
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOlefin cross-metathesis, a mild, modular approach to functionalized cellulose esters†
Xiangtao
Meng
ab,
John B.
Matson
ac and
Kevin J.
Edgar
*ab
aMacromolecules and Interfaces Institute, Virginia Tech, Blacksburg, VA 24061, USA. E-mail: kjedgar@vt.edu; Tel: +1-540-231-0674
bDepartment of Sustainable Biomaterials, Virginia Tech, Blacksburg, VA 24061, USA
cDepartment of Chemistry, Virginia Tech, Blacksburg, VA 24061, USA
First published on 2nd September 2014
Abstract
Olefin cross-metathesis has been demonstrated to be a modular pathway for synthesis of a series of functionalized cellulose esters. As a proof of concept, cellulose acetate was acylated with two terminally olefinic acid chlorides, pent-4-enoyl chloride and undec-10-enoyl chloride, providing olefin-terminated cellulose esters with different side-chain lengths. These ω-unsaturated cellulose esters were then reacted with a variety of cross-metathesis partners, including acrylic acid, methyl acrylate, 2-hydroxyethyl acrylate, poly(ethylene glycol) methyl ether acrylate, and allyl alcohols, using Hoveyda–Grubbs’ 2nd generation catalyst. Complete conversion to cross-metathesis products was achieved in reactions with acrylic acid or acrylates using 3–5 mol% catalyst at 40 °C within 1 h. We further demonstrate successful hydrogenation of these α,β-unsaturated esters and acids, thereby eliminating the potential for radical-induced crosslinking during storage.
Introduction
Derivatives of cellulose and other polysaccharides are important components in a broad range of applications including drug delivery,1 automobile coatings,2 antimicrobials,3,4 and biomedical engineering.5–7 Polysaccharide chemists use chemical modification to enhance processability and to tune polysaccharide properties to meet the requirements of specific applications. It is particularly important to modify cellulose, which is abundant, renewable, and non-toxic, but in its native state has extremely poor solubility and cannot be melt-processed. Cellulose derivatization can reduce interchain hydrogen bonding and crystallinity as a remedy for poor organic and water solubility, and can tailor viscoelastic properties,8 thermal properties, and most importantly add new functional groups to the cellulosic backbone. The ability to broadly and selectively modify cellulose (or other polysaccharides) chemically can not only convey the ability to change physicochemical properties; appending new functional groups can also open doors to various valuable applications. Functionalities including carboxylic acid, hydroxyl,9 amino,10,11 and many others12,13 have been used to enhance the performance of polysaccharides. For example, we have shown that synthetic methods allowing attachment of ω-carboxyalkanoate functionality to cellulose impart the capability for pH-controlled drug release, and for superior performance in generating supersaturated drug solutions from amorphous solid dispersions, due in part to enhanced specific interactions with drug molecules.14,15 Others have pursued chemistries that permit the attachment of “tethers” to the polysaccharide, providing reactive sites (e.g. alcohol groups) that are distant from the main polysaccharide chain and hence more reactive, for easy attachment of targeting and other functional moieties.16–19To date polysaccharide chemistry and in particular cellulose chemistry has depended heavily on classical methods like esterification11,20 and etherification21 for appending functional groups, and indeed virtually all of the important commercial cellulose derivatives are made by either (or combinations) of these methods. While such methods are very useful and have led to entire industries based on the resulting derivatives of renewable cellulose, they are limited in scope. Typically esterification involves strongly acidic catalysts that are incompatible with sensitive functional groups on either cellulose or the acylating reagent, and esterification is also incompatible with difunctional reagents that could crosslink the product. Etherification typically involves strongly basic conditions (NaOH) in an aqueous environment, and so is incompatible with base-sensitive moieties and may be incompatible with reagents that do not possess substantial water solubility. Clearly there is a need to expand the toolkit for those seeking to make functional materials from sustainable polysaccharides; either by identifying new reactions that are mild, broadly useful, flexible, and ideally modular, or finding ways to extend the utility of conventional esterification and etherification to encompass such features. In this context we define “modular” polysaccharide reactions as those which permit the construction of a variety of functionalized moieties via small molecule chemistry, each of which can be attached to the polysaccharide using a reaction that is mild, high-yielding, efficient and dependable; in other words, a reaction bearing considerable similarity to “click” reactions. We will expand upon the differences in our chemistry from modern definitions of polymer click reactions22 in the Conclusions section.
Click chemistry,23 first discovered by the Sharpless group, has enabled the rapid synthesis of molecules with diverse functional appendages. Among these powerful concerted reactions, the azide–alkyne Huisgen cycloaddition24–27 and the thiol–ene click reaction19,28–30 have been used a number of times in polysaccharide chemistry to prepare a variety of functionalized derivatives. These strategies are modular functionalization methods for cellulose derivatives that can enable useful structure–function relationship studies. These methods require use of odorous, toxic, and/or potentially energetic reagents, and necessitate introduction of one or more heteroatom functions (e.g. sulfide or 1,2,3-triazole), limiting the utility of these reactions.31 They are valuable however, and the concepts23 of being modular, wide in scope, providing high yields, and generating only inoffensive by-products can guide our explorations of alternative polysaccharide functionalization pathways.
Olefin cross-metathesis (CM) promises to fulfill the abovementioned criteria in polysaccharide derivative synthesis.32 Grubbs’ rules33 predict that CM can be selective if two partners of differing reactivity are used; for example a type I olefin (e.g. reactive terminal olefin) and a type II or III olefin (e.g. less electron-rich acrylate); results also depend on the catalyst (Fig. 1) used.33 Driven by the loss of the volatile ethylene co-product, full conversion and high yields sometimes can be achieved. Recently, our group published an initial study showing that CM may be a powerful tool for the synthesis of soluble, discrete cellulose ω-carboxyesters.34 This class of polysaccharide derivatives has been shown to have high promise for enhancing drug bioavailability by creating supersaturated drug solutions via amorphous solid dispersion.35,36 These new derivatives were synthesized by CM between cellulose bearing olefin-terminated ester substituents (undec-10-enoate) and acrylic acid employing Hoveyda–Grubbs’ 2nd generation catalyst. This first-ever demonstration of successful CM in polysaccharide chemistry (there were a few previous reports of self-metathesis of cellulose derivatives,37,38 and CM of other olefin-functionalized polymers such as poly(oxazolines)39) hinted at its potential. CM of polysaccharides with differently functionalized partners may be a flexible way to functionalize cellulose and other polysaccharides in a modular manner to efficiently diversify the polysaccharide derivative family. The strategy is to attach a “handle” for CM using relatively conventional esterification chemistry, then carry out modular CM reactions to attach variously functionalized partners to cellulose, exploiting this handle (Fig. 2).
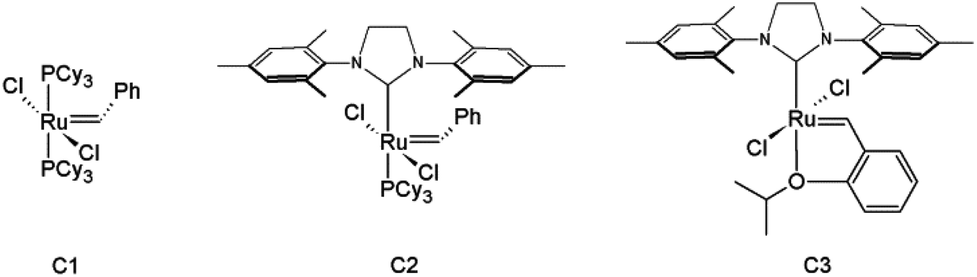 | ||
| Fig. 1 Commonly used Grubbs’ catalysts for olefin metathesis. | ||
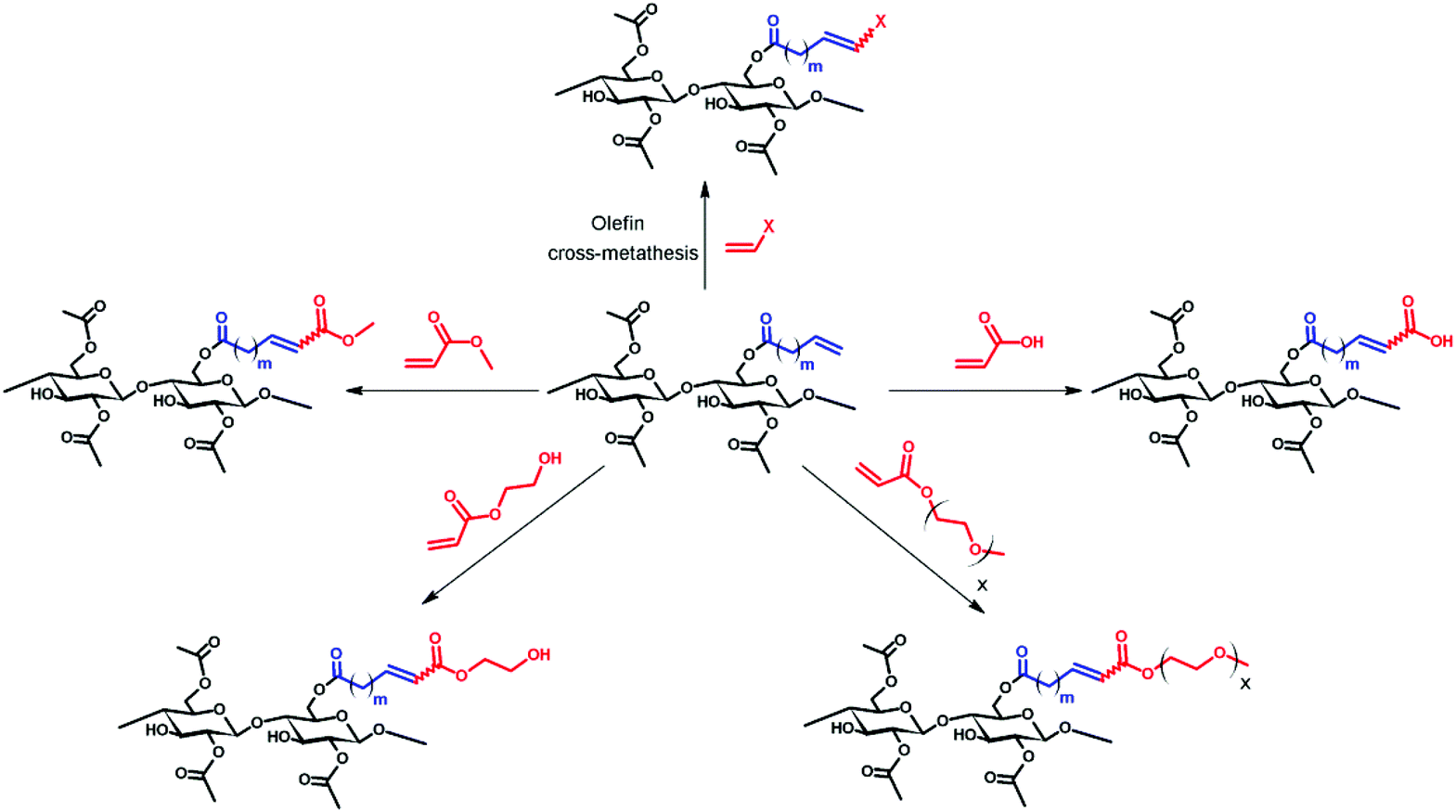 | ||
| Fig. 2 General scheme of olefin CM between terminally olefinic cellulose acetate and different CM partners. | ||
We report herein exploration of such a modular CM approach for synthesis of a group of cellulose derivatives, particularly acrylate esters that would potentially be extremely useful for attaching additional functionality via the pendent ester group. We explore and discuss the scope of CM for cellulose ester functionalization. We provide initial tests of our hypothesis that olefin CM may be a general method for cellulose functionalization and take an important step towards utilization of this chemistry as a flexible, efficient modular strategy for preparation of polysaccharide derivatives that otherwise might remain inaccessible.
Results and discussion
Our previous results34 showed the power of olefin CM for synthesis of carboxylic acid functionalized cellulose derivatives. Using the selective Hoveyda–Grubbs’ 2nd generation catalyst, cellulose alkanoate undec-10-enoate esters (e.g., cellulose acetate propionate undec-10-enoate) were reacted with acrylic acid, resulting in full conversion to cellulose ω-carboxyalkanoates within 1 hour at mild temperatures (40 °C). Not only did this approach provide a new pathway to a broader variety of cellulose ω-carboxyalkanoates, but also raised the possibility that this might be a more widely useful synthetic approach.Cellulose acetate undec-10-enoate with DS 0.67 of undec-10-enoate (2, m = 8 in Scheme 1) was first chosen as starting material. Eight methylene groups separate the terminal olefin and ester carbonyl, providing high olefin electron density and minimal steric hindrance, and making this terminal olefin a reactive substrate for Grubbs’ catalysts.33 We first tested the efficiency of allyl alcohol as a CM partner, but observed only about 60% conversion. Replacing allyl alcohol with 3-buten-2-ol, which has a more sterically hindered olefin and would hence be less prone to self-metathesis, did not improve conversion (data not shown). According to Grubbs’ CM rules33 reaction efficiency depends largely on catalyst type, as well substrate reactivity. Generally, type I olefins (electron rich and/or less sterically hindered, e.g. terminal olefins) are reactive but unselective. In contrast, electron deficient and/or sterically hindered olefins, which can be categorized into type II or III olefins (e.g. acrylic acid and acrylates) are less reactive but more selective. While an olefin metathesis reaction between a type I olefin and a type II or type III olefin tends to give CM products, a reaction between two type I olefins is more likely to generate a mixture of CM and self-metathesis (SM) products. Apparently the olefin electron density of allyl alcohols is sufficient to make them react as type I olefins, leading to competition with allyl alcohol homodimerization and suboptimal conversions.40 Note however that in some cases 60% conversion could be more than adequate, especially since residual double bonds can be readily reduced (vide infra).
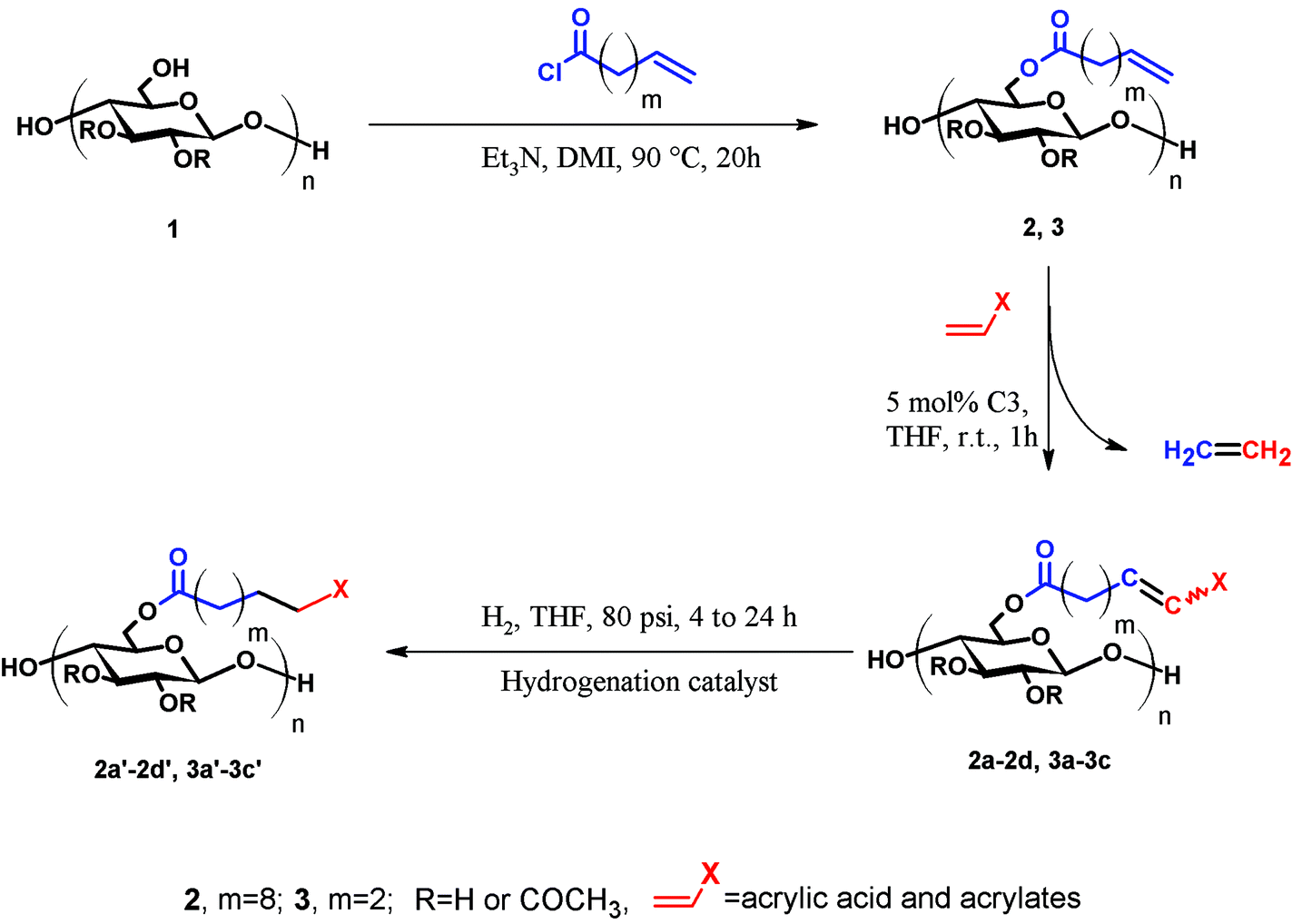 | ||
| Scheme 1 General three-step synthetic method for functionalized cellulose esters via olefin cross-metathesis. Note that structures are not meant to imply regiospecificity; particular positions of substitution in all schemes are only for convenience of depiction and clarity. | ||
Unprotected allyl amine on the other hand provided no evidence of conversion to CM products (data not shown). This is likely due not only to the low selectivity of the electron-rich double bond of allyl amine, but also to the likely coordination of the amine with ruthenium, thereby suppressing its catalytic activity.41,42
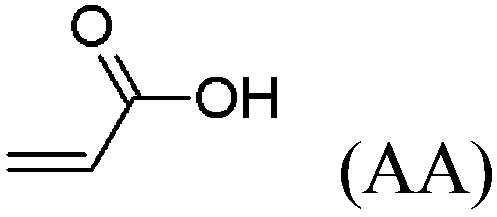
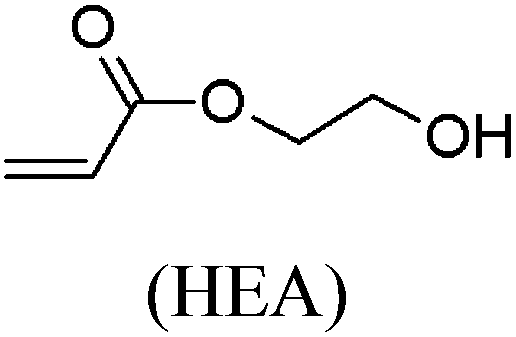
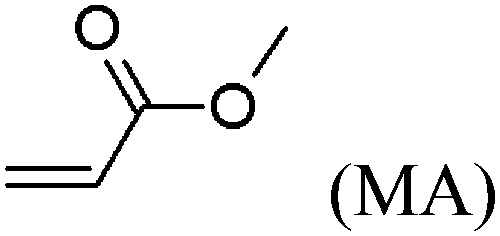
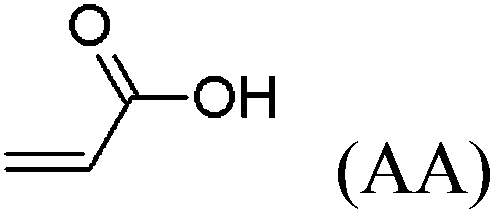
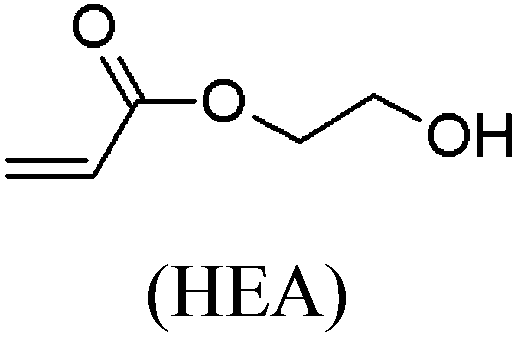
These unsatisfying results led us to acrylates, which are typically type II or III olefins due to the proximity of the electron-withdrawing carbonyl group to the olefin. In this work we studied three representative acrylate partners; the simple ester methyl acrylate (MA), as well as an acrylate ester with a bifunctional alcohol that can be used to append other functionality, 2-hydroxyethyl acrylate (HEA), as well as poly(ethylene glycol) methyl ether acrylate (PEGMEA) (average Mn = 480 Da). We included acrylic acid (AA) for comparison, especially for the cellulose pentenoate examples (Table 1). We were pleased to find that all examined acrylates and acrylate esters gave nearly 100% conversion with perfect selectivity for CM products under mild conditions (40 °C, 1 h, THF, 5 mol% Hoveyda–Grubbs catalyst, 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of acrylate to cellulose ester). CM reaction with HEA will serve to exemplify the results obtained; characterization data for products obtained by reaction with other CM partners can be found in the ESI.† Successful CM was supported by the FTIR spectra (Fig. 3). For example, the peak at 3074 cm−1 in the spectrum of starting cellulose undec-10-enoate 2, assigned to CHR stretch of the terminal olefin RCH
:1 ratio of acrylate to cellulose ester). CM reaction with HEA will serve to exemplify the results obtained; characterization data for products obtained by reaction with other CM partners can be found in the ESI.† Successful CM was supported by the FTIR spectra (Fig. 3). For example, the peak at 3074 cm−1 in the spectrum of starting cellulose undec-10-enoate 2, assigned to CHR stretch of the terminal olefin RCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, is absent from the spectrum of the new CM product 2b. A new absorbance at 1700 cm−1, assigned to the CO stretch of the α,β-unsaturated 2-hydroxyethyl ester, was observed for 2b on the shoulder of the cellulose ester CO stretch peak at 1751 cm−1. Further, the CC stretch signal of the terminal olefin (CHRCH2) in 2 at 1643 cm−1 was shifted to higher frequency 1650 cm−1 (CHRCHR (mostly trans as determined by 1H NMR)).
CH2, is absent from the spectrum of the new CM product 2b. A new absorbance at 1700 cm−1, assigned to the CO stretch of the α,β-unsaturated 2-hydroxyethyl ester, was observed for 2b on the shoulder of the cellulose ester CO stretch peak at 1751 cm−1. Further, the CC stretch signal of the terminal olefin (CHRCH2) in 2 at 1643 cm−1 was shifted to higher frequency 1650 cm−1 (CHRCHR (mostly trans as determined by 1H NMR)).
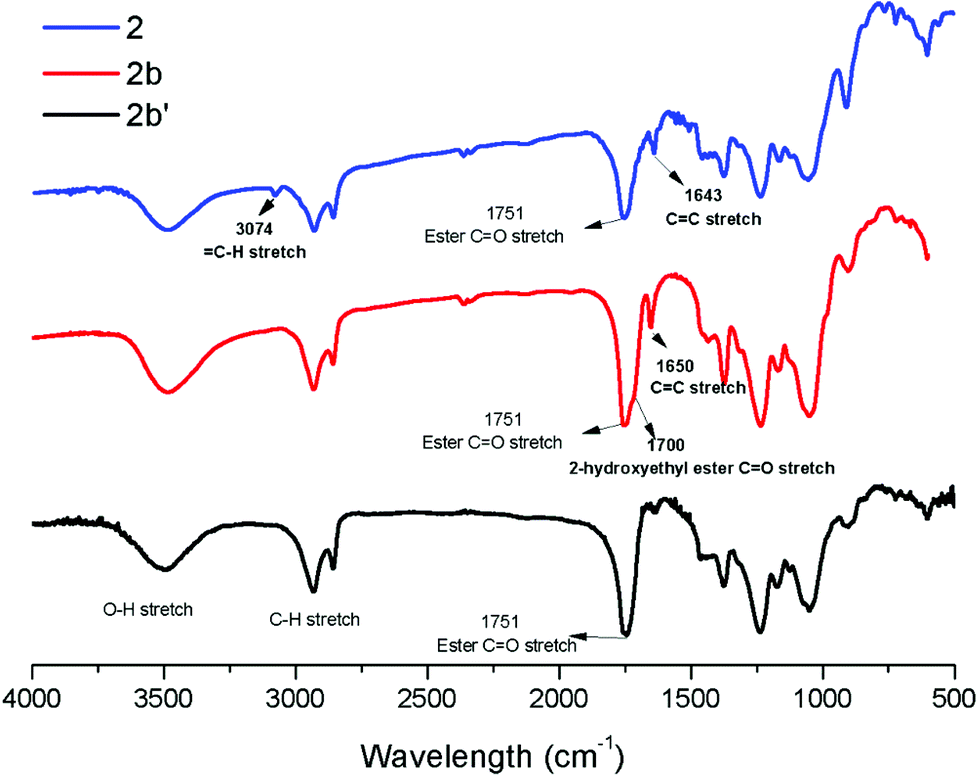 | ||
| Fig. 3 FTIR spectra of terminally olefinic cellulose acetate undec-10-enoate 2, the CM product (with HEA) 2b, and the hydrogenated product 2b′. | ||
| Cpd | Starting cellulose ester | CM partner (abbr.) | E/Z ratioa | Conversiona | Yield % | DS (X)a | DS (Ac)a | Hydrogenation product |
|---|---|---|---|---|---|---|---|---|
| a Determined by proton NMR. b Reported by supplier. | ||||||||
| 1 | — | — | — | — | — | — | 1.82b | — |
| 2 | 1 | — | — | — | 93 | 0.67 | 1.88 | — |
| 2a | 2 |
|
16.7 | ∼100% | 93 | 0.77 | 1.77 | 2a′ |
| 2b | 2 |
|
15.2 | ∼100% | 97 | 0.65 | 1.78 | 2b′ |
| 2c | 2 |
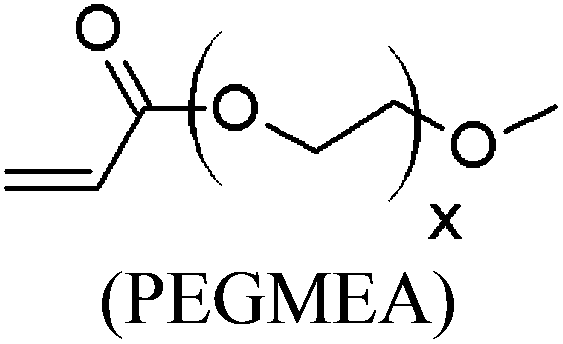
|
33.3 | ∼100% | 84 | NA | NA | 2c′ |
| 2d | 2 |
|
16.7 | ∼100% | 94 | 0.67 | NA | 2d′ |
| 3 | 1 | — | — | — | 89 | 0.56 | 1.80 | — |
| 3a | 3 |
|
15.5 | ∼100% | 94 | 0.56 | 1.85 | 3a′ |
| 3b | 3 |
|
9.7 | ∼100% | 96 | 0.56 | 1.86 | 3b′ |
| 3c | 3 |
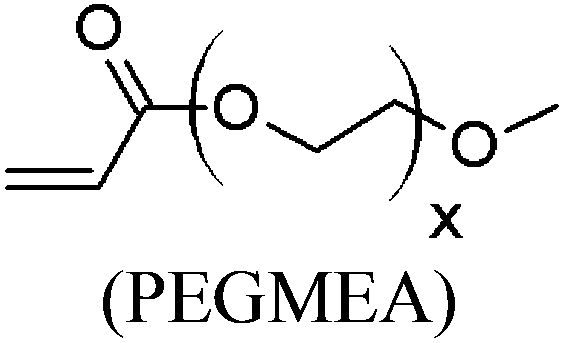
|
8.3 | ∼100% | 88 | NA | NA | 3c′ |
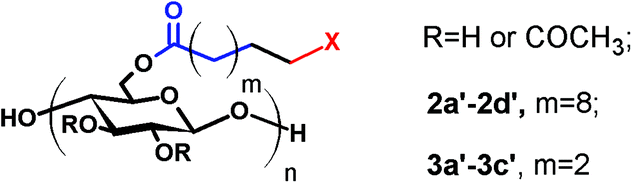
Proton NMR (Fig. 4) is an excellent tool for monitoring the CM reaction; disappearance of the terminal olefin signals of 2 at 4.90 and 5.75 ppm, and emergence of new downfield signals for the α,β-unsaturation (E configuration, 5.86 and 6.89 ppm for 2b), are diagnostic for occurrence and extent of CM, since these resonances are sharp and well-separated from each other and all other resonances of these cellulose derivatives. The corresponding signals of olefinic protons from the minor Z configuration product were observed at 5.77 and 6.28 ppm (Z/E double bond ratios determined by integration of these signals are listed in Table 1). Complete conversion of terminal olefin to α,β-unsaturated ester was thereby confirmed. We did not expect that there would be loss of the acyl groups (e.g. acetate) under these mild reaction conditions, but the 1H NMR spectra allowed us to affirm this hypothesis. Substituent DS was calculated by 1H NMR integration (Table 1), confirming that under these benign CM conditions the ester substituents largely remain untouched. 13C NMR analysis provides further evidence of the clean and complete nature of these CM reactions (Fig. 5). The terminal olefin resonances of compound 2 at 114 and 139 ppm completely disappeared, while new peaks that were assigned to the α, β-unsaturated carbons of 2b appeared downfield at 122 and 148 ppm.
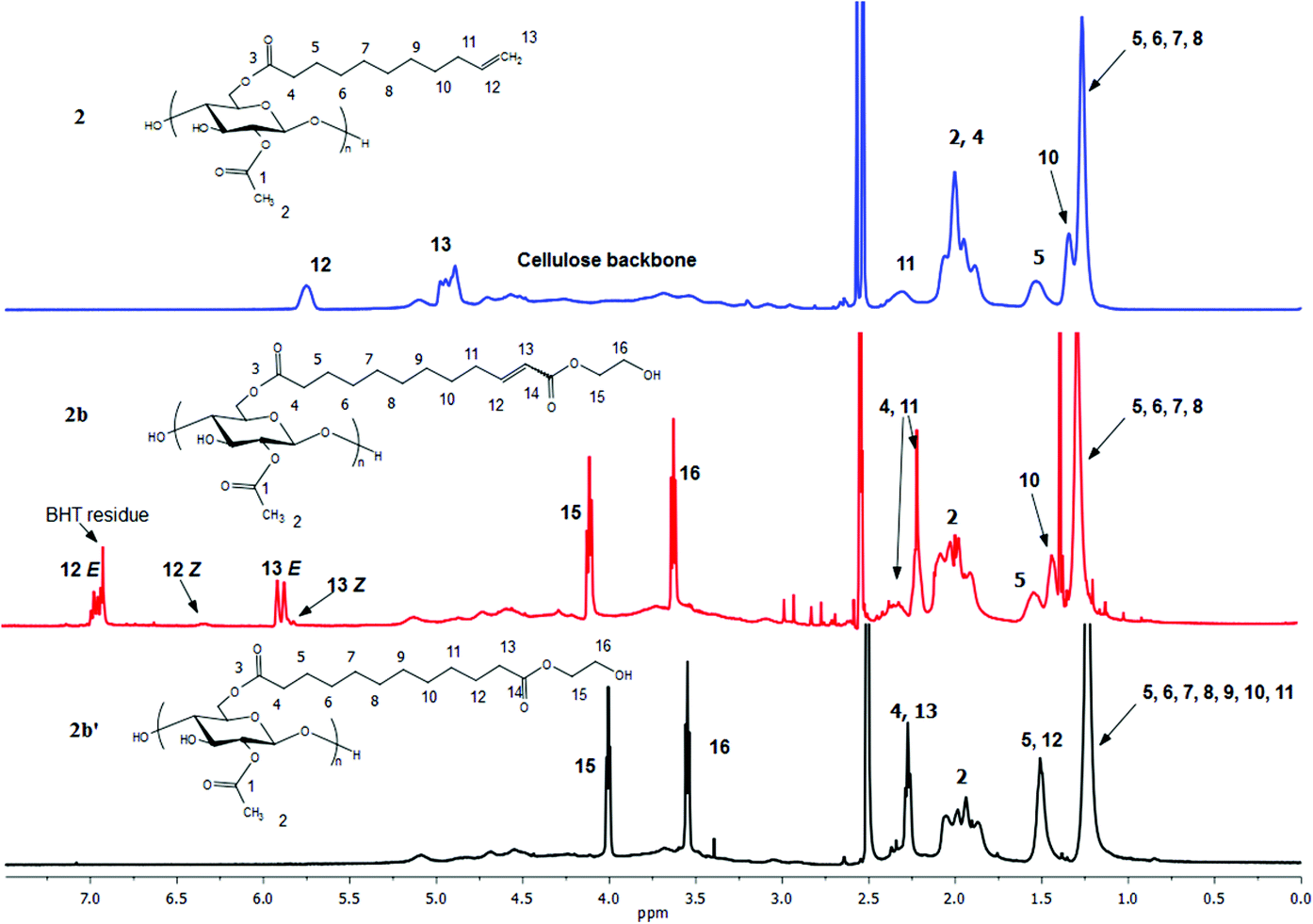 | ||
| Fig. 4 1H NMR spectra of terminally olefinic cellulose acetate undec-10-enoate 2, CM product (with HEA) 2b, and hydrogenated product 2b′. | ||
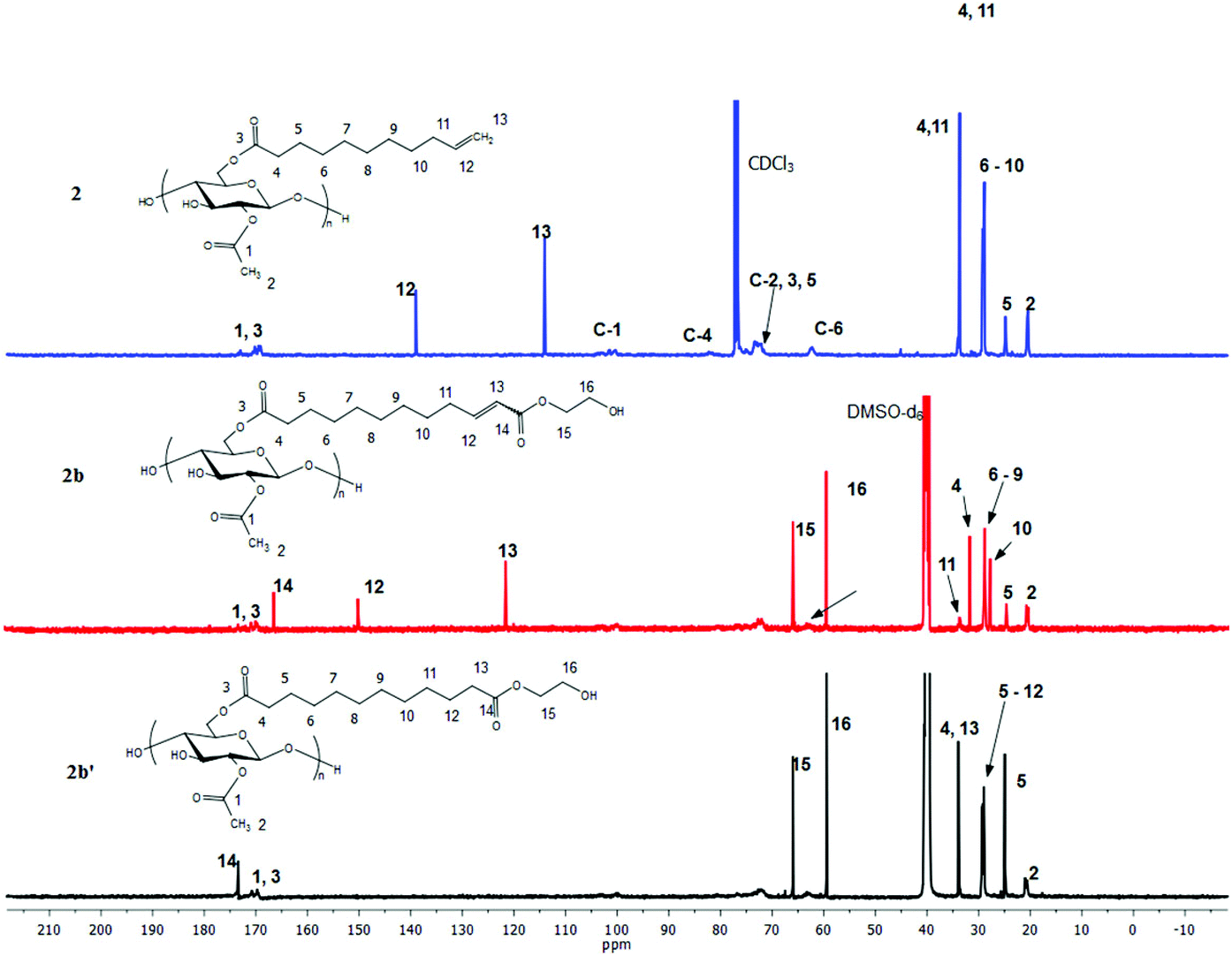 | ||
| Fig. 5 13C NMR of terminally olefinic cellulose acetate undec-10-enoate 2, CM product (with HEA) 2b, and hydrogenated product 2b′. | ||
Similar results (ESI†) were observed in CM reactions between 2 and AA, PEGMEA (average Mn = 480 Da), and MA, giving complete conversion to CM products 2a, 2c and 2d respectively. Although it was not surprising that small acrylates like HEA and MA were effective CM partners, we were pleasantly surprised to observe complete CM conversion with a PEG functionalized acrylate, PEGMEA, considering the potential for steric interference between the cellulose chain and that of the polymeric PEGMEA (average Mn of 480 Da). This invites speculation that a variety of acrylate end-functionalized polymers might also be feasible CM partners for polysaccharide derivatives like these. This modular reaction, like click reactions such as the azide–alkyne Huisgen cycloaddition reaction25,27,43 and “thiol–ene” reaction,28,29,44 may create potential for the synthesis of polysaccharide-based graft copolymers by a “grafting to” approach.
In our previous work,34 we noticed that the CM products of cellulose 10-undecenoate esters and acrylic acid appeared to crosslink during storage. We proposed that free radical oligomerization of the pendant α,β-unsaturated carboxylic acids was responsible for the crosslinking, as supported by the fact that added free radical scavenger (BHT) suppressed the crosslinking. In the current study, similar phenomena were observed for some of our products. Although adding BHT delayed the crosslinking process, the CM products became insoluble after weeks or months of storage, which may be attributed to consumption of the free radical scavenger, and/or the involvement of other crosslinking mechanisms (e.g. secondary olefin metathesis due to residual catalyst, although we consider this unlikely).
Considering that most imaginable crosslinking mechanisms are related to the α,β-unsaturation, it should be possible to eliminate the possibility of crosslinking if one is able to reduce the olefin. Palladium catalyzed hydrogenation has been previously used in carbohydrate and polysaccharide chemistry to reduce double bonds45 without impacting other functional groups. Therefore we pursued palladium catalyzed reduction of CM product olefins. Purified, dried CM products were hydrogenated (H2, Pd/C (10 wt% Pd (dry basis)/C)) at room temperature in THF. For the undec-10-enoate-based derivatives (2a–2d), only ca. 50% olefin hydrogenation was observed using a hydrogen balloon and Pd/C. Higher hydrogen pressure (80 psi) in a Parr reactor was more successful, affording fully hydrogenated products (2a′–2d′) (Table 2). 1H NMR spectra of 2b and 2b′ (Fig. 4 and ESI†) showed that both E olefinic protons at 5.8 and 6.9 ppm and Z olefinic protons at 5.3 and 6.7 ppm were entirely absent from the spectrum of 2b′, indicating complete olefin hydrogenation. The 13C NMR spectrum of 2b provided further confirmation, showing disappearance of the α,β-unsaturated double bond carbon signals at 122 and 148 ppm in 2b after hydrogenation. FTIR spectra of 2b′ (Fig. 3 and ESI†), show disappearance of the previous shoulder peak at 1700 cm−1, which was assigned to the CO stretch of the α,β-unsaturated methyl ester 2b. Moreover, after hydrogenation, the sharp CC stretch (CHRCHR, trans) signal around 1650 cm−1 also disappeared, revealing the previously hidden H2O vibrational peak.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound (abbr. of CM partner useda) | M n (kDa) | DP | Đ | X | DS(X)d | DS(Ac)d | T g (°C) |
| a See Table 1. b Data reported by supplier. c SEC (THF, 40 °C). d Calculated by 1H NMR. e DS cannot be calculated due to peak overlap. f T g not observed by DSC or MDSC. g MW data for 2 are different from corresponding sample in our previous publication (Mn ∼ 36.8 kDa, Đ ∼ 1.98);34 besides being prepared separately the samples were analyzed by different SEC systems. | |||||||
| 1 | 38.0b | 151 | NAb | — | — | 1.82 | 180 |
| 2 | 52.2/49.8c,g | 150/144 | 1.60/1.69c,g | — | 0.67 | 1.88 | 127 |
| 2a′ (AA) | 59.9 | 159 | 1.41 |
|
0.64 | 1.87 | 115 |
| 2b′ (HEA) | 60.4 | 149 | 1.49 |
|
0.69 | 1.76 | 102 |
| 2c′ (PEGMEA) | 96.7 | 148 | 1.52 |
|
NAe | NAe | NAf |
| 2d′ (MA) | 60.6b | 149 | 1.70b |
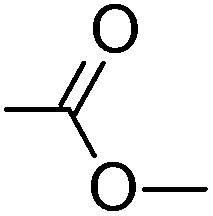
|
0.68 | 1.69 | 111 |
| 3 | 59.9 | 214 | 1.42 | — | 0.56 | 1.80 | 162 |
| 3a′ (AA) | 60.3 | 198 | 1.60 |
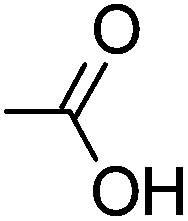
|
NAe | 1.82 | 154 |
| 3b′ (HEA) | 100.8 | 294 | 1.78 |
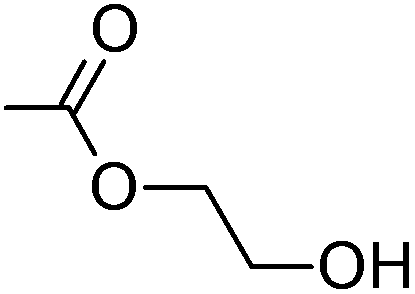
|
0.58 | 1.86 | 141 |
| 3c′ (PEGMEA) | 180.1 | 324 | 1.55 |
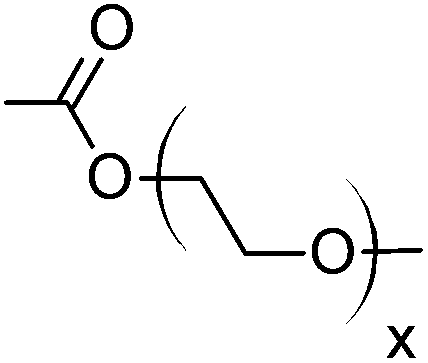
|
NAe | NAe | 76 |
Since THF is a good solvent for both the CM reaction and the subsequent hydrogenation, we wished to explore the potential efficiency of a one-pot reaction. Therefore, hydrogenation catalyst (Pd/C) was added directly to the reaction mixture after completion of CM, and the reaction mixture subjected to hydrogen pressure (80 psi). After hydrogenation, product 1H NMR and FTIR spectra were identical to those of products obtained by the two-step pathway, showing the feasibility of this one-pot synthesis. Considering the possibility of crosslinking (sometimes rapid) of olefin-containing CM products already described (vide supra), the potential for immediate, one-pot olefin reduction is of special importance.
We expected that the length of the tether between the terminal double bond and the ester carbonyl might impact CM efficiency, due to the potential for steric interference of the cellulose main chain with ruthenium complexation in the cases of shorter tethers. To test the potential for such effects, cellulose acetate pent-4-enoate (DS pent-4-enoate 0.56, (CA-Pen, 3)) was synthesized and used in CM reactions. Reaction of CM partners including AA, HEA and PEGMEA with CA-Pen under the same conditions as used for the reactions with the undec-10-enoate esters showed similar results. Full conversion to CM products was achieved (1H NMR, FTIR, ESI†), with essentially no loss of ester substituents during CM. These results show that a 2 carbon spacer between ester carbonyl and terminal double bond is enough for successful, complete CM reaction.
For the pent-4-enoate-based derivatives (3a–3c), however, heterogeneous hydrogenation (Pd/C) reduced less than 30% of the double bonds in an overnight reaction at hydrogen pressure as high as 140 psi. Given the fact that the olefins in 3a–3c are only 3 carbons away from the cellulosic backbone, it is reasonable to attribute the failure of the heterogeneous hydrogenation to steric interference by the cellulose chain which kept the double bonds from proper interactions with the Pd/C surface.46 To overcome this difficulty, homogeneous hydrogenation was performed on the 4-pentenoate-based derivatives using 2 wt% Crabtree's catalyst47 or 3 mol% Wilkinson's catalyst48 under 80 psi H2 in THF. NMR spectra (Fig. 6, 7 and ESI†) of the hydrogenated products of 3a–3c clearly indicated successful hydrogenation in similar fashion as described for 2a–2c. However, for 3a, the resulting hydrogenated products could not be redissolved in THF, and were only partially soluble in DMSO. To obtain soluble hydrogenated product 3a′, three cycles of heterogeneous hydrogenation (Pd/C) were performed (as described in the Methods section). The product obtained in this way was readily soluble, and the double bond was fully reduced as proven by 1H NMR as well as FTIR (ESI†).
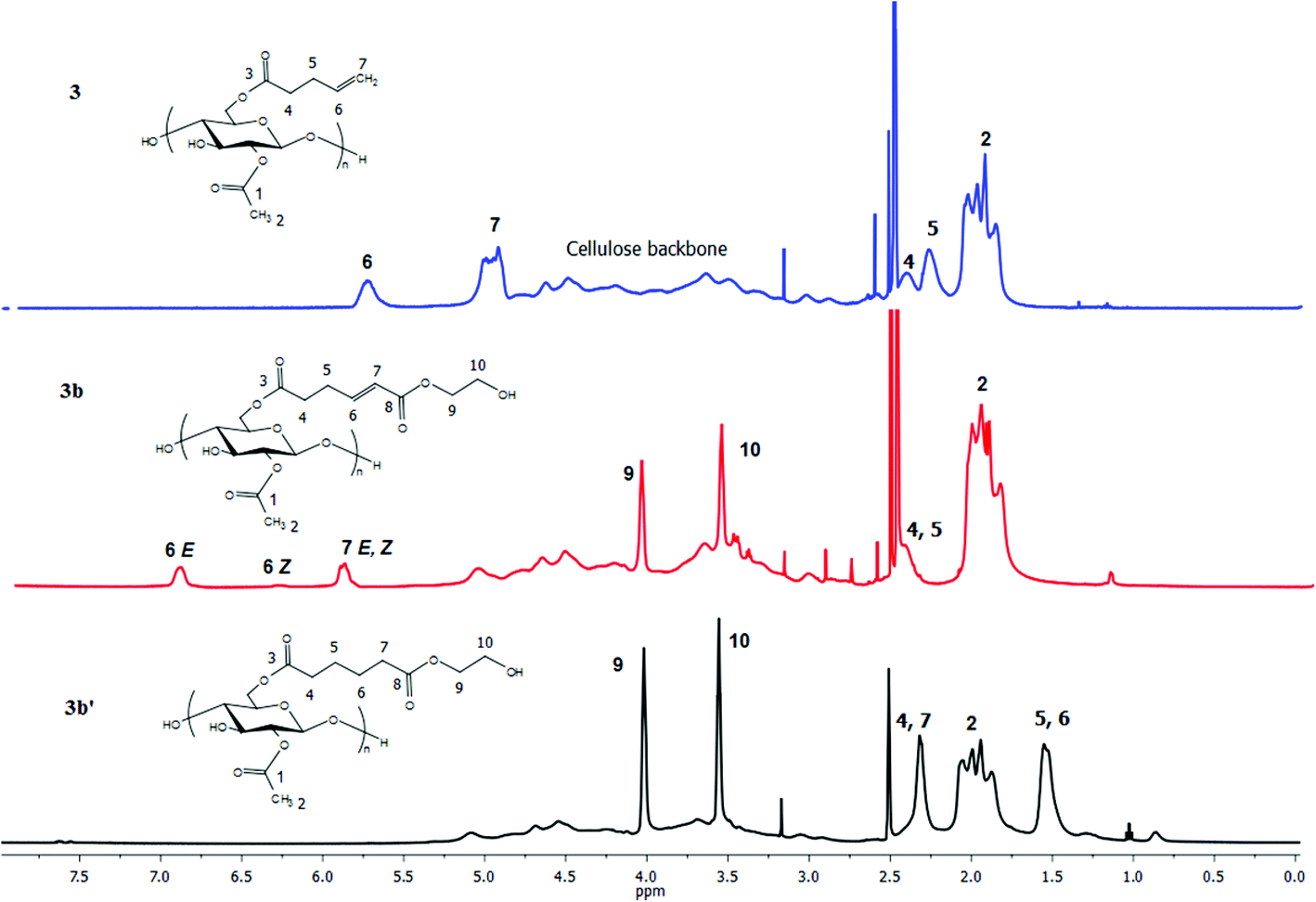 | ||
| Fig. 6 1H NMR spectra of terminally olefinic cellulose acetate pent-4-enoate 3, CM product (with HEA) 3b, and hydrogenated product 3b′. | ||
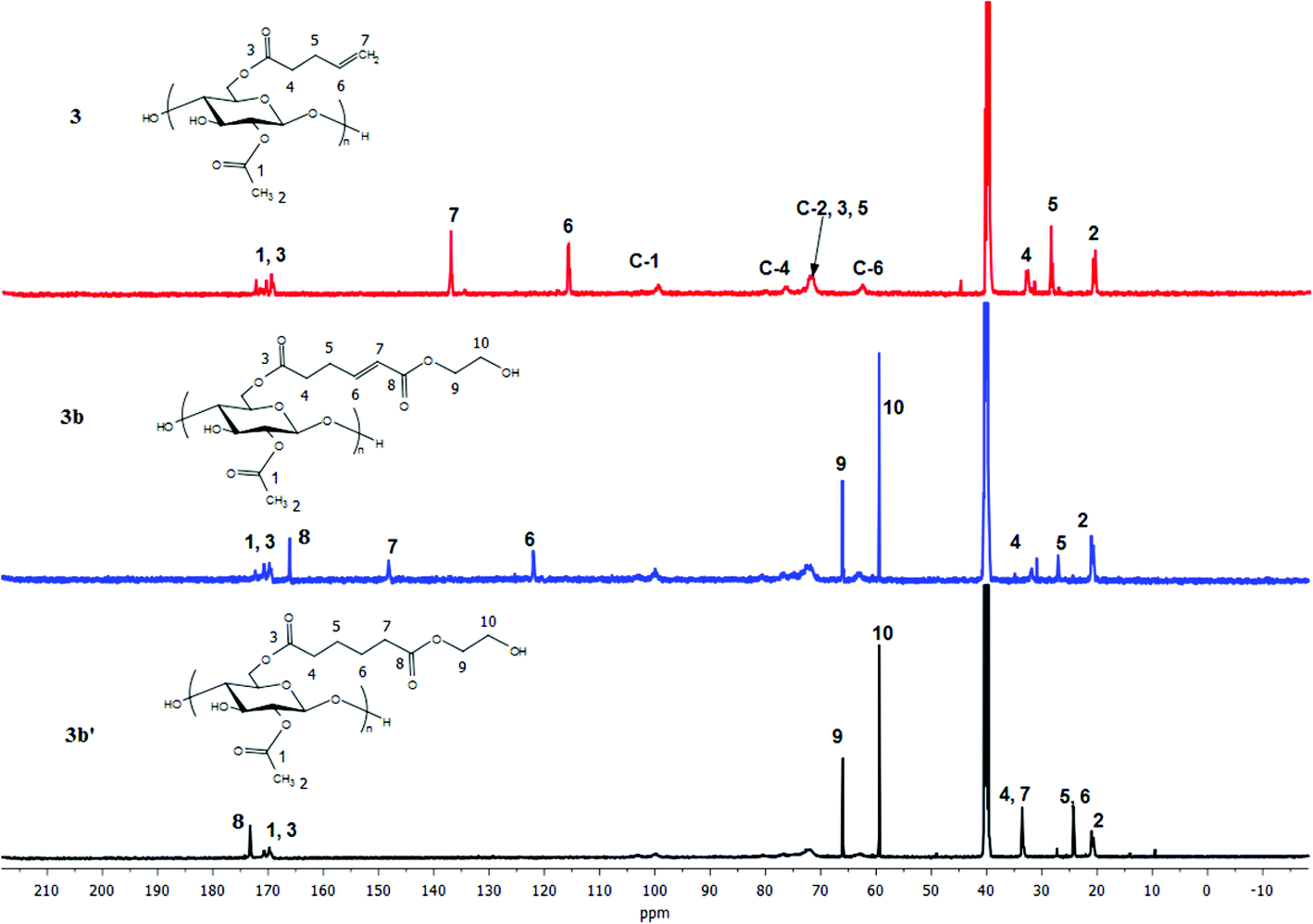 | ||
| Fig. 7 13C NMR of terminally olefinic cellulose acetate pent-4-enoate 3, CM product (with HEA) 3b, and hydrogenated product 3b′. | ||
We used SEC to characterize any change in DP resulting from the CM reaction. Considering that the CM products were prone to cross-link during storage, only hydrogenated samples were analyzed by SEC (Table 2). For 2a′–2d′ and 3a′, both chain scission and chain coupling (due to intermolecular self-metathesis (SM)) were negligible during CM as well as Pd/C catalyzed hydrogenation, indicated by the nearly unchanged DP and dispersity (Đ; IUPAC has discouraged use of the term polydispersity index (PDI), which had been used to describe polymer molecular weight distribution, and replaced it with the term dispersity, represented by the symbol Đ49). These results are consistent with our previous observations.34 The excess of CM partner used (20 equiv.) enhanced the likelihood of the cellulosic terminal double bonds meeting and reacting with a CM partner rather than with another terminal double bond, effectively suppressing SM crosslinking. The mild reaction conditions of both the CM and catalytic hydrogenation reactions, on the other hand, minimized the likelihood of chain scission, and so preserved polymer DP. However, it is noteworthy that 3b′ and 3c′, for which we employed homogeneous hydrogenation catalysts (Wilkinson's or Crabtree's) due to incomplete hydrogenation using Pd/C, had much higher DP than their shared starting material 3. The same phenomenon was observed upon homogeneous hydrogenation of 2b (DP ∼ 345 compared with DP ∼ 149 for 2b′). Combined with the fact that the homogeneous hydrogenation products of 3a lost their solubility, it is possible that the increased DP of 3b′ and 3c′ might be due to undetermined side reactions during homogeneous hydrogenation, or physical crosslinking by the hydrogenation catalysts.
Glass transition temperature (Tg), which reflects the molecular mobility of a polymer, influences the polymer's physicochemical properties including viscoelasticity, brittleness, and physical and chemical stability. It becomes a critical parameter in applications like amorphous solid dispersion formulation,50 where polymers are used as matrices to trap drug molecules in amorphous form, thereby enabling generation of supersaturated aqueous solutions. At temperatures below the formulation Tg, the restricted molecular mobility of the polymer will prevent drug molecule migration and therefore crystallization. For this reason, polymers with Tg at least 50 °C higher than ambient temperature are highly desirable to keep the formulation Tg above ambient temperature in spite of the plasticizing effects of both drug and atmospheric moisture. We employed DSC to determine Tg values of our CM products. Elimination of the double bond by hydrogenation did not significantly affect polymer Tg (ESI†). Although all other polymers exhibited Tg values at least 50 °C above room temperature, 2c′ did not display a detectable glass transition between −40 and 190 °C either by standard or modulated DSC. This may be attributed to the relatively high DS of the PEG tail and its plasticizing effect. Polymers synthesized from 3 cellulose acetate pent-4-enoate (3a′–3c′) had much higher Tg values (≥25 °C higher) than those of their counterparts (2a′–2c′) synthesized from the corresponding cellulose acetate undec-10-enoate (2), which is likely due to internal plasticization by the long (>11 carbons) chains, as well as the slightly lower DS of the pentenoate substituents compared to 2a′–2c′.
Experimental
Materials and instruments
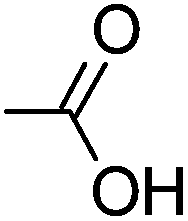
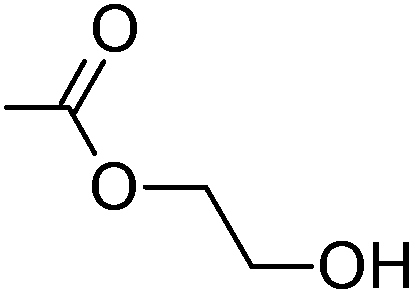
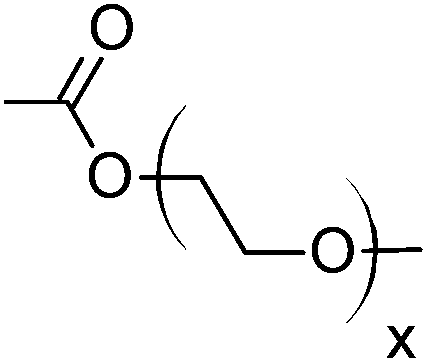
Cellulose acetate (CA-320S, Mn ∼ 38.0 kDa, DP ∼ 151, DS(Ac) ∼ 1.82 (data provided by supplier)) was from Eastman Chemical. Triethylamine and 1,3-dimethyl-2-imidazolidinone (DMI) were purchased from Acros Organics. Anhydrous tetrahydrofuran, acrylic acid, methyl acrylate, 2-hydroxyethyl acrylate, poly(ethylene glycol) methyl ether acrylate, allyl alcohol, 3-buten-2-ol, allylamine, 4-pentenoyl chloride, 10-undecenoyl chloride, ethyl vinyl ether, butylhydroxytoluene (BHT), palladium on carbon (10 wt% loading), Wilkinson's catalyst, Crabtree's catalyst, and Hoveyda–Grubbs’ 2nd generation catalyst were purchased from Sigma Aldrich. Diethylene glycol monovinyl ether was purchased from TCI. DMI were dried over 4 Å molecular sieves before use. All other purchased reagents were used as received. The high pressure reactor used in hydrogenation was mini bench top reactor 4560 purchased from Parr Instrument Company.Measurement
1H NMR spectra were acquired on a Bruker Avance 500 spectrometer operating at 500 MHz. Samples were analyzed as solutions in CDCl3 or DMSO-d6 (ca. 10 mg mL−1) at 25 °C in standard 5 mm o.d. tubes. Three drops of trifluoroacetic acid were added to shift the water peak in DMSO-d6 downfield from the spectral region of interest. To obtain the Tg values of the cellulosic polymers, DSC was performed on a TA Instruments Q2000 apparatus using heat/cool/heat mode. Dry powders (ca. 5 mg) were loaded in Tzero™ aluminum pans. The scanning conditions were set as follows: each sample was equilibrated at 35 °C, and then heated to 150° at 20 °C min−1. The sample was then cooled at 100 °C min−1 to −50 °C. During the second heating cycle the sample was heated to 200 °C at 20 °C min−1. If the heat/cool/heat mode failed to give a clear transition, modulated DSC was performed as follows: each sample was equilibrated at −50 °C, the underlying ramp heating rate was 7 °C, the oscillation amplitude was ±1 °C, and oscillation period was 40s. FTIR spectra were obtained on a Nicolet 8700 instrument. Size exclusion chromatography (SEC), if not otherwise specified, was performed on Agilent 1260 Infinity Multi-Detector SEC using NMP with 0.05 M LiBr as the mobile phase (50 °C) with 3 PLgel 10 μm mixed-B 300 × 7.5 mm columns in series. A system of multiple detectors connected in series was used for the analysis. A multi-angle laser light scattering (MALS) detector (DAWN-HELEOS II, Wyatt Technology Corporation, Goleta, CA), operating at a wavelength of 658 nm, a viscometer detector (Viscostar, Wyatt Technology Corporation, Goleta, CA), and a refractive index detector operating at a wavelength of 658 nm (Optilab T-rEX, Wyatt Technology Corporation, Goleta, CA) provided online results. Data acquisition and analysis was conducted using Astra 6 software (Wyatt Technology Corporation, Goleta, CA). For several samples, SEC was performed in THF as mobile phase (40 °C) on Agilent 1260 Infinity Multi-Detector SEC. For both systems, monodisperse polystyrene standard (Mw ∼ 21k, Đ ∼ 1.02) was run first in every sample series for the purpose of calibration and confirmation.Preparation of cellulose acetate 10-undecenoate (CA-Un, 2)
CA-320S (1, 1.00 g, 4.19 mmol/AGU) was dissolved in DMI (30 mL), and the solution was heated to 90 °C with mechanical stirring under N2. Triethylamine (1.29 mL, 9.22 mmol, 2.2 equiv.) was added; a condenser was used to avoid evaporative loss of the base catalyst. 10-Undecenoyl chloride (1.70 g, 8.36 mmol, 2.0 equiv.) was added dropwise and allowed to react at 90 °C for 20 h. The reaction mixture was then filtered, and the filtrate was precipitated in 300 mL 50:50 water–ethyl alcohol. The precipitate was redissolved in a minimal amount of CH2Cl2 and reprecipitated in hexane. The product was washed with hexane and dried under vacuum at 40 °C.
1H NMR (DMSO-d6): 1.21 (br s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCH2), 1.32 (br s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCH2), 1.50 (br s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCH2), 1.8–2.1(m, COCH2CH2CH2CH2CH2CH2CH2CH2CHCH2, and COCH3), 2.30 (br s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCH2), 3.3–5.3 (m, cellulose backbone), 4.8–5.0 (q, COCH2CH2CH2CH2CH2CH2CH2CH2CHCH2), 5.7 (m, COCH2CH2CH2CH2CH2CH2CH2CH2CHCH2). 13C NMR (CDCl3): 20.4 (COCH3), 24.8 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCH2), 28.8 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCH2), 33.6 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCH2), 114.1 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCH2), 139.0 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCH2), 168.9–173.1 (CO), 62.2 (C-6), 72.0–76.4 (C2, C3, C5), 82.3 (C-4), 100.7 (C-1). Degree of substitution (DS) by 1H NMR: DS(10-undecenoate) (DS(Un)) 0.67, DS(acetate) (DS(Ac)) 1.88; yield: 93%.
Preparation of cellulose acetate 4-pentenoate (CA-Pen, 3)
CA-320S (1, 1.00 g, 4.19 mmol/AGU) was dissolved in DMI (20 mL), and the solution was heated to 90 °C with mechanical stirring under N2. Triethylamine (2.6 mL, 9.22 mmol, 2.2 equiv.) was added; a condenser was used to avoid evaporative loss of the base catalyst. 4-Pentenoyl chloride (1.99 g, 8.38 mmol, 2.0 equiv.) was added dropwise and allowed to react at 90 °C for 20 h. The reaction mixture was then filtered, and the filtrate was precipitated in 300 mL 50:50 water–ethyl alcohol. The precipitate was redissolved in a minimal amount of CH2Cl2 and reprecipitated in hexane. The product was washed with hexane and dried under vacuum at 40 °C.
1H NMR (DMSO-d6): 1.8–2.1(m, COCH3), 2.28 (br s, COCH2CH2CHCH2), 2.42 (br s, COCH2CH2CHCH2), 2.9–5.3 (m, cellulose backbone), 4.9–5.1 (q, COCH2CH2CHCH2), 5.8 (m, COCH2CH2CHCH2). 13C NMR (DMSO-d6): 20.4 (COCH3), 28.6 (COCH2CH2CHCH2), 32.8 (COCH2CH2CHCH2), 116.0 (COCH2CH2CHCH2), 137.3 (COCH2CH2CHCH2), 168.9–173.1 (CO), 62.2 (C-6), 72.0–76.4 (C2, C3, C5), 82.3 (C-4), 100.7 (C-1). Degree of substitution (DS) by 1H NMR: DS(4-pentenoate) (DS(Pen)) 0.56, DS(acetate) (DS(Ac)) 1.80; yield: 89%.
General procedure for olefin cross-metathesis reactions
To a flask charged with cellulose derivative 2 CA-Un or 3 CA-Pen (100 mg, 1.0 equiv. olefin), 5 mg BHT and 5 mL anhydrous THF were added. After the reagents were completely dissolved, cross-metathesis partner (acrylic acid, methyl acrylate, 2-hydroxyethyl acrylate, poly(ethylene glycol) methyl ether acrylate, allyl amine, or allyl alcohol; 20 equiv.) was added followed by the addition of Hoveyda–Grubbs Catalyst 2nd Generation (0.05 equiv. in 2 mL THF) via syringe. After stirring for 1 h under N2 at 40 °C, the reaction was stopped by adding 1–2 drops of diethylene glycol monovinyl ether or ethyl vinyl ether. The product was collected by either dialysis and freeze-drying, or by precipitating in H2O–ethanol followed by sufficient washing by H2O before being dried under vacuum at 40 °C.CHCOOH), 1.38 (br s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOH), 1.50 (br. s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOH), 1.86–2.05(m, COCH3), 2.14 (br s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOH), 2.28 (br s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOH), 2.75–5.25 (m, cellulose backbone), 5.68(d, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOH, Z configuration), 5.74 (d, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOH, E configuration), 6.19 (m, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOH, Z configuration), 6.80 (m, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOH, E configuration). 13C NMR (DMSO-d6): 20.7 (COCH3), 24.8 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOH), 28.0 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOH), 29.0 (COCH2CH2CH2CH2CHCH2CH2CH2CHCHCOOH), 31.8 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOH), 33.8 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOH), 122.4 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOH), 149.0 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOH), 167.4 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOH), 169.1–173.3 (CO), 63.0 (C-6), 72.0–76.4 (C2, C3, C5), 80.4 (C-4), 100.0 (C-1). Conversion by 1H NMR: 100%, E/Z ratio by 1H NMR: 16.7, yield: 93%.
CHCOOCH2CH2OH), 1.37 (br s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH), 1.50 (br. s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH), 1.8–2.1 (m, COCH3), 2.18 (br s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH), 2.28 (br s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH), 2.75–5.25 (m, cellulose backbone), 3.56 (t, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH), 4.06 (t, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH), 5.77(d, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH, Z configuration), 5.86 (d, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH, E configuration), 6.28 (m, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH, Z configuration), 6.89 (m, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH, E configuration). 13C NMR (DMSO-d6): 20.9 (COCH3), 24.8 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH), 27.9 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH), 29.0 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH), 31.8 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH), 33.9 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH), 59.6 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH), 65.9 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH), 121.4 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH), 149.8 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH), 166.2 (COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH2CH2OH), 169.1–173.3 (CO), 63.0 (C-6), 72.0–76.4 (C2, C3, C5), 80.4 (C-4), 100.0 (C-1). Conversion by 1H NMR: 100%, E/Z ratio by 1H NMR: 15.2, yield: 97%.
CHCOO(CH2CH2O)xCH3), 1.41 (br s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOO(CH2CH2O)xCH3), 1.49 (br. s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOO(CH2CH2O)xCH3), 1.8–2.1(m, COCH3), 2.18 (br s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOO(CH2CH2O)xH), 2.28 (br s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOO(CH2CH2O)xCH3), 2.75–5.25 (m, cellulose backbone), 3.23(s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOO(CH2CH2O)xCH3), 3.42, 3.50, 3.61 and 4.17 (m, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOO(CH2CH2O)xCH3), 5.77 (d, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOO(CH2CH2O)xCH3, Z configuration), 5.86 (d, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOO(CH2CH2O)xCH3, E configuration), 6.28 (m, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOO(CH2CH2O)xCH3, Z configuration), 6.89 (m, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOO(CH2CH2O)xCH3, E configuration). Conversion by 1H NMR: 100%, E/Z ratio by 1H NMR: 33.3, yield: 84%.
CHCOOCH3), 1.39 (br s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH3), 1.50 (br. s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH3), 1.8–2.1(m, COCH3), 2.17 (br s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH3), 2.28 (br s, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH3), 2.75–5.25 (m, cellulose backbone), 5.77 (d, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH3, Z configuration), 5.83 (d, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH3, E configuration), 6.27 (m, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH3, Z configuration), 6.87 (m, COCH2CH2CH2CH2CH2CH2CH2CH2CHCHCOOCH3, E configuration). Conversion by 1H NMR: 100%, E/Z ratio by 1H NMR: 16.7, yield: 94%.
CHCOOH), 2.9–5.3 (m, cellulose backbone), 5.78 (m, COCH2CH2CHCHCOOH, E and Z configuration), 6.19 (m, COCH2CH2CHCHCOOH, Z configuration), 6.79 (m, COCH2CH2CHCHCOOH, E configuration). Conversion by 1H NMR: 100%, E/Z ratio by 1H NMR: 15.5, yield: 94%.
CHCOOCH2CH2OH), 2.9–5.3 (m, cellulose backbone), 3.60 (t, COCH2CH2CHCHCOOCH2CH2OH), 4.07 (t, COCH2CH2CHCHCOOCH2CH2OH), 5.88 (m, COCH2CH2CHCHCOOCH2CH2OH, E and Z configuration), 6.28 (m, COCH2CH2CHCHCOOCH2CH2OH, Z configuration), 6.90 (m, COCH2CH2CHCHCOOH, E configuration). 13C NMR (DMSO-d6): 20.6 (COCH3), 27.9 (COCH2CH2CHCHCOOCH2CH2OH), 30.9 (COCH2CH2CHCHCOOCH2CH2OH), 59.4 (COCH2CH2CHCHCOOCH2CH2OH), 66.1 (COCH2CH2CHCHCOOCH2CH2OH), 121.8 (COCH2CH2CHCHCOOCH2CH2OH), 148.0 (COCH2CH2CHCHCOOCH2CH2OH), 166.1 (COCH2CH2CHCHCOOCH2CH2OH), 169.1–173.3 (CO), 63.0 (C-6), 72.0–76.4 (C2, C3, C5), 80.4 (C-4), 100.0 (C-1). Conversion by 1H NMR: 100%, E/Z ratio by 1H NMR: 9.7, yield: 96%.
CHCOO (CH2CH2O)x CH3), 2.75–5.25 (m, cellulose backbone), 3.23(s, COCH2CH2CHCHCOO (CH2CH2O)x CH3), 3.41, 3.50, 3.61 and 4.17 (m, COCH2CH2CHCHCOO (CH2CH2O)x CH3), 5.90(m, COCH2CH2CHCHCOO CH3, E and Z configuration), 6.31 (m, COCH2CH2CHCHCOO CH3, Z configuration), 6.90 (m, COCH2CH2CHCHCOO CH3, E configuration). Conversion by 1H NMR: 100%, E/Z ratio by 1H NMR: 8.3, yield: 88%.
General procedure for reduction of the α,β-unsaturated double bond of the CM products by Pd/C hydrogenation
To a solution of 500 mg CM product dissolved in 50 mL anhydrous THF, 150 mg palladium on carbon (10 wt% loading) was added. The mixture was stirred overnight under 80 psi H2 at room temperature (for compound 3 based sample 3a, after filtering the mixture through Celite, another 150 mg Pd/C was added and reacted under 80 psi H2 for 12 hours. The cycle was repeated once more (total of three hydrogenations) to make sure that all the double bonds were hydrogenated). The mixture was filtered through Celite, concentrated, and then precipitated into hexanes. The precipitate was collected and dried under vacuum at 40 °C.General procedure for olefin cross-metathesis/hydrogenation one-pot reaction
To a Parr Reactor (vessel volume: 600 mL) charged with cellulose derivative 2 or 3 (400 mg, 1.0 equiv. olefin), 20 mg BHT and 40 mL anhydrous THF were added. After the reagents were completely dissolved, the cross-metathesis partner (acrylic acid, methyl acrylate, 2-hydroxyethyl acrylate, poly(ethylene glycol) methyl ether acrylate, or allyl alcohol; 20 equiv.) was added followed by the addition of Hoveyda–Grubbs Catalyst 2nd Generation (0.05 equiv. in 6 mL THF) via syringe. After stirring for 1 hour under N2 at room temperature, 30 wt% Pd/C was added. The mixture was stirred under 80 psi H2 at room temperature. The subsequent reaction and purification followed that in the general procedure above.O), 63.0 (C-6), 72.0–76.4 (C2, C3, C5), 80.4 (C-4), 100.0 (C-1).Yield: 93%.
CHCOOCH2CH2OH), 59.4 (COCH2CH2CH2CH2COOCH2CH2OH), 66.0 (COCH2CH2CH2CH2COOCH2CH2OH), 173.2(COCH2CH2CH2CH2COOCH2CH2OH), 169.1–173.3 (CO), 63.0 (C-6), 72.0–76.4 (C2, C3, C5), 80.4 (C-4), 100.0 (C-1). Yield: 74%.
Conclusions
We provide in this study an expanded vision of a new, modular method for synthesis of cellulose derivatives that can expand the utility of conventional substitution methods like esterification and etherification. We do so by introducing the concept of attaching a handle for olefin cross metathesis by these conventional methods, followed by modular CM to introduce a plethora of new functional groups. We demonstrate that readily available acrylate esters are highly effective CM partners, providing the capability to introduce α,β-unsaturated ester moieties in controlled and selective fashion as side chains of cellulose. We show how the unsaturation can be eliminated by catalytic hydrogenation, even in a one-pot overall reaction, to remove the reactivity and instability introduced by that functional group. At the same time it is clear that the α,β-unsaturation could be used alternatively as a handle for introduction of still other functionality, e.g. by Michael addition of an amine.51 We demonstrate also that such acrylate esters may bear terminal hydroxyl groups and still be effective CM partners (2-hydroxyethyl acrylate, poly(ethylene glycol) methyl ether acrylate). These create still more potential for functionalization by reaction with the hydroxyl group (distant from the main cellulose chain and so relatively unhindered). We predict that such functionalization could be carried out prior to the CM reaction by modifying the CM partner, or after CM and/or after the CM/hydrogenation sequence. Some limitations upon this modular CM method for functionalization of cellulose become apparent as well. The double bonds of allylic alcohols may in some cases be too self-reactive (Type 1 by Grubbs’ rules) to serve as optimal CM partners, although the 60% conversions we achieved might be perfectly useful and acceptable depending on the particular synthetic goal. In contrast, unprotected amines do not appear to be effective CM partners for these terminally unsaturated cellulose esters, most likely due to their propensity to coordinate and thus inactivate the ruthenium catalyst.It is interesting to compare this chemistry to the definition of a polymer click chemistry reaction recently put forth in eloquent fashion by Barner-Kowollik et al.22 Clearly the reaction occurs rapidly and under mild conditions, is chemoselective and has a single reaction trajectory, affords high yields, is modular and wide in scope, and lends itself to easy product purification, as required by the authors’ definition of a polymer click reaction. It does not meet their definition in the sense that the initial products are not fully stable (though they are after the hydrogenation step), and especially in that equimolarity of reagents is not ideal for achieving high yields and selectivity for CM to the exclusion of SM. Therefore we feel that characterizing the reaction as modular and click-like is appropriate, though it does not meet all the click criteria as defined by these authors.
Overall, the mild nature of this CM chemistry, and our growing appreciation of the potential variety of CM partners that can be used, illustrate its high potential for modular modifications of terminally unsaturated cellulose derivatives. Compared with other potential “click” partners, the terminal olefins required for this “click-like” reaction are more readily accessible and can be elaborated with various functional moieties by, for example, simple esterification with acrylic acid. Moreover, the approach is very likely to be applicable to other polysaccharides as well. This example of the marriage of polysaccharide chemistry with organometallic chemistry not only illuminates multiple pathways to novel polysaccharide derivatives, but also creates a valuable platform for structure–activity relationship studies. By modular addition of a variety of CM partners (potentially containing a variety of functional group types) to a single terminally unsaturated polysaccharide derivative, a family of polysaccharide derivatives can be prepared that share identical Mw, DS, substitution pattern, and monosaccharide sequence, differing only in the side-chain functional groups. This synthetic strategy will enable unambiguous investigation of structure–activity relationships with regard, for example, to different appended functional groups, thereby enriching our understanding of these attractive derivatives of natural polysaccharides.
Acknowledgements
We thank the Eastman Chemical Company for their kind donation of the cellulose esters used in this work. We thank the National Science Foundation for partially funding this work through Grant DMR-1308276, the Institute for Critical Technologies and Applied Science at Virginia Tech for facility and financial support, and the Macromolecules and Interfaces Institute of Virginia Tech for educational support. We also thank Evan Margaretta, Dr Sue Mecham and Mark Flynn of Virginia Tech for their help in SEC analyses.Notes and references
- K. J. Edgar, Cellulose, 2007, 14, 49–64 CrossRef CAS.
- K. J. Edgar, C. M. Buchanan, J. S. Debenham, P. A. Rundquist, B. D. Seiler, M. C. Shelton and D. Tindall, Prog. Polym. Sci., 2001, 26, 1605–1688 CrossRef CAS.
- E. I. Rabea, M. E. T. Badawy, C. V. Stevens, G. Smagghe and W. Steurbaut, Biomacromolecules, 2003, 4, 1457–1465 CrossRef CAS PubMed.
- M. Braun and Y. Y. Sun, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3818–3827 CrossRef CAS PubMed.
- S. N. Pawar and K. J. Edgar, Biomaterials, 2012, 33, 3279–3305 CrossRef CAS PubMed.
- M. N. V. R. Kumar, R. A. A. Muzzarelli, C. Muzzarelli, H. Sashiwa and A. J. Domb, Chem. Rev., 2004, 104, 6017–6084 CrossRef PubMed.
- G. G. d'Ayala, M. Malinconico and P. Laurienzo, Molecules, 2008, 13, 2069–2106 CrossRef PubMed.
- C.-W. C. Hsieh and J. F. Kadla, Cellulose, 2012, 19, 1567–1581 CrossRef CAS.
- K. Schumann, A. Pfeifer and T. Heinze, Macromol. Symp., 2009, 280, 86–94 CrossRef CAS PubMed.
- S. C. Fox and K. J. Edgar, Biomacromolecules, 2012, 13, 992–1001 CrossRef CAS PubMed.
- C. S. P. Zarth, A. Koschella, A. Pfeifer, S. Dorn and T. Heinze, Cellulose, 2011, 18, 1315–1325 CrossRef CAS.
- H. Wondraczek, A. Pfeifer and T. Heinze, Cellulose, 2012, 19 Search PubMed.
- S. Ifuku and J. F. Kadla, Biomacromolecules, 2008, 9, 3308–3313 CrossRef CAS PubMed.
- H. Liu, G. A. Ileybare, B. P. Cherniawski, E. T. Ritchie, L. S. Taylor and K. J. Edgar, Carbohydr. Polym., 2014, 100, 116–125 CrossRef CAS PubMed.
- G. A. Ilevbare, H. Y. Liu, K. J. Edgar and L. S. Taylor, Cryst. Growth Des., 2012, 12, 3133–3143 CAS.
- J. Varshosaz, Expert Opin. Drug Deliv., 2012, 9, 509–523 CrossRef CAS PubMed.
- A. Mero and M. Campisi, Polymers, 2014, 6, 346–369 CrossRef CAS PubMed.
- J. O. Zoppe, V. Ruottinen, J. Ruotsalainen, S. Rönkkö, L.-S. Johansson, A. Hinkkanen, K. Järvinen and J. Seppälä, Biomacromolecules, 2014, 15, 1534–1542 CrossRef CAS PubMed.
- W. Z. Xu, X. Zhang and J. F. Kadla, Biomacromolecules, 2012, 13, 350–357 CrossRef CAS PubMed.
- J. Hafren and A. Cordova, Macromol. Rapid Commun., 2005, 26, 82–86 CrossRef CAS PubMed.
- A. Nakagawa, C. Ishizu, V. Sarbova, A. Koschella, T. Takano, T. Heinze and H. Kamitakahara, Biomacromolecules, 2012, 13, 2760–2768 CrossRef CAS PubMed.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- M. Pohl, N. Michaelis, F. Meister and T. Heinze, Biomacromolecules, 2009, 10, 382–389 CrossRef CAS PubMed.
- N. Pahimanolis, A.-H. Vesterinen, J. Rich and J. Seppala, Carbohydr. Polym., 2010, 82, 78–82 CrossRef CAS PubMed.
- M. Bertoldo, S. Nazzi, G. Zampano and F. Ciardelli, Carbohydr. Polym., 2011, 85, 401–407 CrossRef CAS PubMed.
- K. Negishi, Y. Mashiko, E. Yamashita, A. Otsuka and T. Hasegawa, Polymers, 2011, 3, 489–508 CrossRef CAS PubMed.
- G.-L. Zhao, J. Hafren, L. Deiana and A. Cordova, Macromol. Rapid Commun., 2010, 31, 740–744 CrossRef CAS PubMed.
- Q. Zhang, G.-Z. Li, C. R. Becer and D. M. Haddleton, Chem. Commun., 2012, 48, 8063–8065 RSC.
- A. Koschella, M. Hartlieb and T. Heinze, Carbohydr. Polym., 2011, 86, 154–161 CrossRef CAS PubMed.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- S. J. Connon and S. Blechert, Angew. Chem., Int. Ed., 2003, 42, 1900–1923 CrossRef CAS PubMed.
- A. K. Chatterjee, T. L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef CAS PubMed.
- X. Meng, J. B. Matson and K. J. Edgar, Biomacromolecules, 2014, 15, 177–187 CrossRef CAS PubMed.
- G. A. Ilevbare, H. Liu, J. Pereira, K. J. Edgar and L. S. Taylor, Mol. Pharm., 2013, 10, 3392–3403 CrossRef CAS PubMed.
- H. Liu, G. A. Ilevbare, B. P. Cherniawski, E. T. Ritchie, L. S. Taylor and K. J. Edgar, Carbohydr. Polym., 2014, 100, 116–125 CrossRef CAS PubMed.
- N. Joly, R. Granet and P. Krausz, J. Carbohydr. Chem., 2003, 22, 47 CrossRef CAS PubMed.
- N. Joly, R. Granet and P. Krausz, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 407–418 CrossRef CAS PubMed.
- L. M. de Espinosa, K. Kempe, U. S. Schubert, R. Hoogenboom and M. A. R. Meier, Macromol. Rapid Commun., 2012, 33, 2023–2028 CrossRef PubMed.
- Y. A. Lin, J. M. Chalker, N. Floyd, G. J. L. Bernardes and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 9642–9643 CrossRef CAS PubMed.
- T. L. Choi, A. K. Chatterjee and R. H. Grubbs, Angew. Chem., Int. Ed., 2001, 40, 1277–1279 CrossRef CAS.
- C. P. Woodward, N. D. Spiccia, W. R. Jackson and A. J. Robinson, Chem. Commun., 2011, 47, 779–781 RSC.
- A. M. Eissa, E. Khosravi and A. L. Cimecioglu, Carbohydr. Polym., 2012, 90, 859–869 CrossRef CAS PubMed.
- P. Tingaut, R. Hauert and T. Zimmermann, J. Mater. Chem., 2011, 21, 16066–16076 RSC.
- C. R. Reddy, E. Jithender and K. R. Prasad, J. Org. Chem., 2013, 78, 4251–4260 CrossRef CAS PubMed.
- R. F. Heck, Palladium Reagents in Organic Synthesis, Academic Press, New York, 1985 Search PubMed.
- R. Crabtree, Acc. Chem. Res., 1979, 12, 331–337 CrossRef CAS.
- J. A. Osborn, F. H. Jardine, J. F. Young and G. Wilkinson, J. Chem. Soc. A: Inorg., Phys., Theoretical, 1966, 1711–1732, 10.1039/J19660001711.
- R. F. T. Stepto, Pure Appl. Chem., 2009, 81, 351–353 Search PubMed.
- J. A. Baird and L. S. Taylor, Adv. Drug Delivery Rev., 2012, 64, 396–421 CrossRef CAS PubMed.
- C. Hiemstra, L. J. van der Aa, Z. Zhong, P. J. Dijkstra and J. Feijen, Macromolecules, 2007, 40, 1165–1173 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR, 1H NMR, and DSC data of new compounds not included in the body of the manuscript. See DOI: 10.1039/c4py01102c |
| This journal is © The Royal Society of Chemistry 2014 |