
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePLA architectures: the role of branching
Stijn
Corneillie
and
Mario
Smet
*
Polymer Chemistry and Materials, Department of Chemistry, KU Leuven, Belgium. E-mail: stijn.corneillie@chem.kuleuven.be; mario.smet@chem.kuleuven.be
First published on 2nd December 2014
Abstract
Biobased and biodegradable polymers have become more and more interesting in view of waste management and crude oil depletion. Several biopolymers have been researched, among which poly(lactide) (PLA) seems to be the most promising. Although a lot of research has been conducted on this polymer, PLA still displays some serious drawbacks such as limited melt strength, limited toughness, a lack of functional groups etc. One possibility to overcome such problems is by introducing branching. This can be done in several ways, resulting in PLA polymers with different topologies, ranging from star-branched to long chain branched. After a general introduction, several of these topologies will be described in detail.
Introduction
Synthetic plastics play a prominent role in modern life. They have become indispensable and provide a wide range of applications in diverse fields such as packaging, building materials, medical appliances etc. However, a worldwide production of approximately 288 million tons per year supplies the ecosystem with an enormous amount of waste, because synthetic plastics have a high resistance to chemical, physical and biological degradation.1 Moreover, these plastics are mostly derived from non-renewable crude oil and natural gas resources.2 Bioplastics have been introduced in this respect to overcome the necessity of non-renewable resources and at the same time to help decrease the amount of waste produced.3 These plastics can be biobased and/or biodegradable. A plastic is biobased when it is partly or entirely derived from natural resources including starch, cellulose, fatty acids, sugars, proteins etc. Examples of biobased plastics include poly(hydroxyalkanoate) (PHA), poly(lactic acid) (PLA), poly(butylene succinate) (PBS), starch-based polymers etc.3,4 Biodegradation is a process that is able to break down a polymer into H2O, CO2, CH4, and other low molecular weight products and compost by means of light, heat, bacterial activity and moisture. Poly(ε-caprolactone) (PCL) is an example of a synthetic biodegradable polymer, while PLA is biobased and biodegradable.5 An overview of common PLA thermal and mechanical properties can be found in Table 1. PLA is in this respect a very promising material because it combines biodegradability, biocompatibility and an excellent processability, while it is derived from natural resources.6 PLA however shows some disadvantages such as poor melting strength, a low degradation rate, a narrow processing window, limited toughness etc. Solutions for overcoming these disadvantages have been discussed in recent years.7 However, to the best of our knowledge, there has not been an overview regarding changes in architecture. Changing the architecture might improve mechanical properties, such as a reduced brittleness or a lower viscosity, but the resulting macromolecules may also find value in other applications, such as surfactants, carriers for drug delivery etc. Before discussing the strategic improvement of PLA properties, it is necessary to give a brief overview of some important properties of unmodified PLA.| Glass transition temperature (Tg) | 50–70 °C |
| Melting temperature (Tm) | 170–190 °C |
| Crystallinity (χc) | ∼35% |
| Intrinsic viscosity (η, in chloroform) | 3.8–8.2 dL g−1 |
| Tensile strength (film) | 28–50 MPa |
| Tensile modulus (film) | 1200–3000 MPa |
| Shear strength | 54.5 MPa |
| Share modulus | 1210–1430 MPa |
| Bending strength | 132 MPa |
| Bending modulus | 2800 MPa |
| Elongation at break (film) | 2.0–6.0% |
PLA: synthesis and scope
Lactic acid was the first building block to be used in the synthesis of PLA. It was isolated for the first time from sour milk by the Swedish chemist Carl Wilhelm Scheele in 1780. The commercial production of lactic acid started, however, in 1881.8 As can be seen from Fig. 1(a), lactic acid (2-hydroxy propionic acid) has one stereocentre; hence two optically active stereoisomers are possible: L-lactic acid and D-lactic acid. Whilst L-lactic acid is produced in humans and mammals, a combination of both is produced in bacteria. Lactic acid is, however, also produced from corn, beet sugar, cane, etc. by bacterial fermentation of carbohydrates. Through this pathway, sugars are converted to lactic acid by Lactobacilli.8 Lactic acid then undergoes condensation polymerization to form PLA. In this case rather low molecular weight polymers were formed, as can be seen from the Carothers eqn (1):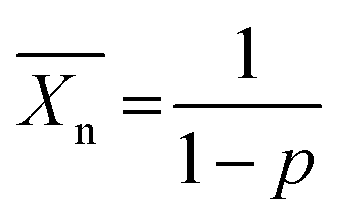 | (1) |
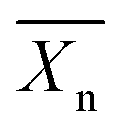 represents the number average value of the degree of polymerization and p the conversion during polymerization.9 It can be seen that a high conversion is needed to obtain a high degree of polymerization (and hence a higher molecular weight). Due to common side-reactions in a condensation polymerisation (such as cyclization due to back-biting, incomplete reaction, the presence of water etc.), only low molecular weight polymers are obtained. These materials tend to be brittle, which limits their usage in industry.
represents the number average value of the degree of polymerization and p the conversion during polymerization.9 It can be seen that a high conversion is needed to obtain a high degree of polymerization (and hence a higher molecular weight). Due to common side-reactions in a condensation polymerisation (such as cyclization due to back-biting, incomplete reaction, the presence of water etc.), only low molecular weight polymers are obtained. These materials tend to be brittle, which limits their usage in industry.
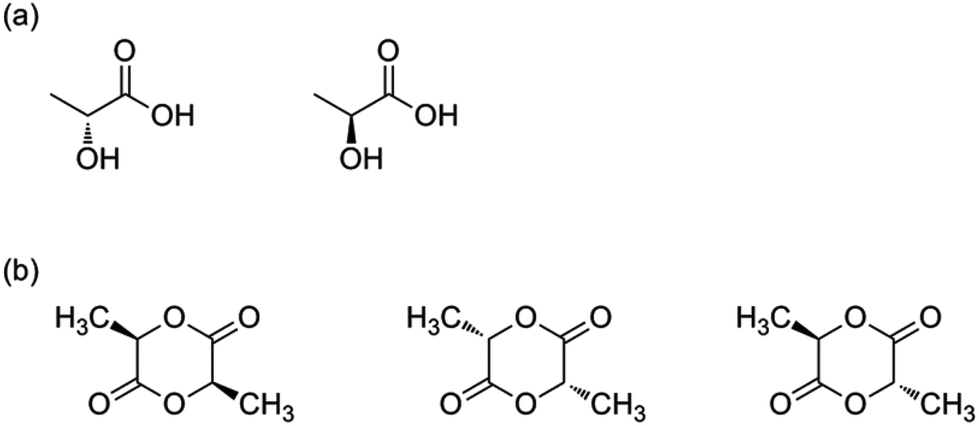 | ||
| Fig. 1 (a) Lactic acid stereoisomers: left: L-lactic acid, right: D-lactic acid. (b) Lactide stereoisomers: from left to right: L-lactide, D-lactide and meso-lactide. | ||
Two molecules of lactic acid can be dehydrated to lactide, a cyclic dimer of lactic acid. Lactide contains two stereocentres, which lead to three optically active stereoisomers: L,L-lactide (or L-lactide); D,L-lactide (or meso-lactide) and D,D-lactide (or D-lactide) (Fig. 1(b)). These lactides can be ring opened to form PLA. The ring opening polymerization (ROP) of lactides was first described by Carothers in 1932, but he only obtained low molecular weight PLA due to impurities.10 In 1954 lactide purification techniques were developed by Dupont, making it possible to obtain high molecular weight PLA.11 Low molecular weight PLA is synthesized on an industrial scale via condensation polymerisation followed by a subsequent depolymerisation under reduced pressure to give a mixture of L-lactide, D-lactide and meso-lactide (depending on lactic acid feedstock, temperature and catalyst).8 These lactides can then undergo the ROP to form high molecular weight PLA.
The ROP of lactides can proceed via an anionic, a cationic or a coordination–insertion pathway. Kricheldorf et al. have shown that trifluoromethanesulfonic acid and methyl trifluoromethanesulfonate are the only appropriate cationic initiators to yield PLA.12 Several initiators for anionic ROP have also been tested in the literature, such as NaH, potassium methoxide etc.: initiators with a higher nucleophilicity tend to be better, because they do not require high temperature conditions.13 When using anionic or cationic ROP, side-reactions (such as back-biting, racemisation) can occur (albeit to a lower extent as compared to the condensation polymerization), giving rise to lower molecular weight polymers and racemisation, impacting in turn the final thermal performances. In this respect it was found that ROP proceeding via a coordination–insertion pathway solves earlier mentioned problems (Fig. 2). Another benefit of the coordination–insertion pathway is that it can be performed in the absence of solvents and hence the reaction can be done in bulk (whilst anionic and cationic ROP often require solvents). Several catalysts have been screened in this respect. Tin, zinc, aluminium and other heavy metal-based catalysts have shown to form high molecular weight PLA.14 The best results were obtained with Al-based catalysts, tin oxide and tin octoate showing more than 90% conversion whilst having less than 1% racemisation. Owing to its greater stability and lower toxicity, combined with a low required amount, tin octoate (tin(II) 2-ethylhexanoate) is the catalyst that is most used in industry.
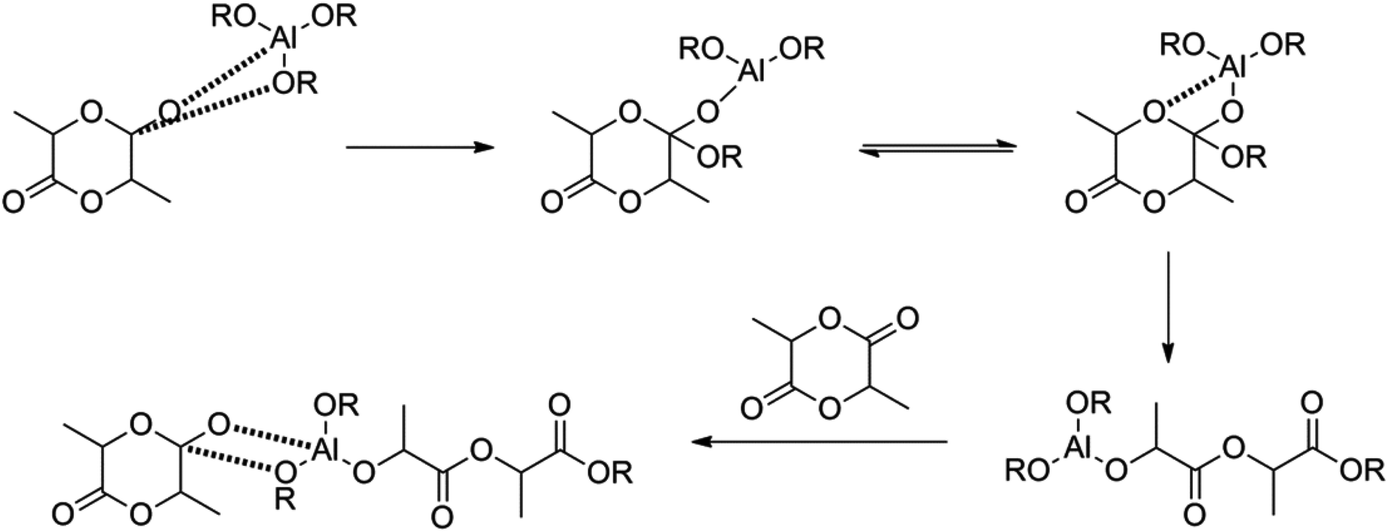 | ||
| Fig. 2 Coordination–insertion mechanism using aluminium alkoxides.14 | ||
In 1994 it was found by Mitsui Toatsu Chemicals that there is another way of making PLA with high molecular weights. They azeotropically dehydrated lactic acid and the catalyst in a refluxing, high-boiling, aprotic solvent under reduced pressure in the presence or absence of a catalyst. The catalysts used include metals, metal oxides and metal salts of group II, III, IV and V. PLA with molecular weight higher than 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da was obtained.15 A schematic representation of the formation of high molecular weight PLA is shown in Fig. 3.
000 Da was obtained.15 A schematic representation of the formation of high molecular weight PLA is shown in Fig. 3.
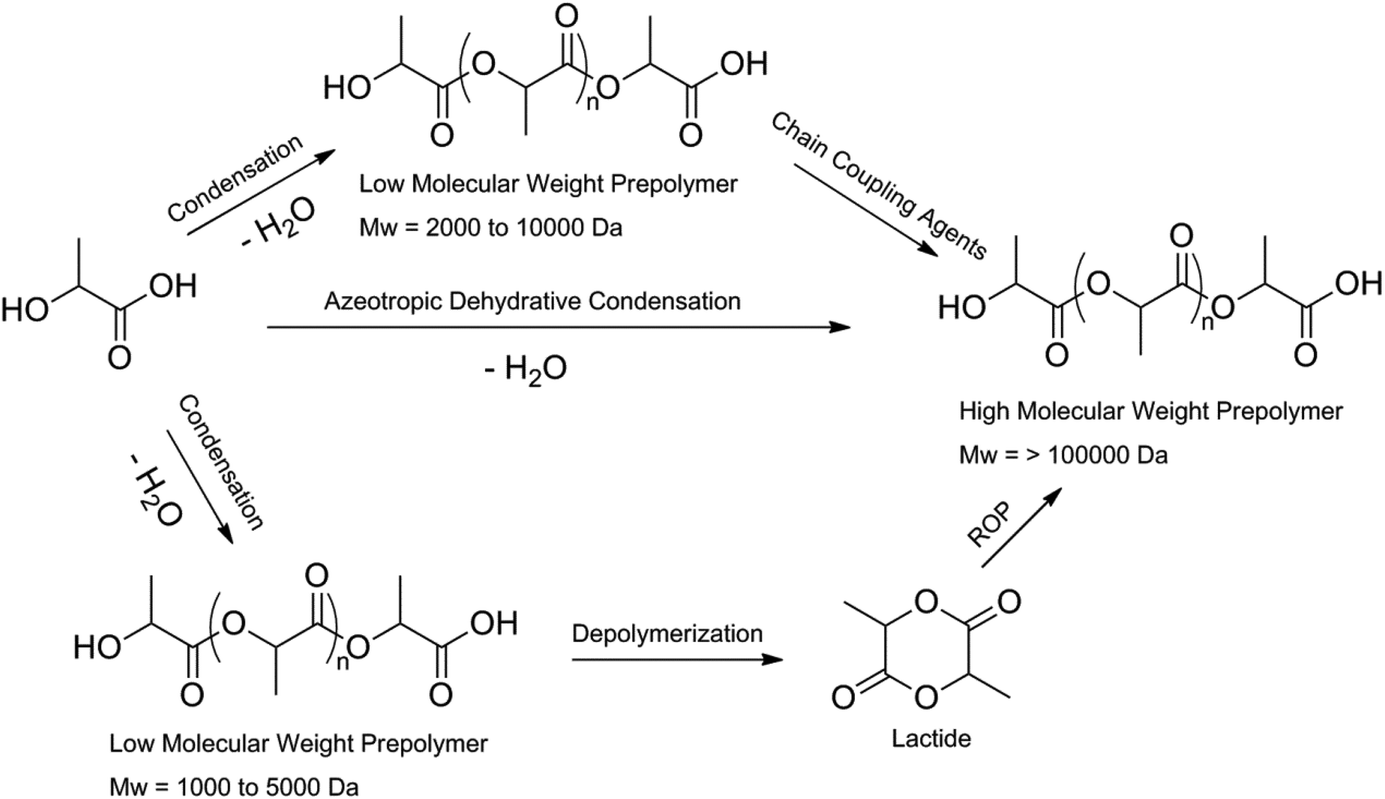 | ||
| Fig. 3 Methods to obtain high molecular weight PLA.8 | ||
In 2001 Hedrick et al. described an organo-catalyzed ROP of lactide, using 4-dimethylaminopyridine (DMAP) and 4-pyrrolidinopyridine (PPY) as catalysts and ethanol as an initiator.16 In this way toxic tin residues are avoided in the product, which is useful for biomedical applications. Several other metal-free catalysts have been investigated, such as phosphines, N-heterocyclic carbenes, guanidines etc. (Fig. 4).17 Notwithstanding the great effort made in this respect, ROP of lactide using Sn(Oct)2 as a catalyst is still the method used in industry.
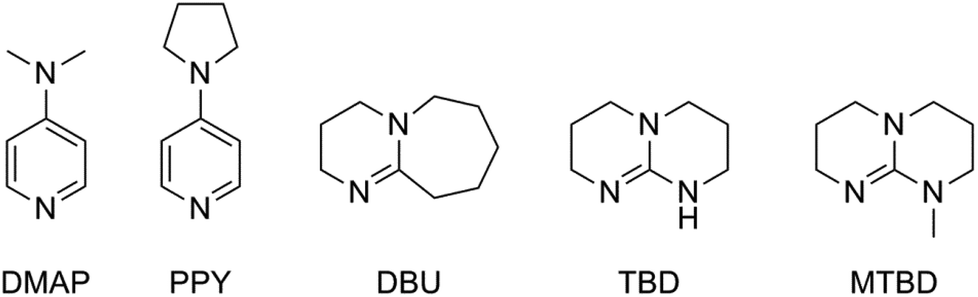 | ||
| Fig. 4 Structures of the most extensively studied metal-free initiators: 4-dimethylaminopyridine (DMAP), 4-pyrrolidinopyridine (PPY), 1,8-diazabicycloundec-7-ene (DBU), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) and its N-methylated analog (MTBD).17 | ||
As discussed before, PLA shows some disadvantages that need to be resolved. Possible ways of aiding PLA modification is by blending, copolymerization, introduction of branching, cross-linking, chain-extension and stereocomplexation. Most of those aids have been reviewed recently; however an overview of branched PLA topologies is lacking in the literature.7
Introduction of branching
Branching is introduced in PLA to obtain more interesting rheological and mechanical properties, such as a lower viscosity or an increase of the polymer strength. This is done by varying the molecular architecture to star-shaped, comb-shaped, dendritic/hyperbranched, H-shaped, long-chain branched or dumbbell-shaped PLA (Table 2). These different PLA topologies could be useful for several applications such as surfactants, pesticide-carriers, and also as carriers for drug delivery. An overview of these different molecular architectures and their possible properties and applications will be given here.| Structure | Properties | References |
|---|---|---|
|
Higher solution viscosity | 18 |
| More prominent shear-thinning behaviour | 18 | |
| Higher storage and loss moduli | 18 | |
| Lower Tg, Tm and crystallinity | 18 | |

|
Higher degradability | 37,53,54 |
| Lower Tg, Tm and crystallinity | 37,58 | |
| Increased hydrophilicity | 41,54,62 | |
| Lower intrinsic viscosity | 58,62 | |
| Better histocompatibility | 53 | |
|
Lower Tg, Tm and melting enthalpy ΔHm | 84,86,89,90 |
| Higher thermal stability | 89 | |
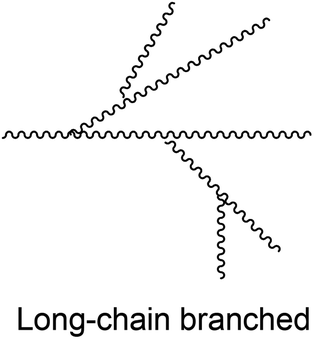
|
Lower crystallinity | 84 |
| Lower contraction factor | 90 | |
| Lower intrinsic viscosity | 90 | |
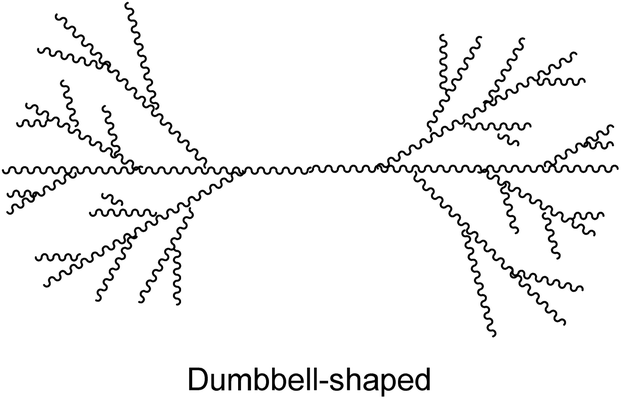
|
Lower Tg, Tm, Td | 96 |
| Lower crystallinity | 96,98 | |
| Decreased hydrophobicity | 96 |
Star-shaped PLA
Star-shaped polymers exhibit lower melting temperatures (Tm), glass transition temperatures (Tg) and crystallization temperatures (Tc) than their linear counterparts. The changes in these properties are related to the introduction of branching points, which makes the material harder to crystallize (lower Tc). Branching makes stacking of the polymers more difficult, resulting a lower temperature necessary to melt the polymers (lower Tm), and also a lower temperature necessary to force the polymers into a glassy state (lower Tg). In addition, star-shaped polymers of PLA exhibit coiling, have lower hydrodynamic volumes and have a higher melt viscosity than linear PLA.18 A stronger correlation between viscosity and temperature is also noted, with the entanglements of arms suppressing longitudinal motion.19 PLA stars can be categorized according to the core: discrete, polymeric and dendritic/hyperbranched cores (Fig. 5). This has been nicely overviewed by Cameron and Shaver.20 A striking effect of the rigidity of the core was noted in the melting temperatures of the star polymers, with increased core rigidity correlating with higher Tm values.21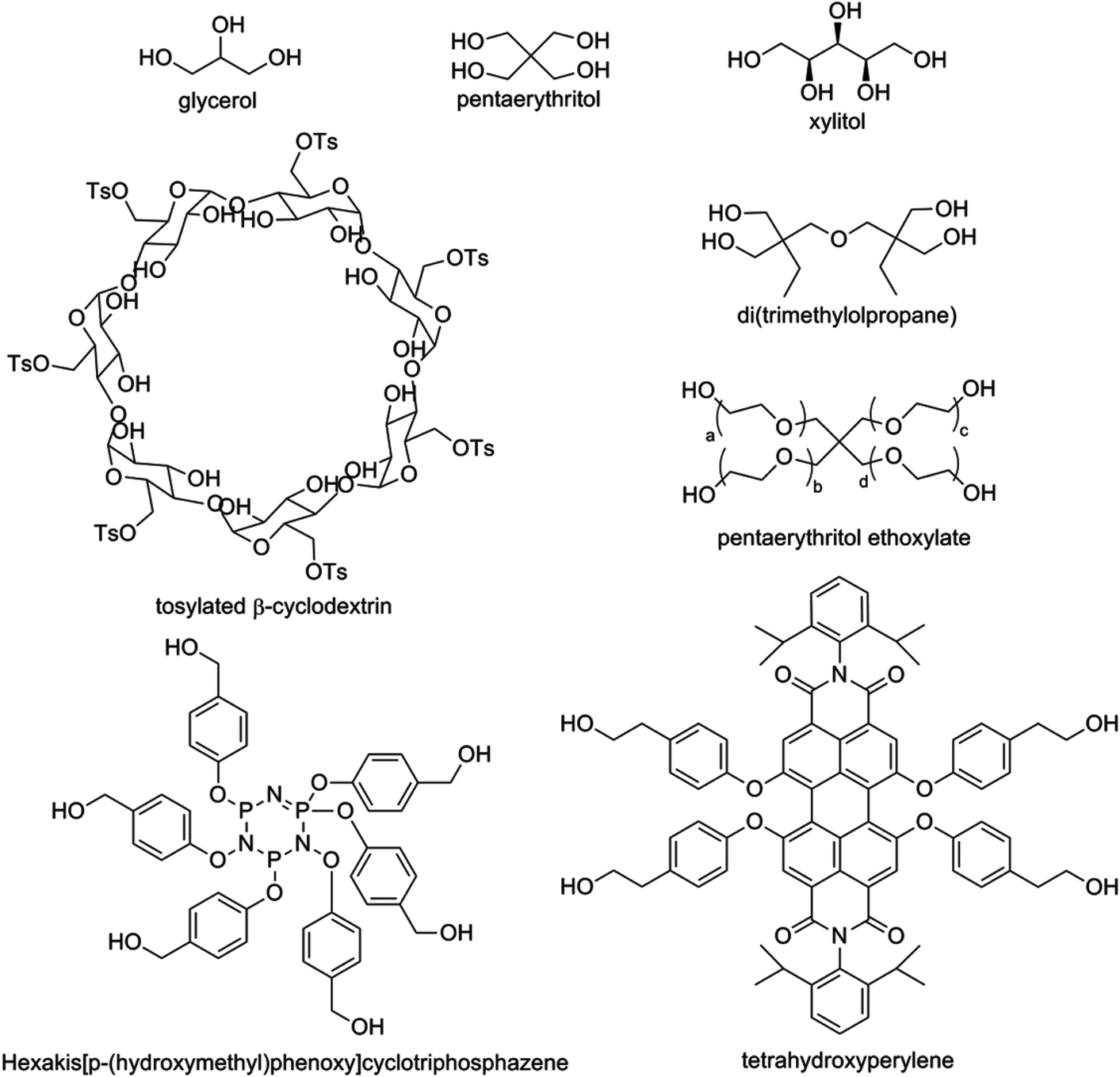 | ||
| Fig. 5 Different polyol cores for the synthesis of star-shaped PLA. Glycerol, pentaerythritol, di(trimethylolpropane) and xylitol are examples of discrete flexible polyol cores used to synthesize three-, four-, or five-arm PLA-stars respectively. Pentaerythritol ethoxylate is a polymeric flexible polyol core used to synthesize a four-arm PLA-star. Tetrahydroxyperylene and hexakis[p-(hydroxymethyl)phenoxy]cyclotriphosphazene are examples of discrete rigid cores used to synthesize four- and six-arm PLA-stars respectively, with an increased melting temperature compared to PLA-stars with a flexible core.20 | ||
The stereospecificity of PLA arms in polymer stars also plays a distinctive role. The first stereocontrolled star was reported by Stanford and Dove.22 Several sterically hindered aluminium salen catalysts were utilized in preparing heterotactic, isotactic or atactic PLA.23 Furthermore, the application of a range of initiating alcohols has been demonstrated to result in the synthesis of PLAs displaying selective α-chain end functionality derived from the initiator, which upon quenching with a protic source results in a ω-hydroxyl chain end.24 Dove et al. used previously mentioned aluminium salen complexes to develop a one-pot synthetic procedure for the production of stereoregular PLAs with controlled functionality at both α- and ω-chain ends. The availability of the chain ends for further functionalization was demonstrated using click-chemistry (Fig. 6).22
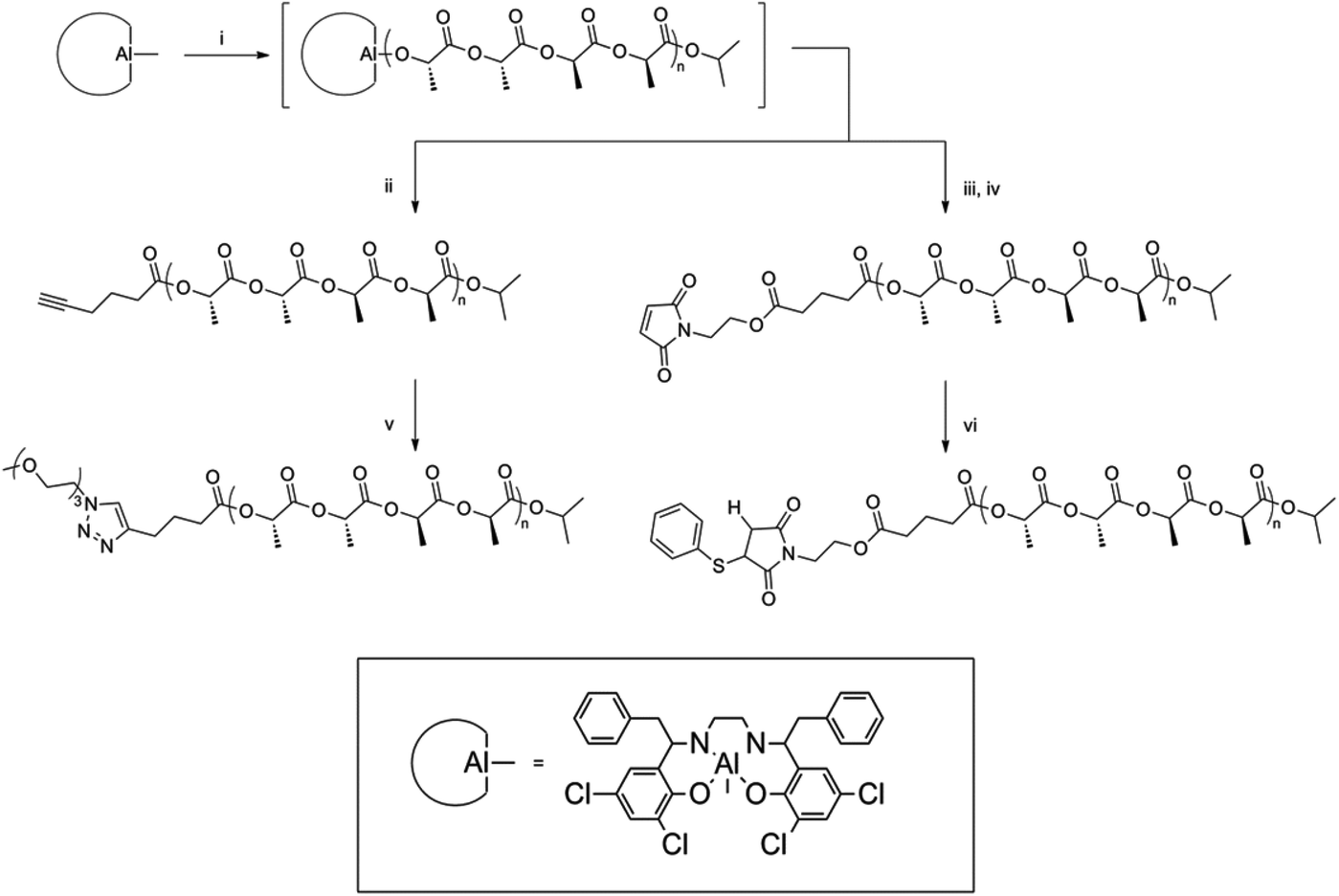 | ||
| Fig. 6 Synthesis and subsequent click-reactions of ω-chain end functionalized PLA.22 | ||
In a subsequent study Shaver and Cameron investigated the effect of the catalyst and tacticity on the physical properties of PLA polymer stars.25 They reported the synthesis of a series of six-armed polymer stars of variable tacticity built on a core molecule of dipentaerythritol (DPE). Because of the low solubility of DPE in toluene (the solvent used in the synthesis of Dove22) the reaction was performed in bulk (that is, in the lactide melt). Three different catalysts were used: tBu[salen]AlMe, Cl[salen]AlMe and Sn(Oct)2.25 In linear PLA systems, the first catalyst showed a bias towards isotacticity, whilst the second shows a bias towards heterotacticity. In the case of PLA star synthesis with DPE as the core, this tacticity control is maintained, even at high temperatures.
The introduction of stereocontrol has a large effect on the thermal stability of the polymers. Atactic PLA stars exhibited the lowest thermal stability, whilst polymers with a specific tacticity showed an enhancement in thermal stability of 40 °C. There was almost no difference shown in thermal stability between isotactic and heterotactic polymers. However, differences were observed in the maximal decomposition temperature (Tmax, the temperature where polymer decomposition has ended). Isotactic-(L) and heterotactic polymers showed a Tmax increase of 30 °C compared to the corresponding atactic polymers. When using rac-lactide, it was found that isotactic-(D/L) polymer stars showed a >50 °C increase in Tmax. The difference between the two isotactic stars might be due to some stereocomplex behaviour of adjacent polymer arms or stereoerrors resulting from imperfect catalyst isospecificity. This results in a broader temperature window over which isotactic-(D/L) stars decompose compared to heterotactic and isotactic-(L) stars.
Tacticity changes also influence the glass transition temperature (Tg). Atactic PLA stars exhibit the lowest Tg, whereas isotactic-(L) stars possess the highest Tg. Heterotactic and isotactic-(D/L) stars possess a Tg between those values. Stereoerrors in the polymers produced from rac-lactide lower the glass transition temperature.26 Similar to Tg transitions, isotactic-(D/L) Tm transitions are lower than isotactic-(L) Tm transitions. This suggests that Tm can be tuned by small variations in monomer composition or by stereoerrors incorporated into the framework.
Cui et al. succeeded in the synthesis of three-armed star-shaped PLAs consisting of isotactic–heterotactic stereoblock PLA arms.27 The ratio between the two distinct stereoregular blocks was accurately adjusted by controlling the polymerization time. Rare-earth-metal-alkyl complexes, which contain O,N,N,O-tetradentate salen ligands, were used as highly active initiators with an excellent heteroselectivity for the ROP of rac-lactide by the same group.28 Adding a three-armed protic compound, triethanolamine, afforded a hydroxyl-ended, purely heterotactic star-shaped PLA with a narrow polydispersity.29 This strategy is based on equilibrium between dormant (hydroxyl-capped) polymers and active (attached to the metal centres that contain the salen ligands) polymers. When the amount of triethanolamine was increased, a higher percentage of the growing polymer chain resided in the dormant state and hence more hydroxyl-capped polymers were found. These hydroxyl-capped polymers were further activated upon addition of another precursor to incorporate rac-lactide, but with a different stereoregularity to afford hard (isotactic) or soft (heterotactic) blocks. When the reaction time was increased from 10 min to 90 min, the conversion increased from 15% to 80%.27 In this way the use of multiple catalytic transformations in one pot could provide a way of creating novel PLA morphologies.
Recently Satoh et al. reported the synthesis of stereo-miktoarm star-shaped PLAs, star-shaped PLAs consisting of both PLLA and PDLA arms in one molecule.30 First, PLLA and PDLA were synthesized using azido- and ethynyl-functionalized initiators having one, two or three initiating sites. In a subsequent step the azido- and the ethynyl-functionalized PLLAs and PDLAs were coupled using a click reaction to give stereo-arm star-shaped PLAs. DSC measurements showed that a stereocomplex crystal was formed without any homochiral crystallization. Increasing the number of PLA arms resulted in a decrease in Tm and the crystallinity of the stereocomplex crystal. Moreover, symmetric stereo-miktoarm star-shaped PLAs showed a higher Tm and crystallinity than those with asymmetric star-shaped architectures.30
Blending linear PLA with a three-arm star-shaped PLA showed excellent flexibility and heat resistance without decreasing the biomass.31 This is due to an increased plasticization and an increased crystallization through mixing PLAs with different architectures. Star-shaped polymers could also be prepared for the purpose of creating micelles, which could be used as drug carriers.32 Polymeric micelles were formed from PLA derivatives, containing a carboxyl end group or the alkali metal salt thereof, in aqueous solution of pH 4 or more.
Comb-shaped or graft-PLA
Graft or comb-shaped polymers in general can be prepared via either of the following pathways: grafting through, grafting onto or grafting from. The ‘grafting through’ process relies on the polymerization of chains end-capped with a polymerizable unit. In the ‘grafting onto’ process end-reactive chains are coupled onto a mutually reactive polymer backbone. In a ‘grafting from’ process graft polymers can be prepared using initiating sites spread along the macromolecular backbone.33 Several comb-shaped and graft polymers have been described using PLA (or derivatives) either as a backbone or as side-chains. Most commonly used backbones are polysaccharides, poly(vinyl alcohol) and poly(acrylates).34To enable a higher solubility of polysaccharides in common organic solvents, Ouchi et al. performed a trimethylsilyl (TMS) protection reaction of the polysaccharides amylose and pullulan.37 These silylated polysaccharides were then used to initiate the ROP of lactide in the presence of potassium tert-butoxide (Fig. 7). However, it was considered that these linear starch polysaccharides were not ideal initiators due to limited control in silylation and because starch is usually a mixture of polysaccharides with various molecular weights and a wide molecular weight distribution.
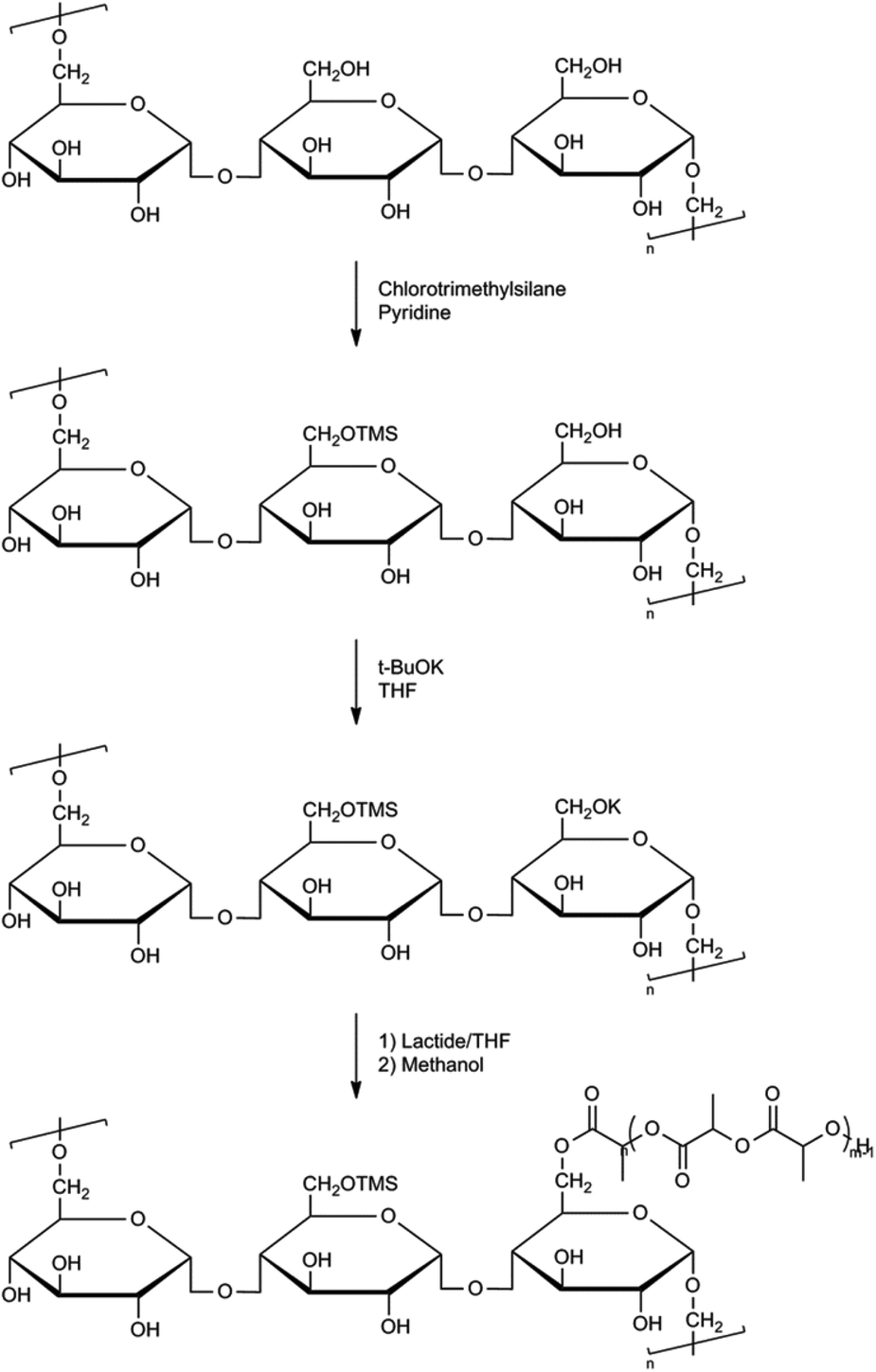 | ||
| Fig. 7 Synthesis of pullulan grafted PLA.37b | ||
Silylated dextran has been proved to overcome the aforementioned problems related to molecular weight distribution and eventually showed control in the silylation reaction. Several groups independently showed similar synthesis procedures to obtain partially or totally silylated dextran as a macroinitiator for the ROP of lactide.38 Ohya et al. used hexamethyldisilazane to silylate dextran and subsequently performed an anionic ROP of lactide using potassium tert-butoxide.38c Further studies showed that the synthesized water-insoluble but swellable dex-g-PLLA with relatively high dextran contents showed a higher water absorption and degradation rate with increasing sugar content.38c,39 PLLA with a dextran content of 25% showed a weight loss of 40% after 30 days. Increasing the sugar content from 25% till 46% reduced the number of days, necessary to degrade the polymer by 40%, by more than 20 days. These water-swollen films lost their mechanical strength and toughness after swelling, making them unusable for implant materials. Wang et al. preferred to use chlorotrimethylsilane for the silylation reaction combined with a coordination–insertion ROP of lactide using Sn(Oct)2.38a The dex-g-PLLA was then used as a compatibilizer to reduce macrophase separation in preparing blend films of PLLA and dextran. This resulted in improved mechanical properties of the PLLA/dextran blends when dex-g-PLLA was introduced (Table 3). It was also demonstrated that foams made of these blends showed good cell affinity. J.-L. Six et al. chose 1,1,1,3,3,3-hexamethyldisilazane (HMDS) for the silylation followed by coordination–insertion ROP of lactide using Sn(Oct)2.38b The same group optimized the silylation reaction by showing the influence of parameters such as the molar mass of initial dextran, the temperature of the medium, the reaction time, the HMDS/OH ratio, the addition of solvent, the influence of several catalysts and the use of a different silylating reagent, N,O-bis(trimethylsilyl)-acetamide (BSA).40 This led to a better control of solubility and allowed polymerization in homogeneous media. The ROP of lactide was then followed by a carefully controlled deprotection reaction to prevent degradation of the dextran backbone and PLA graft.38b The PLA-fraction in the graft polymers defines the solubility in aqueous media. The water-soluble graft copolymers revealed specific organisation at the air–water interface, whilst the water-insoluble graft copolymers showed a similar organisation at the DCM–water interface. These results indicate potential use of the graft copolymers as polymeric surfactants. Polymeric nanoparticles were then prepared from the previously mentioned dex-g-PLA using an emulsion/solvent evaporation procedure.41 The nanoparticles appeared to give surface layers with much lower densities than other hydrophobically modified dextrans.
| Sample | Composition (wt%) | Tensile strength of film (MPa) | Compression modulus of foam (MPa) | ||
|---|---|---|---|---|---|
| PLA | Dextran | PLA-g-dextran | |||
| Blend-1 | 100 | 0 | 0 | 39.2 ± 1.60 | 2.91 ± 0.05 |
| Blend-2 | 90 | 10 | 0 | 13.4 ± 1.10 | 2.25 ± 0.13 |
| Blend-3 | 70 | 30 | 0 | 13.0 ± 1.30 | 2.91 ± 0.05 |
| Blend-4 | 0 | 0 | 100 | 32.6 ± 2.70 | 1.83 ± 0.10 |
| Blend-5 | 0 | 10 | 90 | 26.0 ± 2.28 | 2.80 ± 0.09 |
| Blend-6 | 0 | 30 | 70 | 19.6 ± 0.31 | 2.34 ± 0.14 |
| Blend-7 | 85 | 10 | 5 | 18.6 ± 2.06 | — |
| Blend-8 | 80 | 10 | 10 | 22.6 ± 1.62 | — |
| Blend-9 | 75 | 10 | 15 | 23.1 ± 0.97 | — |
| Blend-10 | 60 | 30 | 10 | 15.5 ± 1.26 | 2.26 ± 0.05 |
| Blend-11 | 50 | 30 | 20 | 16.8 ± 1.92 | 2.39 ± 0.09 |
| Blend-12 | 40 | 30 | 30 | 17.4 ± 2.01 | 2.49 ± 0.06 |
Water-soluble biodegradable graft copolymers of dex-g-PLLA and dex-g-PDLA were prepared and investigated for their ability to form nanometric aggregates in aqueous solution.42 A different synthesis pathway was used to prepare the graft copolymers. First, hydroxyl terminated PLLA and PDLA were prepared by ROP of L- and D-lactide with ethanol as an initiator. The hydroxyl endgroups were then activated using N,N′-carbonyldiimidazole (CDI) followed by a coupling to dextran to yield the desired graft copolymers (Fig. 8). It was also shown that nanogels could be obtained from the enantiomeric dex-g-PLLA and dex-g-PDLA copolymers by intra- and/or intermolecular self-assembly of PLA chains in a dilute aqueous solution. 1H-NMR in D2O showed a broadening effect due to restricted mobility of the PLA chains, indicating aggregate formation. By mixing the aqueous solutions of the graft copolymers, stabilization of the nanogels was observed, because the cores of the PLA-based nanogels could crystallize in a stereocomplex configuration. The dex-g-PLA copolymers were then investigated for their potential use as drug carriers.43 Stereocomplex films created from the blended enantiomeric graft copolymers showed higher physical strength than the separate graft copolymers and retained their mechanically soft and tough characters in the wet state.44 Moreover, a faster degradation behaviour was seen, which is unusual for a polyester having high mechanical toughness.
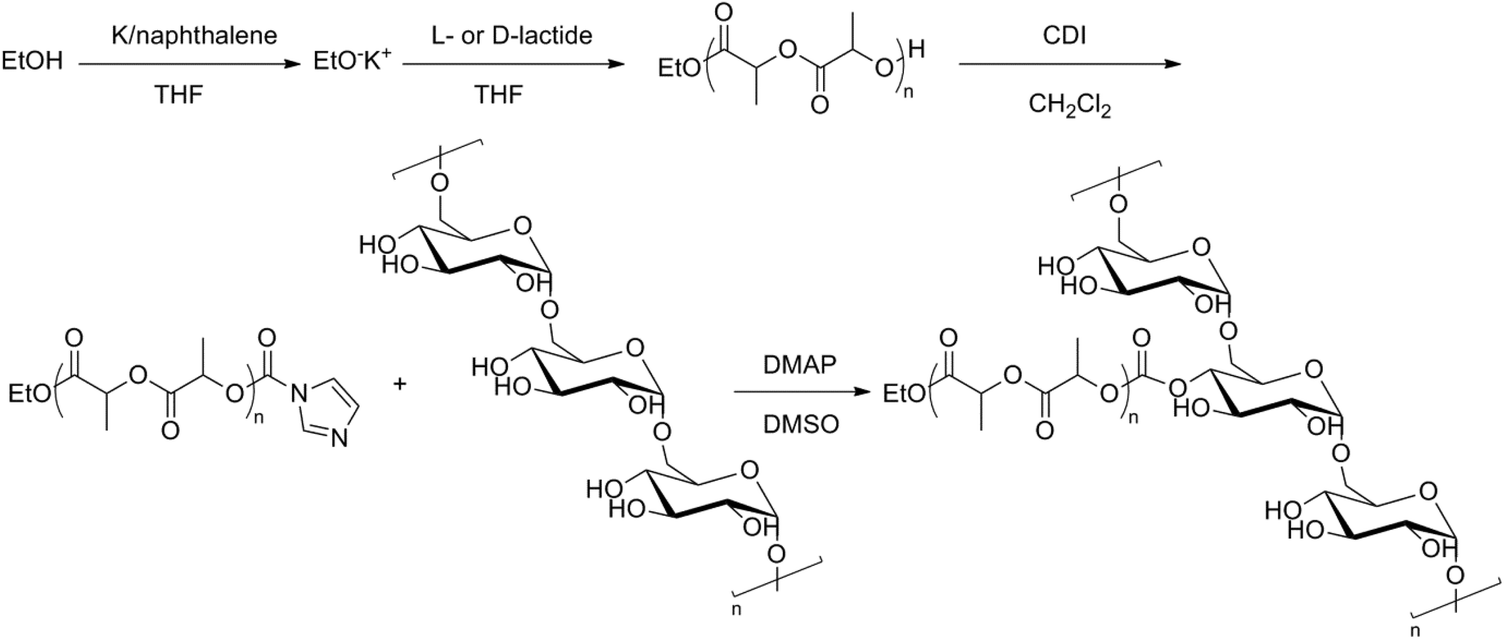 | ||
| Fig. 8 Synthesis pathway to dex-g-PLA.42 | ||
Chitosan (CS) is another polysaccharide, derived from chitin, that can be used as the backbone in graft copolymers. It is biocompatible, biodegradable and almost non-toxic and has amine and hydroxyl groups, which offer the possibility of modification. A graftcopolymer of low molecular weight CS and D,L-lactic acid was prepared without using a solvent.45 Due to aggregation of the hydrophobic side chains a pH-sensitive hydrogel could be formed in aqueous solutions. Further investigations on the swelling mechanism and thermal degradation kinetics revealed hydrogen bonding as pseudocrosslinks and a faster degradation in the case of the grafted chitosan (CS-g-PLA).46
Another amphoteric, biodegradable, pH-sensitive hydrogel was formed by grafting poly(L-lactic acid-co-citric acid) (PLCA) on the chitosan backbone.47 The graftcopolymer was prepared without using a solvent under reduced pressure. The swelling behaviour was also investigated, showing a higher degree of swelling below pH 4 and above pH 8. The same group also synthesized CS-g-PLA containing PLA side chains with a higher molecular weight than that described before.48 First, oligo(L-lactic acid) (OLLA) was prepared under reduced pressure followed by an esterification reaction with 1,6-hexanediol to obtain dihydroxyl OLLA and an oxidation to the subsequent aldehyde. OLLA was then grafted onto chitosan through the reaction of the aldehyde group and the amino group of chitosan, followed by a reduction of the formed imine (Fig. 9).
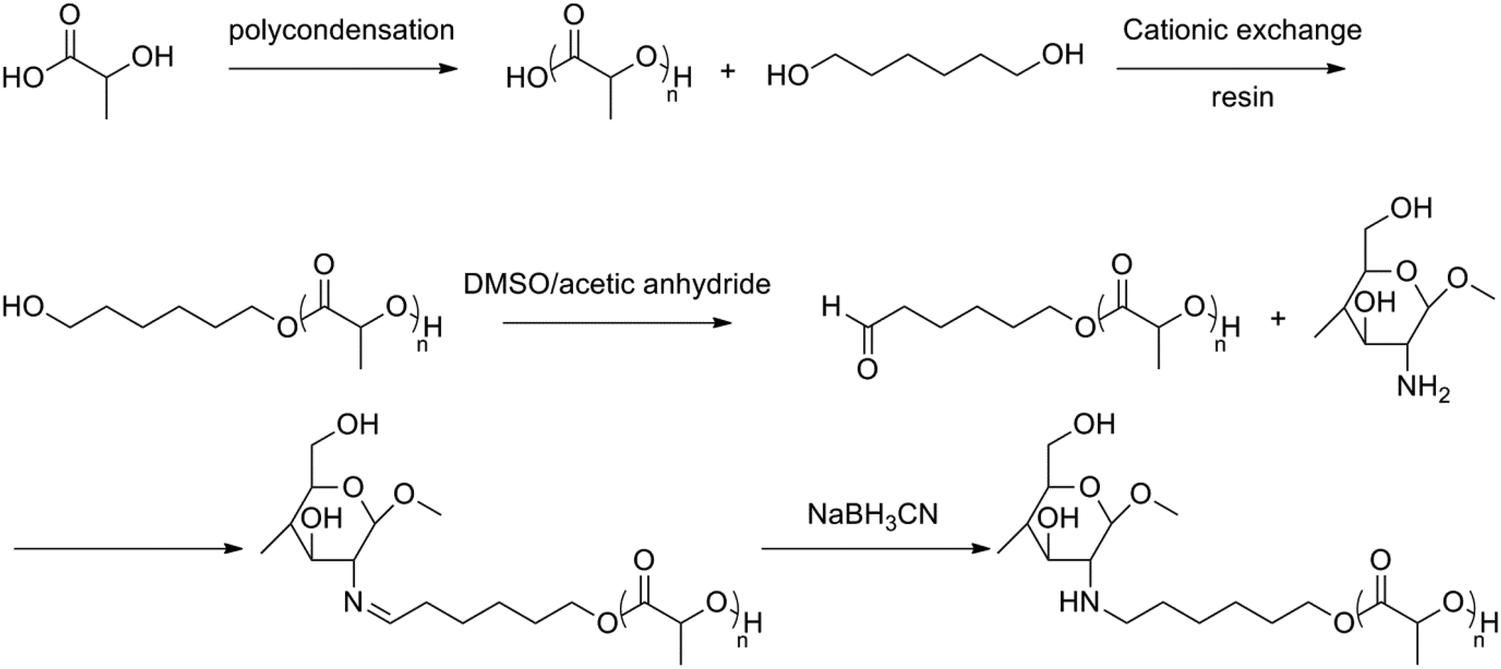 | ||
| Fig. 9 Synthesis of chitosan-graft-oligo(lactic acid).48 | ||
Polymeric micelles were formed from CS-g-PLA, when a higher degree of grafting and longer graft chains were obtained.49 The graft reactions were carried out via ROP of lactide in dimethyl sulfoxide (DMSO) at 80 °C with triethyl amine as a catalyst. A similar strategy was employed by Luckachan and Pillai by making use of Ti(OBu)4 as a catalyst to acquire a graft copolymer.50 Using this catalyst, the risk of racemisation was reduced. Upon exposure to deionised water, the graft copolymers were converted to hydrogels. Longer PLA graft chains gave rise to a higher tendency to self-assemble through hydrogen bonding and dipole–dipole interactions, resulting in a lower swellability and higher thermal stability. A regioselective grafting of PLLA chains to chitosan was accomplished through a protection–graft–deprotection pathway.51 In this way, grafting was selectively done at the hydroxyl groups to maintain the aminosaccharide structure after deprotection, which is helpful for biological activities and cationic polymer properties. In aqueous solution the CS-g-PLA aggregated into both hollow and solid spherical micelles. Increasing the grafting content resulted in decreasing the amount of hollow micelles. This is due to aggregation of the PLLA chains in the inner sphere. A ROP of lactide onto phthaloyl-protected chitosan through self-catalysis resulted in similar graft copolymers.52 Stable complexes were formed with DNA through condensation with the free amino groups of CS-g-PLA, making these complexes potentially useful as drug carriers. A potential use as a pesticide-carrier was also postulated.53
The graft copolymers were spun into a fibrous scaffold using a wet spinning technique.54 The shape and size of the filaments as well as the pore structures of the scaffolds could be controlled by varying several processing conditions, such as the polymer concentration used, the pressure applied during spinning etc. Improved tensile properties and enhanced dimensional stability were obtained, compared to the chitosan scaffolds without PLA graft chains. Submicron and nanofibrous scaffolds were acquired via electro-wet-spinning.54b Improved tensile strength and modulus were found for the hydrated scaffolds compared to the dry ones. Similar scaffolds were obtained using vacuum freeze drying without the addition of a catalyst.55
Two research groups simultaneously used a carbodiimide mediated coupling reaction between the amino group of CS and the carboxyl group of PLA to create CS-g-PLA.56 The first group used 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) as a catalyst with N-hydroxysuccinimide (NHS) to establish grafting.56a The degradation rate compared to CS and PLA increased when a low degree of grafting of the CS-g-PLA was established. Three methods of preparing CS-g-PLA nanoparticles were then investigated: a diafiltration method, an ultrasonication method and a combination of both.57 The combination method gave nanoparticles within a size range of 133–352 nm and a zeta potential range of 36–43 mV, which fulfils the requirements for nanodrug carriers. Differently structured nanoparticles were prepared through different modifications of poly(lactic-co-glycolic acid) (PLGA) with chitosan.58 Nanoparticles created through homogeneous modification by self-assembly displayed a faster hydrolysis compared to the nanoparticles created by surface modification, making them more suitable as drug matrices. The second research group used a three-step synthesis with N,N′-carbonyldiimidazole as a coupling agent to obtain PLA-grafted chitosan.56b Hydrogels were formed due to stereocomplexation between CS-g-PLLA and CS-g-PDLA. A high equilibrium swelling ratio was obtained. Thymopentin (TP5) was added as a model drug to evaluate the potential to use these copolymers as drug carriers. Up to 82% of TP5 was released from the hydrogels over a period of 60 h.
To avoid PLA degradation during reaction, due to a long reaction time and high temperatures, microwave irradiation was used to replace the conventional thermal polymerization to synthesize the graft copolymer.59 When using hydroxyethyl chitosan with Sn(Oct)2 as a catalyst, microwave irradiation improved the polymerization rate and the grafting percentage in comparison to the conventional thermal polymerization. Increasing the grafting density of the copolymer led to a decrease in crystallinity of CS.
Breitenbach and Kissel used PVA as an initiator in the ROP of lactide and Sn(Oct)2 as a catalyst to synthesize brush-like poly(vinyl alcohol)-graft-poly(lactide) (PVA-g-PLA).60 The intrinsic viscosities of the graft copolymers (0.26 dL g−1–0.53 dL g−1) are much lower than the intrinsic viscosity of the homopolymer (1.14 dL g−1). This proves that there is a change in molecular structure. Further investigation via light scattering showed a lower radius of gyration (Rg, decreased by 4.4 nm) for the graft copolymers compared to the linear homopolymer, which is further evidence for a branched structure. A series of these water-insoluble derivatives of PVA-g-PLAs was then evaluated for the preparation of protein-loaded microspheres to determine which polymer properties affect microencapsulation and protein release from the microspheres.61 Increasing the hydrophilicity by insertion of a PVA backbone into PLA chains significantly improved the release of drugs from the microspheres. Increasing the molar mass of the incorporated PVA from 15 × 103 g mol−1 to 20 × 103 g mol−1 showed 80% drug release in 14 days, compared to the same amount of drug release in 55 days in the former case. A correlation between the PLA chain length and the drug release rate was also observed.
Water-soluble PVA-g-PLAs were synthesized by esterification of PVA with D,L-lactic acid in water without a catalyst.62 These graft copolymers showed a lower glass transition temperature (Tg = −16 °C to 35 °C), an improved flexibility (elongation at break = 164–200%), elasticity (92–264 kg cm−2) and tear resistance (4.8–10.1 kg mm−1). A similar strategy was used to create PVA hydrogels.63 Good swelling properties, melt processability and semicrystalline properties were noted, making these hydrogels ideal for medical applications or applications that require high temperatures. Rheological properties were also investigated, exhibiting shear thinning behaviour.64 Blends of these graft copolymers with poly(vinyl pyrrolidone) (PVP) showed stronger intermolecular hydrogen bonding compared to unmodified PVA polymers.65 In this way the miscibility of the graft copolymers could be improved.
Stannous octoate (Sn(Oct)2) is the most commonly used catalyst for the ROP of lactide. However the cytotoxicity of this catalyst against a broad variety of microorganisms is undesirable. Tin impurities still present in the polymer after polymerization might raise problems in designing medical or pharmaceutical applications.66 Schué et al. performed a bulk ROP of lactide with PVA as an initiator and magnesium hydride (MgH2) as a catalyst.67 MgH2 was selected as a catalyst, because magnesium is involved in human metabolism. The reactions were performed in bulk, and good yields were obtained by varying the monomer/initiator-ratio (M/I-ratio) and the temperature. Impurities still present after polymerization were non-toxic, making these polymers useful for biomedical applications. The copolymerization was then optimized to a temperature of 140–160 °C, an M/I-ratio of 3–12 and a reaction time of 16–25 hours.68 The authors also found that isotactic PLA chains were incorporated as graft chains without transesterification and racemisation.
The graft density and the length of PLLA grafts were controlled by varying the feed ratio and the hydrolysis of poly(vinyl acetate-co-vinyl alcohol) (P(Vac-co-VA).69 First, PVAc was partially hydrolyzed to obtain P(Vac-co-VA). The degree of hydrolysis was controlled by varying the reaction time. P(Vac-co-VA) was then used as an initiator in the ROP of lactide with Sn(Oct)2 as a catalyst to form PVAc-g-PLLA. The results showed that increasing the graft density and decreasing the chain length of the graft chains resulted in a lower crystallization rate (a three-fold deceleration of the crystallization rate was noticed when the graft density increased from 5.2% to 9.6%).
Goethals et al. were the first to describe a PLA-grafted poly(acrylate).70 They found that mono- or trisubstituted aluminium alkoxides could be used to synthesize end-functionalized PLAs.71 PLA could be end-functionalised with amino, bromine, allylic and methacrylic end-groups. 2-Hydroxyethyl methacrylate (HEMA) was used to substitute one ethyl group of triethyl aluminium to prepare the mono-functionalized (HEMA-) aluminium oxide. This aluminium oxide was then used an initiator for the ROP of lactide, to obtain HEMA end-capped PLA. Free radical polymerization of HEMA using azobisisobutyronitrile (AIBN) in DMF resulted in poly(2-hydroxyethyl-graft-lactide) PHEMA-g-PLA. DMF was used to prove that no homopolymer of HEMA was formed, as DMF is a nonsolvent of PHEMA.
The group of Román investigated the influence of the degree of substitution of the functionalised aluminium alkoxides.72 The degree of substitution does not alter the coordination–insertion mechanism, but has an influence on the polymerization rate. The polymerization rate decreases when an aluminium trialkoxide is replaced by the corresponding aluminium monoalkoxide. The same group also investigated the reactivity ratios of the macromonomer and the corresponding comonomer, using comonomers with different hydrophilic character.73 These reactivity ratios, which are correlated to the main properties of the graft copolymers, determine the average composition of the systems and the microstructural distribution of the comonomer sequences along the macromolecular backbone. Four different comonomers were examined in the copolymerization of lactide: methyl acrylate (MA), methyl methacrylate (MMA), N,N′-dimethylacrylamide (DMA) and N-vinylpyrrolidone (VP). A drastic decrease of reactivity was noticed when using VP as a comonomer, due to repulsive forces between PLLA and PVP segments.
Possible secondary side-reactions were investigated by Jérôme et al., because they interfere with the propagation step when synthesizing low molar mass macromonomers.74 When the amount of the aluminium alkoxide is high (low monomer/initiator-ratio), free radicals can be generated. These radicals promote the polymerization of methyl methacrylate, rather than the formation of end-functionalised PLAs. Addition of an efficient free radical inhibitor, such as 2,2-di(4-tert-octyl-phenyl)-1-picryl hydrazyl, prevents the free radical polymerization of the acrylate unit and promotes the formation of the end-functionalized PLA.
Le Borgne et al. investigated another way of synthesizing the previously mentioned end-functionalized macromonomers.75 First, α-hydroxyalkanoic acids were subjected to thermal polycondensation under reduced pressure to obtain oligo(α-hydroxyalkanoic acids), followed by a subsequent coupling with methacrylic acid using standard conditions. The same group showed that increasing the proportion of lactic acid units leads to a faster degradation.76
Stereocomplexation between poly(HEMA-g-oligo(L-lactide)) and poly(HEMA-g-oligo(D-lactide)) can act as physical crosslinking for biodegradable hydrogels.77 These hydrogels degrade slower than optically pure crystalline hydrogels. Tailoring the biodegradable hydrogels by varying the degree of polymerization (DP) of the PLA side chains can result in control of the degradation rate.78
A better control of the polymer architecture was achieved by Shinoda and Matyjaszewski using atom transfer radical polymerization (ATRP).79 ATRP is a controlled radical polymerization technique suppressing unwanted side-reactions. Copolymerization of a methacrylate-terminated PLLA macromonomer and an acrylated-terminated PLLA comonomer with methyl methacrylate (MMA) using ATRP produced homogeneously branched PMMA-g-PLLA copolymers with a low polydispersity. A similar strategy was used to obtain a (mini)emulsion of the graft copolymers with a suitable surfactant in water.80 This (mini)emulsion showed elastic properties and hence could be used for coatings. The influence of the PLA chain length on stability in emulsions was also examined.81
Penczek and coworkers used a ‘grafting-from’ technique to synthesize graft copolymers of PLA and poly(acrylates).82 This technique requires a preliminary synthesis of a copolymer containing multiple initiator sites. Atom transfer radical copolymerization of MMA and HEMA using ethyl 2-bromoisobutyrate (EIBBr) as an initiator resulted in such a copolymer containing multiple hydroxyl groups. Subsequent ROP of lactide gave rise to grafting of the acrylate-copolymer. The comb-structure of similar graft copolymers also showed a certain influence on morphology and thermal properties.83
An alternative biodegradable copolymer was acquired using itaconic anhydride as a comonomer.84 This multistep procedure consists of ROP of lactide using HEMA as an initiator, subsequent acetylation of the methacrylate-terminated PLA with acetic anhydride, followed by free radical copolymerization of methacrylate and itaconic anhydride using AIBN. This reaction was tailored for obtaining well-defined biodegradable graft copolymers.85 In the first reaction, the macromonomer approach, the itaconic anhydride-terminated PLA macromonomer undergoes free radical polymerization using AIBN, whereas in the second reaction, the copolymer approach, the copolymer of itaconic anhydride and methacrylate and PLA are made separately and then coupled using Sn(Oct)2.
Hyperbranched and long-chain branched PLA
Another class of branched PLA architectures are the hyperbranched polymers. These polymers have the advantage to possess multiple branching points, making them well-suited for applications in which strain-hardening plays a key role. When only a limited amount of branching is envisaged, the polymer is called long-chain branched. The synthesis of long-chain branched PLA is still in its infancy, and only a few groups have tackled this synthesis.Ouchi et al. were the first to describe a possible synthesis pathway to a hyperbranched PLA polymer.86 Mevalonolactone (ML) was used as a comonomer in the ring opening polymerization of lactide. ML can be seen as a latent AB2 comonomer, where the ‘second’ hydroxyl group is inactive till the lactone ring is attacked (Fig. 10). Branched PLA was prepared via bulk polymerization using Sn(Oct)2 or distannoxane as the catalyst during 1–8 days. Although the GPC chromatogram of the resulting polymer obtained after 1 day was symmetrical and unimodal, GPC chromatograms of polymers obtained after 2 days and 4 days gave additional peaks for low-molecular weight fractions. Alcoholysis or ester exchange could have occurred as side reactions. The GPC chromatogram obtained of the polymer after 8 days showed a new peak at a high-molecular weight fraction. It was suggested that this could be due to ring opening of ML units and this was confirmed via1H-NMR. A new signal was seen at 3.8–3.9 ppm and this was assigned to the OCH2 group of the ring opened ML. A possible reason for the slow formation of branching (and low molar masses, up to 10 × 103 Da) could be due to the lower reactivity of ML compared to lactide for ring opening polymerization.
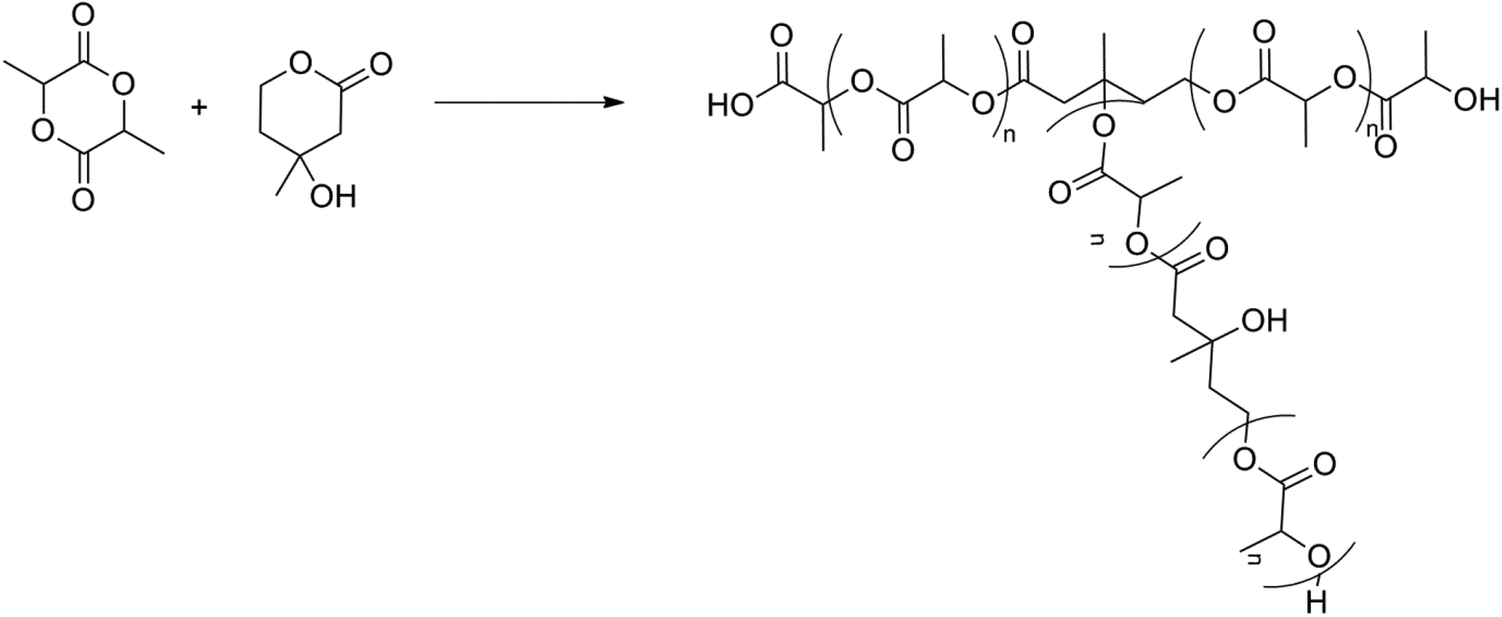 | ||
| Fig. 10 ROP of lactide using mevalonolactone.86 | ||
In 2002, Gottschalk and Frey used 2,2-bis(hydroxymethyl)butyric acid (BHB) as an AB2-comonomer in the ROP of ε-caprolactone, where BHB could give rise to branching due to esterification of the PLA alcohol groups with the acid groups of BHB.87 The idea was to implement the same strategy to synthesize branched PLA.88 The copolyesters were prepared in bulk using Sn(Oct)2 at 120 °C. The degree of branching (DB) could be controlled by varying the AB/AB2 comonomer ratio. However, this has been disproven by Cooper and Storey.89 They used 1,4-butanediol (BDO) as an initiator as a control experiment under the same conditions and found that a similar molecular weight PLA was formed. Further investigations by varying the reaction time and the amount of the catalyst showed a molecular weight difference of only 5%, which proved that only limited or no branching took place. A similar control experiment was also performed in the case of poly(ε-caprolactone), which did show an increase in molecular weight, but the amount of branching could not be controlled. It was concluded that the primary hydroxyl groups of poly(ε-caprolactone) were far more reactive towards further condensation (and branching) than the secondary hydroxyl groups of PLA.
In 2012 Frey et al. tried to synthesize branched PLA using a two-step synthesis involving BHB (Fig. 11).90 The first step was a ROP of lactide using BHB as an initiator, in accordance with the synthesis proven by Storey.89 These macromolecules were self-condensed using standard coupling agents (N,N′-dicyclohexyl carbodiimide (DCC) and 4-dimethylaminopyridine (DMAP)) to form branched structures. These mild conditions were used to prevent etherification as well as epimerization, which occur under acidic conditions used for polycondensation. In this way it is also possible to create long-chain branched (LCB) PLA. This is a high-molecular weight PLA with limited branching points. This reaction pathway is, however, limited to AB2-macromonomers with a molecular weight below 4500 g mol−1. The reactivity of the macromonomers decreases with decreasing the number of chain ends per unit volume.
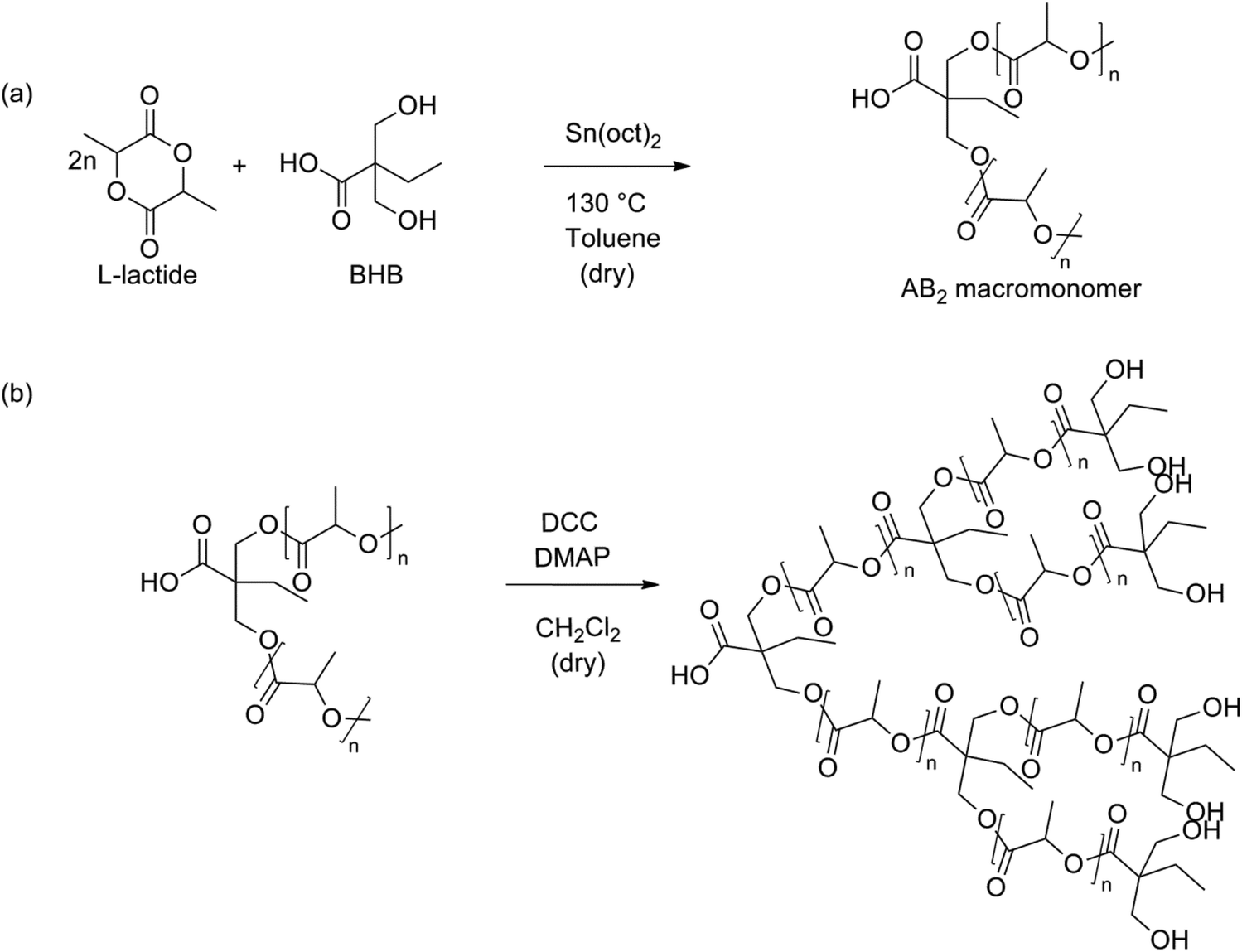 | ||
| Fig. 11 Two-step synthesis of branched PLA: (a) ROP of lactide using BHB and (b) polyesterification of the formed macromonomers.90 | ||
Zhao et al. showed that copolymerization of BHB and lactide could result in a hyperbranched polymer in the presence of a tertiary amine.91 Several amines were used to promote the reaction: aliphatic tertiary amines such as N,N,N,N-tetramethyl-1,3-diaminopropane (TMDAP) and tributylamine (TBA), an aliphatic primary amine (n-hexylamine), an aliphatic secondary amine (piperidine), an aromatic tertiary amine (N,N-dimethylaniline, DMA) and an aromatic heterocyclic tertiary amine (pyridine). From GPC chromatograms it was clearly seen that aliphatic tertiary amines gave the best results. Aromatic amines and aliphatic primary and secondary amines were not so effective as aliphatic tertiary amines due to their weak basicity or high reactivity for the ROP of lactide. Medium molecular-weight polymers were formed when using TMDAP or TBA.
Another possibility of synthesizing branched PLA was found by Knauss et al. using glycidol as a latent AB2 comonomer in the ROP of lactide.92 Epoxide ring opening was prevented in low-temperature solution polymerizations (80 °C), resulting in an essentially linear PLA functionalized with an epoxide group. Polymerizations performed in bulk at high temperatures (130 °C) showed higher molar masses compared to the theoretical molar masses (for an M/I-ratio of 50, Mn,theo = 6.9 × 103 g mol−1, while Mn, GPC = 19.8 × 103 g mol−1) which could indicate that a simultaneous ring opening of lactide and epoxide took place, hence resulting in a branched structure (Fig. 12).
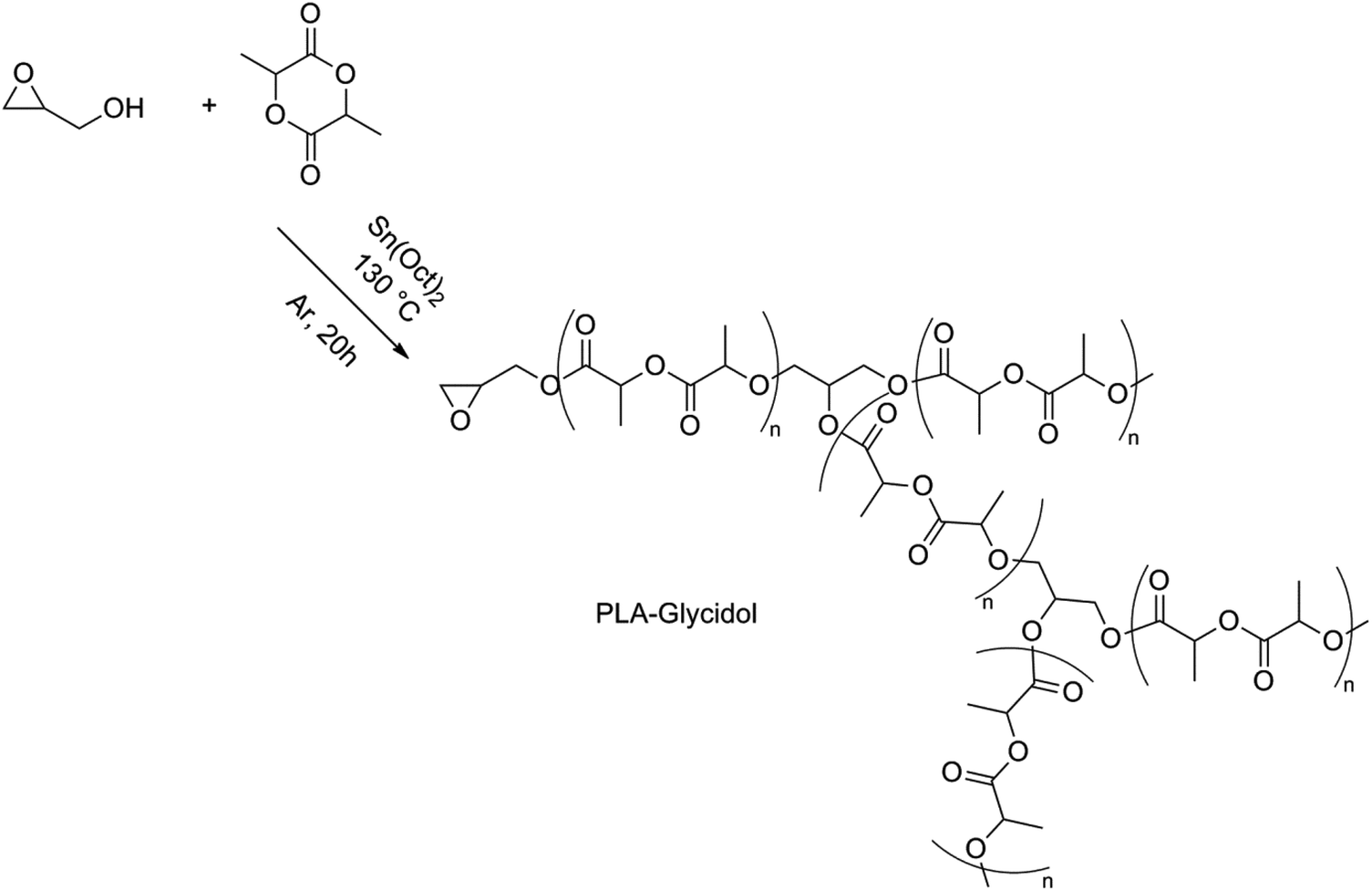 | ||
| Fig. 12 ROP of lactide using glycidol.92 | ||
In 2009 Wolf and Frey proved that 5-hydroxymethyl-1,4-dioxane-2-on (5-HDON) could be used as an inimer to synthesize branched PLA.93 5-HDON can undergo self-condensing ROP using Sn(Oct)2 or 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to form alternating hyperbranched poly(ether ester)s.94 5-HDON can be prepared in a three-pot synthesis, relying on acid–base lactonization from its oligomeric precursor. The copolymerization of lactide and 5-HDON was performed in bulk at 130 °C using Sn(Oct)2 or in DCM using 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD). The average chain length between two branching points gradually increased by a decrease in the inimer fraction; this led to LCB polymers. Increasing the inimer content also resulted in a decrease of both Tg and Tm. The simultaneous decrease in Tg, Tm and melting enthalpy (ΔHm) suggested that branching points were evenly distributed in the polymer.
H-shaped and dumbbell-shaped PLA
H-shaped polymers can be seen as block-copolymers in which two side-arms are attached to every end of a linear polymer chain. When more side-arms are attached to a linear polymer chain the general structure resembles a dumbbell. Because of their special structure, they show unique morphologies and very interesting rheological properties of entangled polymer melts and solutions.95The first example of an H-shaped copolymer containing PLLA chains was reported by Han and Pan in 2006.96 This H-shaped copolymer consisted of a poly(styrene) backbone and PLLA side-chains and was synthesized in three steps using a combination of ATRP and ROP. The molecular weight of the copolymers could be controlled by the molar ratio of the LLA consumed to the macroinitiator added, giving rise to low PDIs (Mw/Mn < 1.2).
In 2007 Waymouth et al. prepared linear poly(ethylene oxide) PEO having either one or two primary amines per chain end and demonstrated the ability of each amine to initiate the ROP of lactide using N-heterocyclic carbenes as the catalyst, thus forming H-shaped and eight-arm block copolymers, respectively.97 These eight-arm block copolymers are also often referred to as Arachne(spider)arm block copolymers.
To overcome problems related to PLA degradation and its high hydrophobicity, hydrophilic PEG units were introduced into the PLA backbone in combination with branched structures. In this way a dumbbell-shaped tri-block copolymer with comb-shaped PLA end arms and a linear PEG center block were connected using aliphatic polyester dendrons.98 These dumbbell-shaped polymers were synthesized via a multi-step synthesis. First, an amine-terminated fourth-generation dendron from 2,2-bis(hydroxymethyl)-propionic acid (bis-MPA) was prepared using a strictly divergent method. This dendron was then conjugated with 1,3-diaminopropane, followed by a coupling reaction with NHS-activated PEG. Deprotection of acetonide groups followed by the ROP of lactide resulted in the dumbbell-shaped polymer (Fig. 13). The Tg (55.2 °C), Tm (174 °C) and crystallinity (χc = 25.7%) of the dumbbell-shaped copolymer were lower than those of linear PLA, because of the plasticization of the PEG blocks and imperfect crystallinity in the branched structure compared to the linear homopolymer. It is believed that a more hydrophobic surface is more favorable for protein adsorption. Increasing the length of the PLA arms (from an M/I-ratio of 10 to an M/I-ratio of 100) results in an increase of the amount of protein (+75 μg mL−1) adsorbed on the surface.
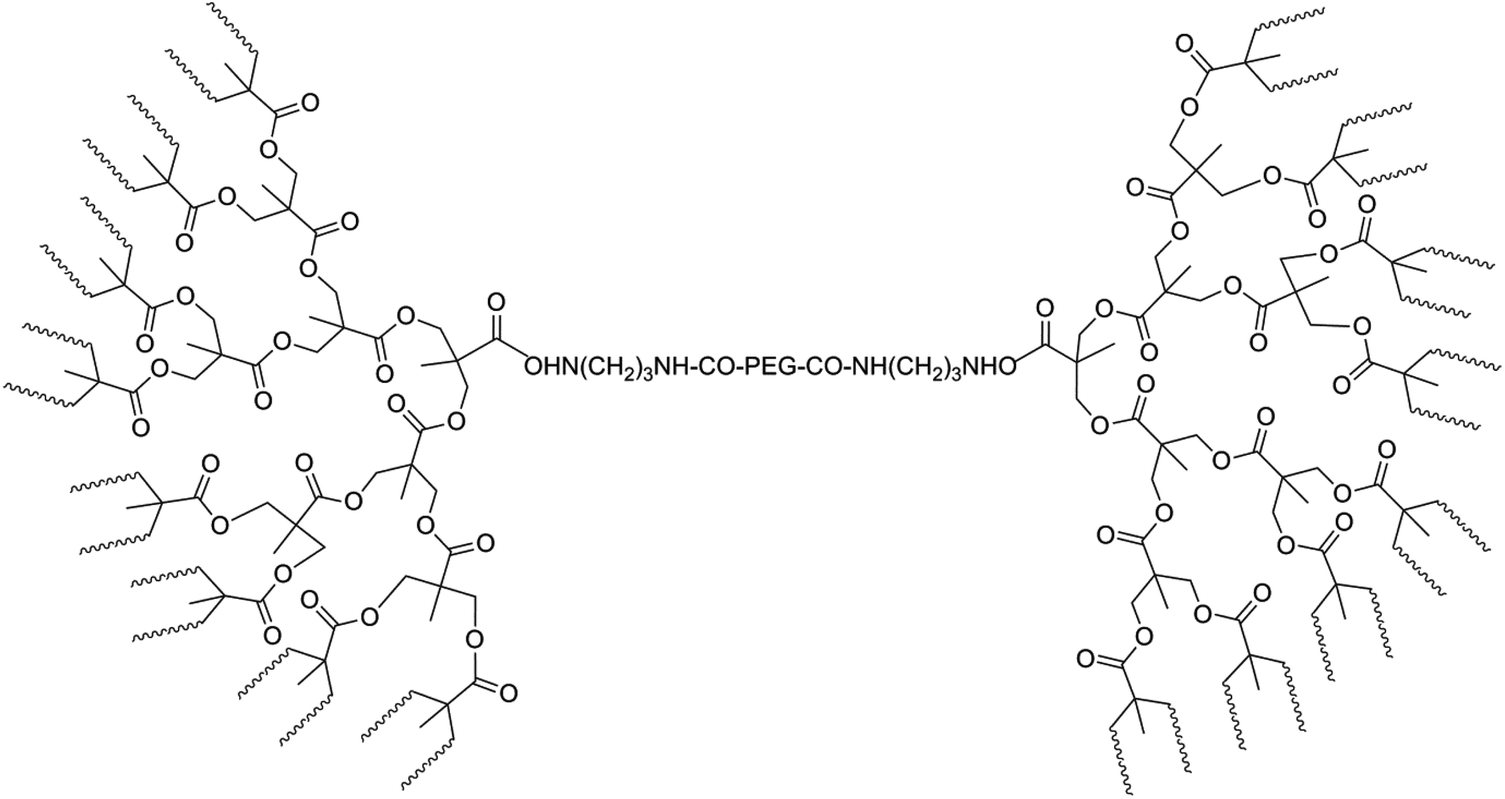 | ||
| Fig. 13 Dumbbell-shaped tri-block copolymer consisting of PLA, bis-MPA and PEG blocks, with waving lines representing PLA-chains.98 | ||
In 2012 Lei et al. found a way to synthesize this dumbbell-shaped tri-block copolymer in a one-pot two-step reaction.99 PEG, bis-MPA and p-toluenesulfonic acid (PTSA) were heated to induce a polycondensation. In a subsequent step lactide was introduced and ring opening polymerized to obtain the tri-block copolymer. By changing the molar ratio of bis-MPA to hydroxyl-terminal groups of PEG, a series of dumbbell-shaped copolymers with different degrees of branching was obtained in one pot and in high yields.
Another dumbbell-shaped tri-block copolymer was obtained by a combination of ROP, atom transfer radical polymerization and click chemistry.100 First, ε-caprolactone was ROP using BDO as an initiator, followed by a coupling reaction with 4-oxo-4-(prop-2-yn-1-yloxy)butanoic acid to achieve a dual-terminated alkynyl poly(ε-caprolactone) (PCL). In a subsequent step, this dual-terminated alkynyl PCL underwent a click reaction with 1-azido-3-chloro-2-propanol (ACP), followed by an azidation of the obtained product. Finally a ROP of lactide followed by click reaction with a monoalkynyl-terminated poly(methyl methacrylate) (PMMA) derivative gave an amphiphilic H-shaped tri-block copolymer (Fig. 15). The thermal properties and the POM analysis investigations show that the branched structure of H-shaped terpolymers and the presence of the amorphous PMMA segments together led to a decrease of the crystallinity of the PCL segments and the complete destruction of the crystallinity of the PLLA segments of the H-shaped terpolymers (Fig. 14). A typical spherulitic morphology and Maltese cross pattern is observed in Fig. 14(a). Fig. 14(b) shows a similar pattern, indicating that a change of terminal groups of PCL had a negligible effect on the crystalline morphology. In Fig. 14(c) it is clearly seen that the crystals adopted a flower-like morphology, instead of the Maltese cross patterns. A decrease of crystallizability of PCL and PLLA due to the mutual influence during the crystallization process might explain the flower-like pattern. The end product showed a rather complex spherulitic morphology, namely, Maltese cross patterns along with band textures as can be seen in Fig. 14(d). The presence of amorphous PDMAEMA and PLLA microregions in the H-shaped terpolymers interferes with the natural growth of spherulites. This leads to a uniform twisting of the lamellae along the radius orientation of the PCL spherulites and hence relates to the formation of banded spherulites.
 | ||
| Fig. 14 Polarized optical micrographs (POM) of (a) HO-PCL-OH, (b) HO-(N3-)PCL(-N3)-OH, (c) PLLA-b-(N3-)PCL(-N3) and (d) PLLA-b-(PDMAEMA-b-)PCL(-b-PDMAEMA)-b-PLLA.100 | ||
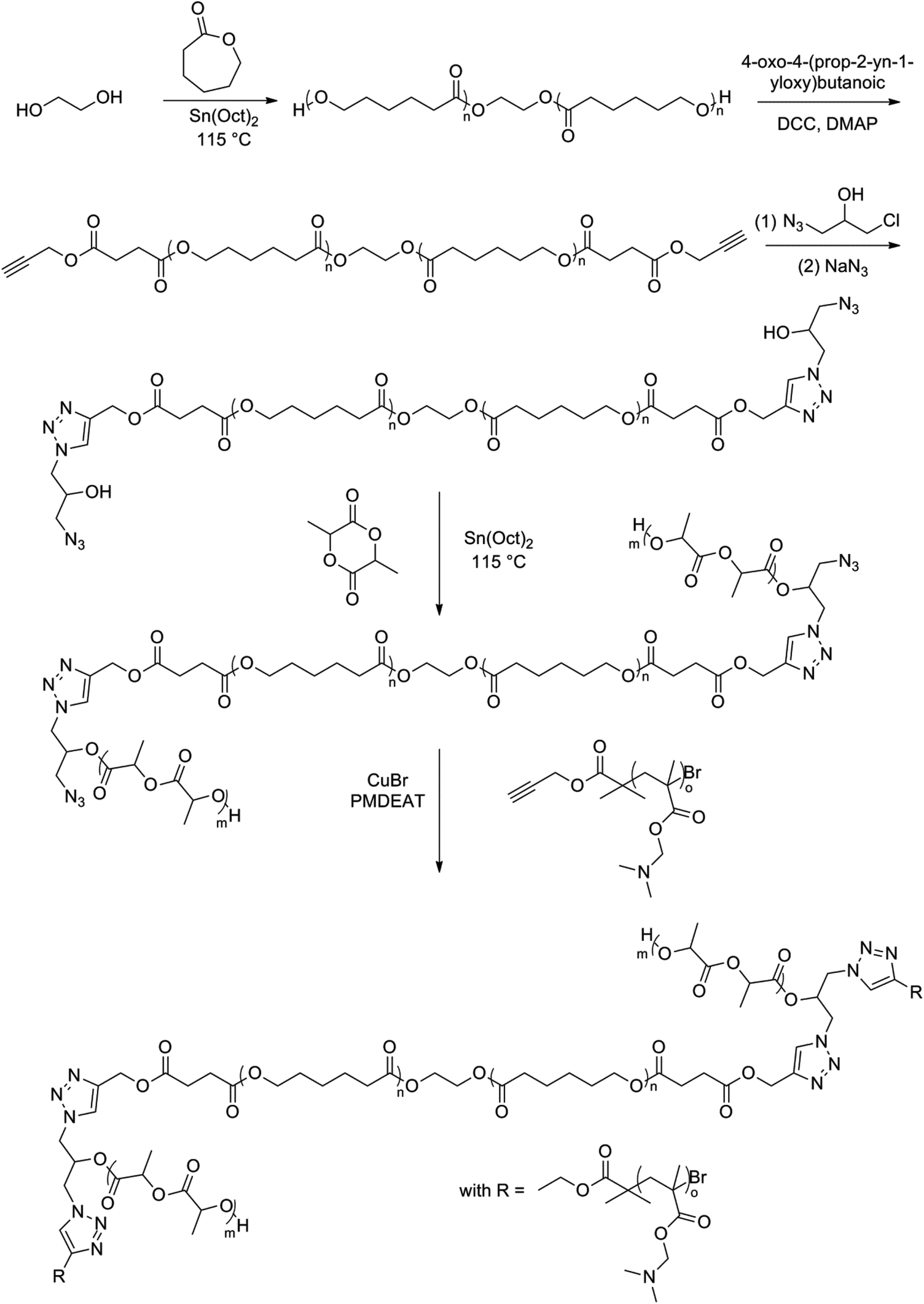 | ||
| Fig. 15 Synthesis of the amphiphilic H-shaped terpolymer PLLA-b-(PDMAEMA-b-)PCL(-b-PDMAEMA-)b-PLLA by the combination of ROP, ATRP and click chemistry.100 | ||
A combination of ring opening metathesis polymerization (ROMP) of cis-cyclooctene and ring opening transesterification polymerization (ROTEP) of D,L-lactide resulted in various architectures.101 Chain transfer agents (CTAs) bearing two, four or eight hydroxyl groups were synthesized and used for the preparation of telechelic poly(cis-cyclooctene) (PCOE) by ROMP. In a subsequent step these prepared macroinitiators were used for ROTEP of lactide to prepare linear, H-shaped and Arachne-arm architectures. In this way soft semicrystalline PCOE was tethered on either side by glassy PLA segments.
Tri-block dumbbell-shaped molecular brushes with well-defined structures were synthesized via a grafting-through ROMP by sequential addition of macromonomers bearing terminal norbornene groups (NB-PLA).102 Changing the macromonomer size and the feed ratio of Grubbs’ catalyst to macromonomers led to a control over the dimensions of the ‘ball’ and ‘bar’. Three PLA macromonomers having terminal norbornene groups, with different degrees of polymerization, were synthesized by DBU-catalyzed ROP of lactide. This was followed by a subsequent ROMP of the norbornene groups using a modified second generation Grubbs’ catalyst: (H2IMes)-(pyr)2(Cl)2RuCHPh. By varying the order of addition of the NB-PLAs, a variation in ‘ball’ and ‘bar’ was obtained.
Conclusion
It is apparent from this review that a lot of research has been conducted for improving PLA properties. Introducing stereospecificity in PLA stars resulted in an optimized control of the thermal stability. Stereocomplexation of PLA-graft copolymers showed a faster degradation behavior, although a high mechanical toughness was seen, which is unusual for a polyester. PLA-graft copolymers also showed a potential use as swellable hydrogels useful for medical applications or as pesticide-carriers. The thermal properties of hyperbranched PLA copolymers could be tailored by changing the inimer-to-monomer-ratio. H-shaped (or dumbbell-shaped) PLAs can combine properties related to linear and (hyper)branched polymers, such as an improved melt strength through entanglements and lower viscosities than their linear counterparts. Notwithstanding the tremendous effort made at optimizing PLA properties, some challenges still remain in competing with petroleum-based plastics. A better biocompatibility is necessary for producing biomedical equipment, a lower permeability is necessary for producing biodegradable bottles for soda, etc. To overcome the still remaining challenges, it might be beneficial to combine the previously described techniques. Another solution might be to introduce functional groups into the PLA backbone for further functionalization. Blending with several (biodegradable) polymers might also be beneficial to improve PLA properties.References
- (a) PlasticsEurope, Plastics – The Facts 2013, An analysis of European plastics production, demand and waste data for 2012; (b) R.-J. Mueller, Process Biochem., 2006, 41, 2124–2128 CrossRef CAS PubMed.
- R. B. Seymour, J. Macromol. Sci., Chem., 1989, 26, 1023–1032 CrossRef.
- (a) G. Chen and M. K. Patel, Chem. Rev., 2012, 112, 2082–2099 CrossRef CAS PubMed; (b) L. Yu, K. Dean and L. Li, Prog. Polym. Sci., 2006, 31, 576–602 CrossRef CAS PubMed; (c) L. Yu, S. Petinakis, K. Dean, A. Bilyk and D. Wu, Macromol. Symp., 2007, 249–250, 535–539 CrossRef.
- (a) A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Caimey, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed; (b) C. Williams and M. Hillmyer, Polym. Rev., 2008, 48, 1–10 CrossRef CAS; (c) S. C. M. Fernandes, C. S. R. Freire, A. J. D. Silvestre, C. P. Neto and A. Gandini, Polym. Int., 2011, 60, 875–882 CrossRef CAS; (d) A. Gandini, Green Chem., 2011, 13, 1061–1083 RSC; (e) K. Yao and C. Tang, Macromolecules, 2013, 46, 1689–1712 CrossRef CAS.
- (a) A. A. Shah, F. Hassan, A. Hameed and S. Ahmed, Biotechnol. Adv., 2008, 26, 246–265 CrossRef CAS PubMed; (b) G. E. Luckachan and C. K. S. Pillai, J. Polym. Environ., 2011, 19, 637–676 CrossRef CAS PubMed; (c) M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC.
- (a) D. J. Sawyer, Macromol. Symp., 2003, 201, 271–282 CrossRef CAS; (b) B. R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef; (c) K. A. Athanasiou, G. G. Niederauer and C. M. Agrawal, Biomaterials, 1996, 17, 93–102 CrossRef CAS; (d) R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed.
- (a) D. Garlotta, J. Polym. Environ., 2002, 9, 63–84 CrossRef; (b) R. M. Rasal, A. V. Janorkar and D. E. Hirt, Prog. Polym. Sci., 2010, 35, 338–356 CrossRef CAS PubMed; (c) L.-T. Lim, R. Auras and M. Rubino, Prog. Polym. Sci., 2008, 33, 820–852 CrossRef CAS PubMed; (d) K. M. Nampoothiri, N. R. Nair and R. P. John, Bioresour. Technol., 2010, 101, 8493–8501 CrossRef PubMed.
- M. H. Hartmann, in Biopolymers from Renewable Resources, ed. D. L. Kaplan, Springer, 1998, 367–411 Search PubMed.
- W. H. Carothers, Trans. Faraday Soc., 1936, 32, 39–49 RSC.
- W. H. Carothers, G. L. Dorough and F. J. van Natta, J. Am. Chem. Soc., 1932, 54, 761–772 CrossRef CAS.
- C. E. Lowe, US Pat., 2668162, 1954 Search PubMed.
- (a) H. R. Kricheldorf and I. Kreiser, Makromol. Chem., 1987, 188, 1861–1873 CrossRef CAS; (b) H. R. Kricheldorf and M. Sumbél, Eur. Polym. J., 1989, 25, 585–591 CrossRef CAS.
- (a) Z. Jedlinski, W. Walach, P. Kurcok and G. Adamus, Makromol. Chem., 1991, 192, 2051–2057 CrossRef CAS; (b) H. R. Kricheldorf and I. Kreiser-Saunders, Makromol. Chem., 1990, 191, 1057–1066 CrossRef CAS; (c) P. Kurcok, A. Matuszowicz, Z. Jedlinski, H. R. Kricheldorf, P. Dubois and R. Jérôme, Macromol. Rapid Commun., 1995, 16, 513–519 CrossRef CAS.
- H. R. Kricheldorf and A. Serra, Polym. Bull., 1985, 14, 497–502 CrossRef CAS.
- (a) K. Enomoto, M. Ajioka and A. Yamaguchi, US Pat., 5310865, 1994 Search PubMed; (b) T. Kashima, T. Kameoka, C. Higuchi, M. Ajioka and A. Yamaguchi, US Pat., 5428126, 1995 Search PubMed; (c) F. Ichikawa, M. Kobayashi, M. Ohta, Y. Yoshida, S. Obuchi and H. Itoh, US Pat., 5440008, 1995 Search PubMed; (d) M. Ohta, S. Obuchi and Y. Yoshida, US Pat., 5444143, 1995 Search PubMed.
- F. Nederberg, E. F. Connor, M. Moller, T. Glauser and J. L. Hedrick, Angew. Chem., Int. Ed., 2001, 40, 2712–2715 CrossRef CAS.
- (a) N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed; (b) D. Bourissou, S. Moebs-Sanchez and B. Martín-Vaca, C. R. Chim., 2007, 10, 775–794 CrossRef CAS PubMed.
- E. S. Kim, B. C. Kim and S. H. Kim, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 939–946 CrossRef CAS.
- V. C. Long, G. C. Berryt and L. M. Ho, Polymer, 1964, 5, 517–524 CrossRef CAS.
- D. J. A. Cameron and M. P. Shaver, Chem. Soc. Rev., 2011, 40, 1761–1776 RSC.
- M. R. Perry and M. P. Shaver, Can. J. Chem., 2011, 89, 499–505 CrossRef CAS PubMed.
- M. J. Stanford and A. P. Dove, Macromolecules, 2009, 42, 141–147 CrossRef CAS.
- (a) P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688–2689 CrossRef CAS PubMed; (b) P. Hormnirun, E. L. Marshall, V. C. Gibson, R. I. Pugh and A. J. P. White, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15343–15348 CrossRef CAS PubMed.
- (a) P. Dubois, R. Jérôme and P. Teyssié, Macromolecules, 1991, 24, 977–981 CrossRef CAS; (b) P. Dubois, R. Jérôme and P. Teyssié, Makromol. Chem., Macromol. Symp., 1991, 42–43, 103–116 CrossRef.
- M. P. Shaver and D. J. A. Cameron, Biomacromolecules, 2010, 11, 3673–3679 CrossRef CAS PubMed.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- W. Zhao, Y. Wang, X. Liu, X. Chen and D. Cui, Chem. – Asian J., 2012, 7, 2403–2410 CrossRef CAS PubMed.
- X. Liu, X. Shang, T. Tang, N. Hu, F. Pei, D. Cui, X. Chen and X. Jing, Organometallics, 2007, 26, 2747–2757 CrossRef CAS.
- W. Zhao, D. Cui, X. Liu and X. Chen, Macromolecules, 2010, 43, 6678–6684 CrossRef CAS.
- T. Isono, Y. Kondo, I. Otsuka, Y. Nishiyama, R. Borsali, T. Kakuchi and T. Satoh, Macromolecules, 2013, 46, 8509–8518 CrossRef CAS.
- H. Uyama, T. Terada and T. Yanagimoto, US Pat., 0275749, 2011 Search PubMed.
- M.-H. Seo, B.-O. Kim, I.-J. Choi and M.-S. Shim, US Pat., 8021652, 2011 Search PubMed.
- P. Lecomte and C. Jérôme, Adv. Polym. Sci., 2012, 245, 173–218 CrossRef CAS.
- Y. Ohya, A. Takahashi and K. Nagahama, in Polymers in Nanomedicine, ed. S. Kunigi and T. Yamaoka, Springer, 2012, pp. 65–114 Search PubMed.
- T. Kissel, Z. Brich, S. Bantle, I. Lancranjan, F. Nimmerfall and P. Vit, J. Controlled Release, 1991, 16, 27–41 CrossRef CAS.
- Y. Li, J. Nothnagel and T. Kissel, Polymer, 1997, 38, 6197–6206 CrossRef CAS.
- (a) Y. Ohya, S. Maruhashi and T. Ouchi, Macromol. Chem. Phys., 1998, 199, 2017–2022 CrossRef CAS; (b) Y. Ohya, S. Maruhashi and T. Ouchi, Macromolecules, 1998, 31, 4662–4665 CrossRef CAS.
- (a) Q. Cai, Y. Wan and S. Wang, Biomaterials, 2003, 24, 3555–3562 CrossRef CAS; (b) C. Nouvel, P. Dubois, E. Dellacherie and J.-L. Six, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2577–2588 CrossRef CAS; (c) T. Ouchi, T. Kontani, T. Saito and Y. Ohya, J. Biomater. Sci., Polym. Ed., 2005, 16, 1035–1045 CrossRef CAS PubMed.
- T. Ouchi, T. Kontani, R. I. E. Aoki, T. Saito and Y. Ohya, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6402–6409 CrossRef CAS.
- (a) C. Nouvel, I. Ydens, P. Degée, P. Dubois, E. Dellacherie and J.-L. Six, Polymer, 2002, 43, 1735–1743 CrossRef CAS; (b) C. Nouvel, P. Dubois, E. Dellacherie and J.-L. Six, Biomacromolecules, 2003, 4, 1443–1450 CrossRef CAS PubMed.
- C. Nouvel, J. Raynaud, E. Marie, E. Dellacherie, J.-L. Six and A. Durand, J. Colloid Interface Sci., 2009, 330, 337–343 CrossRef CAS PubMed.
- K. Nagahama, Y. Mori, Y. Ohya and T. Ouchi, Biomacromolecules, 2007, 8, 2135–2141 CrossRef CAS PubMed.
- K. Nagahama, T. Ouchi and Y. Ohya, Macromol. Biosci., 2008, 8, 1044–1052 CrossRef CAS PubMed.
- K. Nagahama, K. Shimizu, S. Ichimura, A. Takahashi, T. Ouchi and Y. Ohya, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2669–2676 CrossRef CAS.
- X. Qu, A. Wirsén and A. Albertsson, J. Appl. Polym. Sci., 1999, 74, 3193–3202 CrossRef CAS.
- (a) X. Qu, A. Wirsén and A. Albertsson, J. Appl. Polym. Sci., 1999, 74, 3186–3192 CrossRef CAS; (b) X. Qu, A. Wirsén and A. Albertsson, Polymer, 2000, 41, 4841–4847 CrossRef CAS.
- F. Yao, W. Chen, C. Liu and K. D. Yao, J. Appl. Polym. Sci., 2003, 89, 3850–3854 CrossRef CAS.
- F. Yao, C. Liu, W. Chen, Y. Bai, Z. Tang and K. D. Yao, Macromol. Biosci., 2003, 3, 653–656 CrossRef CAS.
- Y. Wu, Y. Zheng, W. Yang, C. Wang, Y. Hu and S. Fu, Carbohydr. Polym., 2005, 59, 165–171 CrossRef CAS PubMed.
- G. E. Luckachan and C. K. S. Pillai, Carbohydr. Polym., 2006, 64, 254–266 CrossRef CAS PubMed.
- G. Li, Y. Zhuang, Q. Mu, M. Wang and Y. Fang, Carbohydr. Polym., 2008, 72, 60–66 CrossRef CAS PubMed.
- L. Liu, A. Shi, S. Guo, Y. Fang, S. Chen and J. Li, React. Funct. Polym., 2010, 70, 301–305 CrossRef CAS PubMed.
- M. Li, Q. Huang and Y. Wu, Pest Manage. Sci., 2011, 67, 831–836 CrossRef CAS PubMed.
- (a) Y. Wan, X. Cao, Q. Wu, S. Zhang and S. Wang, Polym. Adv. Technol., 2008, 19, 114–123 CrossRef CAS; (b) Y. Wan, X. Cao, S. Zhang, S. Wang and Q. Wu, Acta Biomater., 2008, 4, 876–886 CrossRef CAS PubMed.
- Z. Zhang and H. Cui, Molecules, 2012, 17, 3243–3258 CrossRef CAS PubMed.
- (a) J. Li, M. Kong, X. J. Cheng, J. J. Li, W. F. Liu and X. G. Chen, Int. J. Biol. Macromol., 2011, 49, 1016–1021 CrossRef CAS PubMed; (b) Y. Hu, Y. Liu, X. Qi, P. Liu, Z. Fan and S. Li, Polym. Int., 2012, 61, 74–81 CrossRef CAS.
- J. Li, M. Kong, X. J. Cheng, Q. F. Dang, X. Zhou, Y. N. Wei and X. G. Chen, Int. J. Biol. Macromol., 2012, 51, 221–227 CrossRef CAS PubMed.
- F. K. Ma, J. Li, M. Kong, Y. Liu, Y. An and X. G. Chen, Int. J. Biol. Macromol., 2013, 54, 174–179 CrossRef CAS PubMed.
- B. Luo, J. Yang, J. Zhao, C. E. Hsu, J. Li and C. Zhou, J. Appl. Polym. Sci., 2012, 125, 125–131 CrossRef.
- A. Breitenbach and T. Kissel, Polymer, 1998, 39, 3261–3271 CrossRef CAS.
- K. Frauke Pistel, A. Breitenbach, R. Zange-Volland and T. Kissel, J. Controlled Release, 2001, 73, 7–20 CrossRef CAS.
- S. J. Carlotti, O. Giani-Beaune and F. Schué, J. Appl. Polym. Sci., 2001, 80, 142–147 CrossRef CAS.
- J. M. Onyari and S. J. Huang, J. Appl. Polym. Sci., 2009, 113, 2053–2061 CrossRef CAS.
- J. Ding, S.-C. Chen, X.-L. Wang and Y.-Z. Wang, Ind. Eng. Chem. Res., 2011, 50, 9123–9130 CrossRef CAS.
- A. Lejardi, A. Etxeberria, E. Meaurio and J.-R. Sarasua, Polymer, 2012, 53, 50–59 CrossRef CAS PubMed.
- G. Schwach, J. Coudane, R. Engel and M. Vert, Polym. Bull., 1996, 37, 771–776 CrossRef CAS.
- N. Guerrouani, A. Mas and F. Schué, J. Appl. Polym. Sci., 2009, 113, 1188–1197 CrossRef CAS.
- N. Guerrouani, B. Couturaud, A. Mas, F. Schué and J.-J. Robin, Eur. Polym. J., 2013, 49, 1621–1633 CrossRef CAS PubMed.
- N. Ding, B. Shentu, P. Pan, G. Shan, Y. Bao and Z. Weng, Ind. Eng. Chem. Res., 2013, 52, 12897–12905 CrossRef CAS.
- I. Barakat, P. Dubois, R. Jérôme, P. Teyssié and E. Goethals, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 2099–2110 CrossRef CAS.
- I. Barakat, P. Dubois, R. Jérôme and P. Teyssié, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 505–514 CrossRef CAS.
- J. L. Eguiburu, M. J. Fernandez-Berridi and J. San Román, Polymer, 1995, 36, 173–179 CrossRef CAS.
- J. L. Eguiburu, M. J. Fernandez-Berridi and J. San Román, Polymer, 1996, 37, 3615–3622 CrossRef CAS.
- I. Barakat, P. Dubois, C. Grandfils and R. Jérôme, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 497–502 CrossRef CAS.
- C. Juin, V. Langlois, P. Guerin and A. Le Borgne, Macromol. Rapid Commun., 1999, 20, 289–293 CrossRef CAS.
- V. Langlois, K. Vallee-Rehel, J. J. Peron, A. Le Borgne, M. Walls and P. Guerin, Polym. Degrad. Stab., 2002, 76, 411–417 CrossRef CAS.
- D. W. Lim, S. H. Choi and T. G. Park, Macromol. Rapid Commun., 2000, 21, 464–471 CrossRef CAS.
- C. F. van Nostrum, T. F. J. Veldhuis, G. W. Bos and W. E. Hennink, Macromolecules, 2004, 37, 2113–2118 CrossRef CAS.
- H. Shinoda and K. Matyjaszewski, Macromolecules, 2001, 34, 6243–6248 CrossRef CAS.
- K. Ishimoto, M. Arimoto, H. Ohara, S. Kobayashi, M. Ishii, K. Morita, H. Yamashita and N. Yabuuchi, Biomacromolecules, 2009, 10, 2719–2723 CrossRef CAS PubMed.
- K. Ishimoto, M. Arimoto, T. Okuda, S. Yamagushi, Y. Aso, H. Ohara, S. Kobayashi, M. Ishii, K. Morita, H. Yamashita and N. Yabuuchi, Biomacromolecules, 2012, 13, 3757–3768 CrossRef CAS PubMed.
- I. Ydens, P. Degée, P. Dubois, J. Libiszowski, A. Duda and S. Penczek, Macromol. Chem. Phys., 2003, 204, 171–179 CrossRef CAS.
- C. Zhao, D. Wu, N. A. N. Huang and H. Zhao, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 589–598 CrossRef CAS.
- J. A. Wallach and S. J. Huang, Biomacromolecules, 2000, 1, 174–179 CrossRef CAS.
- T. Okuda, K. Ishimoto, H. Ohara and S. Kobayashi, Macromolecules, 2012, 45, 4166–4174 CrossRef CAS.
- F. Tasaka, Y. Ohya and T. Ouchi, Macromol. Rapid Commun., 2001, 22, 820–824 CrossRef CAS.
- S. Skaria, M. Smet and H. Frey, Macromol. Rapid Commun., 2002, 23, 292–296 CrossRef CAS.
- C. Gottschalk and H. Frey, Macromolecules, 2006, 39, 1719–1723 CrossRef CAS.
- T. R. Cooper and R. F. Storey, Macromolecules, 2008, 41, 655–662 CrossRef CAS.
- A. M. Fischer, F. K. Wolf and H. Frey, Macromol. Chem. Phys., 2012, 213, 1349–1358 CrossRef CAS.
- R.-X. Zhao, L. Li, B. Wang, W.-W. Yang, Y. Chen, X.-H. He, F. Cheng and S.-C. Jiang, Polymer, 2012, 53, 719–727 CrossRef CAS PubMed.
- L. M. Pitet, S. B. Hait, T. J. Lanyk and D. M. Knauss, Macromolecules, 2007, 40, 2327–2334 CrossRef CAS.
- F. K. Wolf and H. Frey, Macromolecules, 2009, 42, 9443–9456 CrossRef CAS.
- P. G. Parzuchowski, M. Grabowska, M. Tryznowski and G. Rokicki, Macromolecules, 2006, 39, 7181–7186 CrossRef CAS.
- (a) M. Heinrich, W. Pyckhout-Hintzen, J. Allgaier and D. Richter, Macromolecules, 2002, 35, 6650–6664 CrossRef CAS; (b) T. C. B. McLeish, J. Allgaier, D. K. Bick, G. Bishko, P. Biswas, R. Blackwell, B. Blottière, N. Clarke, B. Gibbs, D. J. Groves, A. Hakiki, R. K. Heenan, J. M. Johnson, R. Kant, D. J. Read and R. N. Young, Macromolecules, 1999, 32, 6734–3758 CrossRef CAS; (c) J. Roovers, Macromolecules, 1984, 17, 1196–1200 CrossRef CAS.
- D. Han and C. Pan, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2794–2801 CrossRef CAS.
- O. Coulembier, M. K. Kiesewetter;, A. Mason, P. Dubois, J. L. Hedrick and R. M. Waymouth, Angew. Chem., Int. Ed., 2007, 46, 4719–4721 CrossRef CAS PubMed.
- F. Gong, X. Cheng, S. Wang, Y. Wang, Y. Gao and S. Cheng, Polymer, 2009, 50, 2775–2785 CrossRef CAS PubMed.
- D. D. Lu, L. Q. Yang, X. L. Shi, Y. Chang, H. Zhang and Z. Q. Lei, Int. J. Polym. Mater., 2012, 61, 384–394 CrossRef CAS.
- W. Yuan, J. Zhang, H. Zou and J. Ren, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2541–2552 CrossRef CAS.
- L. M. Pitet, B. M. Chamberlain, A. W. Hauser and M. A. Hillmyer, Macromolecules, 2010, 43, 8018–8025 CrossRef CAS.
- A. Li, Z. Li, S. Zhang, G. Sun, D. M. Policarpio and K. L. Wooley, ACS Macro Lett., 2012, 1, 241–245 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |