
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of telechelic vinyl/allyl functional siloxane copolymers with structural control†
F. B.
Madsen
,
I.
Javakhishvili
,
R. E.
Jensen
,
A. E.
Daugaard
,
S.
Hvilsted
and
A. L.
Skov
*
Technical University of Denmark, Department of Chemical and Biochemical Engineering, Danish Polymer Centre, Building 227, DK-2800 Kgs. Lyngby, Denmark. E-mail: al@kt.dtu.dk; Tel: +45 4525 2825
First published on 15th September 2014
Abstract
Multifunctional siloxane copolymers with terminal vinyl or allyl functional groups are synthesised through the borane-catalysed polycondensation of hydrosilanes and alkoxysilanes. Copolymers of varying molecular weights (![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) w = 13
w = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200–70300 g mol−1), spatially well-distributed functional groups and high end-group fidelity are obtained in a facile and robust synthetic scheme involving polycondensation, end-group transformation and different functionalisation reactions such as Cu(I)-mediated azide–alkyne cycloaddition. Pendant alkyl chloride, alkyl azide, bromoisobutyryl, 4-nitrobenzene and 1-ethyl-imidazolium chloride fragments with programmable spatial distributions are incorporated in the copolymer backbones. NMR and FTIR spectroscopy as well as size exclusion chromatography corroborate the efficacy and versatility of this modular approach.
200–70300 g mol−1), spatially well-distributed functional groups and high end-group fidelity are obtained in a facile and robust synthetic scheme involving polycondensation, end-group transformation and different functionalisation reactions such as Cu(I)-mediated azide–alkyne cycloaddition. Pendant alkyl chloride, alkyl azide, bromoisobutyryl, 4-nitrobenzene and 1-ethyl-imidazolium chloride fragments with programmable spatial distributions are incorporated in the copolymer backbones. NMR and FTIR spectroscopy as well as size exclusion chromatography corroborate the efficacy and versatility of this modular approach.
Introduction
The development of functional polymers is attracting increasing interest, due to a growing number of applications within nanotechnology-, sustainability-, biomedical- and energy-related fields. Functional polymers with specific control over architecture, polarity, functionality, solubility and reactivity are in especially high demand1 and controlled functionalisation is often used to change bulk and/or surface properties, in order to expand the application range of a given polymer.Polysiloxanes, i.e. polymers containing Si–O–Si repeating units, are a group of polymers with significant industrial importance, since especially polysiloxane elastomers are used widely in advanced applications such as adhesives, membranes, implants and dielectric electro active polymers.2–6
The synthesis of polysiloxanes is generally accomplished through the nucleophilic substitution of chlorosilanes with water, to form low molecular weight linear silanols and cyclic siloxanes which are further reacted into high molecular weight polymers catalysed by acids or bases.7 Ring-opening polymerisation of cyclic siloxanes is very efficient and is extensively used to prepare siloxane homopolymers and random copolymers.8 A convenient method for more well-defined copolymers is the dehydrogenative coupling of organohydrosilanes with organohydrosilanols, due to the high selectivity and easy removal of the hydrogen by-product. However, this type of reaction requires relatively high concentrations (≥0.1 mol%) of precious metal catalysts such as platinum, palladium, ruthenium or rhodium. Furthermore, only moderate molecular weight copolymers have been achieved, and undesired silanol self-condensation can occur and lead to disruption to a perfectly alternating copolymer structure.9–13
The Piers–Rubinsztajn reaction, which uses the tris(pentafluorophenyl)borane catalyst, offers an alternative route to structured siloxane copolymers. Piers et al. reported its use as a catalyst for reactions involving organohydrosilanes,14–16 while Rubinsztajn and Cella used it afterwards as a catalyst for siloxane homo- and copolymers.8,17,18 The Piers–Rubinsztajn reaction therefore refers to a tris(pentafluorophenyl)borane-catalysed reaction between a hydrosilane and an alkoxysilane, forming siloxanes with concomitant loss of an alkane – as seen in Scheme 1.19 The reaction is characterised by being exceptionally rapid, mild and efficient, since it can be carried out at ambient temperatures and uses low amounts of borane catalyst.
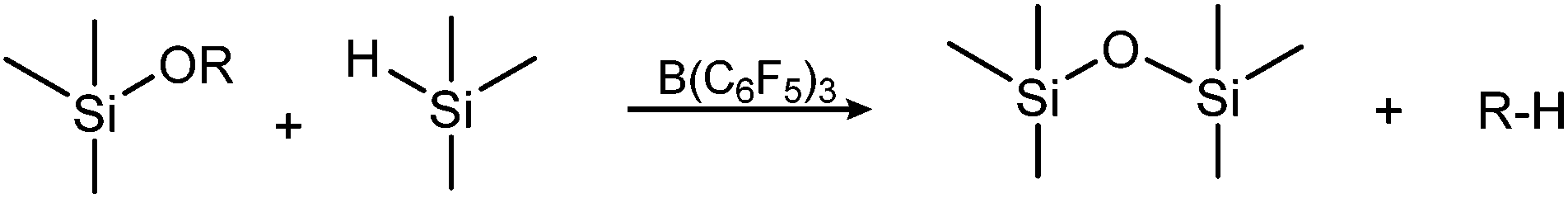 | ||
| Scheme 1 Condensation reaction between hydrosilanes and alkoxysilanes, forming an inert alkane by-product. | ||
While functional polysiloxanes with, for example, amino-, chloro- and mercapto-functional groups are commercially available (e.g. from Gelest), they do not contain functional end-groups that allow them to be used in the synthesis of silicone elastomers. It is therefore difficult to obtain from commercially available raw materials elastomers with well distributed functional groups. Silicone elastomers are frequently prepared by platinum-catalysed hydrosilylation reactions, where vinyl end-groups of polysiloxanes react with hydrosilane groups on siloxane cross-linkers. Functional elastomers can hence be prepared by using one of the components in excess and reacting vinyl- or hydride-functional molecules thereon. This approach, however, compromises network formation, meaning that inferior elastomer properties are thus a consequence.20
The aim of this work is to prepare spatially distributed and controlled functional siloxane copolymers through borane-catalysed polycondensation, which offers a rapid and efficient reaction method. The copolymers will contain vinyl or allyl end-groups, which will allow for platinum catalysed cross-linking reactions with hydrosilane cross-linkers. Furthermore, various functional groups along the backbone of the copolymers open up, for example, Williamson ether syntheses or copper-catalysed cycloaddition reactions of azides and alkynes (CuAAC) forming 1,4-disubstituted-1,2,3-triazoles, a so-called “click” reaction.21–24 Click reactions, which previously have been used to functionalise polymers in general25–28 and polysiloxanes in particular,29–32 are particularly efficient in the functionalization of polymers, as they do not form any by-products and yields are often high.
The prepared copolymers will vary greatly from the siloxane polymers originally prepared by Rubinsztajn and Cella,8,17,18 who prepared non-functional alternating aromatic and non-aromatic copolymers, in that the copolymers presented in this study will contain functional end-groups and reactive functional groups, which are spatially well-distributed through the use of dimethylsiloxane pre-polymer spacer units.
Herein we present the synthesis and characterisation of novel functional siloxane copolymers and describe how to tune the obtained molecular weights and the content of the spatially distributed functional units.
Experimental section
Materials and methods
Hydride-terminated PDMS, DMS-H11 (w ∼ 1200 g mol−1, as determined by 1H-NMR), 3-(chloropropyl)methyldimethoxysilane, vinyldimethylsilane and allyldimethylsilane were acquired from Gelest Inc. Hydride-terminated PDMS (w ∼ 580 g mol−1 as stated by supplier) was purchased from Sigma-Aldrich. All other chemicals were acquired from Sigma-Aldrich and used as received unless otherwise stated.
FTIR spectroscopy was conducted on PerkinElmer Spectrum One model 2000 Fourier Transform Infrared apparatus equipped with a universal attenuated total reflection accessory on a ZnSe/diamond composite. Spectra were recorded in the range of 4000–650 cm−1, with 4 cm−1 resolution and 16 scans. 1H- and 13C-NMR experiments were performed on a Bruker 300 MHz spectrometer in CDCl3, while size-exclusion chromatography (SEC) was performed on a Tosoh EcoSEC HLC-8320GPC instrument equipped with RI and UV detectors and SDV Linear S columns from PSS, Mainz, Germany. Samples were run in toluene at 35 °C at a rate of 1 mL min−1, and molar mass characteristics were calculated using WinGPC Unity 7.4.0 software and linear polydimethylsiloxane (PDMS) standards acquired from PSS, Mainz, Germany.
Syntheses
All reactions were carried out in a nitrogen atmosphere. Structures for 13C-NMR assignment can be found in the ESI.†![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.94 (dd, 2H, 2J = 4.2 Hz, 3J = 15 Hz, CHCH2), 6.12 (dd, 2H, 3J = 16 Hz, 3J = 21 Hz, –CHCH2). 13C-NMR (CDCl3, δC, ppm): −0.52 to 1.53 (c + d + h), 15.04 (e), 26.75 (f), 47.61 (g), 131.86 (a), 139.10 (b).
C stretch); 1410 (Si–CH2 stretch); 1260 (Si–CH3 stretch); 1010 (Si–O stretch). 1H NMR (CDCl3, δH, ppm): −0.05 to 0.09 (m, CH3–Si), 0.62 (m, –Si–CH2–CH2–), 1.50 (d, 4H, 3J = 8.1 Hz, CH2–CHCH2), 1.82 (m, –CH2–CH2–CH2–), 3.50 (t, 3J = 6.9 Hz, Cl–CH2–CH2), 4.83 (m, 4H, CHCH2), 5.77 (m, 2H, CHCH2). 13C-NMR (CDCl3, δC, ppm): −0.53 to 1.54 (d + e + i), 15.04 (f), 23.41 (c), 26.75 (g), 47.61 (h), 112.53 (a), 135.30 (b).
C stretch); 1410 (Si–CH2 stretch); 1260 (Si–CH3 stretch); 1010 (Si–O stretch). 1H NMR (CDCl3, δH, ppm): −0.05 to 0.09 (m, CH3–Si), 0.58 (m, –Si–CH2–CH2–), 1.50 (d, 4H, 3J = 8.1 Hz, CH2–CHCH2), 1.65 (m, –CH2–CH2–CH2–), 3.23 (t, 3J = 7.1 Hz, N3–CH2–CH2), 4.83 (m, 4H, CHCH2), 5.77 (m, 2H, CHCH2). 13C-NMR (CDCl3, δC, ppm): −0.55 to 1.03 (d + e + i), 14.50 (f), 22.77 (g), 23.40 (c), 54.14 (h), 112.51 (a), 135.32 (b).
O stretch); 1630 (CC stretch); 1465 (C–H bend); 1370 (C–H bend); 1410 (Si–CH2 stretch); 1260 (Si–CH3 stretch); 1155 (C–O stretch); 1010 (Si–O stretch). 1H NMR (CDCl3, δH, ppm): −0.06 to 0.13 (m, CH3–Si), 0.58 (m, –Si–CH2–CH2–), 1.54 (d, 4H, 3J = 7.8 Hz, CH2–CHCH2), 1.82 (m, –CH2–CH2–CH2–), 1.91 (s, Br–C(CH3)2), 4.33 (t, 3J = 7.2 Hz, N–CH2–CH2), 4.84 (m, 4H, CHCH2), 5.33 (s, C–CH–O), 5.76 (m, 2H, CHCH2), 7.61 (s, –CCH–N). 13C-NMR (CDCl3, δC, ppm): −0.57 to 1.03 (d + e + q), 14.24 (f), 24.33 (g), 30.62 (p), 52.97 (h), 55.53 (o), 59.22 (k), 123.47 (i), 142.25 (j), 171.46 (l) (a, b and c not visible).
C stretch); 1520 (NO asymmetric stretch); 1410 (Si–CH2 stretch); 1340 (NO symmetric stretch); 1260 (Si–CH3 stretch); 1010 (Si–O stretch). 1H NMR (CDCl3, δH, ppm): −0.45 to 0.13 (m, CH3–Si), 0.55 (m, –Si–CH2–CH2–), 1.57 (d, 4H, 3J = 8.1 Hz, CH2–CHCH2), 2.02 (m, –CH2–CH2–CH2–), 4.41 (t, 3J = 7.2 Hz, N–CH2–CH2), 4.83 (m, 4H, CHCH2), 5.77 (m, 2H, CHCH2), 7.90 (s, –CCH–N), 7.99 (m, Ar–H), 8.27 (m, NO2–Ar–H). 13C-NMR (CDCl3, δC, ppm): −0.55 to 1.03 (d + e + q), 14.23 (f), 24.39 (g), 53.09 (h), 120.68 (i), 124.29 (o), 126.05 (l), 137.04 (k), 145.45 (p), 147.26 (j) (a, b and c not visible).
C stretch); 2965 (C–H stretch); 1630 (CN, CC stretch); 1565 (CN ring stretch); 1455 (C–H bend); 1405 (Si–CH2 stretch); 1260 (Si–CH3 stretch); 1010 (Si–O stretch). 1H NMR (CDCl3, δH, ppm): 0.073–0.16 (m, CH3–Si), 0.54 (m, –Si–CH2–CH2–), 1.60 (m, CH3–CH2–N–), 1.94 (m, –CH2–CH2–CH2–), 4.35 (m, CH3–CH2–N–), 4.43 (m, –N+–CH2–CH2–CH2–), 5.73 (dd, 2H, 2J = 4.2 Hz, 3J = 20 Hz, CHCH2), 5.94 (dd, 2H, 2J = 4.1 Hz, 3J = 15 Hz, CHCH2), 6.13 (dd, 2H, 3J = 15 Hz, 3J = 20 Hz, –CHCH2), 7.42 (d, 3J = 1.5 Hz, N+–CHCH–), 7.60 (d, 3J = 1.5 Hz, N+–CHCH–), 10.9 (s, –N–CHN+–). 13C-NMR (CDCl3, δC, ppm): −0.66 to 1.09 (c + d + m), 13.60 (e), 15.53 (k), 24.24 (f), 45.10 (j), 52.07 (g), 121.34 (i), 122.15 (h), 136.54 (l) (a and b not visible).
CH2), 5.94 (dd, 2H, 2J = 4.2 Hz, 3J = 15 Hz, CHCH2), 6.12 (dd, 2H, 3J = 16 Hz, 3J = 21 Hz, –CHCH2). 13C-NMR (CDCl3, δC, ppm): −0.52 to 1.53 (c + d + h), 15.04 (e), 26.75 (f), 47.61 (g), 131.86 (a), 139.10 (b).
C stretch); 1410 (Si–CH2 stretch); 1260 (Si–CH3 stretch); 1010 (Si–O stretch). 1H NMR (CDCl3, δH, ppm): −0.05 to 0.09 (m, CH3–Si), 0.62 (m, –Si–CH2–CH2–), 1.50 (d, 4H, 3J = 8.1 Hz, CH2–CHCH2), 1.82 (m, –CH2–CH2–CH2–), 3.50 (t, 3J = 6.9 Hz, Cl–CH2–CH2), 4.83 (m, 4H, CHCH2), 5.77 (m, 2H, CHCH2). 13C-NMR (CDCl3, δC, ppm): −0.53 to 1.54 (d + e + i), 15.04 (f), 23.41 (c), 26.75 (g), 47.61 (h), 112.53 (a), 135.30 (b).
C stretch); 1410 (Si–CH2 stretch); 1260 (Si–CH3 stretch); 1010 (Si–O stretch). 1H NMR (CDCl3, δH, ppm): −0.05 to 0.09 (m, CH3–Si), 0.58 (m, –Si–CH2–CH2–), 1.50 (d, 4H, 3J = 8.1 Hz, CH2–CHCH2), 1.65 (m, –CH2–CH2–CH2–), 3.23 (t, 3J = 7.1 Hz, N3–CH2–CH2), 4.83 (m, 4H, CHCH2), 5.77 (m, 2H, CHCH2). 13C-NMR (CDCl3, δC, ppm): −0.55 to 1.03 (d + e + i), 14.50 (f), 22.77 (g), 23.40 (c), 54.14 (h), 112.51 (a), 135.32 (b).
O stretch); 1630 (CC stretch); 1465 (C–H bend); 1370 (C–H bend); 1410 (Si–CH2 stretch); 1260 (Si–CH3 stretch); 1155 (C–O stretch); 1010 (Si–O stretch). 1H NMR (CDCl3, δH, ppm): −0.06 to 0.13 (m, CH3–Si), 0.58 (m, –Si–CH2–CH2–), 1.54 (d, 4H, 3J = 7.8 Hz, CH2–CHCH2), 1.82 (m, –CH2–CH2–CH2–), 1.91 (s, Br–C(CH3)2), 4.33 (t, 3J = 7.2 Hz, N–CH2–CH2), 4.84 (m, 4H, CHCH2), 5.33 (s, C–CH–O), 5.76 (m, 2H, CHCH2), 7.61 (s, –CCH–N). 13C-NMR (CDCl3, δC, ppm): −0.57 to 1.03 (d + e + q), 14.24 (f), 24.33 (g), 30.62 (p), 52.97 (h), 55.53 (o), 59.22 (k), 123.47 (i), 142.25 (j), 171.46 (l) (a, b and c not visible).
C stretch); 1520 (NO asymmetric stretch); 1410 (Si–CH2 stretch); 1340 (NO symmetric stretch); 1260 (Si–CH3 stretch); 1010 (Si–O stretch). 1H NMR (CDCl3, δH, ppm): −0.45 to 0.13 (m, CH3–Si), 0.55 (m, –Si–CH2–CH2–), 1.57 (d, 4H, 3J = 8.1 Hz, CH2–CHCH2), 2.02 (m, –CH2–CH2–CH2–), 4.41 (t, 3J = 7.2 Hz, N–CH2–CH2), 4.83 (m, 4H, CHCH2), 5.77 (m, 2H, CHCH2), 7.90 (s, –CCH–N), 7.99 (m, Ar–H), 8.27 (m, NO2–Ar–H). 13C-NMR (CDCl3, δC, ppm): −0.55 to 1.03 (d + e + q), 14.23 (f), 24.39 (g), 53.09 (h), 120.68 (i), 124.29 (o), 126.05 (l), 137.04 (k), 145.45 (p), 147.26 (j) (a, b and c not visible).
C stretch); 2965 (C–H stretch); 1630 (CN, CC stretch); 1565 (CN ring stretch); 1455 (C–H bend); 1405 (Si–CH2 stretch); 1260 (Si–CH3 stretch); 1010 (Si–O stretch). 1H NMR (CDCl3, δH, ppm): 0.073–0.16 (m, CH3–Si), 0.54 (m, –Si–CH2–CH2–), 1.60 (m, CH3–CH2–N–), 1.94 (m, –CH2–CH2–CH2–), 4.35 (m, CH3–CH2–N–), 4.43 (m, –N+–CH2–CH2–CH2–), 5.73 (dd, 2H, 2J = 4.2 Hz, 3J = 20 Hz, CHCH2), 5.94 (dd, 2H, 2J = 4.1 Hz, 3J = 15 Hz, CHCH2), 6.13 (dd, 2H, 3J = 15 Hz, 3J = 20 Hz, –CHCH2), 7.42 (d, 3J = 1.5 Hz, N+–CHCH–), 7.60 (d, 3J = 1.5 Hz, N+–CHCH–), 10.9 (s, –N–CHN+–). 13C-NMR (CDCl3, δC, ppm): −0.66 to 1.09 (c + d + m), 13.60 (e), 15.53 (k), 24.24 (f), 45.10 (j), 52.07 (g), 121.34 (i), 122.15 (h), 136.54 (l) (a and b not visible).
Results and discussion
Siloxane copolymers with spatially well-distributed functional groups were prepared as illustrated in Scheme 2. Synthesis was accomplished through the tris(pentafluorophenyl)borane-catalysed Piers–Rubinsztajn reaction of 3-chloropropylmethyldimethoxysilane and hydride-terminated dimethylsiloxane pre-polymers to form methoxy-terminated copolymers 1. The borane-catalysed polycondensation of hydrosilanes and methoxysilanes to form 1 involves cleaving C–O and Si–H bonds while forming Si–O and C–H bonds in an exothermic reaction (ΔH ≈ −250 KJ mol−1).8 The reaction is performed at room temperature using low levels of B(C6F5)3 catalyst (<0.5 mol%). At higher catalyst concentrations (∼1–5 mol%), hydrosilylation reactions may compete with the Piers–Rubinsztajn polycondensation reaction.34,35 The reaction is almost instantaneous and is completed within a few minutes, but it was left to stir for 1 hour, in order to ensure the full conversion of reagents. Furthermore, high yields were obtained (>95%). At this point 1H-NMR and FTIR spectroscopy were used to confirm the completeness of the reaction through assessing the disappearance of the hydride-groups on the hydride-terminated pre-polymers. In 1H-NMR, resonance at δH = 4.7 ppm disappeared, which was also corroborated by FTIR, where the distinctive stretch at 2125 cm−1 was no longer present. In order to ensure at this stage that all polymers contained methoxy end-groups, significant amount of excess dimethoxydimethylsilane was added to the reaction mixture so that any remaining hydride groups would react. The excess dimethoxydimethylsilane was easily removed in vacuo.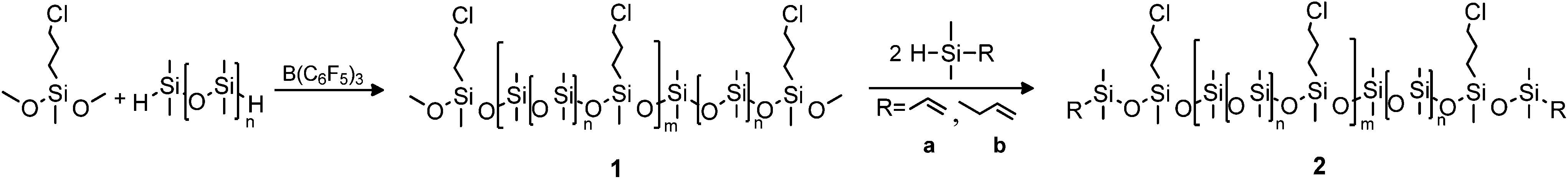 | ||
| Scheme 2 Synthetic route for telechelic vinyl/allyl siloxane copolymers via borane catalysed polycondensation. | ||
The prepared copolymers were characterised by SEC, and molar mass characteristics were calculated from linear polydimethylsiloxane standards. The results are summarised in Table 1. Two dimethylsiloxane pre-polymers of different molecular weights were used, in order to create copolymers with varying mol% of the (chloropropyl)methylsiloxane unit and with different spacer lengths between the functional groups. The molecular weights of the final copolymers were varied furthermore by changing the stoichiometry between the hydrosilane pre-polymers and the methoxysilane compound. According to standard polycondensation theory, the highest molecular weight would be obtained when using stoichiometries closest to unity.36 This is not the case in these experiments, solely due to rough estimates of exact pre-polymer molecular weights, in which case the exact stoichiometry cannot be calculated. In the case of the pre-polymer of w ∼ 1200 g mol−1 the highest molecular weight is obtained when using a stoichiometry of hydrosilane–methoxysilane of 0.95/1, with a molecular weight of w = 70300 g mol−1. For the pre-polymer of w ∼ 580 g mol−1 the highest molecular weight was obtained using the stoichiometry of hydrosilane–methoxysilane of 1/0.95, where w = 56700 g mol−1 was attained.
| Entry |
w pre-polymer [g mol−1] |
Stoichiometry hydrosilane–methoxysilane | Functional group | End-group |
w × 10−3 [g mol−1] |
w/n |
Estimated value of n | Estimated value of m |
|---|---|---|---|---|---|---|---|---|
| 1 | ∼1200 | 1/1 | Chloro | Methoxy | 50.4 | 2.22 | 15 | 34 |
| 2a-1 | ∼1200 | 1/1 | Chloro | Vinyl | 47.9 | 2.20 | 15 | 34 |
| 2a-2 | ∼1200 | 0.95/1 | Chloro | Vinyl | 70.3 | 2.03 | 15 | 49 |
| 2a-3 | ∼1200 | 0.95/1 | Chloro | Vinyl | 69.9 | 2.25 | 15 | 49 |
| 2a-4 | ∼1200 | 1/0.9 | Chloro | Vinyl | 25.9 | 3.23 | 15 | 17 |
| 2a-5 | ∼1200 | 1/0.8 | Chloro | Vinyl | 13.2 | 3.80 | 15 | 8 |
| 2a-6 | ∼580 | 1/1 | Chloro | Vinyl | 37.4 | 3.17 | 7 | 46 |
| 2a-7 | ∼580 | 0.95/1 | Chloro | Vinyl | 20.6 | 3.47 | 7 | 25 |
| 2a-8 | ∼580 | 0.975/1 | Chloro | Vinyl | 22.5 | 2.79 | 7 | 27 |
| 2b-1 | ∼580 | 0.95/1 | Chloro | Allyl | 22.1 | 3.49 | 7 | 26 |
| 2a-9 | ∼580 | 1/0.95 | Chloro | Vinyl | 56.7 | 2.84 | 7 | 71 |
| 3 | ∼580 | 0.95/1 | Azido | Allyl | 18.4 | 3.62 | 7 | 23 |
| 4 | ∼580 | 0.95/1 | 2-Bromoisobutyrate | Allyl | 27.5 | 2.43 | 7 | 26 |
| 5 | ∼580 | 0.95/1 | 4-Nitrobenzene | Allyl | 33.3 | 3.29 | 7 | 34 |
| 6 | ∼580 | 0.975/1 | 1-Ethyl-imidazolium chloride | Vinyl | 32.7 | 4.30 | 7 | 35 |
In order to test the reproducibility of the borane-catalysed polycondensations a reaction with the pre-polymer of w ∼ 1200 g mol−1 was repeated. As seen in Table 1, entries 2a-2 and 2a-3 were prepared using similar reaction conditions, and SEC provided comparable results for both experiments. This shows that the polycondensation reaction is quite robust when using similar reaction conditions.
The aim of this study was to create functional copolymers with molecular weights suitable for silicone elastomer synthesis. The best elastomer properties are usually obtained when using polymers with molecular weights of w ∼ 20000–30000 g mol−1.37 Molecular weights in this range were obtained for copolymer 2a-4 with a stoichiometric ratio of hydrosilane–methoxysilane of 1/0.9 using the pre-polymer of w ∼ 1200 g mol−1, where a molecular weight of w = 25900 g mol−1 was reached, and for 2a-7, 2a-8 and 2b-1 with stoichiometries of hydrosilane–methoxysilane of 0.95/1 and 0.975/1, respectively, using the pre-polymer of w ∼ 580 g mol−1, where molecular weights of w = 20600 g mol−1, w = 22500 g mol−1 and w = 22100 g mol−1 were achieved. Copolymers prepared with the w ∼ 1200 g mol−1 pre-polymer display polydispersity indexes (w/n) from 2.20 to 3.80, whereas copolymers prepared with the lower molecular weight pre-polymer of w ∼ 580 g mol−1 display w/n in the range of 2.84 to 3.62.
End-functionalisation of 1 with vinyl- or allyldimethylsilane produced copolymers 2a and 2b, as seen in Scheme 2. These end-groups were chosen because they allow prepared copolymers to be used in the synthesis of silicone elastomers in platinum-catalysed hydrosilylation reactions. It is also possible to create copolymers with other end-groups, as long as the groups are compatible with the borane-catalysed reaction. Such end-groups, for example, could be, but are not limited to, silanes with aliphatics such as trimethyl-groups, aromatics and halogen-containing compounds.38 The prepared telechelic vinyl/allyl copolymers were characterised by FTIR spectroscopy, 1H- and 13C-NMR spectroscopy and SEC. The end-group conversion was investigated by NMR using low molecular weight copolymers, i.e.2-a5, 2a-7 and 2b-1. In this way it was ensured that the end-groups were clearly visible in the recorded spectra. For the telechelic vinyl copolymers (2a) 1H- and 13C-NMR confirmed the reaction through the disappearance of the O–CH3 protons and a carbon atom at δH = 3.49 ppm and δC = 49.9 ppm, respectively. The presence of –CHCH2 protons as three distinctive doublets of doublets at δH = 5.74–6.12 ppm in the 1H-NMR spectrum of 2a points to the successful formation of vinyl end-groups. Furthermore, the presence of –CHCH2 groups can be detected in the 13C-NMR spectra of 2a with resonances at δC = 131.86 and 139.10 ppm. For telechelic allyl copolymers (2b) the reactions could be followed similarly by 1H- and 13C-NMR through the disappearance of the O–CH3 protons and a carbon atom at δH = 3.49 ppm and δC = 49.9 ppm, respectively. The allyl-groups were detected by 1H-NMR as resonances at δH = 1.5 ppm (–CH2–CHCH2), δH = 4.86–4.92 ppm (–CHCH2) and δH = 5.79 ppm (–CHCH2) and by 13C-NMR as resonances at δC = 26.4 ppm (–CH2–CHCH2), δC = 113.7 ppm (–CHCH2) and 133.8 ppm (–CHCH2).
Converting the methoxy end-groups to vinyl/allyl end-groups did not alter the molecular weight of the copolymers, as illustrated in the SEC traces presented in Fig. 1. This also indicates that no unintended hydrosilylation reactions between the end-groups and vinyl- or allyldimethylsilane occur.
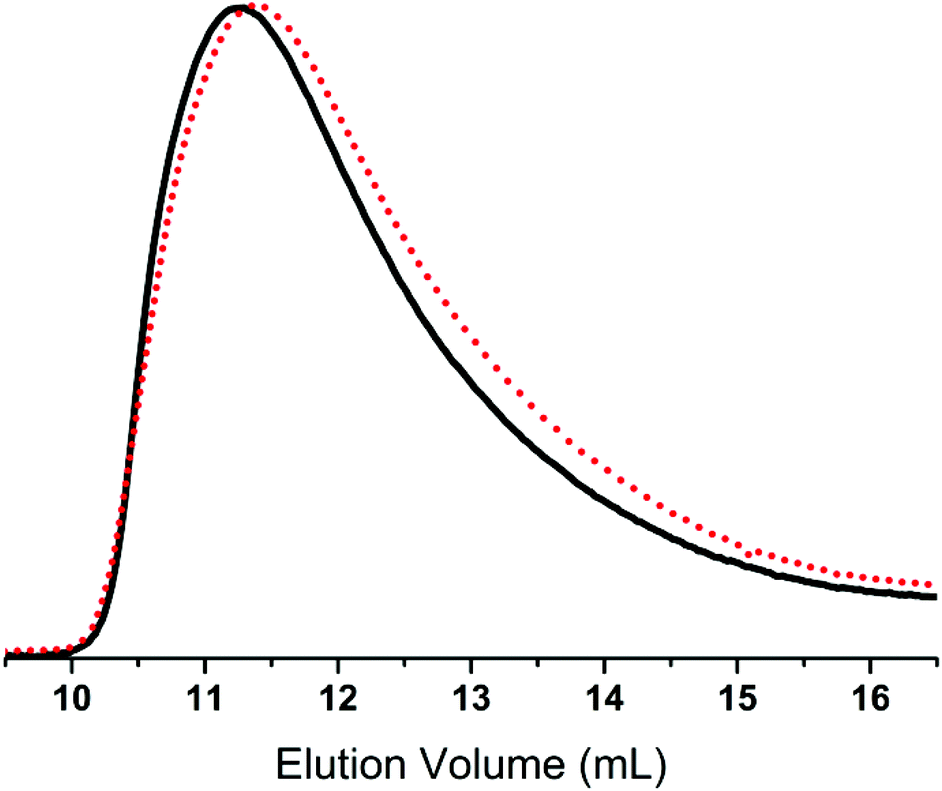 | ||
| Fig. 1 An overlay of the RI SEC traces of methoxy end-functional copolymer 1 (black, ––) and vinyl end-functional copolymer 2a-1 (red, • • •). | ||
The synthesised chloro-functional copolymers were converted to azido-functional (3) through nucleophilic substitution in THF, using tetrabutylammonium azide as a phase-transfer catalyst as seen in Scheme 3. Entry 2b-1 was selected for this purpose, as the obtained molecular weight of w = 22100 g mol−1 lies within the desired range for use in elastomer synthesis. The copolymer was successfully converted from chloro- to azido-functional, and this reaction was followed by FTIR through the appearance of the distinctive –N3 band at 2095 cm−1. Furthermore, 1H- and 13C-NMR corroborated this through a shift in the resonance signals of CH2–Cl (δH = 3.50 ppm, δC = 47.6 ppm) to CH2–N3 (δH = 3.23 ppm, δC = 54.1 ppm). The prepared azido-functional copolymer was characterised by SEC, the results for which are shown in Fig. 2. It is evident that the molecular weight after the azide-substitution reaction is lower than for the corresponding chloro-functional copolymer, which could indicate that degradation has taken place. However, upon examination of the SEC overlays seen in Fig. 2, it is clear that no degradation has taken place, as both chromatograms are broad yet monomodal, with no lower molecular weight fragments appearing at higher elution volumes.
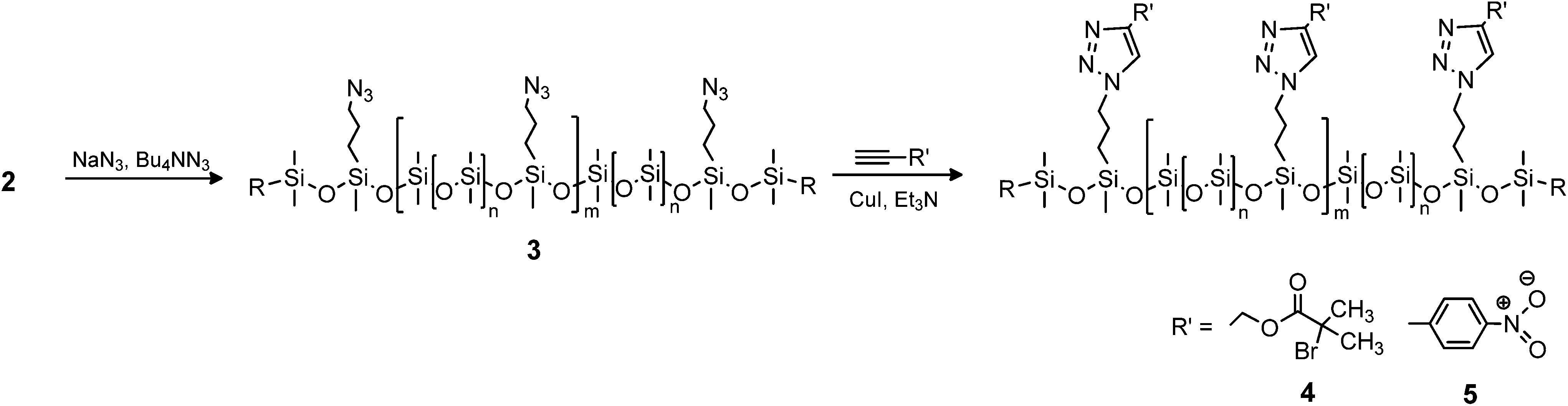 | ||
| Scheme 3 Synthetic route to azido- and CuAAC-functionalised copolymers. | ||
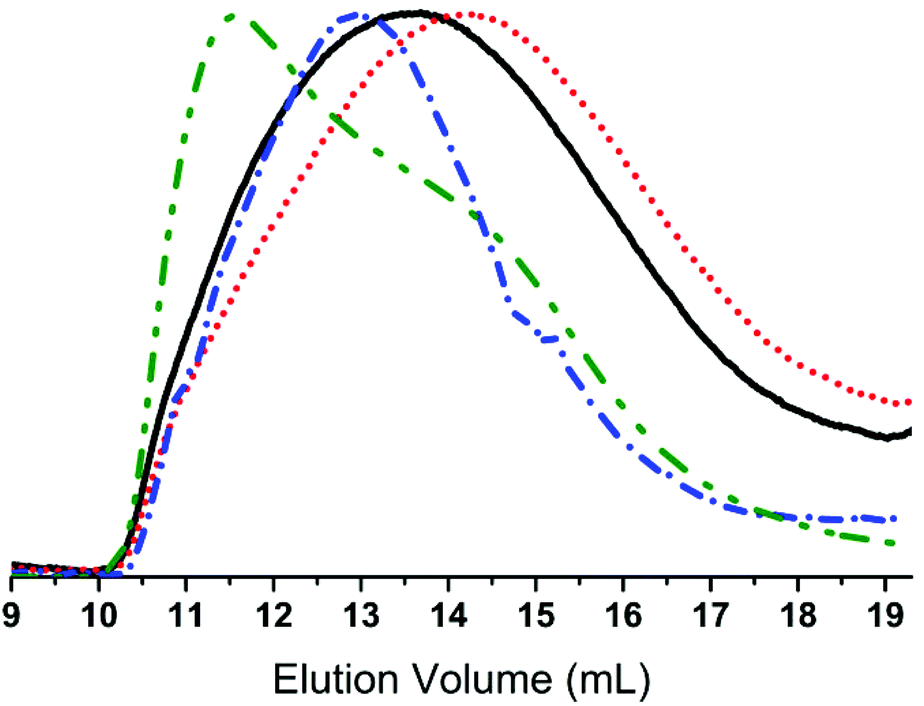 | ||
| Fig. 2 An overlay of the RI SEC traces of chloro-functional copolymer 2b-1 (black, ––), azido-functional copolymer 3 (red, • • •), 2-bromoisobutyrate-functional copolymer 4 (blue, – •) and nitrobenzene-functional copolymer 5 (green, – • •). | ||
The lower obtained w for the azido-functional copolymer must be ascribed to tailing and a shift in the baseline due to enhanced interactions of the latter with the SEC columns.
Functional dimethylsiloxane copolymers were created through a reaction with alkyne-functional molecules using CuAAC. Two different alkyne-containing molecules were chosen, in order to illustrate the versatility of the reaction. An aliphatic ATRP initiator and an alkyne-functional nitrobenzene were utilised as seen in Scheme 3. The ATRP initiator can be used to create polysiloxanes with different polymer side chains, whereas the aromatic compound 4-nitrobenzene can be used to increase dielectric permittivity in dielectric silicone elastomers.32 Click-functionalised copolymers were prepared under similar reaction conditions by means of a CuI-Et3N catalytic system. The reaction products were characterised by FTIR spectroscopy, 1H- and 13C-NMR spectroscopy and SEC. FTIR was used to confirm the completion of the click reaction through the disappearances of the alkyne and azide bands at ∼3300 cm−1 and ∼2095 cm−1, respectively. For the reaction with the ATRP initiator (4), which produced a green-brown polymer, a distinct ester CO band at 1740 cm−1 confirmed the presence of the 2-bromoisobutyrate group. In 5 the presence of 4-nitrobenzene was indicated by the red colour of the obtained polymer confirmed by bands at ∼1605 cm−1 for the aromatic CC bonds and at ∼1520 cm−1 and ∼1340 cm−1 for the NO bonds. FTIR spectra of 3, 4 and 5 can be seen in Fig. 3. The formation of the click products was furthermore confirmed by the presence of the triazole protons in 1H-NMR, which appear at δH = 7.61 ppm and δH = 7.90 ppm for 4 and 5, respectively. An assigned representative 1H-NMR spectrum of 4 can be found in the ESI.†
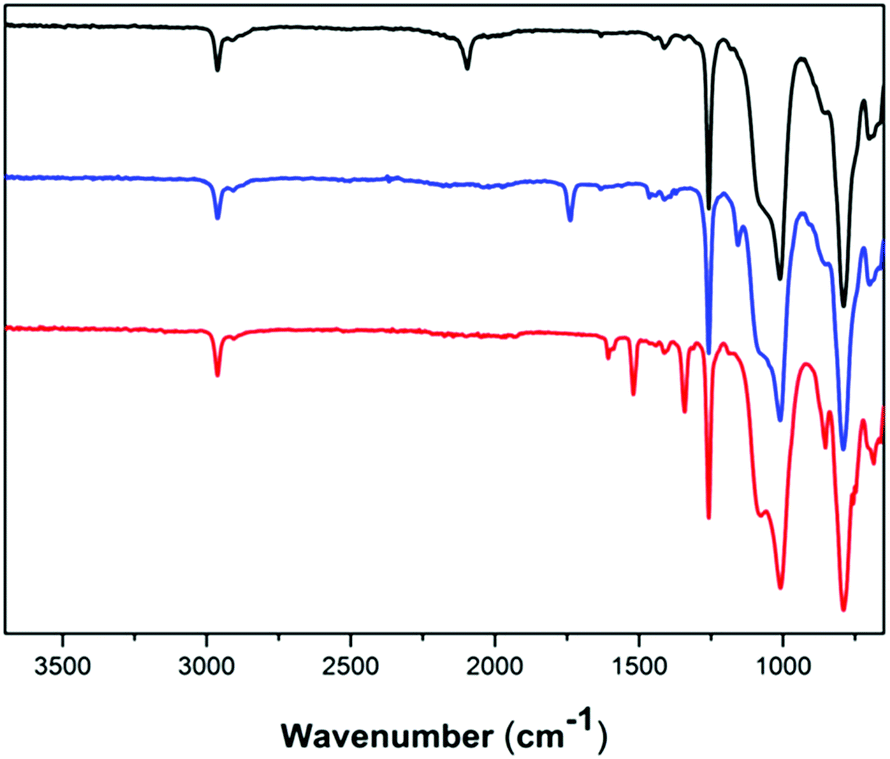 | ||
| Fig. 3 FTIR spectra of azido-functional copolymer 3 (black, top), 2-bromoisobutyrate-functional copolymer 4 (blue, middle curve) and 4-nitrobenzene-functional copolymer 5 (red, bottom curve). | ||
The SEC results for click-products 4 and 5 can be seen in Table 1 and Fig. 2. For both click-products higher molecular weights are obtained, and a slight shift towards higher molecular weight regions can be seen in the SEC traces, pointing to the successful attachment of functional groups onto the copolymers. Furthermore, SEC showed that 5 had a strong UV signal, unlike the starting materials 2b-1 and 3, which proves the attachment of the 4-nitrobenzene chromophore.
Ionic polymers are a rapidly expanding class of materials with interesting and promising properties39–41 which have recently been extended to silicone materials.42 Thus, as an example of another possible post polymerisation modification, the prepared chloro-functional copolymer 2a-8 was used in a substitution reaction with 1-ethylimidazole to form the ionic copolymer seen in Scheme 4. This reaction was followed by 1H-NMR through a shift in the resonance of CH2–Cl (δH = 3.50 ppm) to CH2–N+ (δH = 4.43 ppm). After the reaction, the orange-brown copolymer was less soluble in toluene, thereby demonstrating the increased ionic/polar nature of the copolymer 6. The SEC results for the chloro-functional copolymer 2a-8 and imidazolium-functional copolymer 6 can be seen in Table 1 and Fig. 4. As observed for the click-products, a slight shift towards the higher molecular region is seen in the SEC traces for copolymer 6, which indicates the successful attachment of the functional group.
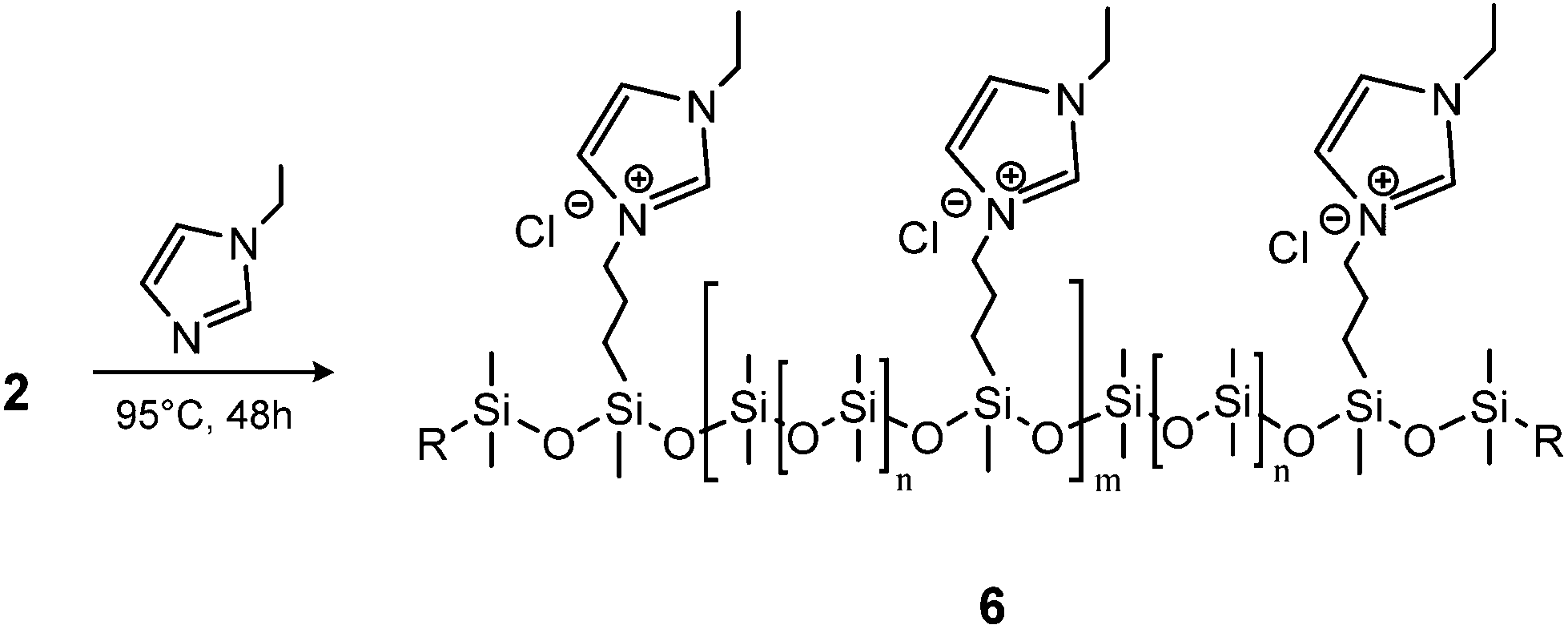 | ||
| Scheme 4 Synthetic route to imidazole-functional copolymer. | ||
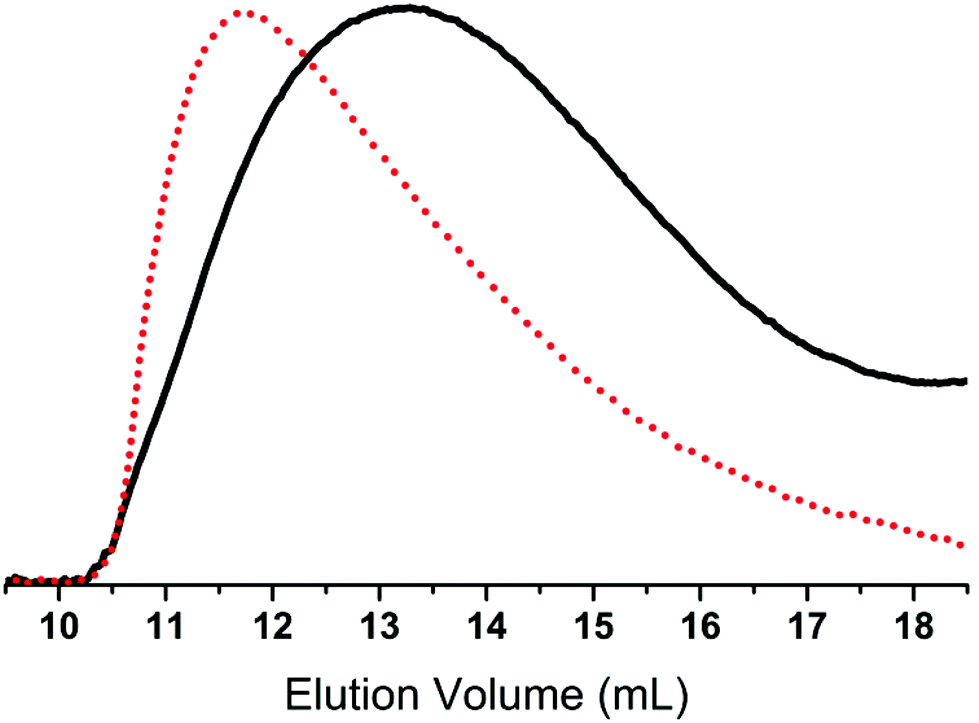 | ||
| Fig. 4 An overlay of the RI SEC traces of chloro-functional copolymer 2a-8 (black, ––) and imidazole-functional copolymer 6 (red, • • •). | ||
The prepared chloro- and azido-functional siloxane copolymers have great potential in the preparation of functional silicone elastomers where the properties of the elastomers can be altered and improved according to the given application and the type of group attached to the copolymer. The specific functional groups could include many different types32 and are not limited to those used in this study. We are currently investigating the use of synthesised functional copolymers for the preparation of functional elastomers, thus elucidating their properties.
Conclusions
In conclusion, we have successfully synthesised telechelic vinyl/allyl chloro-, azido- and CuAAC-functionalised siloxane copolymers and ensured spatial control over the functional groups. The polycondensation reaction proved to be rapid and very robust, with high yields obtained. The method therefore offers a fast and reliable technique for the synthesis of structured functional polysiloxanes. The content of functional groups in the copolymers could be varied by changing the molecular weight of the hydride-terminated dimethylsiloxane pre-polymers used in the polycondensation reactions. Furthermore, the molecular weights of the copolymers could be tuned by varying the stoichiometry of the hydrosilane and methoxysilane starting materials. Polymers with any conceivable concentration of functionality and molecular weight can thereby be obtained.Acknowledgements
The authors wish to acknowledge InnovationsFonden for its financial support.Notes and references
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- J. de Jong, R. G. H. Lammertink and M. Wessling, Lab Chip, 2006, 6, 1125–1139 RSC.
- M. A. Brook, Biomaterials, 2006, 27, 3274–3286 CrossRef CAS PubMed.
- C. Löwe, X. Zhang and G. Kovacs, Adv. Eng. Mater., 2005, 7, 361–367 CrossRef PubMed.
- P. Brochu and Q. Pei, Macromol. Rapid Commun., 2010, 31, 10–36 CrossRef CAS PubMed.
- F. B. Madsen, A. E. Daugaard, S. Hvilsted, M. Y. Benslimane and A. L. Skov, Smart Mater. Struct., 2013, 22, 104002 CrossRef.
- M. A. Brook, Silicon in organic, organometallic, and polymer chemistry, John Wiley & Sons, New York, 2000 Search PubMed.
- S. Rubinsztajn and J. A. Cella, Macromolecules, 2005, 38, 1061–1063 CrossRef CAS.
- Y. Li and Y. Kawakami, Macromolecules, 1999, 32, 6871–6873 CrossRef CAS.
- Y. Li and Y. Kawakami, Macromolecules, 1999, 32, 8768–8773 CrossRef CAS.
- C. L. Homrighausen and T. M. Keller, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1334–1341 CrossRef CAS PubMed.
- Y. Li, M. Seino and Y. Kawakami, Macromolecules, 2000, 33, 1–4 CrossRef CAS.
- R. Zhang, J. E. Mark and A. R. Pinhas, Macromolecules, 2000, 33, 3508–3510 CrossRef CAS.
- D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS.
- J. M. Blackwell, K. L. Foster, V. H. Beck and W. E. Piers, J. Org. Chem., 1999, 64, 4887–4892 CrossRef CAS PubMed.
- D. Parks, J. Blackwell and W. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS PubMed.
- S. Rubinsztajn and J. A. Cella, Polym. Prepr., 2004, 45(1), 635–636 CAS.
- J. Cella and S. Rubinsztajn, Macromolecules, 2008, 41, 6965–6971 CrossRef CAS.
- M. A. Brook, J. B. Grande and F. Ganachaud, Adv. Polym. Sci., 2011, 235, 161–183 CrossRef.
- A. L. Larsen, K. Hansen, P. Sommer-Larsen, O. Hassager, A. Bach, S. Ndoni and M. Jørgensen, Macromolecules, 2003, 36, 10063–10070 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- P. Wu, A. K. Feldman, A. K. Nugent, C. J. Hawker, A. Scheel, B. Voit, J. Pyun, J. M. J. Fréchet, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2004, 43, 3928–3932 CrossRef CAS PubMed.
- M. Meldal, Macromol. Rapid Commun., 2008, 29, 1016–1051 CrossRef CAS PubMed.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS PubMed.
- A. D. Thomsen, E. Malmström and S. Hvilsted, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6360–6377 CrossRef CAS PubMed.
- A. E. Daugaard, S. Hvilsted, T. S. Hansen and N. B. Larsen, Macromolecules, 2008, 41, 4321–4327 CrossRef.
- S. Hvilsted, Polym. Int., 2012, 61, 485–494 CrossRef CAS PubMed.
- A. E. Daugaard and S. Hvilsted, Macromol. Rapid Commun., 2008, 29, 1119–1125 CrossRef CAS PubMed.
- F. Gonzaga, G. Yu and M. A. Brook, Macromolecules, 2009, 42, 9220–9224 CrossRef CAS.
- F. Gonzaga, G. Yu and M. A. Brook, Chem. Commun., 2009, 1730–1732 RSC.
- U. Schmidt, P. C. Zehetmaier and B. Rieger, Macromol. Rapid Commun., 2010, 31, 545–548 CrossRef CAS PubMed.
- F. B. Madsen, I. Dimitrov, A. E. Daugaard, S. Hvilsted and A. L. Skov, Polym. Chem., 2013, 4, 1700–1707 RSC.
- T. Rambarran, F. Gonzaga and M. A. Brook, Macromolecules, 2012, 45, 2276–2285 CrossRef CAS.
- M. Rubin, T. Schwier and V. Gevorgyan, J. Org. Chem., 2002, 67, 1936–1940 CrossRef CAS PubMed.
- C. Xunjun, C. Yingde, Y. Guoqiang and L. Liewen, J. Appl. Polym. Sci., 2007, 106, 1007–1013 CrossRef PubMed.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, Ithaca, New York, 1953, ch. 3, 8 and 9 Search PubMed.
- A. L. Larsen, P. Sommer-Larsen and O. Hassager, e-Polymers, 2004, 50, 1–18 Search PubMed.
- J. B. Grande, D. B. Thompson, F. Gonzaga and M. A. Brook, Chem. Commun., 2010, 46, 4988–4990 RSC.
- M. D. Green, D. S. Cruz, Y. Ye, J. M. Layman, Y. A. Elabd, K. I. Winey and T. E. Long, Macromol. Chem. Phys., 2011, 212, 2522–2528 CrossRef CAS PubMed.
- P. Dimitrov-Raytchev, S. Beghdadi, A. Serghei and E. Drockenmuller, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 34–38 CrossRef CAS PubMed.
- S. Jana, V. A. Vasantha, L. P. Stubbs, A. Parthiban and J. G. Vancso, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3260–3273 CrossRef CAS PubMed.
- L. Yu, L. B. Gonzalez, S. Hvilsted and A. L. Skov, Proc. SPIE-Int. Soc. Opt. Eng., 2014, 9056, 90560C CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Numbered structures for 13C-NMR assignment and 1H-NMR spectrum of 4. See DOI: 10.1039/c4py00919c |
| This journal is © The Royal Society of Chemistry 2014 |