
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-molecular weight star conjugates containing docetaxel with high anti-tumor activity and low systemic toxicity in vivo
T.
Etrych
*a,
J.
Strohalm
a,
M.
Šírová
b,
B.
Tomalová
b,
P.
Rossmann
b,
B.
Říhová
b,
K.
Ulbrich
a and
M.
Kovář
b
aInstitute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic, v.v.i., Heyrovský Sq. 2, 162 06 Prague 6, Czech Republic. E-mail: etrych@imc.cas.cz; Fax: +420-296 809 410; Tel: +420-296 809 224
bInstitute of Microbiology ASCR v.v.i, Vídeňská 1083, 142 20, Prague 4, Czech Republic
First published on 10th September 2014
Abstract
Here we present the polymer conjugates where the core formed by poly(amido amine) dendrimers was grafted with semitelechelic N-(2-hydroxypropyl)methacrylamide (HPMA) copolymers containing docetaxel (DTX) attached by a pH-sensitive hydrazone bond. DTX was derivatized with three different keto acids prior to attachment to the polymer carrier to introduce reactive keto groups into the drug. The therapeutic efficacy of such high-molecular-weight star conjugates is based on: (a) the enhanced permeability and retention (EPR) effect facilitating selective accumulation within solid tumors; (b) pH-controlled release of the drug, thus ensuring faster DTX release in the mildly acidic tumor microenvironment. The star DTX conjugate had a remarkably higher maximum tolerated dose in comparison with free DTX when administered as a single i.v. injection (∼160 mg kg−1vs. 40 mg kg−1 of DTX) in C57BL/6 mice. The star DTX conjugate showed significantly higher antitumor activity than free drug in the EL4 T cell lymphoma growing in syngeneic C57BL/6 mice even when given at the same dose (20 mg kg−1 of DTX eq.). Thus, the star DTX conjugates exert a much higher therapeutic activity and yet a lower systemic toxicity than free DTX.
Introduction
In designing an efficient drug delivery system, proper selection of the polymer carrier and the spacer between the drug and its carrier is crucial.1,2 The tumor-accumulation principle of the enhanced permeability and retention (EPR) effect of macromolecules in solid tumors was originally described in 1986 by Matsumura and Maeda.3 The EPR effect takes advantage of (i) the aberrant architecture of tumor blood vessels, (ii) the higher production of vascular permeability factors and (iii) the lack of functional lymphatic drainage in tumor tissue.3–5 These tumor-specific features lead to the accumulation of biocompatible macromolecular drugs in tumor tissue, which results in their superior anti-tumor activity and decreased adverse effects. Recent progress in designing nanomedicines with anti-tumor activity has utilized the EPR effect as the prime principle of tumor targeting.6,7 Among synthetic polymer drug carriers, the water-soluble HPMA copolymers8–10 represent one of the most intensively studied and most promising systems for enabling actively or passively targeted drug delivery.11–13 HPMA copolymers are nontoxic and nonimmunogenic drug carriers enabling easy control of drug loading. High-molecular-weight (HMW) HPMA copolymers (Mw > 50 g mol−1) circulate longer and preferentially accumulate in tumor tissue via the EPR effect.14 The effect of EPR on accumulation of poly(HPMA) polymers in sarcoma15 and HPMA copolymers (PHPMA) in lymphoma16 and other tumors17 has been demonstrated in mice. Furthermore, the proper selection of the spacer between the drug and the carrier is a crucial step for obtaining a tumor- or tumor cell-specific drug release. The drug can be released from the polymer conjugate by specific enzymatic cleavage18,19 or by pH-controlled chemical hydrolysis20–22 (e.g., a hydrazone bond or cis-aconityl spacer). To improve the tumor accumulation of HPMA copolymer-drug conjugates, the molecular size of the polymer carrier should be increased, either by branching or grafting HPMA copolymers or by their self-assembly to form micellar structures. However, the synthesis of branched polymer chains or graft polymers in a comb-like structure is relatively difficult to control; a high polydispersity index and lower reproducibility become a concern. In designing our drug carrier system we decided to use a dendrimer. Dendrimers are nearly monodisperse and their surface can be freely and explicitly modified, e.g., by attaching semitelechelic PHPMA. Grafting of the dendrimer by PHPMA decreases the toxicity of the dendrimer as well. Similarly, we previously reported that a PHPMA-modified PAMAM dendrimer conjugated with doxorubicin (DOX) showed superior tumor accumulation and anti-tumor activity compared to that of linear HPMA copolymer conjugates of DOX.23,24Taxanes such as paclitaxel and its semisynthetic analog docetaxel (DTX) are potent anti-cancer agents frequently used in treatment regimens for patients with breast, ovarian, prostate, lung, and other cancers. They interfere with microtubule disassembly, thereby inhibiting cell division and inducing cell death.25 As with many other chemotherapeutic agents, metronomic dosing schedules with taxanes have been shown to exert anti-angiogenic activity in solid tumors.26,27 One of the major disadvantages of taxanes is their prominent hydrophobic character. They must be administered in oily formulations using emulsifiers, such as mixtures of ethanol and Cremophor EL or polysorbate 80. The solvents were shown to induce serious adverse reactions in a considerable proportion of patients. A significant decrease in the drug effectiveness due to the presence of Cremophor EL was reported in several tumor models.28 Thus, new strategies for improving the dosage form of taxanes remain highly desirable. In addition, achievement of more favorable pharmacokinetics, better bioavailability, and targeting of the drug to the tumor is required for improvement of the therapeutic effect and a reduction of systemic toxicity. In the literature, there have been described numerous attempts to prepare a drug delivery system for paclitaxel or docetaxel, including nanoparticles, liposomes, micelles or polymer conjugates. Various nanocarriers of paclitaxel are in different stages of clinical evaluation, with some having been commercialized, including Abraxane®, Lipusu, and Genexol PM®.29 Previously, we described conjugates of paclitaxel and docetaxel with a linear HPMA copolymer carrier representing water-soluble prodrugs with tunable drug content and a clear anti-cancer effect in treating experimental murine tumors, such as EL4 T-cell lymphoma and 4T1 breast carcinoma.30
Here we describe the synthesis and physico-chemical properties of HMW star polymer-DTX conjugates designed for efficient tumor accumulation via the EPR effect and macromolecular nature of the star polymer-drug carrier, and present results from the biological evaluation of these new DTX conjugates. The star polymer carriers were prepared by grafting semitelechelic HPMA copolymers (Mw ∼ 27–30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) onto poly(amido amine) (PAMAM) dendrimer cores, which formed HMW star-like structures with a narrow distribution of molecular weights. With the aim of attaching DTX to the polymer carrier via a hydrazone bond, DTX was modified by three different keto acids prior to attachment; thus keto groups suitable for hydrazone bond formation were introduced. Our intention was to keep the hydrodynamic diameter of the star conjugates below 30 nm, as Kataoka's group recently demonstrated that polymer micelles exceeding 30 nm poorly penetrated some types of tumor tissue.31In vitro cytotoxic/cytostatic activity of the polymer DTX conjugates was determined using several murine tumor cell lines. Very good results with the star DTX conjugates were obtained in vivo, including remarkably high maximum tolerated dose (MTD) and an excellent anti-tumor effect demonstrated in mice using the experimental murine EL4 T-cell lymphoma.
000 g mol−1) onto poly(amido amine) (PAMAM) dendrimer cores, which formed HMW star-like structures with a narrow distribution of molecular weights. With the aim of attaching DTX to the polymer carrier via a hydrazone bond, DTX was modified by three different keto acids prior to attachment; thus keto groups suitable for hydrazone bond formation were introduced. Our intention was to keep the hydrodynamic diameter of the star conjugates below 30 nm, as Kataoka's group recently demonstrated that polymer micelles exceeding 30 nm poorly penetrated some types of tumor tissue.31In vitro cytotoxic/cytostatic activity of the polymer DTX conjugates was determined using several murine tumor cell lines. Very good results with the star DTX conjugates were obtained in vivo, including remarkably high maximum tolerated dose (MTD) and an excellent anti-tumor effect demonstrated in mice using the experimental murine EL4 T-cell lymphoma.
Materials and methods
Chemicals
1-Aminopropan-2-ol, methacryloyl chloride, 2,2′-azobis(isobutyronitrile) (AIBN), 4,4′-azobis(4-cyanovaleric acid) (ABIC), 6-aminohexanoic acid (ah), N,N′-dimethylformamide (DMF), N,N′-dicyclohexylcarbodiimide (DCC), N,N′-diisopropylcarbodiimide (DIPC), 1-hydroxy benzotriazole (HOBT), leucylglycine, levulinic acid (LEV), 6-oxohept-2-enoic acid (OHT), glycylphenylalanine, phthalaldehyde (OPA), N-ethyldiisopropylamine (EDPA), dimethyl sulfoxide (DMSO), tert-butyl carbazate, 4,5-dihydrothiazole-2-thiol, ethylenediaminetetraacetic acid (EDTA), ethylenediamine (EDA), cathepsin B and trifluoroacetic acid (TFA) were purchased from Fluka. 5-Cyclohexyl-5-oxovaleric acid (COV) was purchased from Rieke Chemicals. Docetaxel (DTX) was purchased from Aurisco, China and 2,4,6-trinitrobenzene-1-sulfonic acid (TNBSA) from Serva, Heidelberg, Germany. Poly(amido amine) (PAMAM) dendrimers were purchased from Dendritic Nanotechnologies, Inc., USA.Synthesis of monomers and oligopeptide
N-(2-Hydroxypropyl)methacrylamide (HPMA) was synthesized as described in ref. 10 using K2CO3 as a base. M.p. 70 °C; purity > 99.8% (HPLC); elemental analysis: calcd, C 58.72%, H 9.15%, N 9.78%; found, C 58.98%, H 9.18%, N 9.82%.N-(tert-Butoxycarbonyl)-N′-(6-methacrylamidohexanoyl)hydrazine (Ma-ah-NHNH-Boc) was prepared in two-step synthesis as described in ref. 32. M.p. 110–114 °C; purity (HPLC) > 99.5%; elemental analysis: calcd C 57.70, H 8.33, N 13.46; found C 57.96, H 8.64, N 13.25.
The linear tetrapeptide H-Gly-Phe-Leu-Gly-OH (H-GFLG-OH) was prepared as described.30 The product was characterized by MALDI-TOF MS (392.5 M + H) and reverse-phase high-performance liquid chromatography (HPLC) showing a single peak with a retention time of 18.0 min.
The derivative of DTX with levulinic acid (DTX-LEV, see Fig. 1) was synthesized as described in ref. 30. Briefly, LEV (38.1 mg, 0.33 mmol) and DCC (100 mg, 0.51 mmol) were dissolved in 0.5 mL DMF and left at −18 °C for 20 min. Then DTX (200 mg, 0.25 mmol) and DMAP (30 mg, 0.25 mmol) dissolved in 0.5 mL DMF were added, and the reaction mixture was left at 4 °C for 24 h. The reaction mixture was purified by gel filtration on a column filled with silica gel 60 (column 2 × 30 cm, eluent ethyl acetate) using a UV detector (240 nm). DTX-LEV was obtained after evaporation of the solvent, washing with diethyl ether, filtration and drying under vacuum. The yield was 74 mg (68%). HPLC showed 95.3% purity (peak maximum at 12.58 min). MS (APCI): m/z: 904.25 [M − H]−.
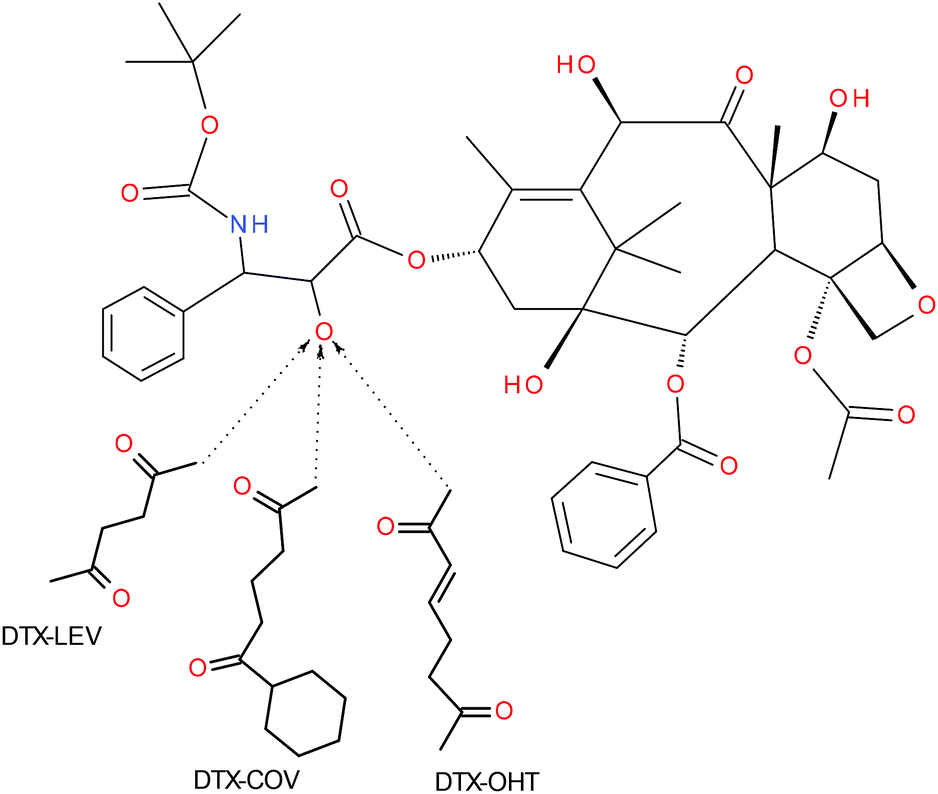 | ||
| Fig. 1 Schematic description of DTX derivatives. | ||
The derivatives of DTX with OHT or COV (DTX-OHT and DTX-COV, see Fig. 1) were synthesized by the same procedure as that described for DTX-LEV. DTX-OHT: the yield was 81 mg (73%). HPLC showed 94.1% purity (peak maximum at 12.78 min). MS (APCI): m/z: 931.30 [M − H]−. DTX-COV: the yield was 84 mg (77%). HPLC showed 92.1% purity (peak maximum at 13.12 min). MS (APCI): m/z: 987.12 [M − H]−.
Purity of all the monomers, oligopeptides and derivatives mentioned above was examined by HPLC (Shimadzu, Japan) using a reverse-phase column (Chromolith Performance RP-18e; 100 × 4.6 mm) with UV detection at 230 nm, the eluent water–acetonitrile with an acetonitrile gradient of 0–100 vol.%, and a flow rate of 0.5 mL min−1.
Synthesis of polymer precursors and polymer-drug conjugates
A semitelechelic HPMA copolymer terminated in the thiazolidine-2-thione (TT) groups containing hydrazide groups protected with tert-butoxycarbonyl groups (Boc) (polymer 1, Table 1) was prepared by radical copolymerization of HPMA (1.0 g, 0.007 mol) with Ma-ah-NHNH-Boc (189 mg, 0.6 mmol) in DMSO (7.4 mL) initiated with the azo-initiator 3,3′-[azobis(4-cyano-4-methyl-1-oxobutane-4,1-diyl)]bis(thiazolidine-2-thione) (ABIC-TT, 0.38 g, 0.76 mmol). Polymerization was carried out in a sealed ampoule at 60 °C (6 h). The polymer was isolated by precipitation in an acetone–diethyl ether mixture (1:1) and purified by re-precipitation from a methanol solution into the same mixture. The polymer was filtered off, washed with diethyl ether, and dried under vacuum. The yield was 850 mg (72.8%). The content of end-chain TT groups was determined spectrophotometrically on a Helios Alfa (Thermochrom) spectrophotometer using ε305 = 10700 L mol−1 cm−1 (methanol).
| Polymer | M w (g mol−1) | M w/Mn | Hydrazide (Hy) content (mol%) | Chain end-group | M n,T (g mol−1) | M n/Mn,Tb |
|---|---|---|---|---|---|---|
| a M n,T is the number-average molecular weight calculated from the GFLG-OH or TT group content. b M n/Mn,T is the functionality of a polymer. c Statistical copolymers of HPMA with Ma-ah-NHNH2. d Semitelechelic copolymers containing the main chain-end reactive group. | ||||||
| 1d | 26900 |
1.81 | 5.3 | TT | 11400 |
1.31 |
| 2d | 27500 |
1.82 | 5.3 | COOH | 14500 |
1.06 |
| 3c | 26900 |
1.77 | 5.8 | — | — | — |
The semitelechelic polymer precursor 2 (Table 1), a HPMA copolymer with chain-terminating carboxyl group of the GFLG oligopeptide, was prepared by the reaction of terminal TT groups of polymer 1 with the amino group of the H-GFLG-OH oligopeptide in DMF as described in ref. 23. The content of end chain GFLG sequences was determined by amino acid analysis (Shimadzu, Japan, pre-column OPA derivatization).
Statistical copolymers of HPMA with Ma-ah-NHNH2 (polymer 3, Table 1) and polymer conjugate 4 (Table 2) bearing DTX-LEV attached via a pH-sensitive hydrazone bond were prepared by the reaction described in detail recently.30
| Polymer or conjugate | Derivative of DTX | M w (g mol−1) | M w/Mn | R h (nm) | DTX (wt%) |
|---|---|---|---|---|---|
| a Biodegradable polymer with GFLG sequence cleavable by lysosomal enzymes. | |||||
| 4 | DTX-LEV | 32000 |
1.65 | 4.3 | 8.9 |
| 5 | — | 192000 |
1.72 | 12.6 | — |
| 6 | DTX-LEV | 240000 |
1.76 | 13.8 | 7.7 |
| 7 | DTX-COV | 222000 |
1.72 | 13.1 | 7.5 |
| 8 | DTX-OHT | 238000 |
1.80 | 13.6 | 7.9 |
| 9a | — | 201000 |
1.71 | 12.9 | — |
| 10a | DTX-LEV | 265000 |
1.68 | 15.2 | 7.6 |
Synthesis of star precursors and polymer conjugates
The star polymer precursors were prepared by grafting the reactive semitelechelic HPMA copolymer precursors 1 or 2 onto the 2nd generation (G2) PAMAM dendrimers containing 16 terminal amino groups and a diaminobutan core.Star polymer precursor 5 containing Boc-protected hydrazide groups was prepared by aminolysis of the TT groups of copolymer 1 with amino groups of the PAMAM dendrimer in methanol. Briefly, polymer 1 (654 mg; 0.06 mmol TT groups) was dissolved in 16 mL of methanol and added into a stirred solution of 16 mg of PAMAM dendrimer in 6.1 mL of methanol. After 2 h, the reaction was completed by adding 5 μL of 1-aminopropan-2-ol. Low molecular weight (LMW) impurities were removed by gel filtration (Sephadex LH-20, methanol solvent). The polymer-modified dendrimer was isolated by precipitation in ethyl acetate.
Star polymer precursor 9 (Table 2) was prepared by the reaction of the carboxyl group of the GFLG-OH terminal sequence of polymer 2 with amino groups of PAMAM dendrimer in DMF using the carbodiimide coupling method. Polymer 2 (200 mg; 12.2 μmol COOH groups) was dissolved in 4 mL of DMF and mixed with 13 μL of DIPC (11.4 mg, 84.7 μmol). After 5 min of stirring, a solution of 4.3 mg PAMAM dendrimer in 550 μL of DMF was added to the reaction mixture. After 20 h of stirring at room temperature, the reaction mixture was diluted with 13 mL of methanol, and LMW impurities were removed by gel filtration (Sephadex LH-20, methanol solvent). The star polymer was isolated by precipitation by acetone.
Free hydrazide groups needed for DTX derivative attachment were obtained in polymer precursors 5 and 9 by removing the protecting Boc groups from the hydrazides with concentrated TFA. Polymer conjugates 6, 7, 8 and 10 (Table 2), which had DTX derivatives attached via a pH-sensitive hydrazone bond, were prepared by the reaction of the precursors 5 or 9 with an appropriate DTX derivative for 2 h in methanol in the dark. The polymer-drug conjugates were purified from LMW impurities (DTX or its degradation products) by gel filtration on a Sephadex LH-20 column using methanol as the eluent.
Characterization of polymers and polymer-DTX conjugates
All the conjugates were characterized and tested for the content of free polymer or free drug using an HPLC (Shimadzu, Japan) equipped with GPC columns Superose™ 6 or TSKgel G3000SWxl and TLC (Kieselgel 60 F254). In addition, the content of free DTX or its derivatives was determined by HPLC after extraction of the respective drug from the aqueous solution of the conjugate by chloroform.The total content of DTX derivatives in the polymer conjugates was determined by HPLC after complete hydrolysis of the polymer conjugates in an HCl solution (pH 2) for 1 h at 37 °C and extraction of derivatives by chloroform. No evidence of by-product formation during analysis was detected.
Determination of the molecular weight and polydispersity of the conjugates was carried out with an HPLC equipped with RI, UV and multiangle light scattering DAWN EOS (Wyatt Co., USA) detectors using a mobile phase of 20% of 0.3 M acetate buffer (CH3COONa/CH3COOH; pH = 6.5; 0.5 g L−1 NaN3) and 80% of methanol and a TSKgel G3000SWxl column.
The content of hydrazide groups in polymer precursors was determined by a modified TNBSA assay as described earlier.33 A molar absorption coefficient ε500 = 17200 L mol−1 cm−1 (λ = 500 nm), estimated for the model reaction of MA-ah-NHNH2 with TNBSA, was used.
The dynamic light scattering (DLS) of aqueous conjugate solutions was measured at a scattering angle of 173° on a Nano-ZS, Model ZEN3600 (Malvern, UK) zetasizer. The hydrodynamic radius (Rh) was determined using the DTS (Nano) program. MS spectra were measured using an LCQ Fleet spectrometer (Thermofisher Scientific).
In vitro release of DTX
The release rate of free or ester-DTX derivatives from the polymer conjugates was investigated by incubation of the conjugate in phosphate buffers at pH 5.0 or 7.4 (0.1 M phosphate buffer with 0.05 M NaCl) or in human plasma at 37 °C. The concentration of the conjugate in solution was equivalent to 1.10–4 mM DTX. The amount of released drugs and their ester derivatives was determined by HPLC analysis after their extraction into an organic solvent. Analysis was performed on an HPLC instrument using a reverse-phase column (Chromolith Performance RP-18e; 100 × 4.6 mm) with UV detection at 230 nm, the eluent water–acetonitrile with an acetonitrile gradient of 0–100%, and a flow rate of 0.5 mL min−1. All drug-release data are expressed as the amounts of free drug relative to the total drug content in the conjugates. All experiments were carried out in triplicate.In vitro degradation of the HMW star polymer conjugates
Polymer precursor 10 was incubated in sodium phosphate buffer (0.05 M sodium phosphate, 0.1 M NaCl, 1 mM EDTA, 3 mM GSH, pH 6) containing 0.5 μM cathepsin B at 37 °C at a final polymer concentration of 15 mg mL−1. The activity of the cysteine proteinase cathepsin B was determined every 24 h using Bz-Arg-NAp as the substrate. The enzyme concentration of cathepsin B was maintained at 5 × 10−7 M, and the decreased enzyme activity was compensated by the addition of a fresh stock solution of cathepsin B. In predetermined time intervals, the molecular weight of the polymers was tested using HPLC.In vitro cytotoxicity
Four murine cancer cell lines were used: EL4 T-cell lymphoma (ATCC TIB-39), 4T1 breast carcinoma (ATCC CRL-2539), CT26 colon carcinoma (ATCC CRL-2638), and LL2 lung carcinoma (ATCC CRL-1642). All cell lines were purchased from ATCC and maintained as recommended by the provider. The cells were cultivated in 96-well flat bottomed culture plates (Nunclon, Denmark) with various concentrations of conjugates and free DTX or DTX-LEV for 3 days. Cell counts at the beginning of cultivation were: 5000 per well for EL4, CT26, and LL2 cells, with 2500 per well for the 4T1 cell line. Cell proliferation was estimated by the standard 3H-thymidine incorporation assay. Alternatively, determination of overall metabolic activity using an MTT assay was used as a parameter of EL4 lymphoma cell proliferation. The IC50 values were calculated as the drug concentrations that inhibit the cell proliferation or metabolic activity by 50% during the 3 day cultivation.Maximum tolerated dose of linear and star polymer-DTX conjugates
Inbred C57BL/6 (H-2b) and BALB/c (H-2d) were obtained from the Institute of Physiology, ASCR, Prague, Czech Republic. The mice were i.v. injected with linear or star polymer-DTX conjugates in 300–400 μL of PBS at doses of 100, 125, 150 and 200 mg kg−1 of DTX eq. of linear conjugate, and 20, 40, 60, 80, 90, 100, 120, 140 and 180 mg kg−1 of DTX eq. of star conjugate. Mice injected with PBS were used as the controls. The number of mice per group ranged between three and nine. Injected mice were monitored daily or every other day until day 21 post administration. We defined the MTD, according to the recent publications, as the highest dose that should elicit minimal signs of toxicity (e.g., ruffled fur, cachexy, and neurotoxicity), no deaths, and the weight loss of less than 15% of body mass.Histology
Three random mice from the groups injected with PBS (control), 200 mg kg−1 of DTX eq. of linear conjugate, or 100, 120 and 140 mg kg−1 of DTX eq. of star polymer-DTX conjugate were sacrificed for the histological analysis on day 7 post injection. Mice injected with free DTX at its MTD (i.e., 40 mg kg−1) were used as a positive control. Tissue samples of spleen, liver, vertebral bone marrow and kidney were collected, fixed in 5% formalin, embedded in paraffin, and stained with hematoxylin/eosin (H&E). The vertebral column had been decalcified in 10% formic acid. Cryostat sections of selected samples were stained with Oil Red O, Sudan Black B and Luxol Fast Blue.Tumor treatment
C57BL/6 mice were transplanted subcutaneously (s.c.) on day 0 with 1 × 105 EL4 cells and treated when palpable tumors (approx. 5–8 mm diameter) developed. The polymer-DTX conjugates were injected i.v. via the tail vein on days 8 and 12, each dose equivalent of 20 mg DTX per kg. For the administration, the conjugates were dissolved in 0.2 mL of PBS. Tumor growth, body weight, and survival of mice were regularly monitored. The tumor volume was calculated as V = a × b2/2, where a = longer diameter and b = smaller diameter. Analysis of statistical significance of the survival times was conducted using the log-rank (Mantel–Cox) test and GraphPad Prism software.In all animal experiments, institutional guidelines for the care and use of laboratory animals were strictly followed under a protocol approved by the Institutional Animal Care and Use Committee of the Academy of Sciences of the Czech Republic, and conducted in compliance with local and European guidelines.
Results and discussion
We have already demonstrated the high efficacy of HMW star HPMA copolymer carriers containing doxorubicin in the treatment of tumors in mice (EL4 T-cell lymphoma).23 We have used the EPR effect as the prime principle of tumor drug targeting in designing anti-tumor nanomedicines. Tumor-specific features causing the EPR effect led to the accumulation of biocompatible macromolecules in tumor tissue, which result in a superior anti-tumor effect with decreased adverse effects. In this study, we focused on improving the properties of the HMW drug carrier system, explored here for DTX delivery, using a new synthesis strategy of the HMW HPMA copolymer-DTX conjugates with well-defined architecture, proper control of molecular weight and with low polydispersity. The HMW polymer carriers were prepared by grafting the semitelechelic HPMA copolymers onto a PAMAM dendrimer core to form the star-like structures depicted in Fig. 2. Molecular weight of the copolymer grafts bearing hydrazide groups enabling subsequent DTX derivative attachment was below the renal threshold and the copolymers were grafted onto the PAMAM dendrimer via a stable amide bond or enzymatically degradable linkages containing GFLG oligopeptide sequences. The molecular weight of the star polymer carrier exceeded the size limit for glomerular filtration of HPMA-based polymers, but the carrier structure allowed elimination of the polymer from the organism after its intracellular degradation. | ||
| Fig. 2 Schematic representation of the synthesis and structures of the star polymer conjugates. | ||
Synthesis of polymer precursors and ester derivatives of DTX
Radical solution copolymerization of HPMA with a monomer bearing Boc-protected hydrazide groups (Ma-ah-NHNH-Boc) was initiated with ABIC-TT. This bifunctional azo-initiator, which contained reactive TT groups, was used for the synthesis of HPMA copolymer precursors terminating with the TT group, here termed semitelechelic copolymers. A previous study has shown that radical HPMA copolymerization can be precisely controlled by the initiator and monomer concentrations and polymerization temperature.34 Therefore, radical polymerization using ABIC-TT was employed here in the synthesis of copolymer 1 (Table 1). The polymerization conditions were chosen to keep the molecular weights of the copolymer under the renal threshold, which was a prerequisite for subsequent elimination of the polymer from the body via glomerular filtration. Despite the preference for a disproportionation termination reaction, the functionality (number of chain-terminating TT functional groups per polymer chain) of the copolymer was higher than 1 (Mn/Mn,T∼ 1.3).The semitelechelic copolymer bearing an end-chain biodegradable oligopeptide (copolymer 2) was prepared by aminolysis of the copolymer 1 with an oligopeptide H-GFLG-OH. The lower yield of the reaction resulted in a decreased functionality of the copolymer 2 (close to 1) without observed changes in molecular weight and polydispersity.
We have developed and published the strategy for the synthesis of polymer-drug conjugates based on DTX derivatives containing the keto group.30 Introduction of the keto group by DTX esterification with keto acids enabled DTX attachment to the polymer carrier via pH-sensitive hydrazone bonds together with more slowly hydrolyzable ester bonds. Three keto acids (levulinic acid, 6-oxohept-2-enoic acid and 5-cyclohexyl-5-oxovaleric acid) were selected as promising candidates for DTX derivatization.
The DTX acid derivatives, DTX-LEV, DTX-OHT and DTX-COV, were prepared by esterification of the primary hydroxyl group at C2′ of DTX with the carboxyl group of the above mentioned acids. The lower yield of the reaction (∼70%) was caused by a competing reaction, i.e., formation of diester derivatives of the acylated drug on the hydroxyls at both the C2′ and C7 positions. The products were purified from free DTX, keto acid diester and other components by preparative chromatography on silica. Approximately 95% product purity was determined by HPLC analysis with UV detection.
Synthesis of star polymer precursors and DTX conjugates
The star polymer conjugates were prepared by grafting semitelechelic copolymers 1 or 2 onto the second generation of PAMAM dendrimer containing 16 amino groups. Two structurally different types of the star polymer conjugates were synthesized. One type contained a stable, non-degradable amide bond between the polymer grafts and the central dendrimer (polymers 5–8). The second type was a biodegradable polymer construct containing the enzymatically degradable oligopeptide GFLG sequence (polymers 9 and 10). In both types, the products of the grafting reactions were HMW polymers with a star structure and a relatively narrow distribution of molecular weights (Mw ∼ 200000 g mol−1; Mw/Mn ∼ 1.7), which made them applicable as drug carriers with passive targeting to solid tumors. Molecular characteristics of all the star copolymers were comparable and the method of synthesis and degradable or non-degradable structure did not significantly influence the molecular weight, polydispersity or Rh of the dendritic polymers derived from the PAMAM dendrimer.
Removing the protective Boc groups from the copolymer hydrazides (precursors 5 and 9) with TFA did not change the molecular weight or polydispersity of the copolymers. Star polymer-DTX conjugates 6, 7, 8 and 10, which contained DTX derivatives bound by a pH-sensitive hydrazone bond, were prepared by the reaction of keto groups of DTX derivatives with hydrazide groups of the polymer precursors 5 or 9 in methanol in the presence of a catalytic amount of acetic acid. In all cases, conversion was nearly complete and attachment of DTX derivatives had no significant influence on molecular weight, polydispersity, or Rh of the polymer conjugates.
The Rh of the polymer coil of the dendritic polymer conjugates in aqueous solution was much higher than that of the linear polymer-drug conjugate 4. Star polymer conjugates showed approximately three times higher Rh than the linear conjugate 4. This fulfilled the prerequisite criteria for enhanced accumulation of polymers in solid tumors due to the EPR effect. The scheme of the grafting reaction is given in Fig. 2.
In vitro drug release
The results of the in vitro release of DTX and its ester derivatives from polymer conjugates 4, 6, 7 and 8 showed that the release rate of the DTX esters or free drug at pH 7.4 (37 °C) was much lower than that at pH 5 (Fig. 3). After 2 h of incubation of the copolymer conjugates at pH 7.4, 7–24% of total DTX and DTX esters were released, while at pH 5, depending on the DTX ester structure, 50% to almost 92% of liberated DTX ester was found. | ||
| Fig. 3 Release of DTX and DTX derivatives from polymer conjugates 4, 6, 7 and 8 incubated in phosphate buffers at pH 5 (A) and pH 7.4 (B) at 37 °C. (◆ —) conjugate 4, DTX-LEV and DTX; (▲—) conjugate 6, DTX-LEV and DTX; (■ —) conjugate 7, DTX-COV and DTX; (● —) conjugate 8, DTX-OHT and DTX; (◆ ----) conjugate 4, DTX; (▲ ----) conjugate 6, DTX; (■ ----) conjugate 7, DTX; (● ----) conjugate 8, DTX; n = 3. | ||
The rate of release at both pHs strongly depended on the specific structure of the DTX ester, with the highest release for the OHT ester, a moderate release for the LEV ester and the lowest release for the COV ester. No significant difference in the rate of release for linear conjugate 4 or HMW star polymer conjugate 6 containing DTX-LEV was found. No effect of the size of the copolymer-drug conjugates or steric hindrance to hydrolysis was observed in this case. At pH 7.4, the hydrazone bond was cleaved faster within the first 8 h of incubation, with the rate decreasing in parallel with the decreasing concentration of hydrazone bonds in the solution. Subsequently, in the case of DTX-LEV, free DTX was slowly released by spontaneous hydrolysis of the ester bond; 10–12% DTX (∼1/3 of total) was released within 24 h of incubation. Contrary to the spontaneous hydrolysis found with DTX-LEV, DTX-OHT and DTX-COV showed no evidence for release of free DTX within 24 h of incubation at pH 7.4. Similarly, no evidence of hydrolysis of the ester bond in all DTX esters was observed during 24 h of incubation at pH 5. Finally, the in vitro release experiment carried out with polymer conjugate 6 in human plasma showed almost the same release profile of total released DTX (free DTX and DTX-LEV) as observed in phosphate buffer at pH 7.4 (see Fig. 4).
 | ||
| Fig. 4 Release of DTX and DTX derivatives from polymer conjugate 6 incubated in phosphate buffer at pH 7.4 or in human plasma at 37 °C. (■ —) buffer, total DTX released; (□ —) buffer, DTX-LEV; (■ ----) buffer, DTX; (● —) plasma, total DTX released; (○ —) plasma, DTX-LEV; (● ----) plasma, DTX; n = 3. | ||
The major difference in DTX release from polymer 6 in plasma and in a buffer with pH 7.4 was free DTX with a two-fold higher free DTX detected after 24 h of incubation of copolymer 6 in plasma. We suppose that the higher rate of hydrolysis of the ester bond in the released DTX-LEV in plasma was caused by the enzymatic activity of esterases present in human plasma. No evidence of enzymatic hydrolysis of the ester bond in DTX-LEV attached to the polymer was observed during 24 h of incubation in human plasma.
A basic criterion for effective drug delivery is the stability of the system during its transport in the blood and release of the drug at the site of action. Polymer conjugates 4, 6 and 10 containing DTX-LEV better fulfilled this criterion than the others, as demonstrated by the higher difference in the stability in model solutions at pH 5 and 7.4. That was why polymer conjugates 4, 6 and 10 were selected for further in vitro and in vivo biological evaluation.
In vitro degradation of star polymer carriers
Recently, we have shown that accumulation of the copolymers in solid tumors via the EPR effect24,35 significantly depends on the molecular weight of the polymer conjugate. Linear HPMA-based copolymers are non-biodegradable polymers with a limited potential to be removed from the organism by glomerular filtration when the molecular weight exceeds the renal threshold (approx. 50000 g mol−1).35 The biodegradable star HPMA-based copolymer 10 consisted of a polymer precursor 2 grafted onto small rigid dendrimer cores via an enzymatically degradable oligopeptide sequence. The degradation of the conjugate was studied in phosphate buffer (pH 6) containing cathepsin B to simulate the intracellular environment (lysosomes).
Star polymer conjugate 10, which contained enzymatically degradable GFLG sequences, was hydrolyzed slowly within 72 h of incubation in the buffer containing 0.5 μM cathepsin B (Fig. 5). As expected, the hydrolysis products were polymers with molecular weights comparable with HPMA copolymers used for the synthesis (∼35000 g mol−1). The stability of the star polymer conjugates in buffers mimicking blood circulation was tested by incubation in a phosphate buffer (pH 7.4) or in human plasma. No changes in molecular weight or polydispersity of the conjugates were observed.
 | ||
| Fig. 5 Decrease in Mw during degradation of polymer 10 in phosphate buffer (pH 6) containing 0.5 μM cathepsin B at 37 °C. | ||
The results of enzymatic degradation demonstrated that the conjugate 10 was stable under conditions modeling the blood stream, but underwent degradation after incubation in an intracellular environment-mimicking media. Thus, the HMW polymer-DTX conjugate derived from the PAMAM dendrimer and HPMA copolymers is expected to accumulate in solid tumors, release DTX and degrade inside tumor cells to form polymer fragments excretable from the body by glomerular filtration.
In vitro cytotoxicity of selected conjugates
Cytostatic/cytotoxic activity of star conjugates, either consisting of a non-degradable carrier (conjugate 6) or of a degradable carrier containing GFLC sequences (conjugate 10), was tested using several murine tumor cell lines and compared with the activity of linear conjugates containing DTX-LEV (conjugate 4), free DTX, and the DTX-LEV derivative. As an alternative to the standard measurement of the proliferative capacity of cells by thymidine incorporation, metabolic activity was used as a relevant parameter because the EL4 cell line is characterized by a very low capacity for thymidine incorporation. The DTX-LEV derivative showed pharmacological activity moderately lower than that of the parent drug in all the tested cell lines (Table 3). Cytostatic/cytotoxic activity of the star polymer conjugates was almost comparable to the activity of DTX-LEV, while the activity of the linear conjugate 4 was slightly lower than that observed in star conjugates. A similar activity relationship between the free drug and the polymer conjugate was observed in cell lines more sensitive to DTX (EL4, 4T1, and LL2), and in less DTX-sensitive cells (CT26). We conclude from these data that the derivatization of DTX by levulinic acid followed by binding of the derivative to the polymer carrier led to the preparation of a conjugate possessing cytotoxic/cytostatic activity in various tumor cell lines in vitro. The shape of the polymer carrier only slightly influenced the pharmacological activity of the conjugates in vitro, as the star-shaped polymer carriers linking DTX-LEV induced merely higher cytostatic activity than the linear one.| Cell line | EL4 | 4T1 | LL2 | CT26 | ||||
|---|---|---|---|---|---|---|---|---|
| Drug/conjugate | IC50b |
SD | IC50c |
SD | IC50c | SD | IC50c |
SD |
| a IC50 value is expressed as ng of DTX or DTX eq. mL−1. Each IC50 value is the mean of data obtained from several independent experiments. b Cytotoxic activity detected using the MTT assay c Cytostatic activity detected by 3H-TdR incorporation. | ||||||||
| DTX | 2.5 | 0.6 | 1.7 | 0.4 | 5.5 | 1.5 | 23 | 6.9 |
| DTX-LEV | 4.3 | 1.2 | 3.0 | 0.5 | 9.9 | 3.4 | 32 | 10.1 |
| Conjugate 4 | 6.6 | 1.6 | 3.8 | 1.3 | 11.1 | 3.3 | 36 | 7.6 |
| Conjugate 6 | 5.0 | 2.6 | 3.6 | 0.5 | 9.4 | 3.0 | 26 | 3.5 |
| Conjugate 10 | 4.9 | 1.1 | 2.8 | 0.8 | 7.8 | 2.6 | 26 | 8.5 |
MTD determination of linear and star docetaxel-loaded HPMA copolymers in healthy mice
To determine the in vivo maximum tolerated dose (MTD) of linear and star polymer-DTX conjugates, we injected them i.v. into mice at titrated doses up to 200 mg kg−1 of DTX eq. of linear conjugate and up to 180 mg kg−1 of DTX eq. of star conjugate.Initially, we determined the approximate MTD on smaller groups of C57BL/6 mice. In these preliminary experiments, we found that MTD of the linear conjugate (conjugate 4) is much higher than 150 mg kg−1 of DTX eq. as we observed no signs of toxicity and minimum weight loss (data not shown). In the case of the star conjugate (conjugate 6), we assumed its MTD to lie higher than 100 mg kg−1 of DTX eq. because all experimental mice appeared healthy and no morbidity or mortality was observed among groups (data not shown). Simultaneously, we tested the MTD also on BALB/c mice with no differences observed between strains (data not shown).
In accordance with the above mentioned preliminary testing, we injected larger groups of C57BL/6 mice (9 mice per group; 3 random mice were used for histological analysis) with titrated doses of the linear or star conjugate. The linear conjugate (conjugate 4) was administered at a dose of 200 mg kg−1 of DTX eq., which was the highest dose that we was able to solubilize and inject into mice as a bolus. Nevertheless, it still appeared to be safe with minimum weight loss and no signs of toxicity (Fig. 6A).
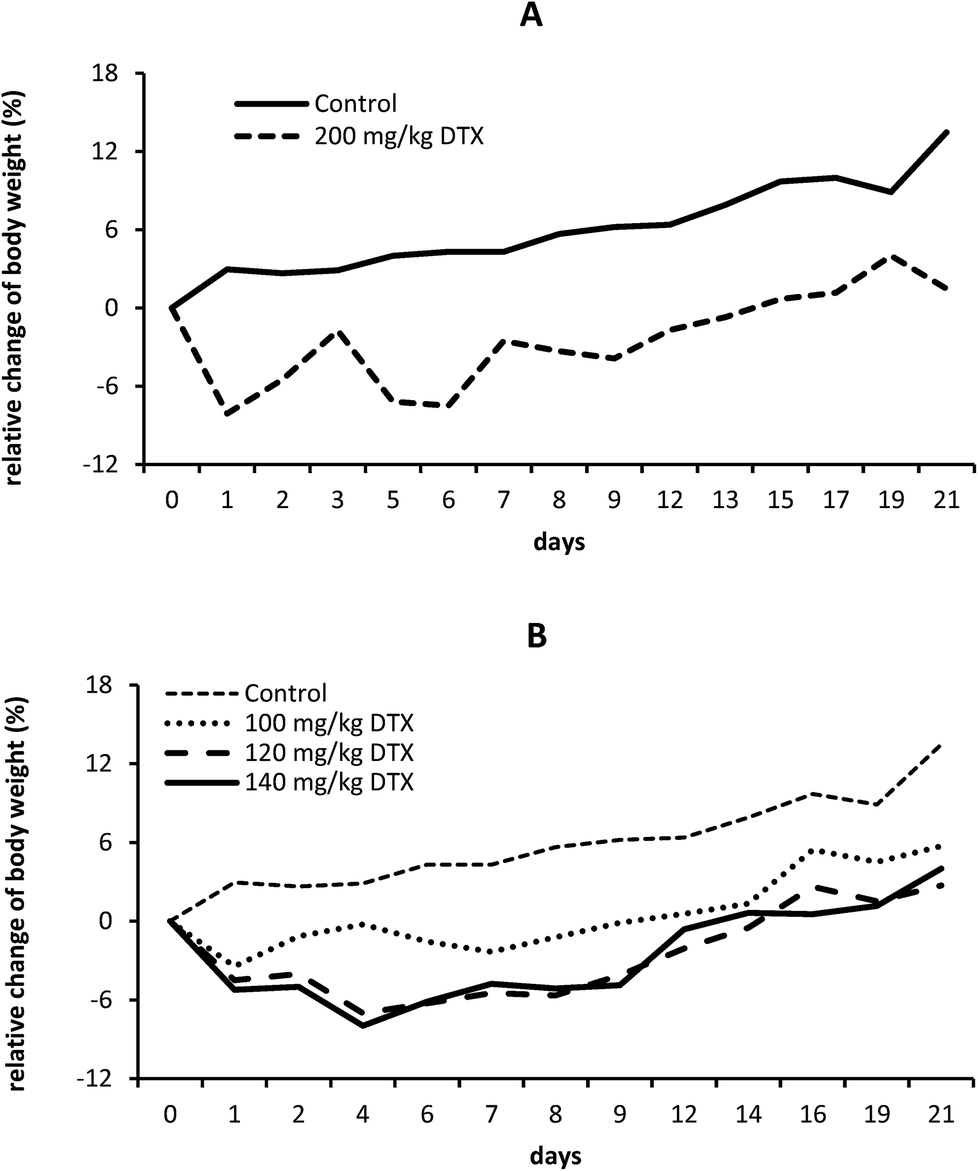 | ||
| Fig. 6 Determination of MTD of linear polymer-DTX and star polymer-DTX. C57BL/6 mice were i.v. injected with 200 mg kg−1 of DTX eq. of linear polymer-DTX conjugate (A); 100, 120 and 140 mg kg−1 of DTX eq. of star polymer-DTX conjugate (B) and their body weight was monitored until day 21 post administration. Mice i.v. injected with PBS were used as controls. Each experimental group contained 6 mice. | ||
Thus we suspect that the MTD is much higher and unreachable in a single dose injection (due to the high viscosity of the polymer solution) leaving a wide therapeutic window for this application of the conjugates. In the case of the star conjugate (conjugate 6), it was injected at doses of 100, 120 and 140 mg kg−1 of DTX eq. We observed no pathology or weight loss (Fig. 6B); therefore, we injected a small group of mice (3 mice per group) with 180 mg kg−1 of DTX eq., i.e., the highest dose that we were able to solubilize. Mice showed an approx. 15% weight reduction with mild signs of toxicity (data not shown), which led us to the assumption that MTD of the star conjugate lies between 140 and 180 mg kg−1 of DTX eq. In comparison with free DTX, which has an MTD of approximately 40 mg kg−1,36 both conjugates have significantly lowered toxicity and a wide therapeutic window.
Histological analysis of selected organs
Histology of selected organs from 3 random experimental mice was performed on day 7 post injection. Spleen, kidney, liver and vertebral bone marrow were analysed, and no pathological differences or anomalies were observed compared with naïve control mice. Similar results were observed in mice injected with free DTX at its MTD. Therefore, we assume that the tested conjugates cause no observable damage to tissues and organs including bone marrow and are safe up to the maximum dose applicable in a bolus.In vivo anti-tumor activity
The HMW star conjugates were designed to achieve enhanced tumor accumulation, leading to an improved anti-tumor effect for the treatment of solid tumors. In an EL4 lymphoma model, we observed an anti-tumor effect of conjugate 6 that was significantly better than the effect of the linear polymer-DTX conjugate (conjugate 4) or free DTX-LEV derivative, even at low dosing. Administration of 20 mg DTX eq. per kg on days 8 and 12 post tumor transplantation significantly reduced the tumor growth, prolonged survival time (median survival 100.5 days vs. 26 days in untreated controls), and finally led to complete cure in 50% of the mice treated with the star conjugate (conjugate 6). The linear polymer-DTX conjugate (conjugate 4) also significantly reduced the tumor growth (Fig. 7A) and prolonged survival when compared with untreated controls (median 39 days vs. 26 days), but only 25% of the mice remained tumor-free (Fig. 7B).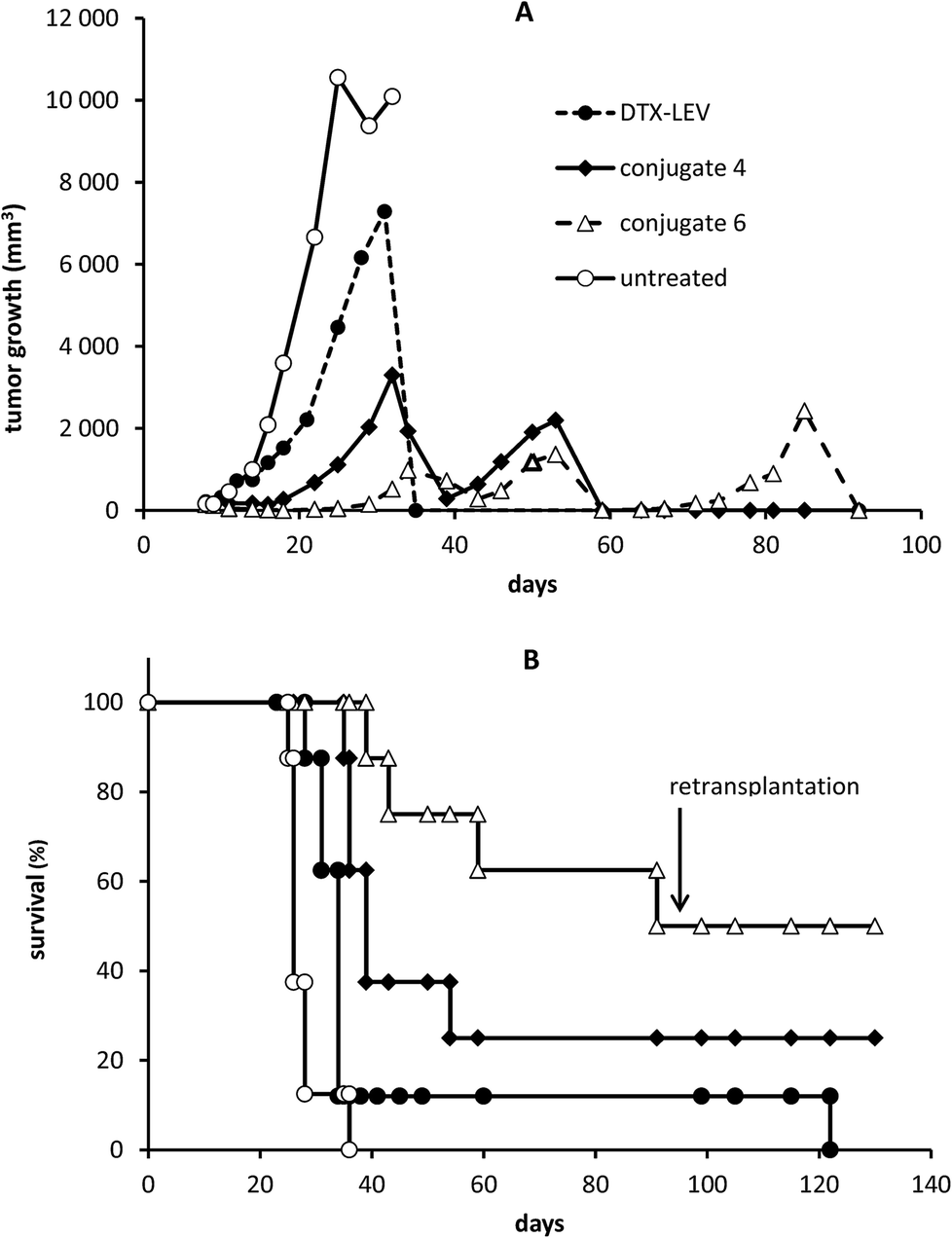 | ||
| Fig. 7 Comparison of anti-tumor activities of linear DTX conjugate (conjugate 4), star non-degradable DTX conjugate (conjugate 6) and free DTX-LEV derivative in the murine EL4 lymphoma model. C57BL/6 mice were transplanted with 1 × 105 EL4 cells s.c. and treated with 20 mg DTX eq. per kg or DTX-LEV injected i.v. on days 8 and 12. The long-term survivors, in which the tumors completely regressed, were challenged with 1 × 105 EL4 cells s.c. on day 95 after the first tumor transplantation and left without any treatment. Tumor growth (A) and survival time (B) were monitored. | ||
Indeed, treatment with free DTX-LEV led only to a slightly prolonged survival (median 34 days vs. 26 days in untreated controls) and only 1 of 8 mice was alive after the treatment. The body weight of the treated mice remained stable at least until day 25, documenting that the treatment with the conjugates was not associated with systemic toxicity (data not shown). Upon the second tumor challenge, the animals cured with the polymer conjugates (conjugates 4 and 6) showed tumor resistance, manifested by the absence of tumor growth even without any treatment. In contrast, the resistance was not apparent in the DTX-LEV-cured animal (Fig. 7B).
The enhanced anti-tumor activity of the HMW star conjugate could be directly related to increased accumulation of the drug in the tumor tissue due to the EPR effect. This is concordant with our previous data concerning a similar star conjugate containing doxorubicin.23,35 This star DOX conjugate caused larger drug accumulation in the tumor than a linear one, peaking at 24 hours post administration. The enhanced drug accumulation is predominantly dependent on the size of the polymer carrier.35 We hypothesize that similar to DOX conjugates, enhanced accumulation of DTX could be achieved with the star conjugate, leading to an increased anti-tumor effect. It should be noted that the size of the star DTX conjugate is approximately 26 to 30 nm (Table 2); thus, we assume that these conjugates would have a good ability to penetrate into the tissue.
Combined, the high MTD and the low dose anti-tumor effects of the DTX star demonstrate their considerable therapeutic potential and low/undetectable systemic toxicity. We have already shown that reduced toxicity of drug conjugates toward the immune system is a prerequisite for obtaining safe cancer treatments, which may lead to immunologically mediated tumor resistance, which has been observed in experimental tumor treatments and even in patients.30,37,38 The cancer resistance resulting from the treatment with HPMA copolymer conjugates serves as a safeguard against the cancer recurrence. Currently, consensus exists that modern cancer therapy not only kills the cancer but also initiates anti-cancer immune responses. The way the cancer cells die upon treatment is essential for initiating anti-cancer immune responses that support the therapy and is responsible for the final outcome of therapy. Cancer cell killing and anti-cancer immune responses are inseparable components of successful cancer therapy.39,40 Intact immune mechanisms were shown to be a prerequisite for the complete experimental tumor regression induced by HPMA-based polymer therapeutics.41 In DOX-containing HPMA conjugates, induction of immunogenic cancer cell death was documented.37,41–43 Some modulatory effects at low concentrations were also observed in DTX treatment, such as a reduction in immunosuppressive capacity of myeloid-derived suppressor cells that dampen the anti-cancer immune responses of the host organism in tumor-bearing animals and patients.44 We speculate that a similar activity could manifest also in treatment with DTX bound to the HPMA copolymer carrier. In addition to the previously described star polymer-DOX conjugates,23,35 we can summarise that HMW star polymer carriers could be used not only for enhanced drug delivery of soluble drugs as DOX, but also for solubilisation, elimination of systemic toxicity, controlled drug delivery and the related increase in the anti-tumor effect of highly hydrophobic, non-water soluble drugs such as DTX.
Conclusions
In the present study we described the synthesis, physicochemical characterization, in vitro drug release, biodegradation and in vivo anti-cancer activity of high-molecular-weight DTX conjugates based on star HPMA copolymer carriers. The increased size of the star conjugates as compared with the linear conjugates makes them excellent candidates for solid tumor treatment based on the enhanced passive accumulation driven by the EPR effect. Three types of star conjugates were synthesized with different hydrazone-containing spacers between the DTX and HPMA copolymer to enable controlled drug release in solutions modeling intracellular or intratumoral environments. The rate of release of DTX and/or its ester derivative is strongly dependent on the specific structure in the vicinity of the hydrazone bond, being the highest for the spacer containing the OHT ester of DTX, moderate for the LEV ester and the lowest for the COV ester. No significant difference was found in the rate of release when a linear conjugate or HMW star polymer conjugate was studied. In vivo evaluation of the MTD and anti-cancer activity of the star DTX-polymer conjugates confirmed their very low toxicity and superior activity over the linear polymer conjugate or free DTX in the treatment of EL4 T-cell lymphoma in mice. We have found that the star DTX conjugate had a remarkably higher MTD compared with DTX (single dose 140–180 mg DTX eq. kg−1vs. 40 mg kg−1 stated for DTX), thus the conjugation of DTX to the HMW polymer carriers led to a significant decrease in systemic toxicity and opened a wide therapeutic window for the treatment of selected solid tumors. A remarkably high anti-tumor effect was induced in treating experimental murine EL4 lymphoma even at suboptimal doses of the star DTX conjugate (20 mg kg−1 DTX eq., below 14% of MTD) resulting in a 50% cure rate, significantly exceeding the effect induced by the linear DTX conjugate or free DTX-LEV. In conclusion, we illustrated that the star pH-sensitive polymer-DTX conjugates have the potential to induce an excellent in vivo anti-tumor effect without apparent adverse effects.Acknowledgements
This work was supported by the Czech Science Foundation (project no. P301/11/0325 and P207/12/J030) and the Ministry of Education, Youth and Sports of the Czech Republic (project CZ.1.07/2.3.00/20.0055), and Institutional Research Concept RVO 61388971. The authors thank Pavlina Jungrova and Helena Misurcova for technical assistance.Notes and references
- R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198–1215 CrossRef CAS PubMed.
- J. Kopecek, P. Kopeckova, T. Minko, Z. R. Lu and C. M. Peterson, J. Controlled Release, 2001, 74, 147–158 CrossRef CAS.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- H. Maeda, Proc. Jpn. Acad. B: Phys., 2012, 88, 53–71 CrossRef CAS.
- T. Lammers, J. Controlled Release, 2012, 161, 151–151 CrossRef CAS PubMed.
- V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131–135 CrossRef CAS PubMed.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS PubMed.
- J. Kopecek, P. Kopeckova, T. Minko and Z. R. Lu, Eur. J. Pharm. Biopharm., 2000, 50, 61–81 CrossRef CAS.
- K. Ulbrich, V. Subr, J. Strohalm, D. Plocova, M. Jelinkova and B. Rihova, J. Controlled Release, 2000, 64, 63–79 CrossRef CAS.
- T. Etrych, P. Chytil, T. Mrkvan, M. Sirova, B. Rihova and K. Ulbrich, J. Controlled Release, 2008, 132, 184–192 CrossRef CAS PubMed.
- K. Ulbrich, T. Etrych, P. Chytil, M. Jelinkova and B. Rihova, J. Controlled Release, 2003, 87, 33–47 CrossRef CAS.
- D. Wang, P. Kopeckova, T. Minko, V. Nanayakkara and J. Kopecek, Biomacromolecules, 2000, 1, 313–319 CrossRef CAS.
- Y. Noguchi, J. Wu, R. Duncan, J. Strohalm, K. Ulbrich, T. Akaike and H. Maeda, Jpn. J. Cancer Res., 1998, 89, 307–314 CrossRef CAS PubMed.
- L. W. Seymour, Y. Miyamoto, H. Maeda, M. Brereton, J. Strohalm, K. Ulbrich and R. Duncan, Eur. J. Cancer, 1995, 31A, 766–770 CrossRef CAS.
- T. Etrych, T. Mrkvan, P. Chytil, C. Konak, B. Rihova and K. Ulbrich, J. Appl. Polym. Sci., 2008, 109, 3050–3061 CrossRef CAS.
- P. S. Steyger, D. F. Baban, M. Brereton, K. Ulbrich and L. W. Seymour, J. Controlled Release, 1996, 39, 35–46 CrossRef CAS.
- D. Putnam and J. Kopecek, Adv. Polym. Sci., 1995, 122, 55–123 CrossRef CAS.
- K. Ulbrich, J. Strohalm, V. Subr, D. Plocova, R. Duncan and B. Rihova, Macromol. Symp., 1996, 103, 177–192 CAS.
- F. Kratz, U. Beyer and M. T. Schutte, Crit. Rev. Ther. Drug, 1999, 16, 245–288 CrossRef CAS.
- P. C. A. Rodrigues, T. Roth, H. H. Fiebig, C. Unger, R. Mulhaupt and F. Kratz, Bioorgan Med. Chem., 2006, 14, 4110–4117 CrossRef CAS PubMed.
- K. Ulbrich and V. Subr, Adv. Drug Delivery Rev., 2004, 56, 1023–1050 CrossRef CAS PubMed.
- T. Etrych, L. Kovar, J. Strohalm, P. Chytil, B. Rihova and K. Ulbrich, J. Controlled Release, 2011, 154, 241–248 CrossRef CAS PubMed.
- T. Etrych, J. Strohalm, P. Chytil, P. Cernoch, L. Starovoytova, M. Pechar and K. Ulbrich, Eur. J. Pharm. Sci., 2011, 42, 527–539 CrossRef CAS PubMed.
- J. F. Diaz and J. M. Andreu, Biochemistry, 1993, 32, 2747–2755 CrossRef CAS.
- D. Belotti, V. Vergani, T. Drudis, P. Borsotti, M. R. Pitelli, G. Viale, R. Giavazzi and G. Taraboletti, Clin. Cancer Res., 1996, 2, 1843–1849 CAS.
- D. S. Grant, T. L. Williams, M. Zahaczewsky and A. P. Dicker, Int. J. Cancer., 2003, 104, 121–129 CrossRef CAS PubMed.
- S. S. W. Ng, A. Sparreboom, Y. Shaked, C. Lee, S. Man, N. Desai, P. Soon-Shiong, W. D. Figg and R. S. Kerbel, Clin. Cancer Res., 2006, 12, 4331–4338 CrossRef CAS PubMed.
- P. M. Ma and R. J. Mumper, J. Nanomed. Nanotechnol., 2013, 4, 1–16 Search PubMed.
- T. Etrych, M. Sirova, L. Starovoytova, B. Rihova and K. Ulbrich, Mol. Pharmaceutics, 2010, 7, 1015–1026 CrossRef CAS PubMed.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815–823 CrossRef CAS PubMed.
- K. Ulbrich, T. Etrych, P. Chytil, M. Jelinkova and B. Rihova, J. Drug Targeting, 2004, 12, 477–489 CrossRef CAS PubMed.
- T. Etrych, P. Chytil, M. Jelinkova, B. Rihova and K. Ulbrich, Macromol. Biosci., 2002, 2, 43–52 CrossRef CAS.
- V. Subr and K. Ulbrich, React. Funct. Polym., 2006, 66, 1525–1538 CrossRef CAS PubMed.
- T. Etrych, V. Subr, J. Strohalm, M. Sirova, B. Rihova and K. Ulbrich, J. Controlled Release, 2012, 164, 346–354 CrossRef CAS PubMed.
- U. Vanhoefer, S. Cao, A. Harstrick, S. Seeber and Y. M. Rustum, Ann. Oncol., 1997, 8, 1221–1228 CrossRef CAS.
- M. Sirova, M. Kabesova, L. Kovar, T. Etrych, J. Strohalm, K. Ulbrich and B. Rihova, Curr. Med. Chem., 2013, 20, 4815–4826 CrossRef CAS.
- M. Sirova, J. Strohalm, V. Subr, D. Plocova, P. Rossmann, T. Mrkvan, K. Ulbrich and B. Rihova, Cancer Immunol. Immun., 2007, 56, 35–47 CrossRef CAS PubMed.
- A. M. M. Eggermont, G. Kroemer and L. Zitvogel, Eur. J. Cancer, 2013, 49, 2965–2967 CrossRef CAS PubMed.
- A. J. M. Thomas, Expert Opin. Biol. Ther., 2014, 14, 1–4 Search PubMed.
- B. Rihova and M. Kovar, Adv. Drug Delivery Rev., 2010, 62, 184–191 CrossRef CAS PubMed.
- B. Rihova, J. Strohalm, K. Hoste, M. Jelinkova, O. Hovorka, M. Kovar, D. Plocova, M. Sirova, M. Stastny, E. Schacht and K. Ulbrich, Macromol. Symp., 2001, 172, 21–28 CrossRef CAS.
- B. Rihova, J. Strohalm, J. Prausova, K. Kubackova, M. Jelinkova, L. Rozprimova, M. Sirova, D. Plocova, T. Etrych, V. Subr, T. Mrkvan, M. Kovar and K. Ulbrich, J. Controlled Release, 2003, 91, 1–16 CrossRef CAS.
- K. N. Kodumudi, K. Woan, D. L. Gilvary, E. Sahakian, S. Wei and J. Y. Djeu, Clin. Cancer Res., 2010, 16, 4583–4594 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |