
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of thermoresponsive nanogels by thermo-nanoprecipitation and in situ encapsulation of bioactives†
Michael
Giulbudagian‡
a,
Mazdak
Asadian-Birjand‡
a,
Dirk
Steinhilber
a,
Katharina
Achazi
a,
Maria
Molina
a and
Marcelo
Calderón
 *ab
*ab
aFreie Universität Berlin, Institute of Chemistry and Biochemistry, Takustr. 3, 14195 Berlin, Germany
bHelmholtz Virtuelles Institut – Multifunctional Biomaterials for Medicine, Helmholtz-Zentrum Geesthacht, Teltow, Germany. E-mail: marcelo.calderon@fu-berlin.de; Web: http://www.bcp.fu-berlin.de
First published on 15th September 2014
Abstract
A synthetic method for thermoresponsive, glycerol based nanogels has been developed. The nanogels were synthesized by nanoprecipitation of the orthogonally functionalized macromonomers and their gelation in water. The crosslinking points were generated by strain promoted azide–alkyne cycloaddition which enabled the in situ encapsulation of Doxorubicin HCl. The mild and surfactant free reaction conditions make these nanogels ideal candidates for biomedical applications.
The term nanogel (NG) refers to nanometer sized crosslinked polymeric networks. Such networks reveal intrinsic properties, such as high water content, soft nature, cell and tissue compatibility, and excellent water dispersability/solubility.1–3 Therefore NGs are mainly developed as drug carriers which shrink or swell significantly by expelling or absorbing large amounts of water. NGs that respond to environmental stimuli like temperature, pH, and light facilitate the controlled release of the encapsulated drug cargo in a specific site of action upon environmental triggers.1,4 Among such stimuli, temperature is attractive due to its simple accessibility and applicability both in vitro and in biological environment.5,6
The building blocks of such nanostructures are of main importance when considering biomedical applications. Polyethers like linear thermoresponsive polyglycerol (tPG) and dendritic polyglycerol (dPG) are good candidates due to their biocompatible profile7–9 and their tunable character.10–13 tPG, based on glycidyl methyl ether (GME) and ethyl glycidyl ether (EGE), exhibits cloud point temperatures (Tcp) and undergoes a coil-to-globule phase transition. Their Tcp, which depends on the incorporated GME/EGE monomer ratio, is adjustable in the range between 15–57 °C.14–16 Additionally, the tunable size and the multifunctional surface of dPG enable diverse modifications to control their properties in terms of chemical reactivity, solubility, charge, and biocompatibility.10,17
The synthesis of NGs is typically based on templates which afford the generation of a crosslinked network in a confined space. Such network formation is accomplished by the polymerization of monomers in the presence of a crosslinker or alternatively by crosslinking of pre-synthesized polymer chains.2 Emulsion and dispersion/precipitation polymerizations have been widely utilized for the synthesis of thermoresponsive NGs (tNGs).18,19 Such techniques may require the use of surfactants and high shear stress. Our group has demonstrated that radical polymerization can be used for the synthesis of tNGs based on dPG.18,20 However, radical crosslinking procedures may be unfavorable for the in situ incorporation of sensitive bioactives. Recently a method was developed by Haag et al. which allowed the surfactant free synthesis of degradable NGs by the so-called inverse nanoprecipitation method using a variety of gelation chemistries.21,22 This technique showed high encapsulation properties and a release of active biomacromolecules in acidic environments. Furthermore, a hydrogel construction kit has been developed for the bioorthogonal encapsulation and pH-controlled release of living cells.23
In this paper we describe for the first time a new concept to easily generate tNGs while remaining mild, surfactant free, and with the potential to precisely tune the length between the crosslinking points by choosing a polymer of a certain molecular weight. The crosslinked network could be generated by nanoprecipitation through orthogonal strain-promoted azide–alkyne cycloaddition (SPAAC), which enabled the selective ligation with minimum purification steps and in situ incorporation of bioactives. The nanoprecipitation method refers to a simple procedure to form polymeric nanoparticles by the precipitation of a dissolved material upon an exposure to a non-solvent.24
The water soluble macro-crosslinker, cyclooctyne modified dendritic polyglycerol (dPG-Oct), and the thermoresponsive macromonomer, azide functionalized tPG (N3-tPG-N3), were solubilized in dimethylformamide (DMF) and precipitated in water at 45 °C (Fig. 1). Considering the excellent solubility of the tPG in a wide variety of solvents, we took advantage of its thermoresponsive behaviour to precipitate it in water, above its transition temperature. Since the precipitation of the polymer occurs due to its thermoresponsive behaviour rather than by using a typical non-solvent, we call this novel approach thermo-nanoprecipitation (TNP). Moreover, to enhance the hydrophobicity of the dPG and induce its co-precipitation with tPG, 7 to 28 free hydroxyl groups were functionalized with cyclooctyne functionalities.
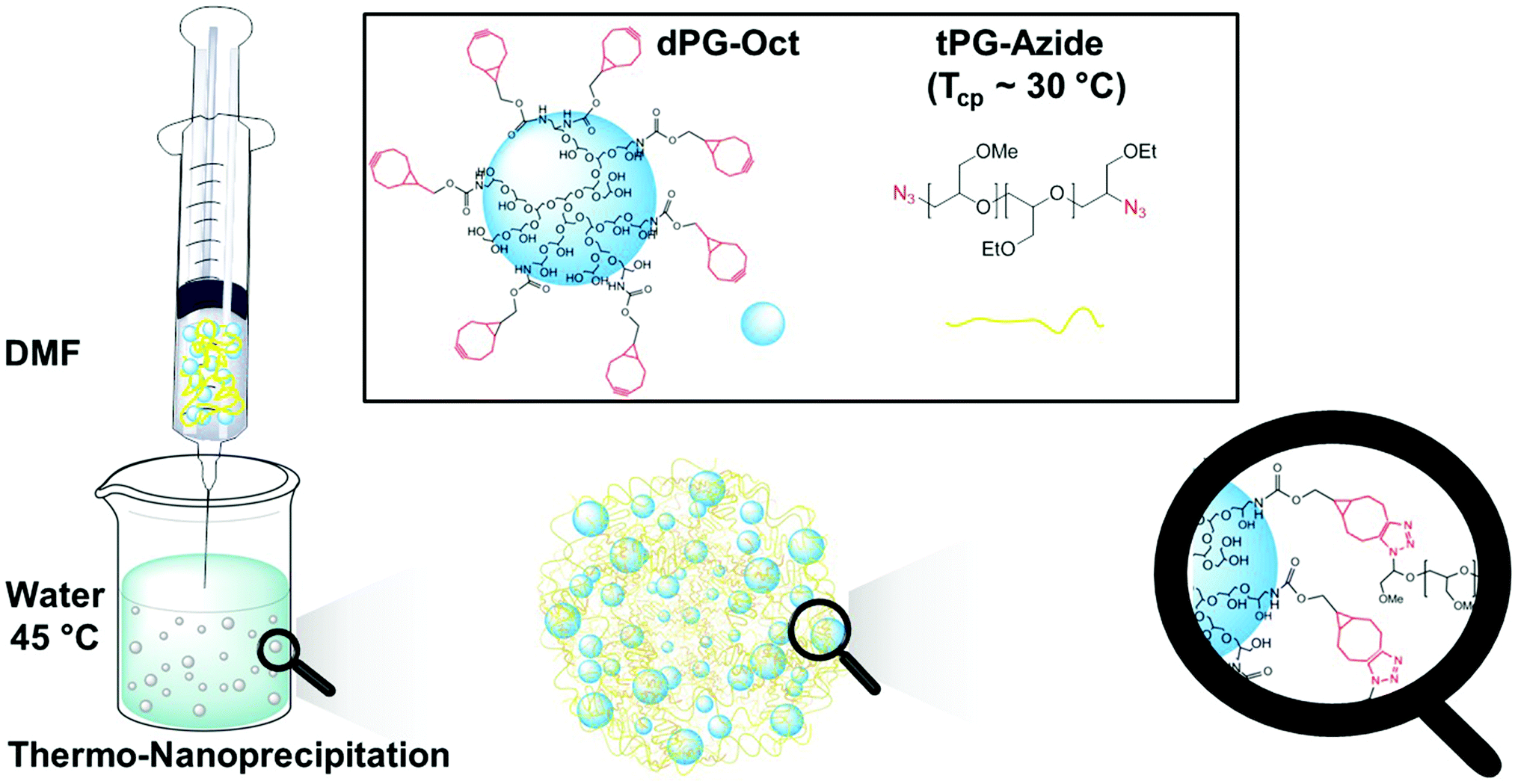 | ||
| Fig. 1 Schematic representation of nanogel synthesis by thermo-nanoprecipitation (TNP) of N3-tPG-N3 and dPG-Oct. | ||
The selection of the solvent/non-solvent on the other hand plays a critical role when one intends to encapsulate drugs in situ particle formation. The careful selection of the solvent for such systems dictates which drugs can be encapsulated within the tNGs as the drug itself has to be soluble in the selected solvent. In such manner the drug would interact with the macro-molecules when injected into the non-solvent and be entrapped in the formed crosslinked network.
The tNG's responsive behaviour, polydispersity, and potential for biomedical applications were studied. The developed method can potentially serve as a universal template for preparing tNGs from different thermoresponsive polymers.
A rational screening of the synthetic conditions was performed to explore the potential of the technique to yield particles with different sizes and transition temperatures. Table 1 summarizes the synthetic conditions. Particles with a narrow size distribution could be obtained by TNP in water, which served as a non-solvent. Generally, dPG-Oct with an average molecular weight of 10 kDa with 7, 14, or 28 functional groups was coprecipitated with N3-tPG-N3 of 5 kDa or 15 kDa. DMF was used as solvent and water at 45 °C as non-solvent, while the concentration of the macromonomers and the ratio between the solvent and the non-solvent were optimized to afford the formation of a stable dispersion. The stability of the dispersion, in terms of coalescence and Ostwald ripening, was critical allowing the crosslinking reaction to proceed to completion. The remaining unreacted cyclooctyne functionalities were quenched with azidopropanol or alternatively, with an azide functionalized cyanin dye (Cy5) as fluorescent label for further biological studies. Detailed synthetic conditions are described in the ESI.†
| Solvent (S) | Non-solvent (NS) | S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NS :NS |
tPG
|
dPG
|
Total macromonomer conc.b (mg mL−1) | tNG diameter (nm), (PDI)c |
|---|---|---|---|---|---|---|
| a Determined by GPC. b Total concentration of the macromonomers in the solvent (DMF). c Polydispersity index (PDI) determined by DLS at 25 °C, average of 3 measurements from the intensity distribution curves. | ||||||
| DMF | H2O (T > Tcp) | 1:20 |
5, 15 | 10 | 2–24 | 91–331, (0.06–0.26) |
The crosslinking reaction could be monitored by FT-IR spectroscopy which confirmed the disappearance of the azide signal at 2099 cm−1 indicating triazole formation through strain promoted cycloaddition of tPG-Oct and tPG-azide. To verify the chemical crosslinking, a control experiment using macromonomers without crosslinking functionalities was performed. After the TNP the reaction crude was dissolved in DMF, and measured by DLS. Sizes of about 5–10 nm could be observed by DLS, which correspond to the polymeric precursors and indicate that no tNGs were formed.
Moreover, tNGs were characterized by dynamic light scattering (DLS), atomic force microscopy (AFM), transmission electron microscopy (TEM), and nanoparticle tracking analysis (NTA). These techniques confirmed the formation of spherical particles with narrow size distribution. As an example, AFM image of tNG is shown in Fig. 2a. While by DLS particles of ∼100 nm of diameter could be determined, by AFM the mean particle size was determined to be smaller (∼60 nm) due to the partial deswelling of the particles on the mica surface at ambient conditions. However, TEM measurement of the tNGs in terms of size and morphology confirmed the formation of spherical particles of ∼100 nm (Fig. 2b). In order to complement the DLS measurements tNG was also analysed by NTA and diffusion-ordered NMR spectroscopy (DOSY). The diameter measured at 25 °C by DLS for a tNG with diameter of 180.3 nm (PDI 0.07) correlated with the one measured by NTA (173.3 nm, standard deviation 4.23 nm, PDI 0.006). Both techniques showed monomodal distributions and low size dispersity. In DOSY measurements a range of sizes was observed, however diffusion coefficients corresponding to particles with diameters of about 130 nm were predominantly found. The size dispersity did not allow to explicitly determine a diameter value for the tNGs. Additionally, NTA enabled evaluating the particle concentration and therefore, estimating the molecular weight of the tNG. A concentration of 1.69 × 108 particles mL−1 was measured which corresponds to an apparent molecular weight of 71.24 MDa.
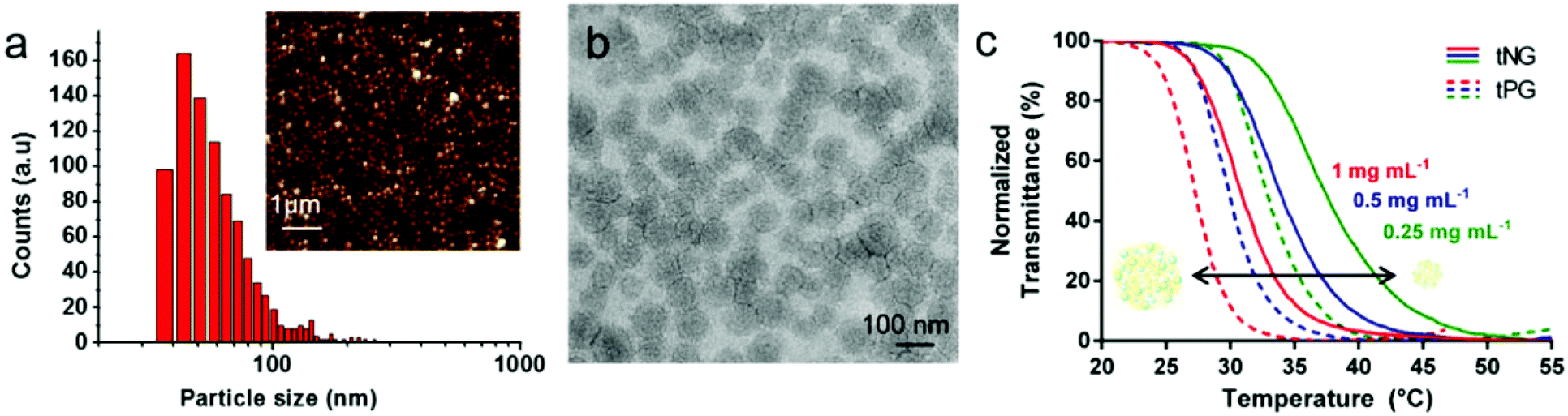 | ||
| Fig. 2 Characterization of tNGs. (a) AFM statistical analysis of the inserted AFM image. (b) TEM image of tNGs. (c) Normalized transmittance of tNG solution (solid lines) and corresponding thermoresponsive precursors, tPG (dashed lines), in PBS as a function of temperature at 0.25 mg mL−1 (green), 0.5 mg mL−1 (blue), and 1 mg mL−1 (red). | ||
Previous reports of particles formed by the nanoprecipitation technique showed that smaller sized particles were obtained when low polymer concentrations were used and that the size increased with increasing concentration of the polymers in the solvent.21,25 Contrary to this observation, our concentration dependent analysis, performed by DLS measurements, showed no significant size change for the tNGs by varying the macromonomer concentration from 2 up to 24 mg mL−1 while keeping the dPG-Oct feed ratio constant (Table S1,† tNG04–tNG09). This interesting feature would allow the tailoring of NGs, which incorporate bioactives in situ, without losing the desired nanometric sizes and responsiveness. In addition, no tendency in size was observed for tNGs prepared with tPG of 5 kDa while increasing the dPG-Oct feed ratio, or modifying the molecular weight of the tPG. It is noteworthy that the synthetic method was found to be reproducible under same conditions (±7 nm, out of 3 preparations), however the physico-chemical properties of the tNGs and the control over them are currently under investigation. The tNGs remained stable in a solution kept over a period of at least 3 month at 6 °C.
The thermoresponsive behaviour was analyzed by UV-Vis transmittance measurements for full characterization of the tNGs. Tcp values of about 30 °C were observed for all tNGs at 1 mg mL−1, when a thermoresponsive polymer of 1:1 ratio of GME and EGE was used for the synthesis. As it was expected, these values are about 3 °C higher when compared with the Tcp values of the corresponding tPG as shown in Fig. 2c (solid lines compared with dashed lines). This phenomenon can be attributed to higher hydrophilic character induced by the dPG upon crosslinking. Furthermore, comparison of polymer and tNG solutions of different concentrations revealed concentration dependent phase transition behaviour. A decrease of 3 °C was observed upon elevated concentrations, when the transmittance was measured for 0.25, 0.5, and 1 mg mL−1 solutions (Fig. 2c, red, blue, and green lines). Such behaviour can be related to enhanced interactions between the polymer chains at higher concentrations. Thus, both the concentration dependent Tcp, as well as the adjustable nature of the tPG through tailorable GME/EGE ratios in the polymer chain, can be easily applied to finely tune the Tcp of the tNGs for a desired application.
To demonstrate the encapsulation ability of the tNG Doxorubicin HCl (Dox) was chosen as an anti-cancer drug model. Dox was dissolved in DMF together with the polymeric precursors and co-precipitated in water to form the tNG. The loaded tNGs were purified from the free Dox using a centrifugal filter device and the loading was determined by UV-Vis (λ = 488 nm) to be 52.40 wt%. Such an encapsulation technique may ensure the entrapment of the encapsulated moiety within the nanocarrier rather than being adsorbed on its surface. Moreover, it enables high encapsulation efficiencies and a homogenous distribution of the guest within the nanocarrier.26 Moreover, the size of the loaded carrier (104 nm, PDI 0.29, mean of two experiments), analysed by DLS, was found to be similar to the size of tNG prepared by the corresponding route.
As the tNGs show potential as drug delivery systems, e.g. for cancer treatment, the cytotoxicity of tNG was exemplarily evaluated in adenocarcinomic human alveolar basal epithelial (A549) tumor cells by real time cell analysis (RTCA) measurements and MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Up to a concentration of 2 mg mL−1 no cytotoxicity could be observed in the MTT assay as well as in the RTCA measurements after 72 h treatment (Fig. 3a). Furthermore, RTCA measurement showed no decrease in viability for 24 and 48 h post treatment (data not shown).
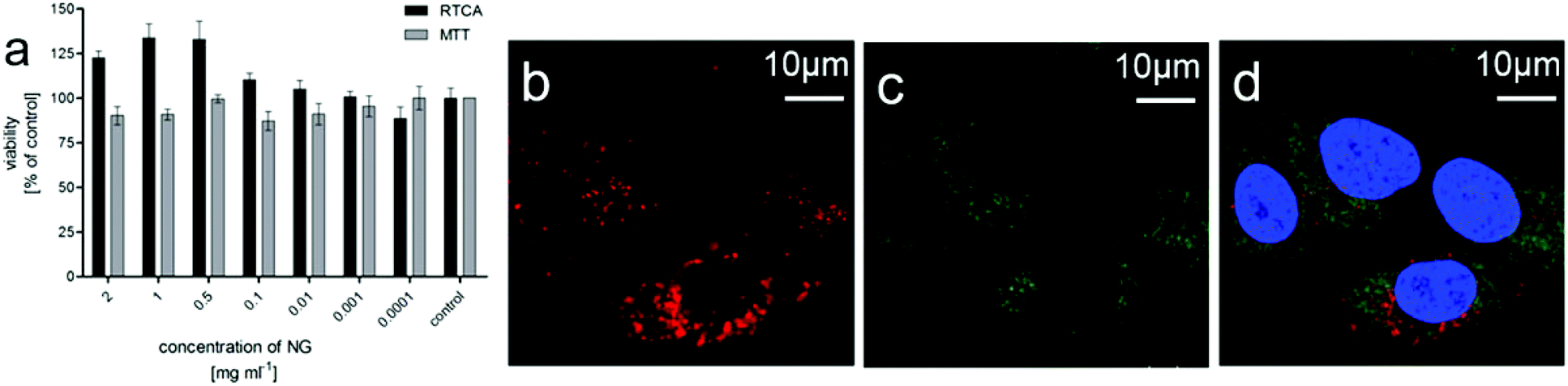 | ||
| Fig. 3 Biological evaluation of tNGs in A549 tumor cells. (a) Cytotoxicity profile of tNGs determined by real time cell analysis (RTCA) and MTT assay (n = 3; error bars: SEM (standard error of the mean)). (b) Cellular uptake was observed for Cy5 labeled tNG. (c) Green areas represent FITC (fluorescein isothiocyanate) labeled early endosome antibody EEA1 stained endosomes. (d) Blue areas represent the DAPI (4′,6-diamidin-2-phenylindol) stained nucleus of the cells as shown in the merge image. | ||
Internalization of tNGs by tumor cells, which is important for later use as a drug carrier, was verified by confocal microscopy (Fig. 3b–d). Cy5-labeled tNGs could be observed in cellular compartments of A549 cells treated for 18 h. The tNGs particularly accumulated in the perinuclear region. However, no tNG was found in the nucleus and no co-localization of tNG-Cy5 with early endosomes has been observed.
Conclusions
In summary, we have presented a versatile, surfactant-free, and mild synthetic method, which is named TNP and is used for the preparation of tNGs crosslinked by copper-free click chemistry. These tNGs have shown low cytotoxicity with excellent cellular penetration properties. The use of pre-synthesized polymers allowed us to precisely control the transition temperature of the tNGs by controlling the GME/EGE ratio of the tPG. The conditions of this synthetic method provide an effective approach to yield a library of thermoresponsive nanocarriers via a simple and biocompatible route. Dox could be successfully encapsulated in situ particle formation with a high loading efficiency. The ability of the tNGs to release the encapsulated payload upon a thermal trigger in biological models is currently under investigation. Furthermore, the potential of tuning the size of the carriers by modifying the polymer chain length and the crosslinking density are being evaluated as a tool to achieve desired release kinetics.Acknowledgements
The authors acknowledge support from Deutsche Forschungsgemeinschaft (DFG)/German Research Foundation via SFB1112, Project A04. We gratefully acknowledge financial support from the Bundesministerium für Bildung und Forschung (BMBF) through the NanoMatFutur award (13N12561), and the Helmholtz Virtual Institute, Multifunctional Biomaterials for Medicine. Dr Maria Molina acknowledges financial support from the Alexander von Humboldt Foundation. The authors acknowledge Dr P. Winchester for proof reading.Notes and references
- A. V. Kabanov and S. V. Vinogradov, Angew. Chem., Int. Ed., 2009, 48, 5418–5429 CrossRef CAS PubMed.
- R. T. Chacko, J. Ventura, J. Zhuang and S. Thayumanavan, Adv. Drug Delivery Rev., 2012, 64, 836–851 CrossRef CAS PubMed.
- M. Asadian-Birjand, A. Sousa-Herves, D. Steinhilber, J. C. Cuggino and M. Calderon, Curr. Med. Chem., 2012, 19, 5029–5043 CrossRef CAS.
- J. Zhuang, R. Chacko, D. F. Amado Torres, H. Wang and S. Thayumanavan, ACS Macro Lett., 2013, 3, 1–5 CrossRef PubMed.
- T. Kawano, Y. Niidome, T. Mori, Y. Katayama and T. Niidome, Bioconjugate Chem., 2009, 20, 209–212 CrossRef CAS PubMed.
- W. H. Blackburn, E. B. Dickerson, M. H. Smith, J. F. McDonald and L. A. Lyon, Bioconjugate Chem., 2009, 20, 960–968 CrossRef CAS PubMed.
- M. Weinhart, T. Becherer, N. Schnurbusch, K. Schwibbert, H.-J. Kunte and R. Haag, Adv. Eng. Mater., 2011, 13, B501–B510 CrossRef.
- R. K. Kainthan, J. Janzen, E. Levin, D. V. Devine and D. E. Brooks, Biomacromolecules, 2006, 7, 703–709 CrossRef CAS PubMed.
- M. Calderon, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 22, 190–218 CrossRef CAS PubMed.
- J. Khandare, M. Calderon, N. M. Dagia and R. Haag, Chem. Soc. Rev., 2012, 41, 2824–2848 RSC.
- C. Tonhauser, C. Schüll, C. Dingels and H. Frey, ACS Macro Lett., 2012, 1, 1094–1097 CrossRef CAS.
- M. Schömer, J. Seiwert and H. Frey, ACS Macro Lett., 2012, 1, 888–891 CrossRef.
- B. Schulte, A. Walther, H. Keul and M. Möller, Macromolecules, 2014, 47, 1633–1645 CrossRef CAS.
- S. Aoki, A. Koide, S.-i. Imabayashi and M. Watanabe, Chem. Lett., 2002, 31, 1128–1129 CrossRef.
- S. Reinicke, J. Schmelz, A. Lapp, M. Karg, T. Hellweg and H. Schmalz, Soft Matter, 2009, 5, 2648–2657 CAS.
- M. Weinhart, T. Becherer and R. Haag, Chem. Commun., 2011, 47, 1553–1555 RSC.
- A. L. Sisson and R. Haag, Soft Matter, 2010, 6, 4968–4975 RSC.
- J. C. Cuggino, C. I. Alvarez I, M. C. Strumia, P. Welker, K. Licha, D. Steinhilber, R.-C. Mutihac and M. Calderon, Soft Matter, 2011, 7, 11259–11266 RSC.
- H. Gao, A. Miasnikova and K. Matyjaszewski, Macromolecules, 2008, 41, 7843–7849 CrossRef CAS.
- M. Molina, M. Giulbudagian and M. Calderón, Macromol. Chem. Phys., 2014 DOI:10.1002/macp.201400286.
- D. Steinhilber, M. Witting, X. Zhang, M. Staegemann, F. Paulus, W. Friess, S. Küchler and R. Haag, J. Controlled Release, 2013, 169, 289–295 CrossRef CAS PubMed.
- X. Zhang, K. Achazi, D. Steinhilber, F. Kratz, J. Dernedde and R. Haag, J. Controlled Release, 2014, 174, 209–216 CrossRef CAS PubMed.
- D. Steinhilber, T. Rossow, S. Wedepohl, F. Paulus, S. Seiffert and R. Haag, Angew. Chem., Int. Ed., 2013, 52, 13538–13543 CrossRef CAS PubMed.
- S. Schubert, J. J. T. Delaney and U. S. Schubert, Soft Matter, 2011, 7, 1581–1588 RSC.
- M. Chorny, I. Fishbein, H. D. Danenberg and G. Golomb, J. Controlled Release, 2002, 83, 389–400 CrossRef CAS.
- T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112, 2853–2888 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, methods, detailed experimental procedures, and cellular studies. See DOI: 10.1039/c4py01186d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |