmström and co-workers.38 Filter papers (2 × 3 cm2) were washed in ethanol, acetone, and THF by ultrasonication in each solvent for 2 min. Each filter paper was then immersed in a solution of S–S BiB (93 mg, 0.22 mmol), TEA (24.5 mg, 0.24 mmol), and catalytic amounts of DMAP in THF (5 mL). The reaction proceeded for 1 h at room temperature on a shaking device. The initiator immobilised filter papers were thoroughly washed in THF and EtOH to remove any remaining reactants or byproducts formed. Finally the filter papers were dried in a vacuum oven (50 °C) overnight.Grafting of MA from BiB immobilised filter paper
A typical procedure for the photoinduced CRP of MA was adopted from Haddelton and co-workers.22 MA (6.0 g, 70 mmol), Cu(II)Br2 (1.0 mg, 4.6 μmol), Me6TREN (6.4 mg, 28 μmol), EBiB (45.3 mg, 0.232 mmol), BiB immobilised filter paper, and DMSO (6.0 g) were added to a 30 mL jar, equipped with a magnetic stirrer. The cream jar was sealed with a septum, purged with argon for 15 min, and placed under a UV-nail gel curing lamp (36 W, λmax ≈ 360 nm) for 90 min. The polymer formed from the sacrificial initiator (EBiB) was purified by diluting the samples with small amounts of acetone followed by precipitation of the polymer into cold methanol (MeOH). The modified papers were carefully washed with acetone, THF and EtOH. The washed filter papers, and the polymer formed from the sacrificial initiator, were dried in a vacuum oven (50 °C) overnight. To modify the target DP of the grafted polymer, the amount of monomer added was altered. The amount of solvent was regulated to maintain 50 wt%. All other parameters remained unchanged throughout the reactions. Two control experiments were performed. First, a filter paper without an immobilised initiator was subjected to the same reaction conditions as the grafted papers. This ensured that the free polymer formed from the sacrificial initiator in bulk was removed by the selected washing procedure, and that no polymerisation was initiated from filter paper if no immobilised initiator was present. Second, a BiB modified filter paper was placed in DMSO and monomer and subjected to the same conditions in terms of temperature, time and UV light exposure, as the grafted filter paper to ensure that no polymerisation occurred without the presence of the catalyst complex.Grafting of MA from S–S BiB immobilised filter paper
The procedure used for the grafting of the S–S BiB immobilised filter paper is identical to the procedure was used for the grafting from the BiB immobilised filter paper, as described above.Cleavage of PMA from S–S BiB grafted filter paper
The polymer grafts were cleaved from the filter paper in a similar manner as previously described by Malmström and co-workers.38 A PMA grafted S–S BiB filter paper (2 × 3 cm2) was added to a reaction mixture of DTT (160 mg, 1.04 mmol) and TEA (211 mg, 2.08 mmol) in THF (20 mL) in a glass vial and left to react for 5 days on a shaking device. The filter paper was then thoroughly washed with THF, acetone, and EtOH and left to dry in a vacuum oven (50 °C) overnight. The polymer solution was concentrated and the cleaved polymer was analysed by SEC.Surface initiated ring opening co-polymerisation (SI-ROP) of αClεCL and εCL from filter paper
A typical procedure used for the SI-ROP from filter paper was as follows: filter papers (1 × 1.5 cm2) were washed in acetone and EtOH and dried in a vacuum oven (50 °C) overnight. Two filter papers were placed in a vial equipped with a magnetic stirrer and αClεCL (1.0 g, 6.7 mmol), εCL (2.3 g, 20 mmol), toluene (2 mL) and benzyl alcohol (9.7 mg, 90 μmol) were added. The vial was sealed with a rubber septum and degassed by three vacuum/argon cycles (5 + 5 min). During the third argon cycle, SnOct2 (66 mg, 0.16 mmol) was added. The vial was immersed in an oil bath (110 °C) and flushed with argon for 15 min. The polymerisation was allowed to proceed for 15 h and was terminated by dilution with an excess amount of THF. The polymer formed from the sacrificial initiator was purified by precipitation into cold MeOH. The grafted filter papers were washed by ultrasonication (THF, 3 min), followed by Soxhlet extraction (THF, 24 h). The washed filter papers, and the polymer formed from the sacrificial initiator, were dried in a vacuum oven (50 °C) overnight. To vary the composition of αClεCL and εCL, the amount of εCL added to the mixture was varied. The amount of Sn(Oct)2 and benzyl alcohol were altered to maintain 2 wt% and give a target DP of 300 respectively. All other parameters remained unchanged throughout the reactions.Grafting of DEGA from poly(αClεCL-co-εCL) grafted filter paper
A typical procedure used for the grafting of DEGA from poly(αClεCL-co-εCL) grafted filter paper was as follows: DEGA (1.5 g, 8.0 mmol), Cu(II)Br2 (0.4 mg, 1.6 μmol), Me6TREN (2.2 mg, 9.6 μmol), poly(αClεCL24-co-εCL76) (41 mg, corresponding to 0.08 mmol Cl, i.e. initiating groups), poly(αClεCL24-co-εCL76) grafted filter paper (1 × 1.5 cm2), deionised H2O (250 μL), and DMSO (12.0 g) were added to a 30 mL cream jar, equipped with a magnetic stirrer. The cream jar was sealed with a septum and purged with argon for 15 min, and placed in a UV-nail gel curing box (36 W, λmax ≈ 360 nm) for 90 min. The polymer formed from the sacrificial initiator was purified by precipitation into deionised H2O (T = 30 °C). The modified papers were carefully washed with acetone, THF and EtOH. The washed filter papers and the polymer formed from the sacrificial initiator were dried in a vacuum oven (50 °C) overnight. Filter papers with different compositions of the poly(αClεCL-co-εCL) were utilised for the grafting of DEGA, and the amount of macroinitiator added was then altered, to maintain the number of Cl units constant at 0.08 mmol. All other parameters remained unchanged throughout the reactions.Results and discussion
A photoinduced CRP system, adapted from Haddleton and co-workers,22 which utilises UV-light to generate Cu(I) catalyst complexes from Cu(II) without the use of a reducing agent, has been successfully employed to graft MA and DEGA from Whatman 1 filter paper. The reactions were easily conducted by adding the reactants to the reaction vessel, purging with argon, and performing the reaction in a UV-nail curing box (36 W, λmax ≈ 360 nm) for 90 min.Grafting of PMA from BiB-immobilised filter paper
In all polymerisations, a sacrificial initiator was added to the solution, resulting in a free, unattached polymer, formed in parallel to the grafted chains. Conversions, molecular weights, and molar dispersities of the free polymers were determined by 1H NMR and SEC (Table 1, Fig. S3†). The targeted DPs for the grafting were 300 and 600, respectively. All reactions were performed in duplicate. The monomer conversion and the SEC results for the polymer formed from the sacrificial initiator show that the reactions were consistent and reproducible. The molecular weights determined by the SEC were slightly higher than the theoretical molecular weights calculated from the monomer conversion (as determined by 1H-NMR). The dispersities of the polymers formed were low, showing the formation of well-defined polymers during the polymerisations. The increase in target DP from 300 to 600 did not affect the final conversion and a high conversion was reached within 90 minutes of irradiation.| Sample namea | DPtarget | Conversionb (%) | M n, theo (g mol−1) | M n, SEC (g mol−1) | Đ |
|---|---|---|---|---|---|
| a Samples have been denoted as follows: BiB-PMAx(y) where x represents the reactions target DP and y = 1 or 2 denotes duplicates. S–S before the sample name indicates that the free polymer has been formed in the reaction with a cleavable initiator attached to the filter paper. Cleaved S–S before the sample name represents cleaved polymer for the respective reactions. b Monomer conversion calculated from 1H NMR. c M n, theo calculated from the conversion according to 1H NMR assuming 100% initiator efficiency. d Results obtained from DMF-SEC. | |||||
| BiB-PMA300(1) | 300 | 88 | 22700 |
24100 |
1.1 |
| BiB-PMA300(2) | 300 | 84 | 21700 |
29000 |
1.1 |
| BiB-PMA600(1) | 600 | 91 | 47000 |
61300 |
1.1 |
| BiB-PMA600(2) | 600 | 88 | 45500 |
59600 |
1.1 |
| S–S BiB-PMA300(1) | 300 | 89 | 23000 |
28800 |
1.1 |
| Cleaved S–S BiB-PMA300(1) | 300 | — | — | 64000 |
1.3 |
| S–S BiB-PMA300(2) | 300 | 78 | 20100 |
27200 |
1.1 |
| Cleaved S–S BiB-PMA300(2) | 300 | — | — | 38400 |
1.4 |
| S–S BiB-PMA600(1) | 600 | 73 | 37700 |
41300 |
1.1 |
| Cleaved S–S BiB-PMA600(1) | 600 | — | — | 47900 |
1.5 |
| S–S BiB-PMA600(2) | 600 | 81 | 41800 |
49700 |
1.1 |
| Cleaved S–S BiB-PMA600(2) | 600 | — | — | 66600 |
1.4 |
The appearance of the carbonyl peak (1730 cm−1) in the FT-IR spectrum (Fig. 1) of the grafted filter papers clearly showed that filter papers had been grafted with the polymer. The carbonyl peak intensity in the spectrum revealed that all the papers had been grafted with relatively large amounts of the polymer. The agreement in the intensity of the peaks from duplicate samples demonstrated that there was more polymer grafted on the filter papers grafted in the reactions with target DP 600, showing that it was possible to regulate the amount of grafted polymer by altering the target DP using a sacrificial initiator. The intensity of the carbonyl peak in the spectrum was significantly higher than the typical intensities reported for polymer grafted filter paper,28,29,40–42 showing the efficiency of the photoinduced CRP method. CAM of the grafted papers showed that they were hydrophobic with contact angles above 120°. However, it should be noted that the analysed filter paper is a rough inhomogeneous material and the contact angles should only be considered as indications of the change in hydrophobicity and not as absolute values. The structures of the grafted filter papers were also compared to BiB modified filter papers treated in the same manner as the grafted filter papers (Fig. 2). This treatment caused no change in the FT-IR spectra or CAM and hence behaved similar to untreated filter paper. Similarly, unmodified filter paper present during polymerisation from a sacrificial initiator showed no carbonyl peak after being subjected to the same washing procedure as the grafted filter papers. Grafted and unmodified filter papers were analysed by FE-SEM (Fig. 2) and it appeared that the grafting of PMA from the filter papers created an open fibre structure. However, this was believed to be caused by the DMSO solvent.
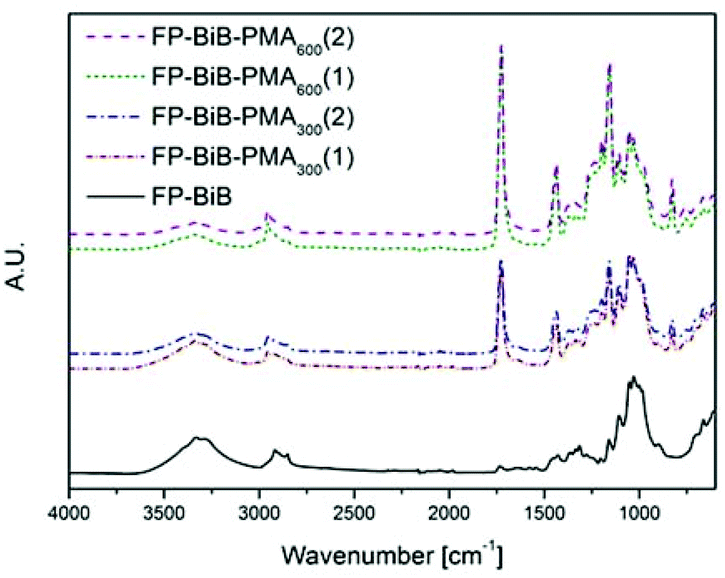 | ||
| Fig. 1 FT-IR spectra of PMA grafted BiB modified filter paper. | ||
 | ||
| Fig. 2 Unmodified sample swollen in DMSO and dried before FE-SEM analysis: (a) ×100 magnification and (b) ×5.00 k magnification and grafted sample BiB-PMA600: (c) ×100 magnification and (d) ×5.00k magnification. | ||
DMSO is known to cause swelling of cellulose fibres and the theory is that the swelling facilitates the possibility for larger amounts of polymer to be grafted due to a larger available surface area, and as the polymerisation proceeded it started to disintegrate the fibre network. This idea was supported by investigating traditional ATRP for the grafting of MA in DMSO from filter paper, which also resulted in a large amount of grafted polymer compared to previously reported results for MA grafting in other organic solvents.28,29
Grafting of PMA from S–S BiB-immobilised filter paper
The reaction scheme for the grafting of S–S BiB modified filter paper is shown in Scheme 1. The reactions were performed similar to the BiB immobilised filter paper, with the only difference being the structure of the immobilised initiator. Conversions, molecular weights, and dispersities of the free polymers and the cleaved PMA grafts were determined from 1H NMR and SEC (Table 1). The target DPs chosen for the grafting were the same as for the grafting from the BiB modified filter papers (DP of 300 and 600). Reactions were performed in duplicate and showed good reproducibility. The slightly lower monomer conversion in comparison with the BiB modified filter papers may suggest that fewer initiating groups were available on the surface or that the S–S BiB initiator was not as efficient in initiating polymerisation as the BiB initiator. This was supported by the FT-IR measurements which showed lower intensities of the carbonyl peak for the S–S BiB grafted filter papers than for the BiB grafted filter papers (Fig. S1†). Comparing the SEC results for the polymer formed from the sacrificial initiator during the grafting of BiB modified filter papers to the free polymer formed during the grafting of S–S BiB modified filter papers showed that the Mn of the polymers with target DP 300 were similar. The Mn for the polymers with target DP 600 was lower for the S–S BiB modified filter papers. For the S–S BiB grafted filter papers, the Mn reported for the DP 600 polymers were closer to the theoretical Mn, possibly suggesting a higher efficiency of the sacrificial initiator in these reactions. The dispersities for the polymers formed from the sacrificial initiator were 1.1 for all reactions, showing the formation of well-defined polymers.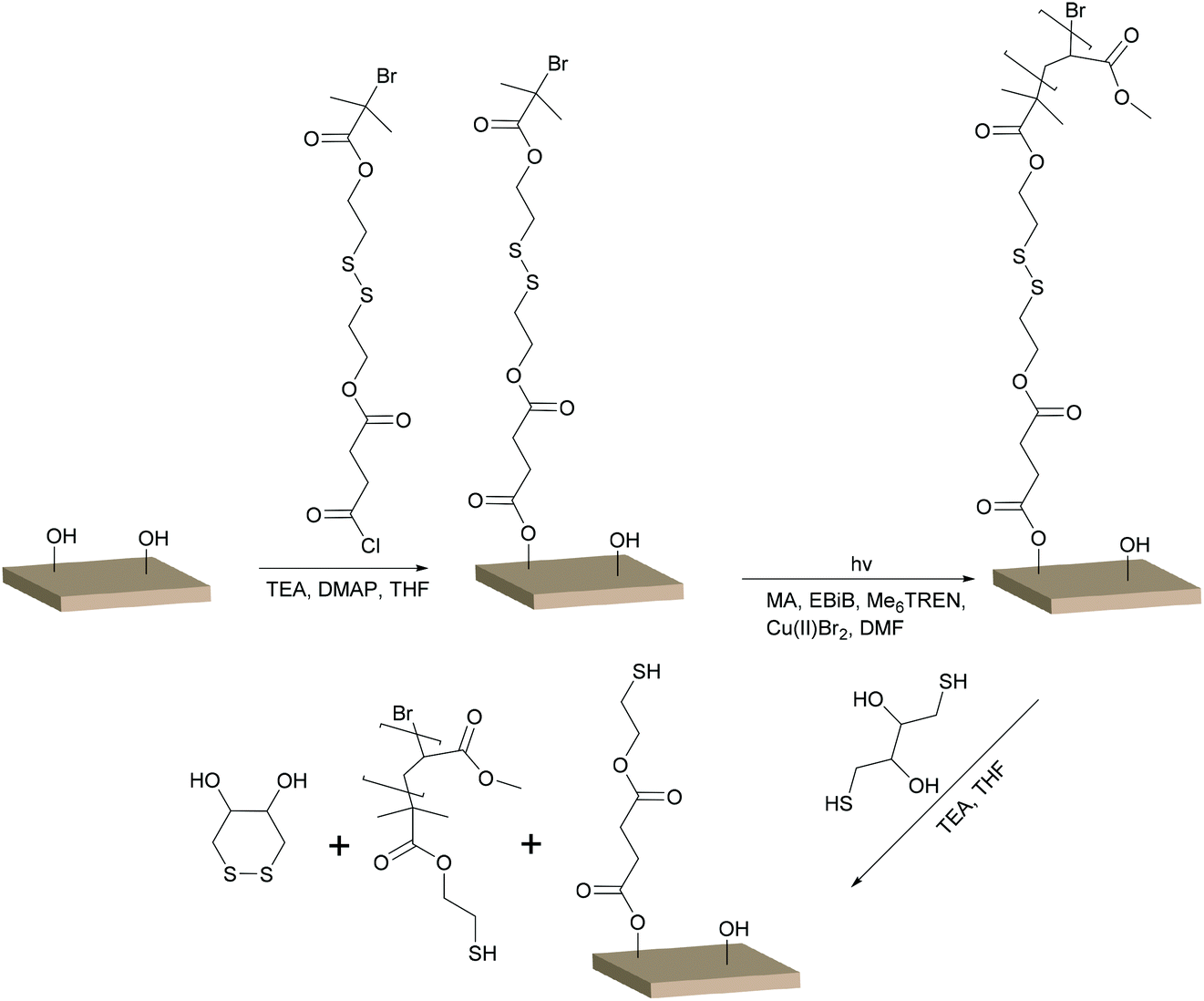 | ||
| Scheme 1 UV induced controlled radical polymerisation of MA from S–S BiB modified filter paper and subsequent cleavage of the grafted chains with DTT. | ||
The chains grafted from the S–S BiB modified filter papers were cleaved by the utilisation of DTT, as previously reported.38 FT-IR spectra (Fig. S1†) of the filter papers before and after cleavage revealed that the intensity of the carbonyl peak was strongly reduced after cleavage indicating that a majority of the polymer had been cleaved off the surface. However, a small signal from the carbonyl remained which suggested that some polymers remained on the substrates. Additional cleavage attempts were performed with TCEP and also DTT in DMSO, but it was not possible to remove any additional polymer. A plausible explanation to this could be that some of the grafting occurred inside of the swollen fibre wall, and that the total removal of this polymer was not achieved under the chosen experimental conditions, even when the polymer had been cleaved from the surface. FE-SEM images of the S–S BiB modified filter papers (Fig. S2†), before and after cleavage, support the result from the FT-IR that most of the polymer was removed from the filter paper during the cleaving reactions.
SEC analysis of the cleaved polymer showed that the polymerisation from the surface did not result in well-defined polymers as the bulk polymerisations. The polymer cleaved from the surface had a higher dispersity and also a higher molecular weight than the polymer formed from the sacrificial initiator. This result was not in agreement with the earlier study performed using ARGET-ATRP where there was a good correlation between the grafted polymer and the polymer formed from a sacrificial initiator.38 A difference between the previous study and this study was that the polymer in this study was not fully cleaved from the surface. However, as the grafted polymer showed a higher dispersity, with a higher molecular weight than the polymer formed from the sacrificial initiator (Fig. S4 and S5†), it seems unlikely that the removal of all polymers would decrease the dispersity. A possible explanation for the difference between the studies could be that the Cu(I) species, which in this study was created due to UV-light exposure, reacted quickly after formation. Effectively, this would have caused the species to react more readily on the areas of the filter papers most exposed to the UV-light. Another possible explanation for the higher dispersity of the grafted polymer chains could be that the swelling, followed by disintegration of the fibres, continuously caused more initiating sites to be exposed on the filter paper surface. This in turn would have resulted in a continuous initiation of new polymer chains from the surface.
CAM of the grafted and cleaved filter papers showed that the grafted papers had a hydrophobic surface with stable contact angles above 120°. The cleaved filter papers were highly hydrophilic and adsorbed water drops instantly (Table S1†) which further corroborates the cleavage of the PMA chains from the surface.
Ring opening co-polymerisation (ROP) of αClεCL and εCL from filter paper
Rather than immobilising an initiator function on the OH-groups of cellulose, a polymer containing an initiator function for ATRP could be grafted from the surface and conversely increase the density of the initiating groups off the surface. In earlier work by our group, glycidyl methacrylate (GMA) was grafted from a cellulose surface followed by the opening of the epoxy-groups, resulting in two hydroxyl groups per repeating unit. The hydroxyl groups were then subsequently reacted with BiB to introduce two initiating functions per repeat unit of PGMA.43 However, this reaction demanded four reaction steps, including the immobilisation of BiB on the filter paper. A more elegant and straightforward approach was utilised herein by the ROP of a cyclic monomer containing a halogen in the α-position, which initiates directly from the OH-groups and hence requires only one step. This is the first study in which an α-functionalised monomer has been a ring-opened form of a cellulose substrate and utilised in a “graft-on-graft” approach for surface modification. αClεCL was copolymerised with εCL from the surface of filter paper and benzyl alcohol was utilised as a sacrificial initiator, creating a free polymer as in the case of MA. Different ratios of αClεCL and εCL were utilised, resulting in varied densities of the initiating Cl-groups.The reaction scheme for the copolymerisation grafting of αClεCL and εCL, and the subsequent grafting of DEGA, from filter paper can be seen in Scheme 2. Conversions, molecular weights and dispersities of the free polymers were determined from 1H NMR and SEC (Table 2, Fig. S6–S8†). The target DP for all three monomer compositions for the copolymerisation of αClεCL and εCL was 100. The final molecular weights of the polymers formed from the sacrificial initiator were significantly lower for all three polymerisations than the theoretical molecular weight calculated from 1H NMR. This indicated that water may have been present and initiated polymerisation or caused hydrolysis, despite careful drying of all reactants prior to the polymerisations. The rather broad dispersities of the final polymers from the ROP indicated that the polymerisations were not well controlled. However, a good control of the ROP was not the scope of this study, and was therefore not of major concern. The monomer composition of the polymers, determined by 1H NMR, was in good agreement with the ratio of the co-monomer feed, showing that it was possible to regulate the ratio of Cl units incorporated in the final polymers. This result was important, as the incorporated Cl units were further used as initiators for the polymerisation of DEGA. The grafted filter papers were analysed by FT-IR (Fig. 3). As shown from the peak of the carbonyl in the spectra, polymers have been grafted from all three filter papers. Despite the relatively low intensity of the carbonyl peak, the grafted papers were hydrophobic when analysed with CAM. In FE-SEM images of the P(αClεCL-co-εCL) grafted filter papers (Fig. 4) the fibrillar structure of the fibres is still visible after grafting, also indicative of only small amounts of grafted polymer. Unfortunately, it was not possible to determine the monomer ratio in the polymer grafted from the filter papers, as the grafted polymer could not be separated from the cellulose. It was assumed that the monomer ratio of the grafted polymer was the same as for the polymer formed in bulk.
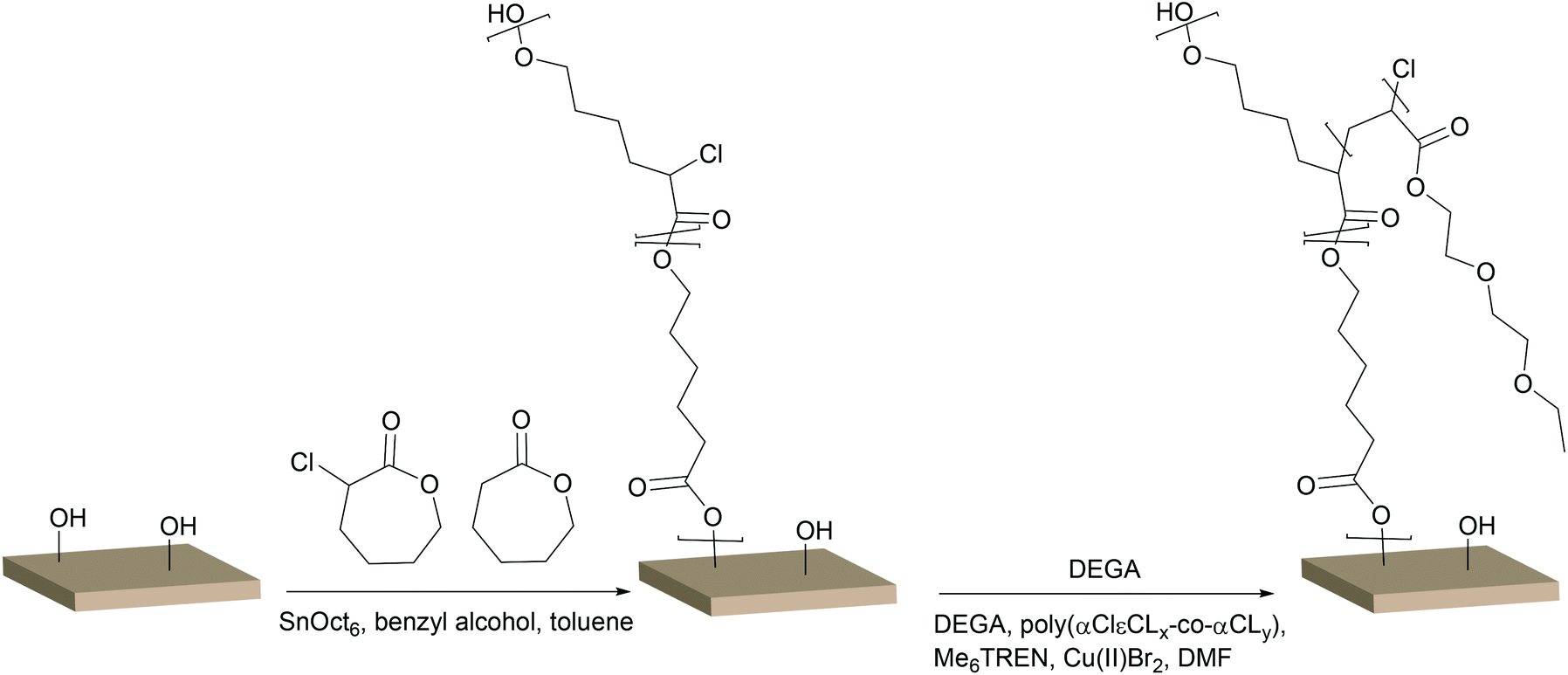 | ||
| Scheme 2 Surface initiated ring opening polymerisation of αClεCL and εCL followed by UV induced controlled radical polymerisation of DEGA from Whatman 1 filter paper. | ||
 | ||
| Fig. 3 FT-IR spectra of poly(αClεCL24-co-εCL76) grafted filter paper; before and after grafting with DEGA. | ||
 | ||
| Fig. 4 (a) Poly(αClεCL24-co-εCL76) and (b) poly(αClεCL24-co-εCL76)-g-PDEGA. | ||
| Sample namea | DPtarget | Conversionb (%) | M n, theo (g mol−1) | M n, SEC (g mol−1) | Đ |
|---|---|---|---|---|---|
| a Samples have been named accordingly poly(αClεCLx-co-εCLy), where x and y equal the molar composition in the polymer. The addition of -g-PDEGA(z) indicates that the poly(αClεCLx-co-εCLy) has been used as a macro initiator for the polymerisation of DEGA, and z = 1 or 2 has been given to differentiate duplicate samples. b Monomer conversion calculated from 1H NMR. c M n, theo. calculated from the conversion according to 1H NMR assuming 100% initiator efficiency. d Results obtained from CHCl3-SEC. | |||||
| Poly(αClεCL24-co-εCL76) | 300 | >99 | 36700 |
13700 |
1.6 |
| Poly(αClεCL40-co-εCL60) | 300 | >99 | 38400 |
23100 |
1.5 |
| Poly(αClεCL70-co-εCL30) | 300 | >99 | 41500 |
9200 | 1.6 |
| Poly(αClεCL24-co-εCL76)-g-PDEGA(1) | 100 | 71 | 999000 |
174000 |
1.6 |
| Poly(αClεCL24-co-εCL76)-g-PDEGA(2) | 100 | 71 | 999000 |
13000 |
1.6 |
| Poly(αClεCL40-co-εCL60)-g-PDEGA(1) | 100 | 75 | 1732000 |
192000 |
1.5 |
| poly(αClεCL40-co-εCL60)-g-PDEGA(2) | 100 | 74 | 1709000 |
208000 |
1.5 |
| Poly(αClεCL70-co-εCL30)-g-PDEGA(1) | 100 | 59 | 2374000 |
301000 |
1.4 |
| Poly(αClεCL70-co-εCL30)-g-PDEGA(2) | 100 | 60 | 241300 |
304000 |
1.4 |
PDEGA grafted poly(αClεCL-co-εCL) filter paper
The Cl-functionality of the grafted P(αClεCL-co-εCL) was subsequently utilised for the photoinduced CRP of DEGA. A free, sacrificial macroinitiator (i.e. free poly(αClεCL-co-εCL) was added to the reaction mixture to form a free, unbound graft-on-graft copolymer in solution which could be further analysed by conventional, solution based methods. The unbound polymer was assumed to have similar characteristics as the graft-on-graft copolymer on the surface.Conversions, molecular weights and dispersities of PDEGA-grafted poly(αClεCLx-co-εCLy), used as a sacrificial macroinitiator, were determined by 1H NMR and SEC (Table 2, Fig. S6–S8†). The target DP, calculated from the ratio of CL containing repeating units in the polymers, was 100 for all reactions. The Mn of the polymers, as determined by SEC, was significantly lower than the theoretical Mn. This difference in Mn was to a large extent caused by the comb structure of the polymers, which has a smaller hydrodynamic volume than the linear polymers used for calibration. Since macroinitiators with several initiating groups were used for the polymerisation, it was difficult to draw any conclusion regarding the dispersity of the PDEGA grafts. However, it is clear that the dispersity does not increase, which indicates the formation of relatively well-defined PDEGA grafts during the reactions.
FT-IR spectra of the grafted filter papers (Fig. 3) showed a large increase in the intensity of the carbonyl peak, showing that the grafting of DEGA from the poly(αClεCLx-co-εCLy) grafted filter papers was successful. FE-SEM images of the graft-on-graft filter papers (Fig. 4) showed a smoothening of the fibre surface also indicative of a successful polymer grafting. The synthesis of graft-on-grafts from cellulose in a two-step reaction, compared to the previous study involving a total of five steps, enabled a higher grafting density than when a BiB modified cellulose was utilised. We believe this modification to be an attractive method for creating high grafting densities as well as complex architectures on cellulosic and other bio-fibre based surfaces containing hydroxyl groups.
PDEGA is a thermoresponsive polymer with an LCST in the range 9.0–16.5 °C.44–46 Previous work has shown that cellulose fibres grafted with types of polymers produced thermoresponsive materials.47,48 The filter paper modified in the graft-on-graft approach with PDEGA grafted from the poly(αClεCL-co-εCL) was tested for thermo-responsive behaviour in the form of switchable hydrophobicity. CAM showed that the PDEGA grafted filter papers adsorb a water drop instantly at low temperature (approx. 4 °C). When the temperature was increased above the LCST of PDEGA, to approx. 50 °C, the water contact angle increased to around 90°. The adsorption of water at the lower temperature shows that the properties of the grafted PDEGMA had a larger influence on the hydrophilic/hydrophobic properties of the papers than the grafted poly(αClεCL-co-εCL) and that the thermoresponsive properties of PDEGMA were transferred to the cellulose surface. This was in agreement with earlier results reported.29
Conclusions
The photoinduced CRP technique has successfully been employed for the surface-initiated polymer grafting of cellulose surfaces. The polymerisations proceeded in an efficient manner, resulting in large amounts of grafted polymers. Cleavage of the grafted chains revealed that the polymerisation from the cellulose surface proceeded with control; however it proceeded with less control than the bulk polymerisation and the cleaved polymer possessed a higher molecular weight. Furthermore, it was shown that a graft-on-graft copolymerisation from the surface of filter paper could be performed in a simple two step polymerisation. The combination of a surface-initiated ROP of monomers containing a halogen in the α-position, followed by the photoinduced controlled radical polymerisation effectively reduced the number of steps as compared to previous synthetic routes. Utilising a monomer which resulted in a thermoresponsive polymer for the second polymerisation in the graft-on-graft polymerisation produced a thermo-responsive cellulosic material. Overall, the photoinduced CRP proved to be a highly versatile technique for the grafting of cellulose surfaces, resulting in large grafting amounts in short reaction periods, with good control over the grafted layer.Acknowledgements
The BiMaC Innovation Centre and the KAMI foundation are greatly acknowledged for financial support.Notes and references
- D. Roy, M. Semsarilar, J. T. Guthrie and S. Perrier, Chem. Soc. Rev., 2009, 38, 2046–2064 RSC.
- D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- A. Carlmark, E. Larsson and E. Malmström, Eur. Polym. J., 2012, 48, 1646–1659 CrossRef CAS PubMed.
- M. N. Belgacem and A. Gandini, Compos. Interfaces, 2005, 12, 41–75 CrossRef CAS PubMed.
- K. Missoum, M. N. Belgacem and J. Bras, Materials, 2013, 6, 1745–1766 CrossRef CAS PubMed.
- G. Siqueira, J. Bras and A. Dufresne, Polymers, 2010, 2, 728–765 CrossRef CAS PubMed.
- E. Malmström and A. Carlmark, Polym. Chem., 2012, 3, 1702–1713 RSC.
- H. Kang, X. Gao, R. Liu and Y. Huang, ACS Symp. Ser., 2012, 1107, 109–131 CrossRef CAS PubMed.
- F. Joubert, O. M. Musa, D. R. W. Hodgson and N. R. Cameron, Chem. Soc. Rev., 2014, 43, 7217–7235 RSC.
- S. Hansson, V. Trouillet, T. Tischer, A. S. Goldmann, A. Carlmark, C. Barner-Kowollik and E. Malmström, Biomacromolecules, 2013, 14, 64–74 CrossRef CAS PubMed.
- S. Hansson, T. Tischer, A. S. Goldmann, A. Carlmark, C. Barner-Kowollik and E. Malmstroem, Polym. Chem., 2012, 3, 307–309 RSC.
- T. Tischer, T. K. Claus, K. K. Oehlenschlaeger, V. Trouillet, M. Bruns, A. Welle, K. Linkert, A. S. Goldmann, H. G. Boerner and C. Barner-Kowollik, Macromol. Rapid Commun., 2014, 35, 1121–1127 CrossRef CAS PubMed.
- T. Tischer, C. Rodriguez-Emmenegger, V. Trouillet, A. Welle, V. Schueler, J. O. Mueller, A. S. Goldmann, E. Brynda and C. Barner-Kowollik, Adv. Mater., 2014, 26, 4087–4092 CrossRef CAS PubMed.
- S. Kalia, S. Boufi, A. Celli and S. Kango, Colloid Polym. Sci., 2014, 292, 5–31 CAS.
- Z. Guan and B. Smart, Macromolecules, 2000, 33, 6904–6906 CrossRef CAS.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- J. E. Poelma, B. P. Fors, G. F. Meyers, J. W. Kramer and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52, 6844–6848 CrossRef CAS PubMed.
- J. Mosnacek and M. Ilcikova, Macromolecules, 2012, 45, 5859–5865 CrossRef CAS.
- D. Konkolewicz, K. Schroder, J. Buback, S. Bernhard and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1219–1223 CrossRef CAS.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 2271–2275 CrossRef CAS.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Rapid Commun., 2011, 32, 58–62 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou, Q. Zhang, J. Burns, S. R. Samanta, C. Waldron, A. J. Haddleton, R. McHale, D. Fox, V. Percec, P. Wilson and D. M. Haddleton, J. Am. Chem. Soc., 2014, 136, 1141–1149 CrossRef CAS PubMed.
- M. E. Levere, I. Willoughby, S. O'Donohue, A. de Cuendias, A. J. Grice, C. Fidge, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1086–1094 RSC.
- A. Anastasaki, C. Waldron, P. Wilson, R. McHale and D. M. Haddleton, Polym. Chem., 2013, 4, 2672–2675 RSC.
- P. M. Wright, G. Mantovani and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7376–7385 CrossRef CAS.
- B. Wenn, M. Conradi, A. D. Carreiras, D. M. Haddleton and T. Junkers, Polym. Chem., 2014, 5, 3053–3060 RSC.
- A. Anastasaki, V. Nikolaou, A. Simula, J. Godfrey, M. Li, G. Nurumbetov, P. Wilson and D. M. Haddleton, Macromolecules, 2014, 47, 3852–3859 CrossRef CAS.
- A. Carlmark and E. Malmström, J. Am. Chem. Soc., 2002, 124, 900–901 CrossRef CAS PubMed.
- A. Carlmark and E. E. Malmström, Biomacromolecules, 2003, 4, 1740–1745 CrossRef CAS PubMed.
- G. Morandi, L. Heath and W. Thielemans, Langmuir, 2009, 25, 8280–8286 CrossRef CAS PubMed.
- G. Li, H. Yu and Y. Liu, Adv. Mater. Res., 2011, 221, 90–94 CrossRef CAS.
- M. Li, G. Shan, Y. Bao and P. Pan, J. Appl. Polym. Sci., 2014, 131, 41115 Search PubMed.
- S. Lenoir, R. Riva, X. Lou, C. Detrembleur, R. Jerome and P. Lecomte, Macromolecules, 2004, 37, 4055–4061 CrossRef CAS.
- C. Detrembleur, M. Mazza, O. Halleux, P. Lecomte, D. Mecerreyes, J. L. Hedrick and R. Jerome, Macromolecules, 2000, 33, 14–18 CrossRef CAS.
- S. El Habnouni, V. Darcos and J. Coudane, Macromol. Rapid Commun., 2009, 30, 165–169 CrossRef CAS PubMed.
- P. Olsen, J. Undin, K. Odelius and A.-C. Albertsson, Polym. Chem., 2014, 5, 3847–3854 RSC.
- M. Ciampolini and N. Nardi, Inorg. Chem., 1966, 5, 41–44 CrossRef CAS.
- S. Hansson, P. Antoni, H. Bergenudd and E. Malmström, Polym. Chem., 2011, 2, 556–558 RSC.
- S. Lenoir, R. Riva, X. Lou, C. Detrembleur, R. Jerome and P. Lecomte, Macromolecules, 2004, 37, 4055–4061 CrossRef CAS.
- C. Tyagi, L. K. Tomar, P. Kumar, V. Pillay and H. Singh, Anal. Methods, 2014, 6, 7374–7383 RSC.
- S. Hansson, E. Östmark, A. Carlmark and E. Malmström, ACS Appl. Mater. Interfaces, 2009, 1, 2651–2659 CAS.
- D. Roy, J. T. Guthrie and S. Perrier, Macromolecules, 2005, 38, 10363–10372 CrossRef CAS.
- D. Nyström, J. Lindqvist, E. Östmark, P. Antoni, A. Carlmark, A. Hult and E. Malmström, ACS Appl. Mater. Interfaces, 2009, 1, 816–823 Search PubMed.
- K. Skrabania, J. Kristen, A. Laschewsky, O. Akdemir, A. Hoth and J.-F. Lutz, Langmuir, 2007, 23, 84–93 CrossRef CAS PubMed.
- C. Boyer, M. R. Whittaker, K. Chuah, J. Liu and T. P. Davis, Langmuir, 2010, 26, 2721–2730 CrossRef CAS PubMed.
- D. Popescu, R. Hoogenboom, H. Keul and M. Moeller, Polym. Chem., 2010, 1, 878–890 RSC.
- J. Lindqvist, D. Nyström, E. Östmark, P. Antoni, A. Carlmark, M. Johansson, A. Hult and E. Malmström, Biomacromolecules, 2008, 9, 2139–2145 CrossRef CAS PubMed.
- E. Larsson, C. Cobo Sanchez, C. Porsch, E. Karabulut, L. Wågberg and A. Carlmark, Eur. Polym. J., 2013, 49, 2689–2696 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional FT-IR, FT-SEM, CAM and SEC results. See DOI: 10.1039/c4py01618a |
| This journal is © The Royal Society of Chemistry 2015 |