DOI:
10.1039/C4PY00763H
(Paper)
Polym. Chem., 2014,
5, 6003-6011
Thermal and living anionic polymerization of 4-vinylbenzyl piperidine†
Received
31st May 2014
, Accepted 1st July 2014
First published on 2nd July 2014
Abstract
Elevated temperatures that are often required for controlled radical polymerization processes lead to the thermal autopolymerization of 4-vinylbenzyl piperidine. In situ FTIR spectroscopy monitored 4-vinylbenzyl piperidine autopolymerization, and pseudo-first-order thermal polymerization kinetics provided observed rate constants (kobs). Arrhenius analysis determined the thermal activation energy (Ea) for 4-vinylbenzyl piperidine, revealing an activation energy requirement 80 kJ mol−1 less than styrene due to the presence of the piperidine ring. The similarities in chemical structure of styrene and 4-vinylbenzyl piperidine suggested a thermally initiated polymerization according to the Mayo mechanism; however, the piperidine substituent enabled a proposed cationic polymerization to enhance overall polymerization rates. In the absence of thermal polymerization, living anionic polymerization of 4-vinylbenzyl piperidine provided a viable strategy for achieving piperidine-containing polymers with predictable molecular weights and narrow polydispersities. This study also reports piperidine-containing polymeric precursors for subsequent alkylation to form novel piperidinium ionomers and polyelectrolytes.
Introduction
Ionomers and polyelectrolytes are functional macromolecules that incorporate cationic or anionic sites either pendant to or within the polymer backbone.1–3 These compositions enable beneficial properties such as ionic conductivity, thermal and chemical stability, and anion exchange capability. Ion-containing polymers are versatile materials that continuously enable emerging technologies such as water purification,4,5 gas separation,6,7 gene delivery,8,9 biosensors,10 fuel cells,11,12 and electromechanical actuators.13 Chemical and thermal stability remain a concern for charged polymers in these diverse technologies. Numerous studies widely address ammonium-based polyelectrolytes containing imidazolium,6,14 triazolium,15 and pyridinium16 rings given high alkylation efficiency, thermal stability, and limited base stability. Polyelectrolytes functionalized with cyclic amines, such as pyrolidine and piperidine, offer comparable performance.17 In fact, earlier reports revealed that piperidinium cations are electrochemically more stable than imidazolium analogues and exhibit higher lithium ion transference.18
Living anionic polymerization offers an efficient route for producing well-defined ammonium-based polymeric architectures with tunable molecular weights, narrow molecular weight distributions, and stereochemical control at modest polymerization temperatures.19–23 Controlled radical polymerization also advantageously provides several synthetic strategies for achieving controlled linear chain propagation, and these methods encompass stable free radical (SFRP),24–27 atom transfer (ATRP),28–30 and reversible addition-fragmentation transfer (RAFT) polymerizations.14,31–33 Synthetic protocols for controlled radical polymerization processes often require elevated temperatures, high monomer concentrations, and long reaction times. These conditions are also suitable for thermal autopolymerization of styrenic monomers, which impede polymerization control. Traditional living anionic strategies provide an alternative approach to promote linear chain propagation in polar or nonpolar solvents at ambient temperatures in short reaction times.34–37 Moreover, subsequent block copolymer formation with 1,3-dienes in nonpolar solvents favors the desirable 1,4-configuration and the associated low glass transition temperature for the cis-isomer.
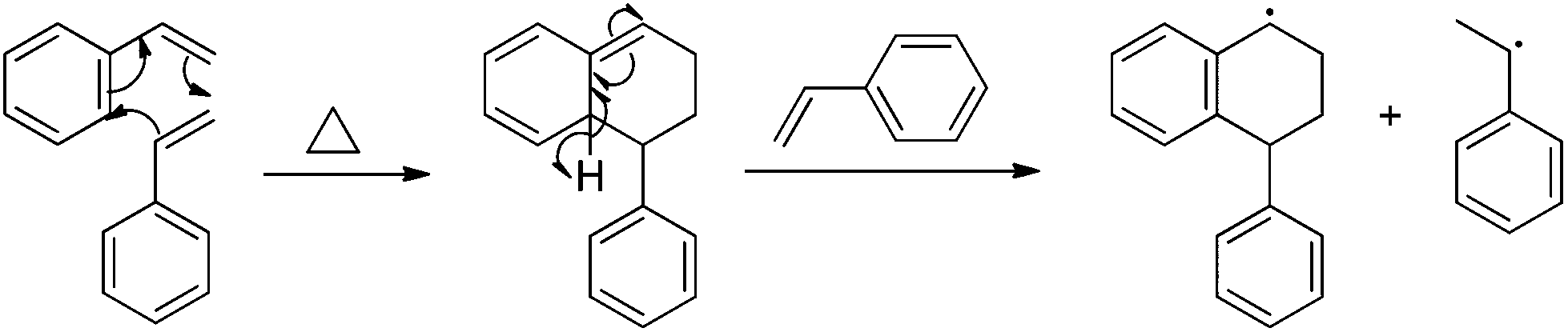
Numerous studies describe styrene autopolymerization and its implication in controlled radical polymerization processes.38,39 Self-initiation often follows the notable Mayo thermal mechanism (Scheme 1),39,40 where styrenic monomers initially react to form a [4 + 2] Diels–Alder adduct. The adduct reacts with available monomer and produces two free radicals for polymerization. Adjacent aromatic resonance stabilizes both radical intermediates. Styrenic derivatives with additional polarity and resonance stabilization augment radical and thermal polymerization. In particular, the addition of electron-withdrawing groups to styrenic monomers increases SFRP rates,41 and activated 2,2,6,6-tetramethylpiperidinyl-1-oxy (TEMPO) enables SFRP for styrene and styrenic derivatives.24 Further work also highlights fused ring effects on styrenic monomers. In particular, 2-vinylnapthalene has an observed thermal polymerization rate constant that exceeds styrene by one order-of-magnitude.25,38 Solvent and organic additives also exhibit strong effects on styrenic thermal polymerization. Strong acids such as camphor sulfonic acid (CSA) and hydroiodic acid protonate the Diels–Alder adduct and inhibit thermal polymerization.
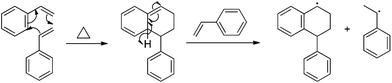 |
| | Scheme 1 Mayo thermal-initiation mechanism for styrene. | |
4-Vinylbenzyl piperidine (4-VBP) is an appealing styrenic derivative that possesses a piperidinyl ring in the para position, an ideal site for alkylation and production of novel piperidinium polymers for potential ionomer and polyelectrolyte applications. Unfortunately, limited investigations address the effects of piperidine substituents on deleterious thermal autopolymerization. Herein, we use in situ Fourier transform infrared (FTIR) spectroscopy to monitor 4-VBP and styrene autopolymerization, reporting observed rate constants for both monomers from pseudo-first-order kinetic treatment of the thermal polymerization data and determination of the corresponding activation energies using an Arrhenius analysis. Given the similarities in chemical structure between styrene and 4-VBP, we propose that thermally initiated polymerization follows the Mayo mechanism (Scheme 1) and the amino substituent induces a parallel cationic polymerization for enhanced autopolymerization rates. As a means to avoid higher polymerization temperatures but retain a controlled process, we describe for the first time the living anionic polymerization of 4-VBP. We employ in situ FTIR spectroscopy to monitor the anionic polymerization and invoke pseudo-first-order kinetics to determine observed rate constants for propagation. Living anionic polymerization is an efficient method for achieving controlled polymerization of novel piperidine-containing macromolecules with well-defined structures and low degrees of compositional heterogeneity.42 Subsequent alkylation with bromoalkanes additionally produces novel, well-defined piperidinium-containing polymers that exhibit high thermal stability. As ion-containing polymers emerge for many electro-active membrane applications, designing novel charged polymers with controlled architectures will further expand their commercial potential.
Experimental section
Materials
Bis(trifluoromethane)sulfonimide lithium salt (99%), bromoethane (98%), pyridine (99%), 4-vinylbenzyl chloride (≥90%), 4-(bromomethyl)-benzyltriphenylphosphonium bromide (99%), styrene (99%), and 1.4 M sec-butyllithium solution in cyclohexane were purchased from Sigma Aldrich and used as received unless otherwise noted. Styrene, containing 10–15 ppm of t-butyl catechol, was distilled from calcium hydride and dibutyl magnesium. N-benzyl piperidine (99.9%) was purchased from Acros and used as received. 4-Vinylbenzyl piperidine43 was synthesized according to the previous literature, and experimental characterization is detailed in the ESI.†
Instrumentation
In situ FTIR analysis employed a Mettler Toledo ReactIR 45M attenuated total reflectance reaction apparatus equipped with a light conduit and DiComp (diamond composite) insertion probe. 1H NMR spectroscopy was performed on a Varian Unity 400 at 400 MHz in deuterated chloroform. Thermogravimetric analysis (TGA) was performed using a TA Instruments TGA 2950 at a 10 °C min−1 heating ramp. Differential scanning calorimetry (DSC) was performed using a TA Instruments Q1000. Scans were obtained under N2 with heating at 10 °C min−1 and cooling at 100 °C min−1; Tg's were recorded on the second heating cycle. Size-exclusion chromatography (SEC) was used to determine the molecular weights of piperidinyl-containing polymers at 40 °C in THF at 1 mL min−1. THF SEC was performed on a Waters SEC equipped with two Waters Styragel HR5E (THF) columns, a Waters 717 plus autosampler, a Wyatt MiniDAWN, and a Waters 2414 differential refractive index detector. An Optilab T-rEX refractometer (λ = 658) was used to measure dn/dc values offline for determination of absolute weight-average molecular weights.
In situ FTIR monitoring of thermal polymerization
In situ FTIR spectroscopy monitored the thermal polymerization of styrene and 4-vinylbenzyl piperidine at 80 °C, 100 °C, and 120 °C. A typical polymerization was performed as follows: 4-vinylbenzyl piperidine (10.0 g) was added to a two-necked, 25 mL, round-bottomed flask with a small magnetic stir bar. One neck was sealed with a rubber septum, and a DiComp probe was inserted into the second neck and tightly sealed. The probe tip was submerged below the monomer surface and the ReactIR FTIR spectrometer was programmed to collect a spectrum every 1 min for 24 h. The flask was purged with nitrogen for 15 min and placed in an oil bath heated at 120 °C. After 24 h, the thermally polymerized monomer was readily dissolved with THF or chloroform and precipitated into methanol.
Thermal polymerization of styrene with N-benzyl piperidine
The following protocol describes a typical thermal solution polymerization of styrene with N-benzyl piperidine. Styrene (3.5 g, 33.6 mmol, 3.85 mL) and N-benzyl piperidine (5.89 g, 33.6 mmol, 6.20 mL) were combined in a 25 mL, two-necked, round-bottomed flask with a magnetic stir bar. One neck was sealed with a rubber septum and the other was sealed with the DiComp probe. The probe tip was inserted below the surface of the reaction mixture and the ReactIR spectrometer was programmed to collect a spectrum every 1 min for 24 h. The mixture was sparged with nitrogen for 15 min and submerged in a 120 °C oil bath. After the 24 h FTIR experiment, the reaction mixture was diluted with chloroform and precipitated into methanol. The procedure was repeated, varying the styrene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BP molar ratio from 1:1 to 1:0.1.
:BP molar ratio from 1:1 to 1:0.1.
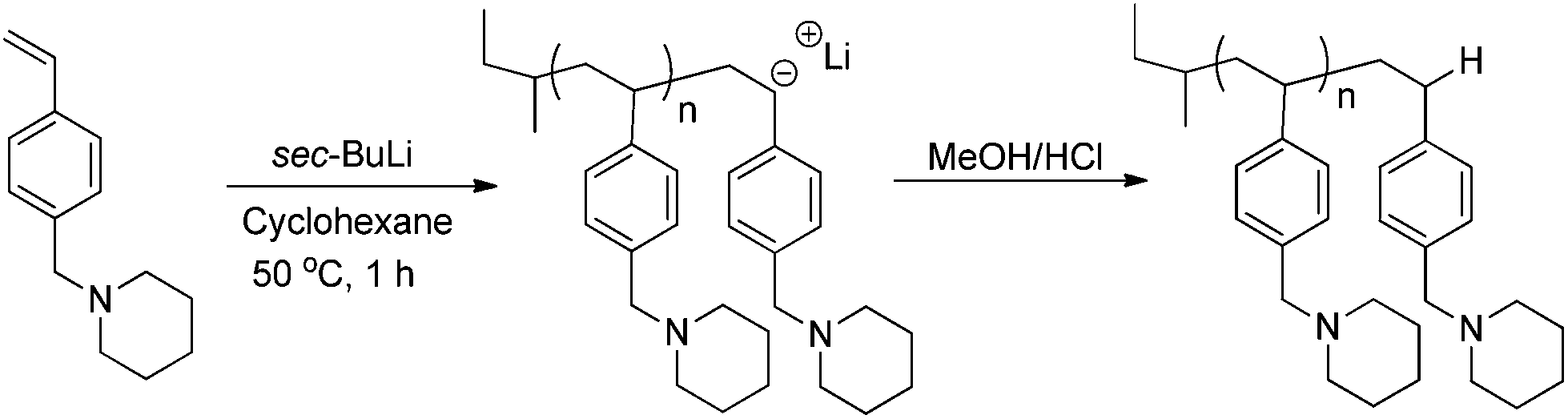
Anionic polymerization of 4-vinylbenzyl piperidine
To a 100 mL flame dried, nitrogen purged, and sealed round-bottomed flask containing a stir bar, 2.00 mL (1.98 g) 4-vinylbenzyl piperidine and 60 mL of cyclohexane were added. The reaction flask was heated to 23 °C and sec-butyllithium (0.35 mL, 0.5 mmol) was injected to the reaction solution with a syringe. The living poly(4-VBP) anions in cyclohexane exhibited a deep orange color that appeared homogeneous at 23 °C. The polymerization was terminated after 1 h with degassed methanol (1.0 mL), and the resulting polymer solution was precipitated into methanol and dried at 23 °C under reduced pressure (0.5 mmHg) for 48 h to obtain a white powder (85–90% yield). 1H NMR (400 MHz, CDCl3, 25 °C, δ): 0.53 (–CH3– sec-butyl protons) 1.5 (–CH2– piperidine ring protons, –CH2– polymer backbone, –CH– polymer backbone), 2.4 (–CH2–N piperidine protons), 3.5 (–CH2–N methylene protons), 6.2–7.3 (aromatic protons).
In situ FTIR monitoring of the anionic polymerization of 4-vinylbenzyl piperidine
2.0 mL 4-vinylbenzyl piperidine (1.98 g) and 8 mL of dry cyclohexane were added to a two-necked, 25 mL, flame-dried, round-bottomed flask with a magnetic stir bar. One neck was sealed with a rubber septum, and the DiComp probe was inserted into the second neck and sealed. The probe tip was submerged below the monomer surface, and the ReactIR spectrometer was programmed to collect a spectrum every 1 min for 5 h. The flask was purged with nitrogen for 15 min and placed in an oil bath heated at 50 °C. sec-Butyllithium (0.02 mL) initiated growth of a 10000 g mol−1 polymer. After 5 h with FTIR analysis, the product was precipitated into methanol.
Synthesis of poly(4-vinylbenzyl piperidine-b-styrene)
A series of poly(4-VBP-b-S) block copolymers with varying molecular weights were synthesized in an anionic polymerization reactor. A typical synthesis was performed as follows: A 600 mL-capacity anionic polymerization reactor was filled with cyclohexane (250 mL) and maintained under constant nitrogen pressure (40 psi) at 23 °C. The glass anionic reactor consisted of a 600 mL glass bowl, a stainless steel top plate, and stainless steel magnetically coupled mechanical stirrer. In addition, a heat-exchanged coil, a thermocouple, a septum sealed port, various stainless steel transfer lines to introduce solvent (cyclohexane), and inlet/vent for purified nitrogen are directly connected to the anionic reactor. 16 mL 4-vinylbenzyl piperidine (15.84 g) was syringed into the reactor and 2.26 mL sec-butyllithium (3.16 mmol) was then added to the solution to initiate growth of a 5000 g mol−1 polymer. The first reaction was allowed to proceed for 1 h and the second monomer, 18 mL styrene (19.80 g) was sequentially added to the reaction mixture. The polymerization was terminated after 1 h with degassed methanol (1.0 mL), and the resulting block copolymer was precipitated into methanol and dried at 23 °C under reduced pressure (0.5 mmHg) for 24 h to obtain a white powder (80–90% isolated yield).
Alkylation of piperidine-containing copolymers
The following protocol describes a typical alkylation on 4-VBP-containing polymers. 2.00 g poly(4-VBP) and 2.00 molar ratio of 4-(bromomethyl)-benzyltriphenylphosphonium bromide was dissolved in 25 mL 20/80 (v/v%) chloroform–dimethyl sulfoxide mixture. The reaction components were allowed to reflux at 85 °C for 24 h. The resulting piperidinium polymer precipitated from the reaction solution. The final product was isolated and re-precipitated into diethyl ether. The final polymer was allowed to dry at 30 °C under reduced pressure (0.5 mmHg) for 48 h. 1H NMR of a reaction aliquot confirmed quantitative alkylation and the final structure (see ESI†). 1H NMR (400 MHz, CD3OD, 25 °C, δ): 0.53 (–CH3– sec-butyl protons) 1.5 (–CH2– piperidine ring protons, –CH2– polymer backbone, –CH– polymer backbone), 2.4 (–CH2–N piperidine protons), 3.5 (–CH2–N methylene protons), 4.7 (–CH2–N methylene protons), 5.2 (–CH2–P methylene protons), 6.5–8.0 (–CH– aromatic protons).
Anion-exchange reaction of piperidinium homopolymers
The following protocol describes a typical anion exchange of bromide with bis(trifluoromethane)sulfonimide lithium salt (LiTf2N) in piperidinium homopolymers. 6.00 g (excess) of LiTf2N was dissolved in 200 mL of dichloromethane, and 1 g of piperidinium homopolymer was dissolved in 10 mL of dichloromethane. The dissolved polymer was added drop-wise to the LiTf2N solution and was allowed to stir overnight at room temperature. The final solution was filtered and washed with 100 mL deionized water 3 times. The organic phases were collected and titrated with silver nitrate. Silver bromide precipitate was not observed, indicating complete anion exchange. Dichloromethane was roto-evaporated, and the product was dried at 30 °C under reduced pressure (0.5 mmHg) for 48 h.
Results and discussion
Thermal autopolymerization of 4-vinylbenzyl piperidine
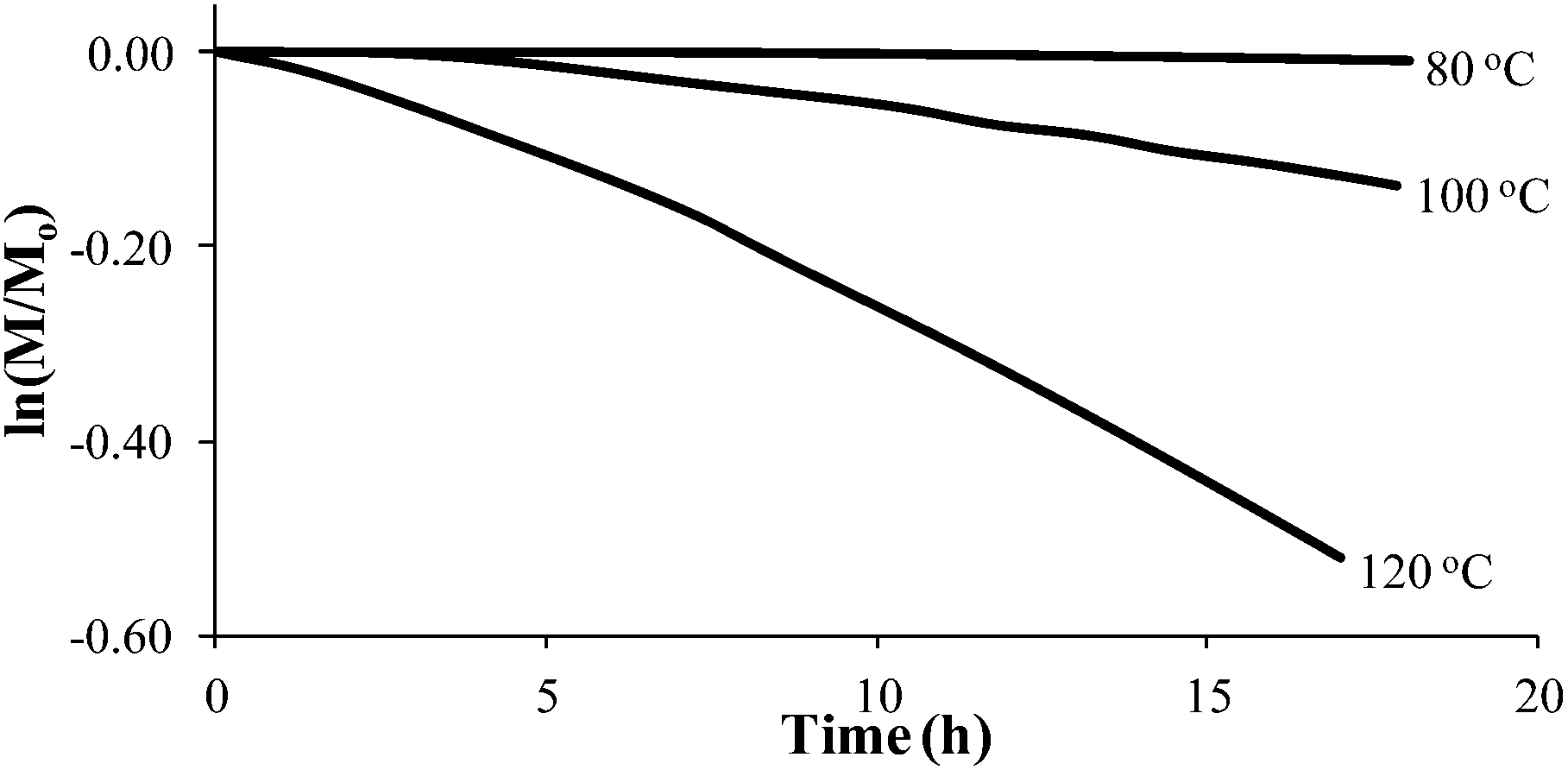
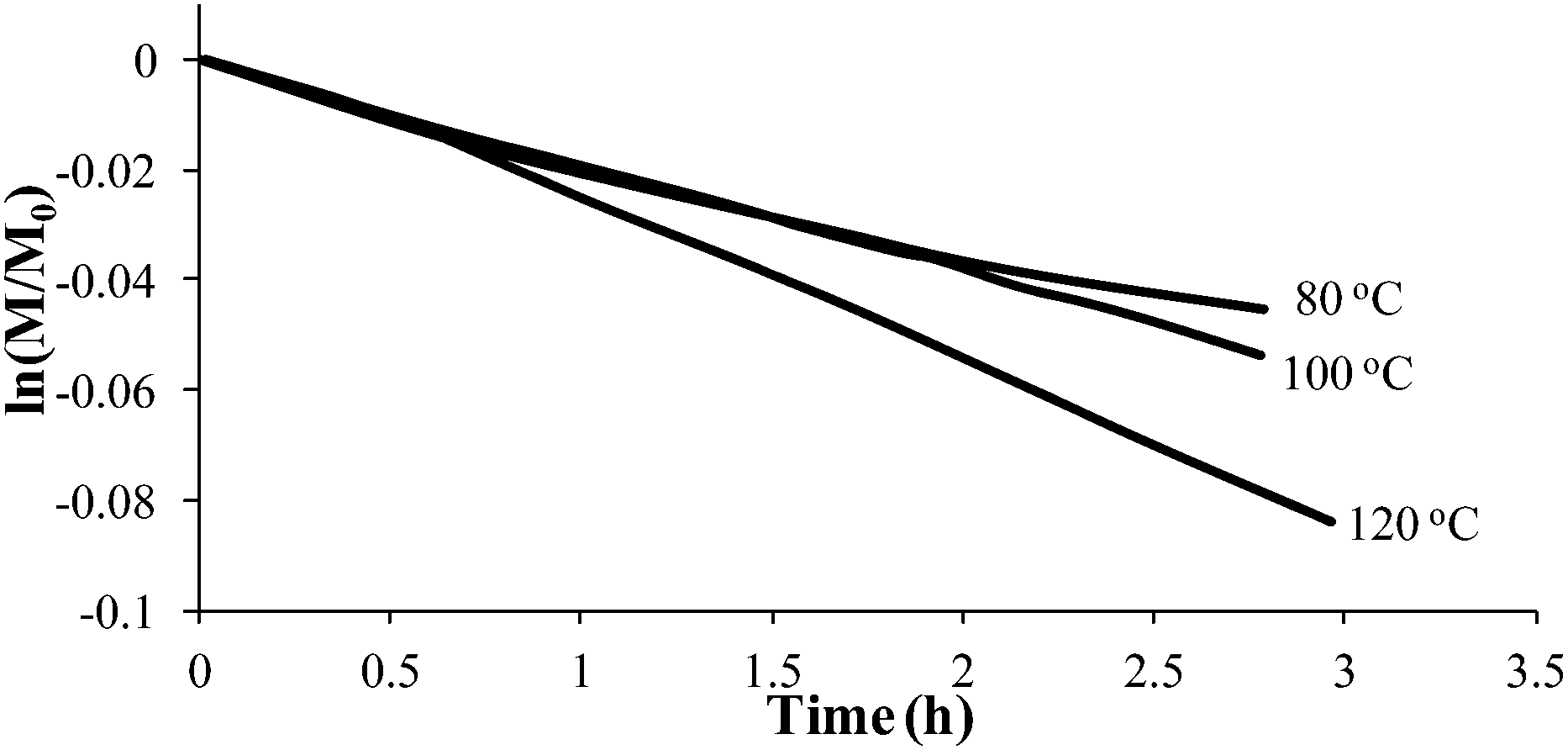
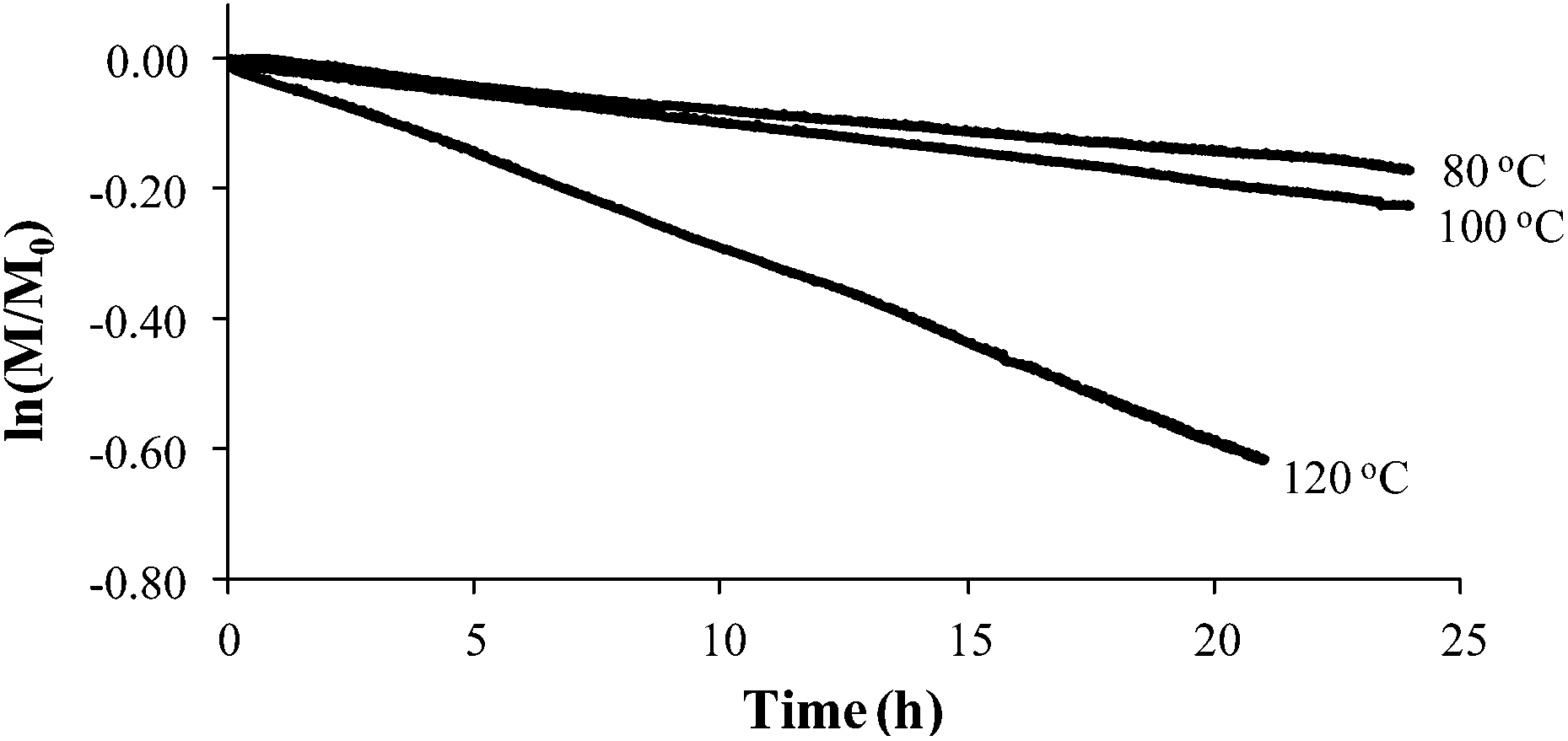
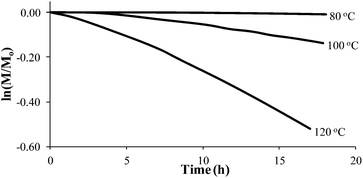
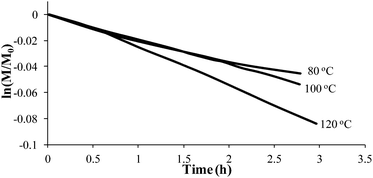
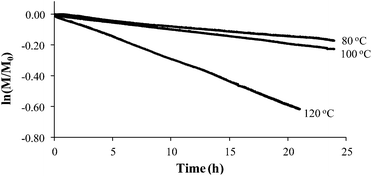
Styrene readily undergoes thermal polymerization above 100 °C,39,44,45 and amine based additives are known to influence the polymerization rate.44,45In situ FTIR spectroscopy elucidated the effects of piperidine on thermal polymerization kinetics by monitoring styrene and 4-VBP monomer consumption at 80 °C, 100 °C, and 120 °C (Fig. 1 and 2). The change in monomer concentration (M/M0) at these temperatures were deemed to be within experimental error, and plotting M/M0versus time revealed thermal polymerization rate constants (kobs) for the various temperatures according to pseudo-first-order polymerization kinetics. Relating the observed rate constants according to the Arrhenius relationship enabled the determination of thermal activation energies for both monomers.
 |
| | Fig. 1 Pseudo-first-order kinetic analysis of styrene thermal polymerization. | |
 |
| | Fig. 2 Pseudo-first-order kinetic analysis of 4-VBP thermal polymerization. | |
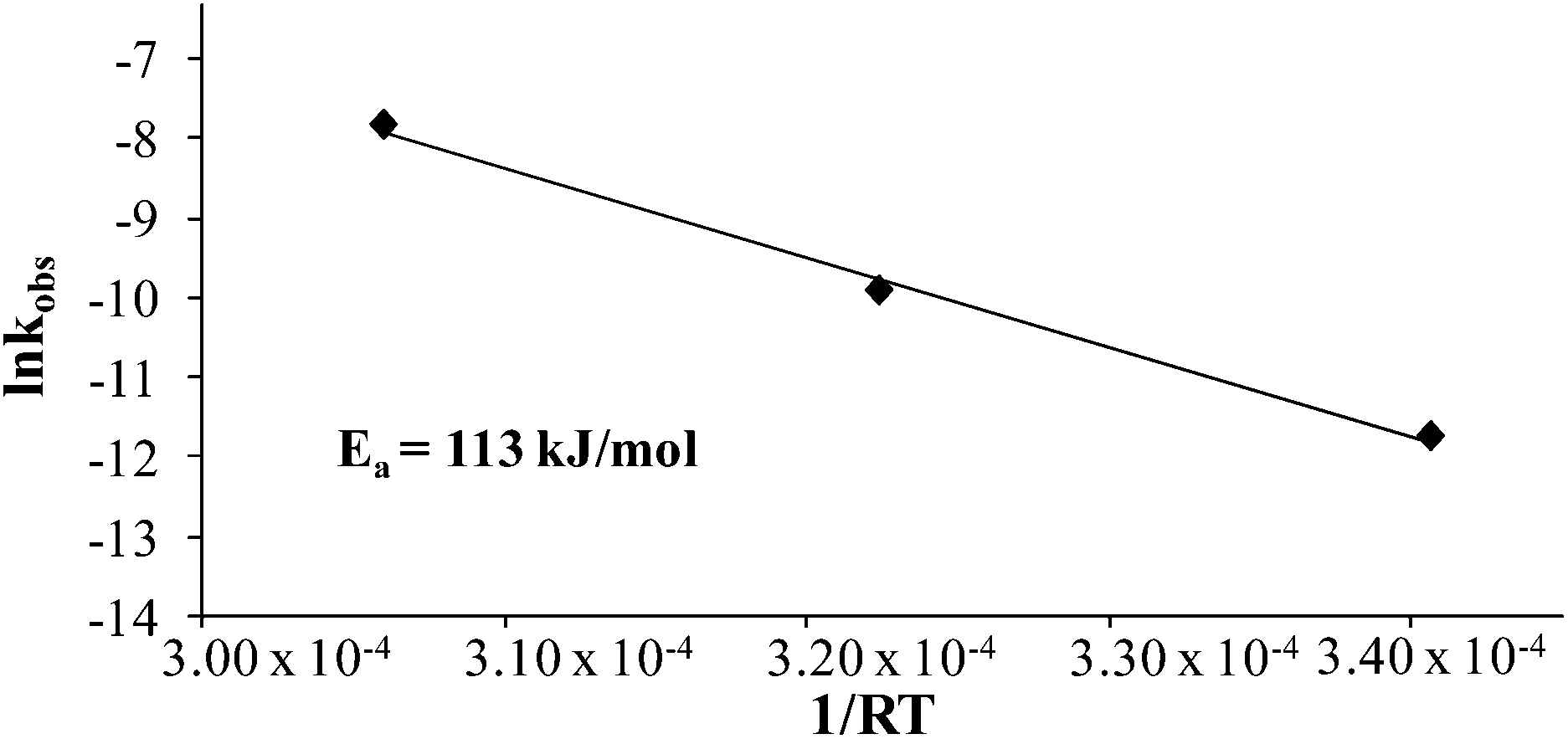
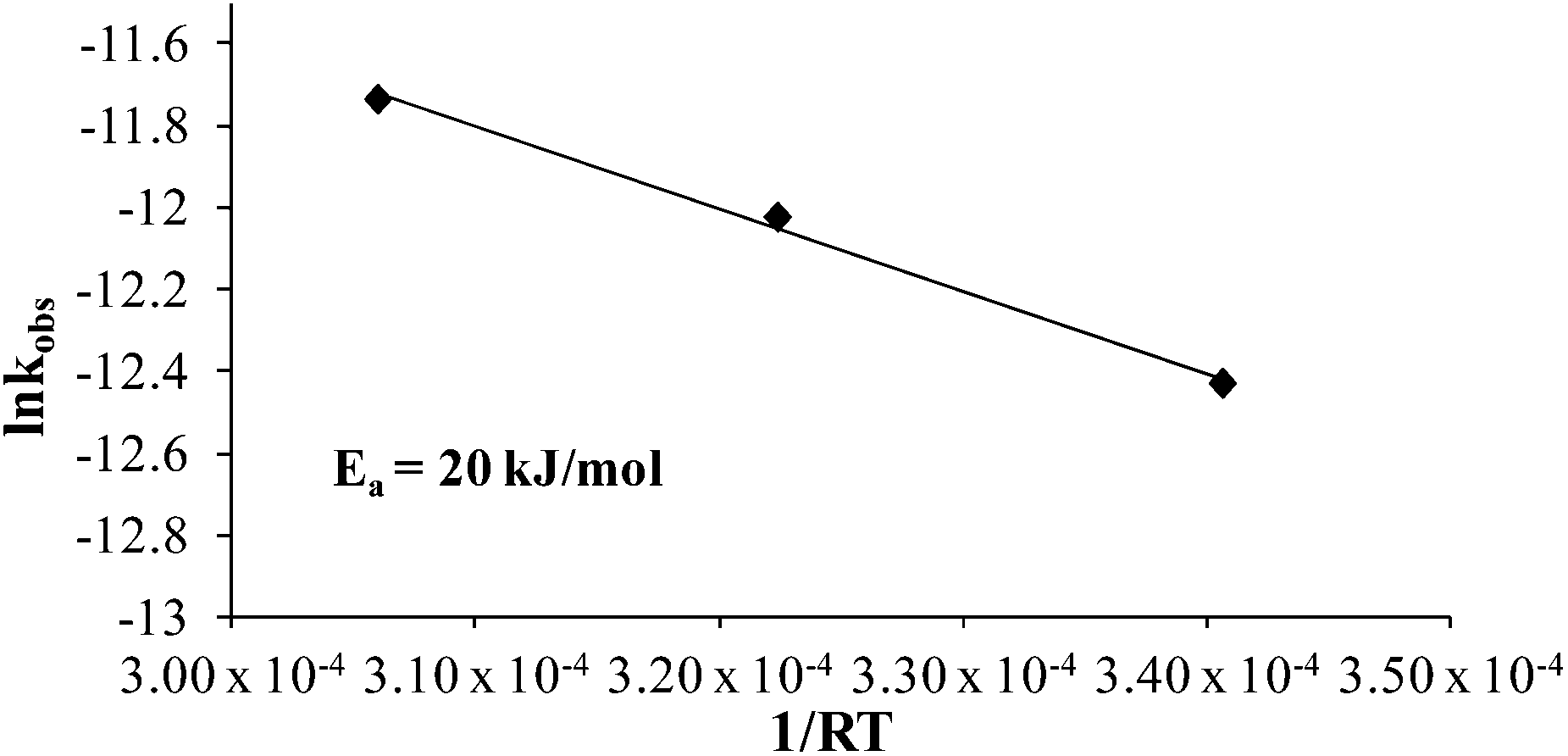
Comparing monomer consumption behavior in both studies showed that styrene exhibited significant polymerization rates over the temperature range with minimal monomer consumption at 80 °C. Arrhenius analysis related the observed rate values for styrene as a function of lnkobsvs. 1/T and derived a thermal activation energy of 113 kJ mol−1 (Fig. 3), which is in good agreement with previous reports.46 4-VBP exhibited significantly increased polymerization rates relative to styrene, and observed rate constants exceeding the value for styrene by roughly one order-of-magnitude. 4-VBP also revealed a markedly lower activation energy compared to styrene, requiring only 20 kJ mol−1 of activation energy. The unexpected, low activation energy accounts for the ease of thermal polymerization (Fig. 4).
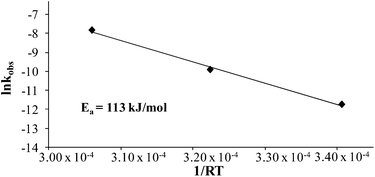 |
| | Fig. 3 Arrhenius analysis of the thermal polymerization of styrene at 80 °C, 100 °C, and 120 °C. | |
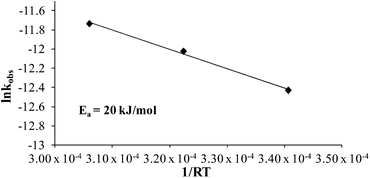 |
| | Fig. 4 Arrhenius analysis of the thermal polymerization of 4-VBP at 80 °C, 100 °C, and 120 °C. | |
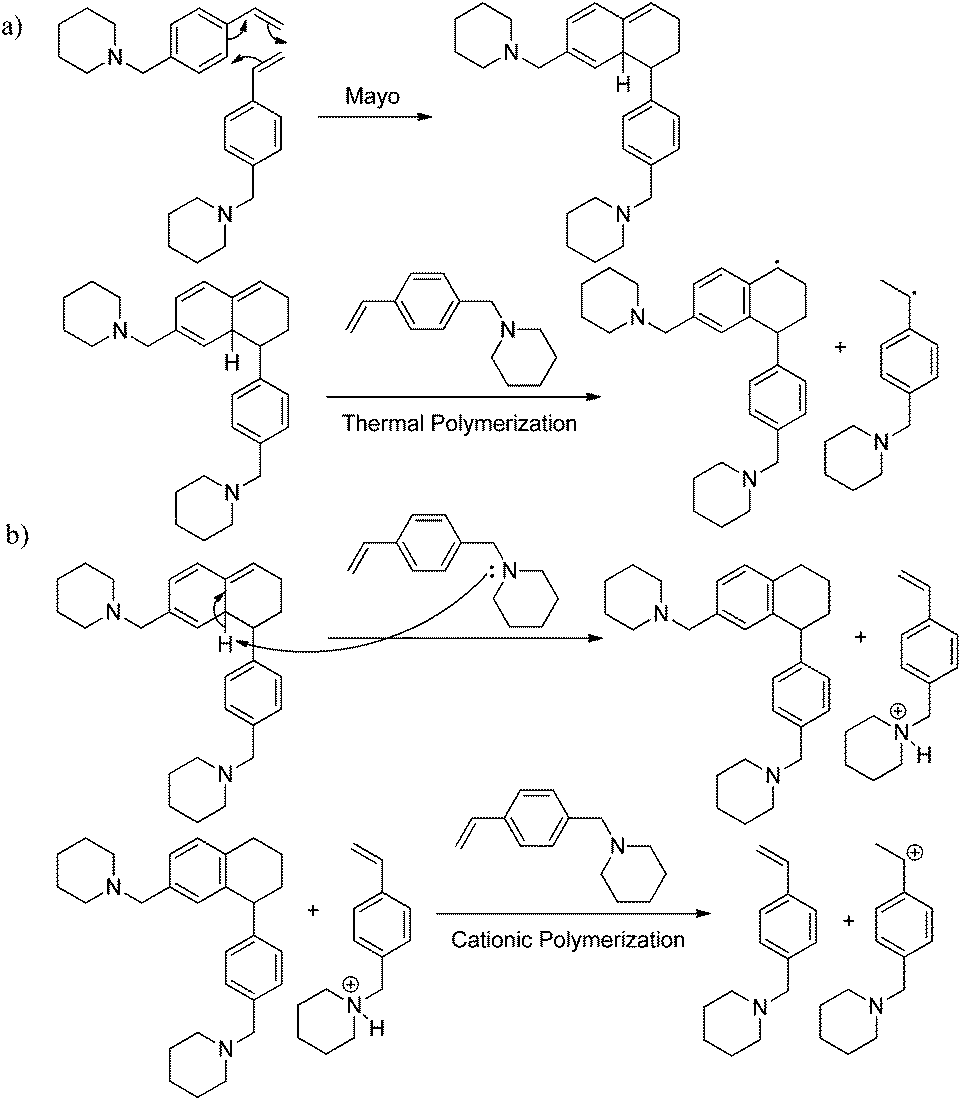
4-VBP is structurally comparable to styrene and thus undergoes the proposed Mayo thermal polymerization mechanism. The cyclic amine provides a basic site, allowing deprotonation of the Mayo adduct, which presumably induces a parallel cationic polymerization (Scheme 2). This acceleration in thermal polymerization rate is analogous to a previous report highlighting the catalytic effects of thermally labile esters, which form acids at high temperatures and thereby induce the cationic polymerization of styrene during thermal autopolymerization.47 4-VBP induces cationic polymerization with an acidified piperidine site in the absence of additional acid catalyst.
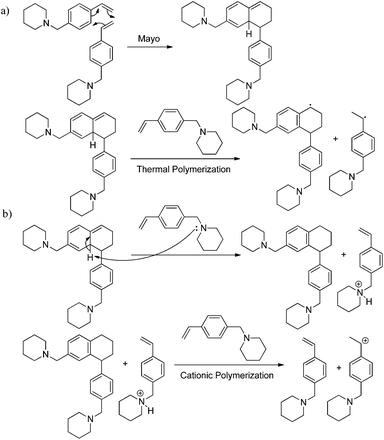 |
| | Scheme 2 (a) Proposed Mayo thermal polymerization of 4-VBP with (b) parallel cationic polymerization. | |
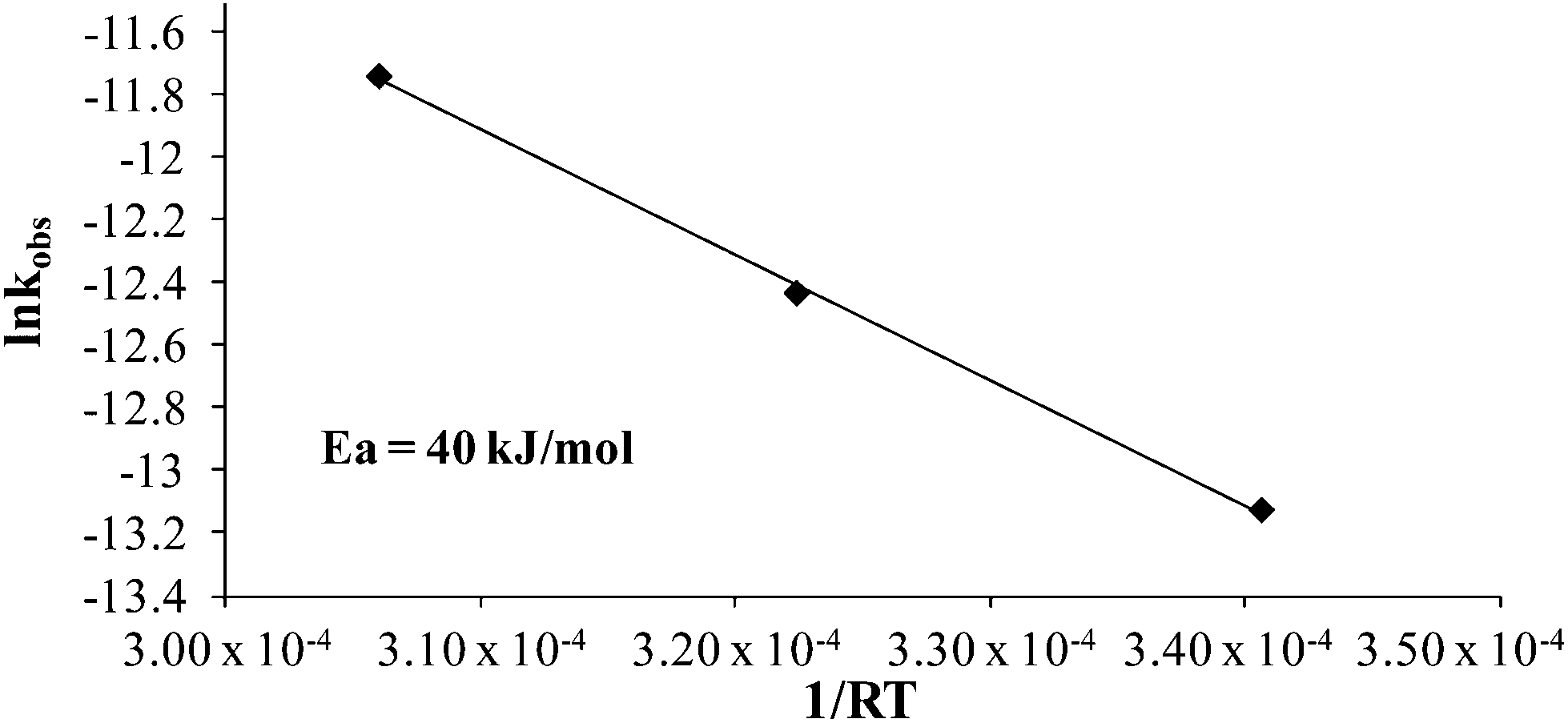
Various additives alter styrene thermal polymerization rates,39,44 and given the increased polymerization rates of 4-VBP over styrene, BP was probed as an additive. In situ FTIR spectroscopy monitored a 1:1 molar ratio of styrene:BP, at 80 °C, 100 °C, and 120 °C (Fig. 5), and revealed a significantly increased thermal polymerization rate compared to the corresponding styrene control. Pseudo-first-order kinetics determined a thermal polymerization activation energy value of 40 kJ mol−1 for styrene with the BP additive (Fig. 6). The significant drop in activation energy from 113 kJ mol−1 for neat styrene suggests an accelerating behavior of the piperidinyl additive. Additional thermal studies at 120 °C at 1.0:0.1 molar equivalents styrene:BP and styrene:N-methyl piperidine further confirmed the accelerating effects of the piperidine ring, revealing comparable kobs values of 8.0 × 10−6 s−1 and 4.0 × 10−6 s−1. These observations further support the accelerating effects of piperidinyl additives on styrene thermal polymerization and support the proposed mechanism involving an induced cationic polymerization.
 |
| | Fig. 5 Pseudo-first-order kinetic plot for the thermal polymerization of 1:1 molar ratio styrene:BP. | |
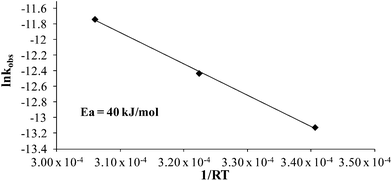 |
| | Fig. 6 Arrhenius plot for the thermal polymerization of 1:1 molar ratio styrene:BP at 80 °C, 100 °C, and 120 °C. | |
Living anionic polymerization of 4-vinylbenzyl piperidine
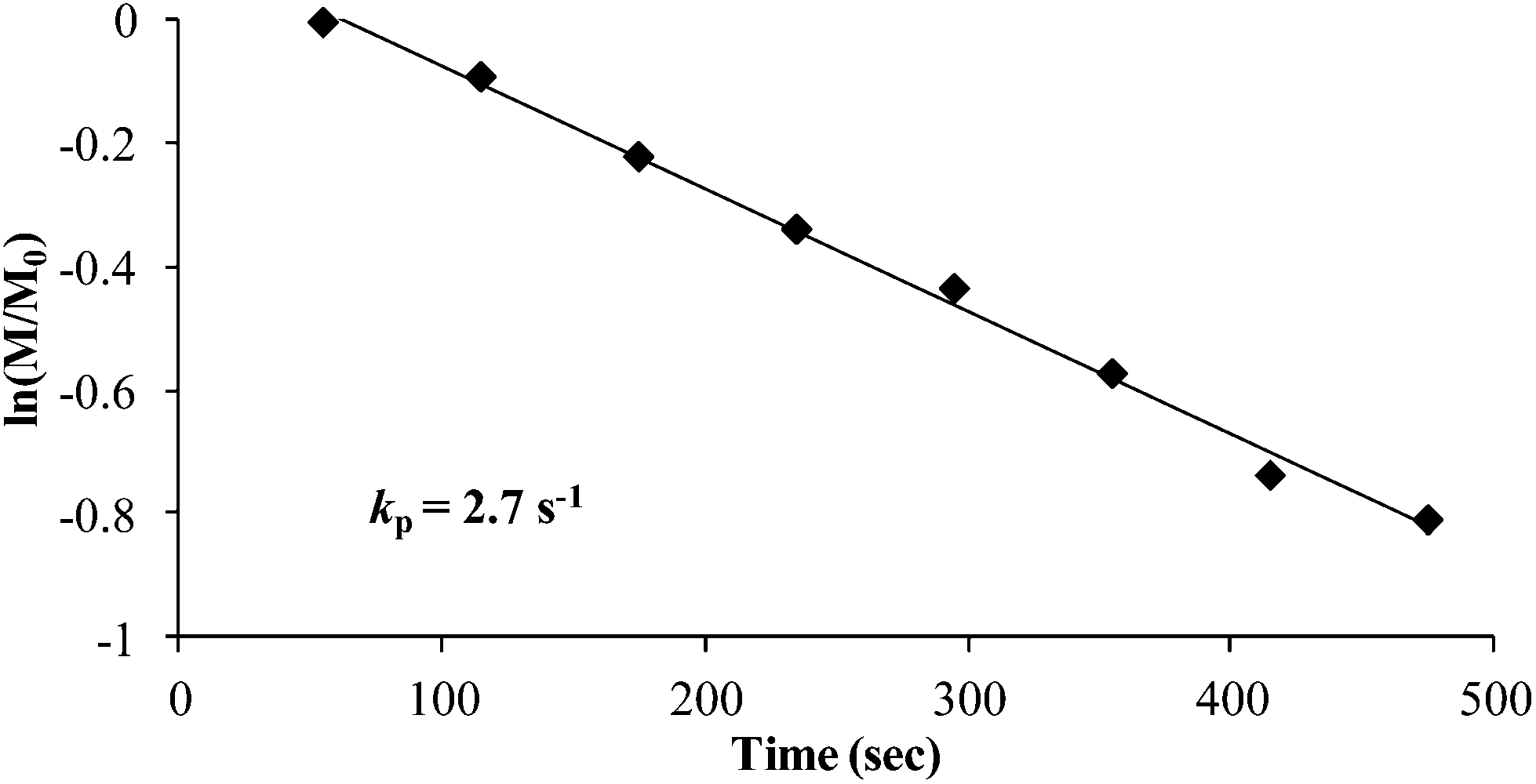
Once the thermal polymerization behavior was defined, in situ FTIR spectroscopy probed a controlled anionic polymerization strategy for synthesizing poly(4-vinylbenzyl piperidine). As depicted in Scheme 3, poly(4-VBP) was synthesized using a 10 wt% monomer–cyclohexane solution at 50 °C and sec-butyllithium as the initiator. Overall, this synthetic strategy offers a novel route for polymerizing 4-vinylbenzyl piperidine and serves as an attractive approach to circumvent undesirable, thermal autopolymerization. A waterfall plot of the polymerization reveals the decrease in the absorbance of the monomer peak found at 909 cm−1 attributed to the vibration of the vinyl component (S1). Tracking the disappearance of vinyl concentration over time indicated that complete monomer consumption occurred within 28 min. The spectral data was utilized further in the generation of a pseudo-first order kinetic plot with monomer concentration (M/M0) plotted as a function of time. In situ FTIR spectroscopy monitored the anionic polymerization of 4-VBP and pseudo-first-order kinetics analysis revealed a propagation rate constant, kp, of 2.7 s−1 (Fig. 7), which is an agreeable value in comparison to styrene (2.0 s−1) and isoprene (1.5 s−1) at identical reaction conditions.48 As expected, 4-VBP exhibited a faster propagation rate than a competing thermal polymerization process and comparable to the anionic propagation of styrene.
 |
| | Scheme 3 General reaction scheme for the anionic polymerization of 4-VBP. | |
 |
| | Fig. 7 Pseudo first-order kinetic plot for 4-VBP anionic polymerization. | |
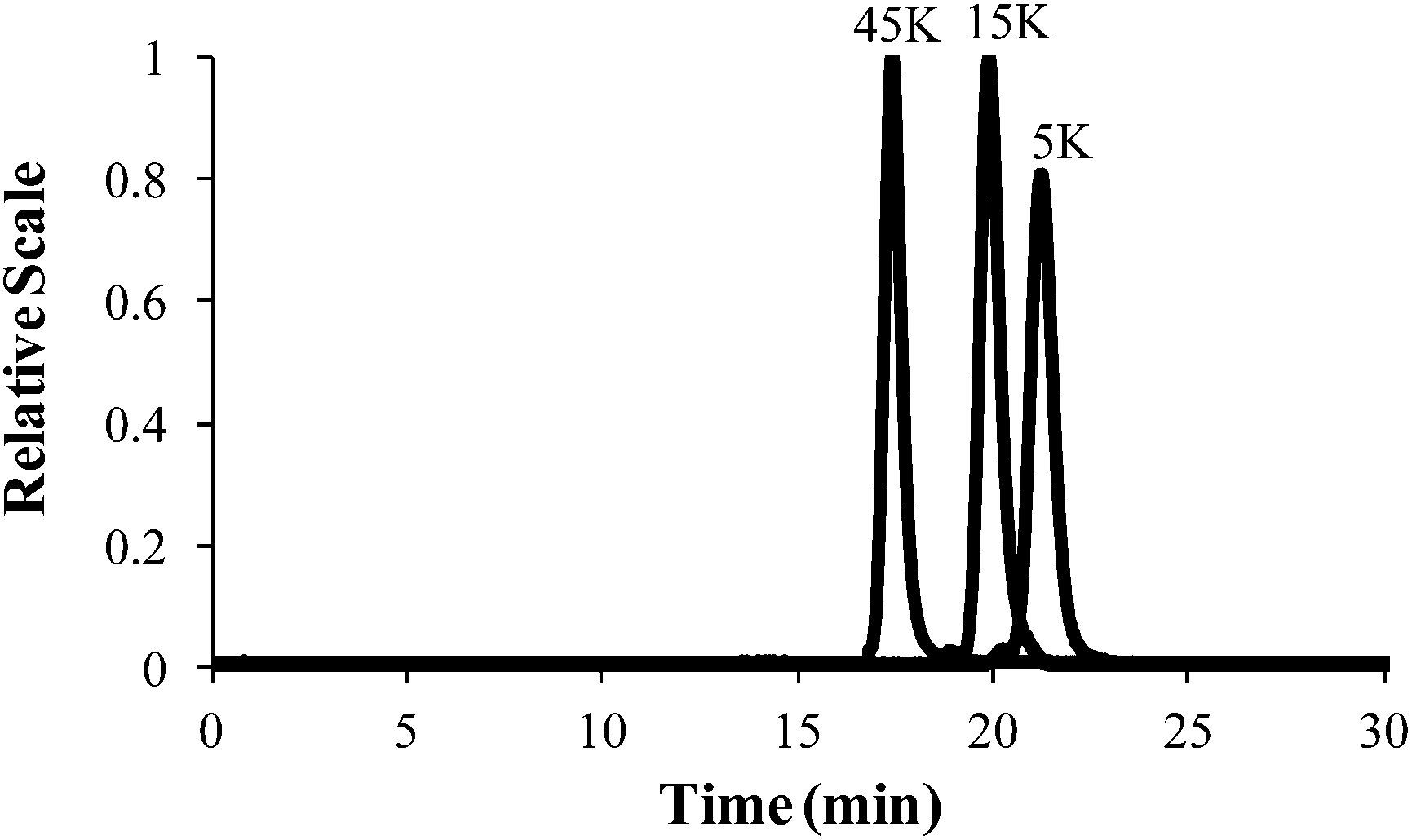
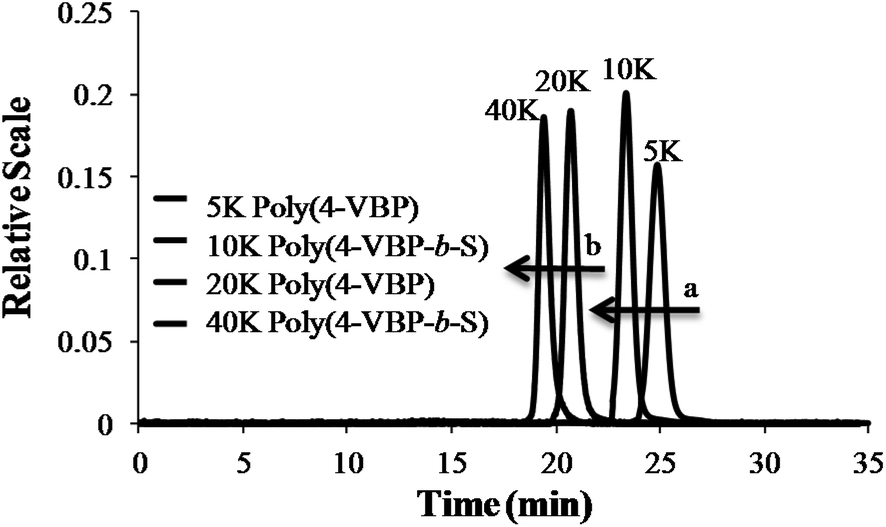
Controlled anionic polymerization also occurred successfully at room temperature to obtain a series of increasing molecular weight homopolymers, with target Mn values of 5000 g mol−1, 15000 g mol−1 and 45000 g mol−1. The polymerization studies occurred in nonpolar cyclohexane, which facilitated ambient temperature conditions while ensuring stereochemical control for achieving narrow molecular weight distributions. Size exclusion chromatography (SEC) highlighted the controlled shift in increasing target Mn values, monomodality in the chromatograms, and narrow polydispersity for the homopolymer series (Fig. 8 and Table 1). Sequential monomer addition studies with styrene further verified the controlled polymerization of 4-VBP, revealing characteristics of a truly “living” polymerization. In these studies, sec-butyllithium initiated the anionic polymerization of 4-VBP and produced living propagating chains with target Mn of 5000 g mol−1 and 20000 g mol−1. Sequential monomer addition enabled the anionic copolymerization with styrene, resulting in two poly(4-VBP-b-S) block copolymers with final Mn of 12000 g mol−1 and 45000 g mol−1. SEC chromatograms in Fig. 10 show the molecular weight shift during the sequential addition from poly(4-VBP) to poly(4-VBP-b-S) with narrow molecular weight distributions, which is indicative of living polymerization (Fig. 9 and Table 1).
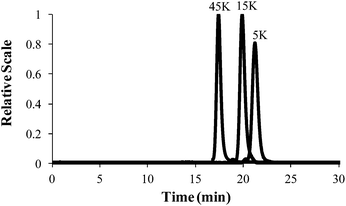 |
| | Fig. 8 SEC characterization of poly(4-VBP) with various molecular weights. | |
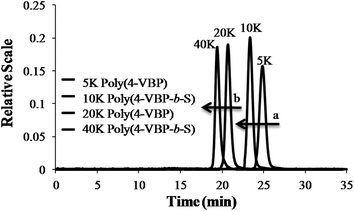 |
| | Fig. 9 SEC characterization of poly(4-VBP) and poly(4-VBP-b-S) before and after sequential monomer addition with styrene. (a) SEC shift in Mn from 5 K poly(4-VBP) to 10 K poly(4-VBP-b-S). (b) SEC shift in Mn from 20 K poly(4-VBP) to 40 K poly(4-VBP-b-S). | |
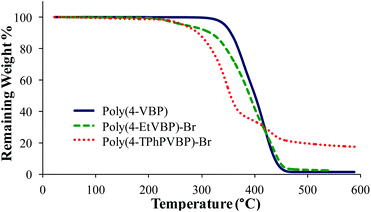 |
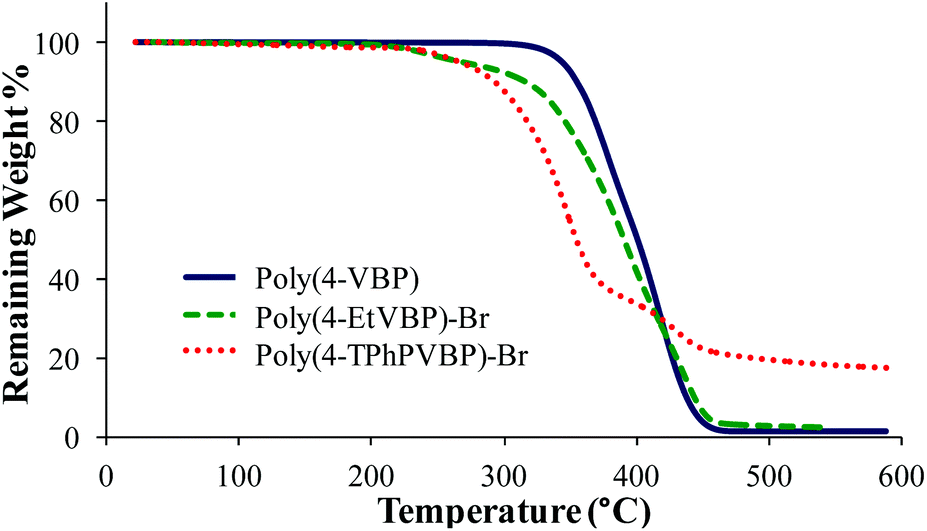
| | Fig. 10 TGA characterization of poly(4-VBP), poly(4-EtVBP)-Br, and poly(4-TPhPVBP)-Br. | |
Table 1 SEC characterization for poly(4-VBP) and poly(4-VBP-b-S)
| Target poly(4-VBP) Mn (g mol−1 × 103) |
SEC Mn (g mol−1 × 103) |
M
w/Mn |
| 5.0 |
6.2 |
1.03 |
| 15.0 |
13.4 |
1.05 |
| 20.0 |
21.2 |
1.03 |
| 45.0 |
42.3 |
1.04 |
| Target poly(4-VBP-b-S) Mn (g mol−1 × 103) |
SEC Mn (g mol−1 × 103) |
M
w/Mn |
| 10.0 |
12.2 |
1.03 |
| 40.0 |
45.3 |
1.05 |
Post-polymerization alkylation and thermal analysis
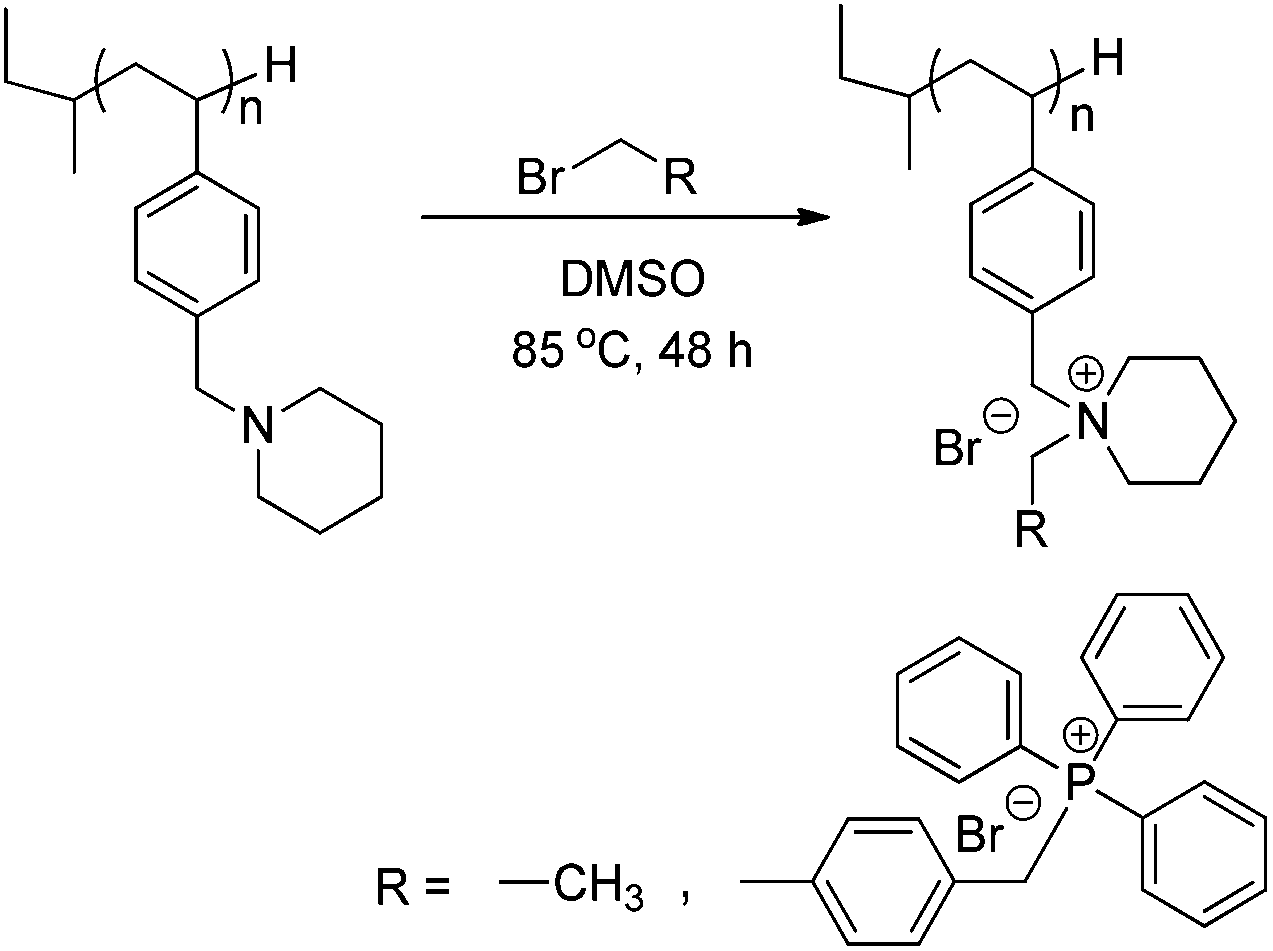
Post-polymerization alkylation achieved novel piperidinium-containing polymers,49 which are becoming increasingly important for many energy and biomedical applications. For example, ammonium and phosphonium containing polymers are highly recognized for their antibacterial properties.50–54 Recent studies show structures containing both salts further exhibit improved bacterial resistance,46 an ideal feature for applications in water purification55,56 and gene delivery.8,46Scheme 4 depicts the general alkylation reaction on poly(4-VBP) homopolymers for yielding novel poly(piperidinium)s. Post-polymerization alkylation occurred with ethyl bromide (EtBr) and the larger 4-(bromomethyl)-benzyltriphenylphosphonium bromide (TPhPBr) to demonstrate the versatility of the alkylation method (Scheme 4).
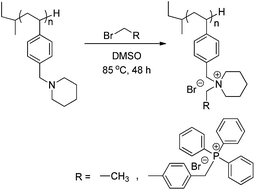 |
| | Scheme 4 Alkylation of poly(4-VBP) with various bromoalkanes to develop new classes of ammonium-containing charged polymers. | |
Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) probed the thermal properties for the various polymers (Table 2). Post-polymerization alkylation imparted charge association along the piperidinium backbone, leading to restricted polymer mobility and increased glass transition temperature (Tg) values. DSC analysis revealed increased Tg values for poly(4-EtVBP)-Br (219 °C) and poly(4-TPhPVBP)-Br (176 °C), in comparison to the neutral poly(4-VBP) (82 °C). Cationic site, alkyl substituent length, and counterion structural variations influenced thermal transitions. These variables also affect thermal stability, as seen in the TGA transitions for poly(4-VBP), poly(4-EtVBP)-Br and poly(4-TPhPVBP)-Br (Fig. 10). Both piperidinium samples exhibited a two-step degradation profile with the initial step (Td, 5% wt loss) occurring at approximately 310 °C, in contrast to poly(4-VBP), which showed a one-step degradation profile (Td, 5% wt loss) of 373 °C. The experimental weight loss for both piperidinium samples correlated with the theoretical weight loss for nucleophilic degradation at the benzylic position, suggesting that both failed to undergo Hofmann elimination degradation due to the absence of accessible antiperiplanar leaving groups within the 4-vinylbenzyl structure. The piperidinium ring structure contains β-hydrogens susceptible for Hofmann elimination, yet requires high-energy input to disrupt ring stability.17,18 Consequently, the piperidinium ring provides enhanced thermal stability and facilitates nucleophilic degradation pathways. Previous reports detailed similar results for poly(4-vinylbenzyl imidazolium)s, which also degrade through a nucleophilic substitution pathway.57,58
Table 2 Thermal properties for poly(4-VBP), poly(4-EtVBP)-Br, and poly(4-TPhPVBP)-Br
| Sample |
T
d, 5% wt loss
(°C) |
T
g
(°C) |
Experimental weight lossa (%) |
Theoretical weight lossc (%) |
|
TGA, 10 °C min−1, N2 atmosphere.
DSC, 10 °C min−1, second heat, N2 atmosphere.
Calculated weight loss % from a nucleophilic substitution degradation pathway.
|
| Poly(4-VBP) |
373 |
82 |
— |
— |
| Poly(4-EtVBP)-Br |
310 |
219 |
10 |
9 |
| Poly(4-TPhPVBP)-Br |
311 |
176 |
62 |
63 |
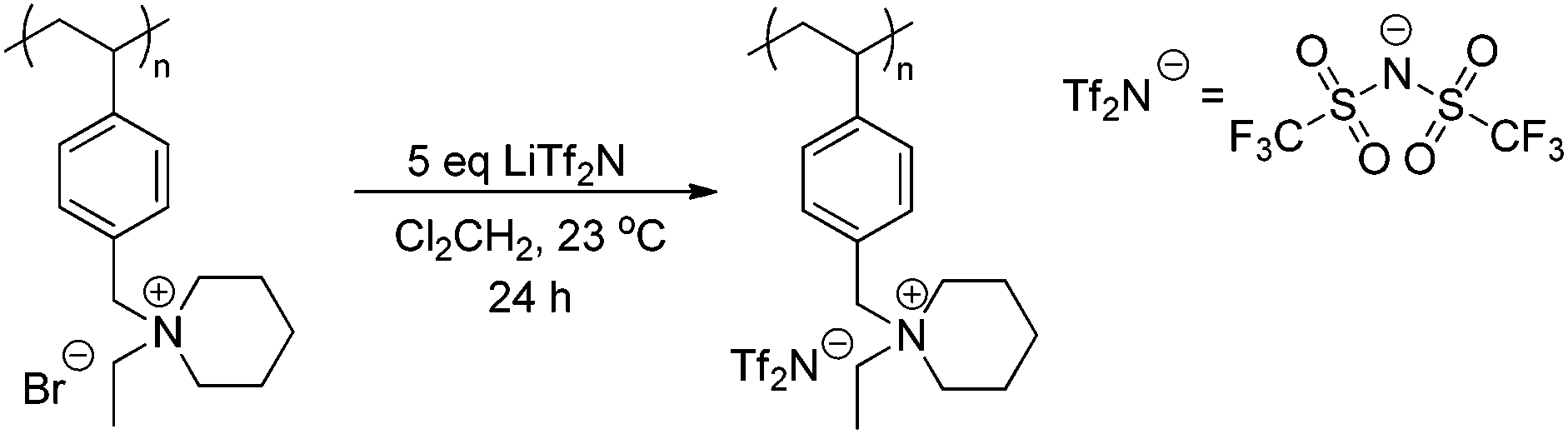
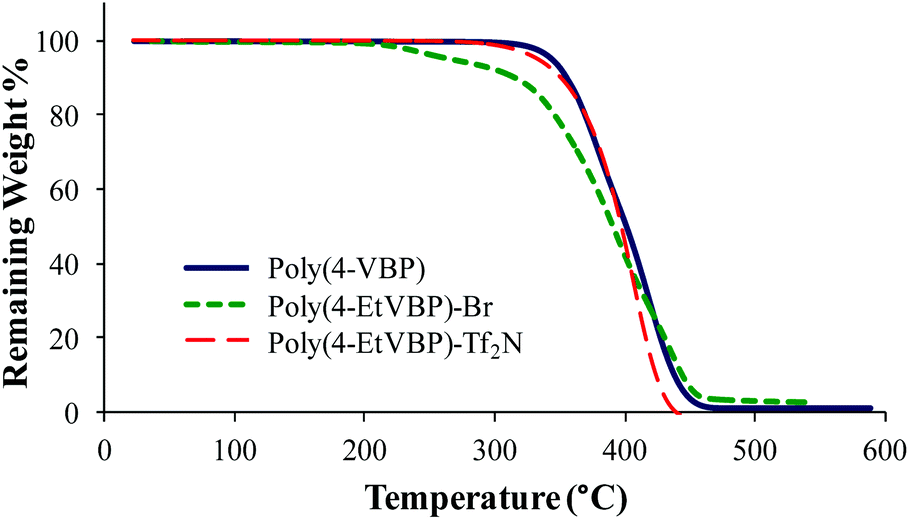
Anion exchange of the piperidinium bromide-containing polymer with LiTf2N offers an opportunity to achieve counteranion variation, while maintaining architectural integrity. Scheme 5 depicts the LiTf2N exchange reaction with poly(4-EtVBP)-Br. Exchanging bromide with LiTf2N improved the thermal stability of the piperidinium-containing polymers (Fig. 11). Previous reports detailed similar results for other polyelectrolytes where exchange to less basic anions improved thermal stability.57,59 Delocalized Tf2N counteranions also hindered ionic association and facilitated segmental motion, resulting in a depressed Tg value by approximately 100 °C.
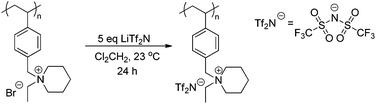 |
| | Scheme 5 Anion exchange of piperidinium homopolymers. | |
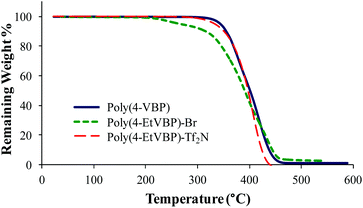 |
| | Fig. 11 TGA characterization of poly(4-VBP), poly(4-EtVBP)-Br, and poly(4-TPhPVBP)-Tf2N. | |
Conclusions
These investigations demonstrated the high propensity for 4-vinylbenzyl piperidine to undergo thermal autopolymerization, and revealed living anionic polymerization as a facile and controlled approach toward producing novel piperidinyl-containing polymers. In situ FTIR spectroscopy monitored 4-vinylbenzyl piperidine autopolymerization, demonstrating that 4-vinylbenzyl piperidine exhibited a thermal activation energy 80 kJ mol−1 less than styrene. Additional in situ FTIR spectroscopic studies involving a 1:1 molar ratio of styrene:N-benzyl piperidine resulted in an activation energy 60 kJ mol−1 less than styrene. A proposed cationic polymerization mechanism accounts for the accelerating effects of piperidine derivatives on thermal polymerization. Living anionic polymerization provided a successful strategy for overcoming thermal autopolymerization challenges, and enabled the first reported living anionic polymerization of 4-vinylbenzyl piperidine. Homopolymers with controlled number-average molecular weights and narrow molecular weight distributions were synthesized, and sequential addition with styrene produced well-defined poly(4-vinylbenzyl piperidine-b-styrene) block copolymers. Lastly, post alkylation studies resulted in novel piperidinium ionomers and polyelectrolytes that will provide scientific insight into ion transport properties. Current efforts are focused on incorporating 4-vinylbenzyl piperidine in well-defined block copolymers with tunable compositions, morphologies, and ion transport behavior. These endeavors also highlight varying block copolymer compositions, including experimental details that elucidate the effects of piperidine ring structure on diene microstructure. These studies will be reported in a future publication.
Acknowledgements
This material is based on work supported by the U.S. Army Research Laboratory and the U.S. Army Research Office under contract/grant number W911NF-07-1-0452 Ionic Liquids in Electro-Active Devices Multidisciplinary University Research Initiative (ILEAD MURI). We thank Dr Carl Willis of Kraton Polymers for insightful discussions. We also thank Dr Asem I. Abdulahad for contributions to the anionic polymerization reactor design.
Notes and references
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123 RSC.
- S.-W. Wang, W. Liu and R. H. Colby, Chem. Mater., 2011, 23, 1862 CrossRef CAS.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535 CrossRef CAS.
- E. S. Hatakeyama, H. Ju, C. J. Gabriel, J. L. Lohr, J. E. Bara, R. D. Noble, B. D. Freeman and D. L. Gin, J. Membr. Sci., 2009, 330, 104 CrossRef CAS PubMed.
- H. B. Park, B. D. Freeman, Z.-B. Zhang, M. Sankir and J. E. McGrath, Angew. Chem., Int. Ed., 2008, 47, 6019 CrossRef CAS PubMed.
- M. S. Shannon, M. S. Hindman, S. P. O. Danielsen, J. M. Tedstone, R. D. Gilmore and J. E. Bara, Sci. China Chem., 2012, 55, 1638 CrossRef CAS.
- D. F. Sanders, Z. P. Smith, R. Guo, L. M. Robeson, J. E. McGrath, D. R. Paul and B. D. Freeman, Polymer, 2013, 54, 4729 CrossRef CAS PubMed.
- S. T. Hemp, A. E. Smith, J. M. Bryson, M. H. Allen and T. E. Long, Biomacromolecules, 2012, 13, 2439 CrossRef CAS PubMed.
- S. Monge, B. Canniccioni, A. Graillot and J.-J. Robin, Biomacromolecules, 2011, 12, 1973 CrossRef CAS PubMed.
- E. B. Anderson and T. E. Long, Polymer, 2010, 51, 2447 CrossRef CAS PubMed.
- H. Ghassemi, D. J. Riley, M. Curtis, E. Bonaplata and J. E. McGrath, Appl. Organomet. Chem., 1998, 12, 781 CrossRef CAS.
- S. Gu, R. Cai and Y. Yan, Chem. Commun., 2011, 47, 2856 RSC.
- R. Gao, D. Wang, J. R. Heflin and T. E. Long, J. Mater. Chem., 2012, 22, 13473 RSC.
- M. H. Allen, S. T. Hemp, M. Zhang, M. Zhang, A. E. Smith, R. B. Moore and T. E. Long, Polym. Chem., 2013, 4, 2333 RSC.
- P. Dimitrov-Raytchev, S. Beghdadi, A. Serghei and E. Drockenmuller, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 34 CrossRef CAS.
- V. Sambhy, B. R. Peterson and A. Sen, Angew. Chem., Int. Ed., 2008, 47, 1250 CrossRef CAS PubMed.
- G. Couture, A. Alaaeddine, F. Boschet and B. Ameduri, Prog. Polym. Sci., 2011, 36, 1521 CrossRef CAS PubMed.
- W. Ogihara, S. Washiro, H. Nakajima and H. Ohno, Electrochim. Acta, 2006, 51, 2614 CrossRef CAS PubMed.
- M. J. Monteiro, M. Sjoberg, d. V. J. Van and C. M. Gottgens, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4206 CrossRef CAS.
- N. A. Listigovers, M. K. Georges, P. G. Odell and B. Keoshkerian, Macromolecules, 1996, 29, 8992 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7901 CrossRef CAS.
- R. D. Allen, I. Yilgor and J. E. McGrath, ACS Symp. Ser., 1986, 302, 79 CrossRef CAS PubMed.
- D. T. Williamson, J. F. Elman, P. H. Madison, A. J. Pasquale and T. E. Long, Macromolecules, 2001, 34, 2108 CrossRef CAS.
- W. Devonport, L. Michalak, E. Malmstroem, M. Mate, B. Kurdi, C. J. Hawker, G. G. Barclay and R. Sinta, Macromolecules, 1997, 30, 1929 CrossRef CAS.
- J. R. Lizotte and T. E. Long, Macromol. Chem. Phys., 2003, 204, 570 CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS PubMed.
- M. K. Georges, R. A. Kee, R. P. N. Veregin, G. K. Hamer and P. M. Kazmaier, J. Phys. Org. Chem., 1995, 8, 301 CrossRef CAS.
- E. M. W. Tsang, Z. Zhang, A. C. C. Yang, Z. Shi, T. J. Peckham, R. Narimani, B. J. Frisken and S. Holdcroft, Macromolecules, 2009, 42, 9467 CrossRef CAS.
- Y. P. Borguet and N. V. Tsarevsky, Polym. Chem., 2012, 3, 2487 RSC.
- J. Lu, F. Yan and J. Texter, Prog. Polym. Sci., 2009, 34, 431 CrossRef CAS PubMed.
- M. J. Monteiro, M. Hodgson and B. H. De, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3864 CrossRef CAS.
- R. Wang and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2468 CrossRef CAS.
- T. Wu, D. Wang, M. Zhang, J. R. Heflin, R. B. Moore and T. E. Long, ACS Appl. Mater. Interfaces, 2012, 4, 6552 CAS.
- O. W. Webster, Science, 1991, 251, 887 CrossRef CAS PubMed.
- R. Faust and J. P. Kennedy, Polym. Bull., 1986, 15, 317 CrossRef CAS.
- M. Sawamoto, Prog. Polym. Sci., 1991, 16, 111 CrossRef CAS.
- M. Szwarc, M. Levy and R. Milkovich, J. Am. Chem. Soc., 1956, 78, 2656 CrossRef CAS.
- J. R. Lizotte, B. M. Erwin, R. H. Colby and T. E. Long, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 583 CrossRef CAS.
- F. R. Mayo, J. Am. Chem. Soc., 1968, 90, 1289 CrossRef CAS.
- Y. K. Chong, E. Rizzardo and D. H. Solomon, J. Am. Chem. Soc., 1983, 105, 7761 CrossRef CAS.
- P. M. Kazmaier, K. Daimon, M. K. Georges, G. K. Hamer and R. P. N. Veregin, Macromolecules, 1997, 30, 2228 CrossRef CAS.
- M. Morton and L. J. Fetters, Rubber Chem. Technol., 1975, 48, 359 CrossRef CAS.
- H. Miyamura, M. Morita, T. Inasaki and S. Kobayashi, Bull. Chem. Soc. Jpn., 2011, 84, 588 CrossRef CAS.
- R. A. Gregg and F. R. Mayo, J. Am. Chem. Soc., 1953, 75, 3530 CrossRef CAS.
- J. Katzer, W. Pauer and H.-U. Moritz, Macromol. React. Eng., 2012, 6, 213 CrossRef CAS.
- E.-R. Kenawy, F. I. Abdel-Hay, A. E.-R. R. El-Shanshoury and M. H. El-Newehy, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2384 CrossRef CAS.
- Y. Kim, H. J. Harwood and D. Priddy, J. Appl. Polym. Sci., 2002, 83, 1786 CrossRef CAS.
- T. E. Long, H. Y. Liu, B. A. Schell, D. M. Teegarden and D. S. Uerz, Macromolecules, 1993, 26, 6237 CrossRef CAS.
- J. S. Parent, A. Penciu, S. A. Guillen-Castellanos, A. Liskova and R. A. Whitney, Macromolecules, 2004, 37, 7477 CrossRef CAS.
- A. Popa, G. Ilia, S. Iliescu, G. Dehelean, A. Pascariu, A. Bora and C. M. Davidescu, Mol. Cryst. Liq. Cryst., 2004, 418, 195 CrossRef CAS.
- N. Beyth, I. Yudovin-Farber, R. Bahir, A. J. Domb and E. I. Weiss, Biomaterials, 2006, 27, 3995 CrossRef CAS PubMed.
- M. B. Harney, R. R. Pant, P. A. Fulmer and J. H. Wynne, ACS Appl. Mater. Interfaces, 2009, 1, 39 CAS.
- K. Takai, T. Ohtsuka, Y. Senda, M. Nakao, K. Yamamoto, J. Matsuoka and Y. Hirai, Microbiol. Immunol., 2002, 46, 75 CrossRef CAS PubMed.
- T. Tashiro, Macromol. Mater. Eng., 2001, 286, 63 CrossRef CAS.
- A. C. Sagle, J. Membr. Sci., 2009, 340, 92 CrossRef CAS PubMed.
- E. S. Hatakeyama, H. Ju, C. J. Gabriel, J. L. Lohr, J. E. Bara, R. D. Noble, B. D. Freeman and D. L. Gin, J. Membr. Sci., 2009, 330, 104 CrossRef CAS PubMed.
- S. T. Hemp, M. Zhang, M. H. Allen Jr., S. Cheng, R. B. Moore and T. E. Long, Macromol. Chem. Phys., 2013, 214, 2099 CrossRef CAS.
- R. L. Weber, Y. Ye, S. M. Banik, Y. A. Elabd, M. A. Hickner and M. K. Mahanthappa, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1287 CrossRef CAS.
- S. Cheng, M. Zhang, T. Wu, S. T. Hemp, B. D. Mather, R. B. Moore and T. E. Long, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 166 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectroscopy. See DOI: 10.1039/c4py00763h |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence