
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Network formation mechanisms in conjugated microporous polymers†
Andrea
Laybourn
a,
Robert
Dawson
ab,
Rob
Clowes
a,
Tom
Hasell
a,
Andrew I.
Cooper
a,
Yaroslav Z.
Khimyak
c and
Dave J.
Adams
*a
aDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: d.j.adams@liverpool.ac.uk
bInstitute of Chemistry, Functional Materials, Technische Universität Berlin, Hardenbergstraße 40, Berlin, 10623, Germany
cSchool of Pharmacy, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK
First published on 21st July 2014
Abstract
We discuss in detail the mechanism of formation of a highly microporous polymer, CMP-1, formed mainly via Sonogashira–Hagihara coupling. We demonstrate how the microporosity evolves with time, and discuss the importance of alkyne homo-coupling on the microporosity.
Introduction
Microporous polymers are useful for heterogeneous catalysis, separations, sensing, and energy applications.1,2 For example, there has been much focus on the capture and separation of gases such as CO2 in microporous materials.1,3–5 Porous polymers are a growing platform for such applications.1,5–7 The broad family of microporous polymers now comprises several different classes of materials, including polymers of intrinsic microporosity (PIMs), hypercrosslinked polymers, and covalent organic frameworks (COFs), to name but a few.6,8One sub-class of microporous polymers is conjugated microporous polymers (CMPs),6,9,10 first reported in 2007.9 CMPs are amorphous polymers consisting of monomers linked together in a π-conjugated manner. The rigid polymer structure and the three dimensional nature in CMPs results in an extended structure that is permanently microporous. The first reported CMP, CMP-1, was synthesised by the palladium catalysed cross-coupling of 1,3,5-triethynylbenzene with 1,4-diiodobenzene (Scheme 1). Later, we showed that 1,4-dibromobenzene could also be used.11,12
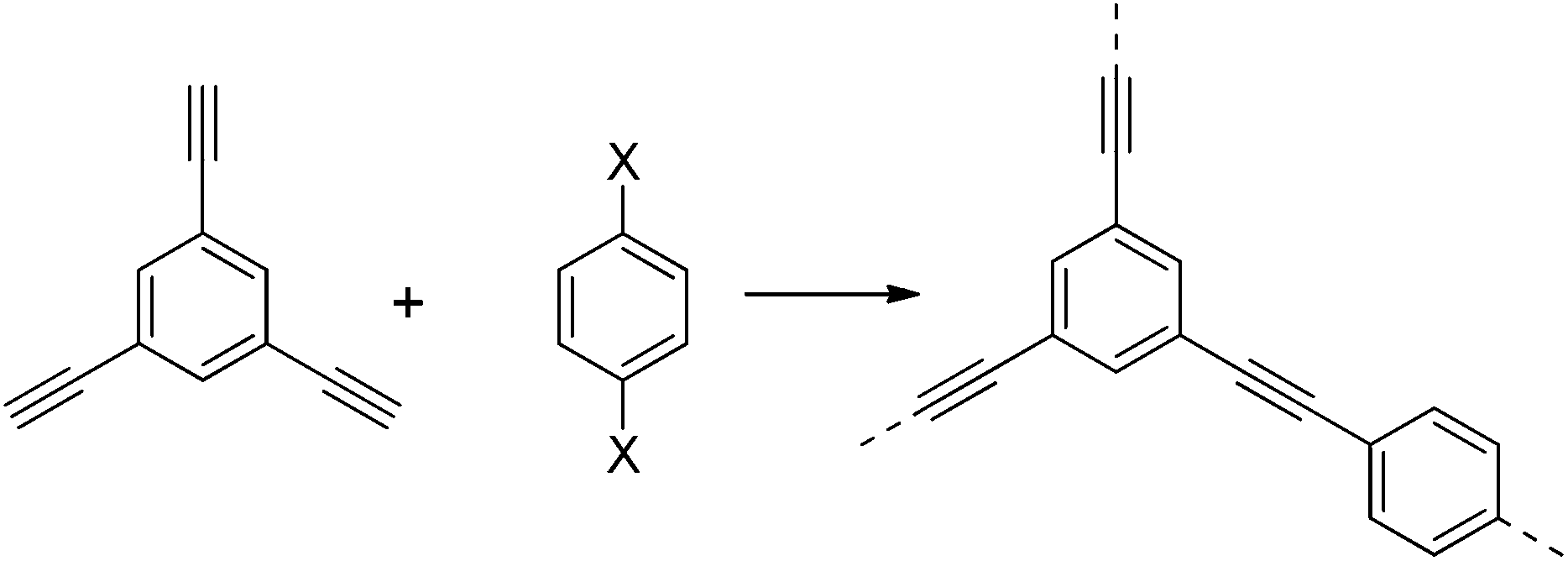 | ||
| Scheme 1 Synthesis of CMP-1 (X = Br or I). | ||
This cross-coupling strategy for the formation of CMPs is highly versatile. A range of functionalised dibromobenzenes can be used, allowing the formation of microporous polymers containing different chemical groups, including primary amines, carboxylic acids, fluorinated groups, nitro groups, and so on.11,13 We have used this strategy to prepare polymers with specific targeted properties such as favourable interactions with gases14 and controllable hydrophobicity,11 and also to prepare polymers that function as heterogeneous catalysts.15 Since the initial introduction of CMPs,9 there has been an explosion of interest in the area,6,16–26 with many research groups working on CMPs and related polymers such as porous aromatic frameworks (PAFs).27 Despite this, there is a real lack of data on how these CMP networks are actually formed. This is surprising, since the rapid phase separation that occurs in these step growth polymerisation reactions raises the question of why it is possible to achieve good yields and, apparently, high molar mass extended networks.
The first CMPs were synthesised in toluene,9,28 but we later showed that the choice of the reaction solvent is important,12 with DMF generally providing polymers with higher surface areas. Tan et al. have also emphasised the importance of solvent for these reactions.29 During the synthesis, often carried out for many hours if not days, precipitation and gelation are typically observed, often at a quite early stage. It is therefore perhaps unsurprising that the choice of solvent is important, since this might dictate the progress of this phase separation, as well as affecting the morphology of the phase separated network – for example, by swelling the network to a greater or lesser degree. To date, however, there has been a very limited mechanistic understanding of the way in which these polymers form.
Mechanistic studies for these networks and full characterisation of the resultant CMPs are challenging due to the total insolubility of these materials in all solvents tested. As a result, analysis is dominated by infra-red (IR) spectroscopy, solid-state NMR (ssNMR), and elemental analysis. Here, we examine in detail the synthesis of CMP-1 in DMF, with the aim of understanding the evolution of molecular structure and microporosity in the network-forming reaction.
Experimental
Materials and methods
1,3,5-Triethynylbenzene (98%) was obtained from ABCR and used as received. All other chemicals and solvents were obtained from Sigma-Aldrich and used as received. Anhydrous grade N,N-dimethylformamide (DMF) was used. All chemicals had a purity of 98% or greater.Characterisation
Results and discussion
We focus here on the synthesis of CMP-1 (Scheme 1) in DMF since we have shown that this solvent generally leads to polymers with higher BET surface areas (SABET).12 As in all of our work so far, we use a ratio of 1,3,5-triethynylbenzene to 1,4-dibromobenzene that gives a 1.5 molar excess of alkynyl groups. This ratio, which is perhaps counterintuitive, was chosen on purely empirical grounds9 because it was found to be the molar ratio that leads to polymers with the highest SABET.12,28Previously, we used extended reaction times of 24 or 72 hours for the synthesis of CMP-1.9,11,12 Again, these reaction times were chosen somewhat empirically, rather than on the basis of an understanding of the reaction kinetics. Here, we investigate the reaction kinetics of the polymerisation from 10 minutes to 42 hours. We first consider the chemical composition of the isolated insoluble materials, before discussing the physical properties and in particular the porosity of the polymers.
At early times (<40 minutes), no insoluble polymer was collected. Solution state 1H NMR spectroscopy (see Fig. S1, ESI†) of the quenched reaction medium at these early times showed evidence of the formation of oligomers, but specific assignment of their structure was not possible. The absolute combined peak intensity decreased over this time period, implying that phase-separated, insoluble material was being formed that was not detectable by solution NMR spectroscopy, however this oligomeric material was not collected effectively on the filter paper (mesh size = 11 μm31) at this early stage in the reaction. From 40 minutes onwards, the reaction mixture gelled visibly, accompanied by the formation of a brown precipitate. The gravimetric yield of the intermediate precipitate increases after 40 minutes and then becomes relatively constant after about 300 minutes reaction time (Table 1), whereupon the isolated yield is close to the yield predicted for full network conversion. The theoretical yields are based on complete conversion. The higher than expected yields at longer reaction times are in line with our previous reports,11,12 and can be ascribed to residual end groups, catalyst residues and entrained water vapour or solvent residues.32
| Reaction time (min) | Yield (mg) | Yield (% theoretical) | % Ca | % Ha | % Na |
|---|---|---|---|---|---|
| a Elemental analysis data. b Predicted values, calculated assuming complete reaction and full network conversion. | |||||
| Predictedb | 226 | 100 | 95.2 | 4.8 | 0 |
| 10 | 0 | 0 | — | — | — |
| 20 | 0 | 0 | — | — | — |
| 30 | 0 | 0 | — | — | — |
| 40 | 109.1 | 48.2 | 78.8 | 3.2 | 0 |
| 60 | 142.0 | 62.8 | 82.0 | 3.2 | 0 |
| 120 | 163.5 | 72.1 | 83.0 | 3.3 | 0 |
| 300 | 239.6 | 106 | 84.4 | 3.4 | 0 |
| 420 | 248.0 | 110 | 85.7 | 3.4 | 0 |
| 1080 | 244.9 | 108 | 79.5 | 3.9 | 0.6 |
| 2520 | 264.8 | 117 | 81.7 | 3.4 | 0.3 |
Elemental analysis showed that the percentage of carbon and hydrogen remains fairly consistent at 80 ± 4% and 3.3 ± 0.5%, respectively, for the polymers collected at different reaction times (Table 1). These values are in agreement with those reported previously for other CMP networks,11,12 and as observed before, the results deviate significantly from the predicted ideal values. There are many examples now of deviations between experimental and predicted microanalyses for porous materials. Explanations for this include poor combustion of polymeric materials,33,34 trapped solvent and gases,35 catalyst retention,36 and the presence of unreacted end groups.11,12,37 Palladium residues would also affect these measurements, and these microporous polymers undoubtedly physisorb atmospheric water vapour, which might further distort microanalyses. All of these explanations are in principle possible for the CMP materials here. In particular, unreacted end groups are a likely contributor, since the presence of bromine would lead to lower carbon contents, especially for the intermediate materials collected at short reaction times. Also, the materials collected at 1080 and 2520 minutes contain low levels of nitrogen (see Table 1), possibly indicating trapped triethylamine or residual DMF, despite extensive Soxhlet extraction.
EDX analysis of two polymers that formed after 60 minutes and after 1080 minutes, respectively, indicated the presence of palladium and copper due to residual catalyst (Table 2). The presence of bromine indicates residual end groups. The percentage of bromine decreases between 60 and 1080 minutes, from 4.8% to 2.9%. A reduction in bromine content with increasing reaction time suggests fewer end groups in the final material and therefore a more extended polymeric structure compared with materials collected at earlier stages in the reaction. However, the low bromine content at 60 minutes shows that a significant degree of cross-linking has already occurred.
This suggests that the CMP networks are relatively highly condensed after 60 minutes reaction time. In terms of degree of condensation, models of hypothetical networks generated on the basis of matching the bromine contents (Fig. S2 and S3, ESI†) were constructed. To match a bromine content of 4.8%, the network has one bromine end group per 14 aromatic rings (Fig. S2†), corresponding to a molecular weight of 1650 g mol−1. To match a bromine content of 2.9%, the hypothetical network has one bromine end group per 24 aromatic rings and a molecular weight of 2770 g mol−1. Hence, these data imply that the network condensation continues in the solvent-swollen, phase-separated state after polymer precipitation has occurred.
This synthesis of CMP-1 involves reaction between a halogenated monomer and an alkyne-containing monomer which is in excess. The resulting networks should therefore also contain alkyne end groups. Indeed, alkyne end groups have been reported for CMP networks previously.12 The presence of terminal alkyne functionalities was demonstrated by FTIR (Fig. 1). The peak at ca. 2200 cm−1 corresponds to a polymerised, internal alkyne (R–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–R) and the peak at ca. 2100 cm−1 can be ascribed to alkyne end groups (R–CC–H).9 The ratio of the peak at 2200 cm−1 to the peak at 2100 cm−1 clearly increases as the reaction time progresses, with the data at later times showing almost complete consumption of the terminal alkyne; this is in good agreement with the NMR data (discussed later). As noted above, for the synthesis of CMP-1, we use a 1.5 molar excess of alkynyl groups. Hence, the complete disappearance of the terminal alkyne groups implies that the network is not simply formed via Sonogashira coupling with the bromine monomer, as discussed further below.
C–R) and the peak at ca. 2100 cm−1 can be ascribed to alkyne end groups (R–CC–H).9 The ratio of the peak at 2200 cm−1 to the peak at 2100 cm−1 clearly increases as the reaction time progresses, with the data at later times showing almost complete consumption of the terminal alkyne; this is in good agreement with the NMR data (discussed later). As noted above, for the synthesis of CMP-1, we use a 1.5 molar excess of alkynyl groups. Hence, the complete disappearance of the terminal alkyne groups implies that the network is not simply formed via Sonogashira coupling with the bromine monomer, as discussed further below.
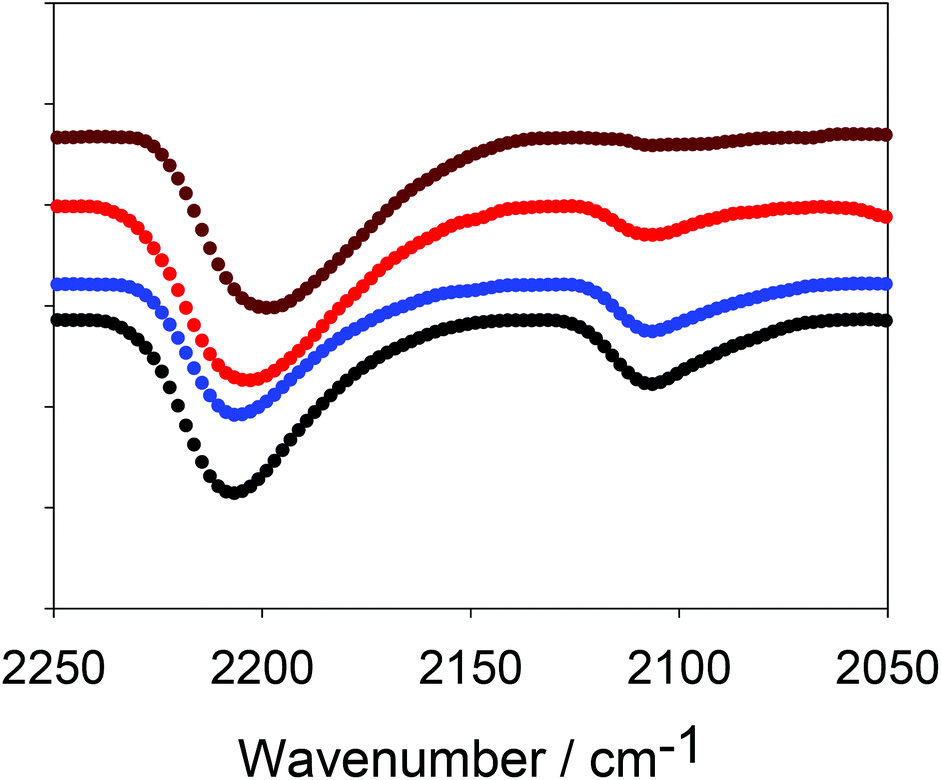 | ||
| Fig. 1 FTIR spectra of CMP-1 at different reaction times (black data = 40 min; blue = 60 min, red = 120 min and brown = 1080 min) showing consumption of terminal alkyne groups (peak at 2100 cm−1). Data are offset on the y-axis for clarity. | ||
A more accurate method of quantifying the level of end groups involves the use of ssNMR. Previously, the extent of polymerisation for CMP materials was determined using single pulse excitation (SPE) 13C NMR with high power 1H decoupling (HPDEC).9,11,12 The quantitative nature of these measurements for CMP networks has been verified by CP kinetics experiments.9 The structures of the CMP-1 intermediates were elucidated by 13C{1H} MAS NMR (Fig. 2). All reaction intermediates show aromatic peaks at 131.9 ppm (–CAr–H) and 123.9 ppm (–CAr–CC–CAr) and an alkyne peak at 91.5 ppm (–CAr–CC–), confirming that polymerisation has been successful.9 A resonance at 82.4 ppm, ascribed to alkyne end groups (–CC–H) is also present in the NMR spectra, in agreement with the FTIR data. All peaks are consistent with the spectra of CMP networks reported previously.9,11,12
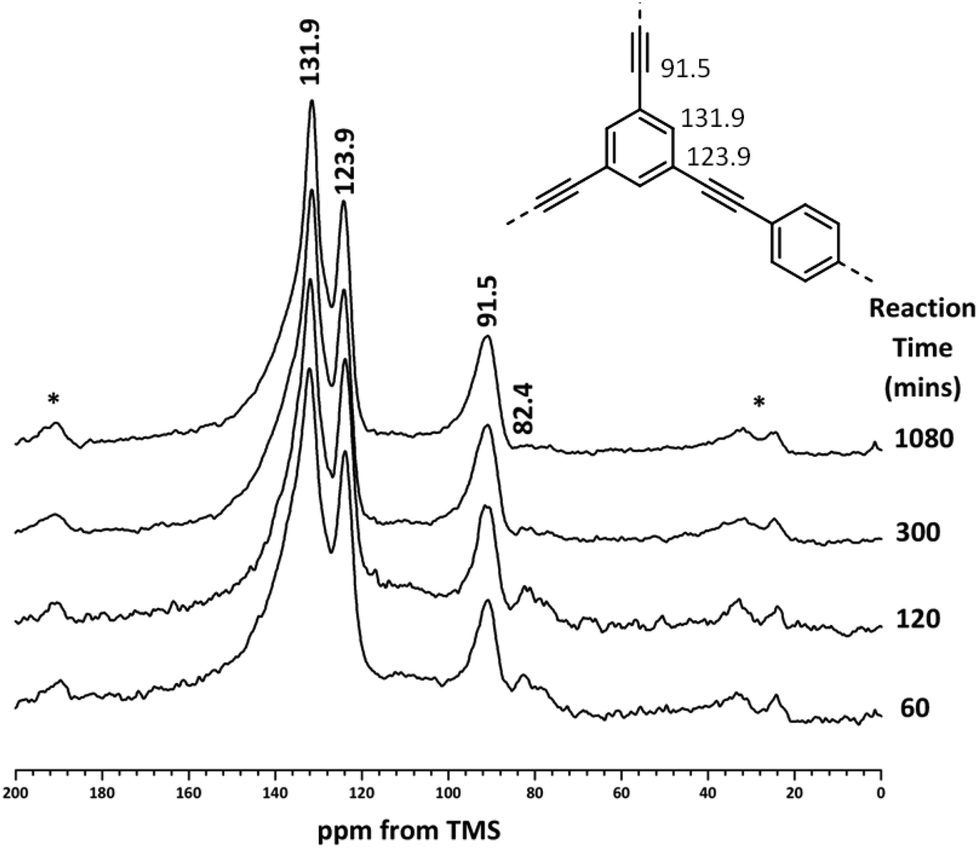 | ||
| Fig. 2 13C{1H} HPDEC MAS NMR spectra of CMP-1 materials recorded at an MAS rate of 10 kHz. Structure of CMP-1 labelled with peak assignments (inset). Asterisks denote spinning sidebands. | ||
As with previous CMP networks, we also observe a large shoulder resonance at ca. 137 ppm.9,11,12,28,38 Based upon the spectra of the monomers (see Fig. S4, ESI†), we can confidently assign the peak at ca. 137 ppm to an aromatic carbon (–CAr–H) adjacent to an alkyne group. This peak is also observed in homocoupled CMP networks (HCMPs),39 and suggests that homocoupling between alkyne groups occurs at later stages in the reaction once the bromine monomers have been consumed. This assumption is validated by the low percentage of bromine found in the EDX (Table 2) and the low levels of alkyne end groups in both FTIR and NMR data (Fig. 1 and 2, respectively).
In order to further probe the presence of terminal and polymerised alkyne groups, the ratio of peak intensities in the SPE 13C{1H} MAS NMR spectra were examined using two methods. The first involves calculating the ratio of peak intensities of aromatic to polymerised alkyne, i.e. the ratio of peaks at (131.9 + 123.9)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :91.5 ppm.9,28 The second method requires determination of the ratio of peak intensities for polymerised alkyne to end group alkyne, i.e. the ratio of peaks at 91.5:82.4 ppm.11,12
:91.5 ppm.9,28 The second method requires determination of the ratio of peak intensities for polymerised alkyne to end group alkyne, i.e. the ratio of peaks at 91.5:82.4 ppm.11,12
For a fully polymerised, idealised CMP-1 network (Fig. 3a; that is, not taking into account the actual experimental stoichiometry), a ratio of 3 dibromobenzenes and 2 triethynylbenzenes would be required for an ideal structure. In this idealized case, an aromatic:internal alkyne ratio of 1:0.40, and an internal alkyne:terminal end group alkyne ratio of 1:0 would be expected. However, we use a reaction stoichiometry so that triethynylbenzene is in excess (Fig. 3b). In this case, the expected aromatic:internal alkyne ratio would be 1:0.42 for the experimental stoichiometry without any homocoupling between terminal alkynes. As shown in Fig. 3b, with residual pendant terminal alkyne end groups, one possible permutation for this polymer would in fact be linear. While a perfect linear structure is highly improbable, a significant quantity of terminal alkyne groups is certainly to be expected. Indeed, if the Sonogashira–Hagihara coupling were to occur ‘perfectly’, with no side reactions, then one would expect a ratio of 1:0.20 for the internal alkyne:terminal end group alkyne ratio. The actual evolution of the experimental peak ratios with time are summarised in Table 3.
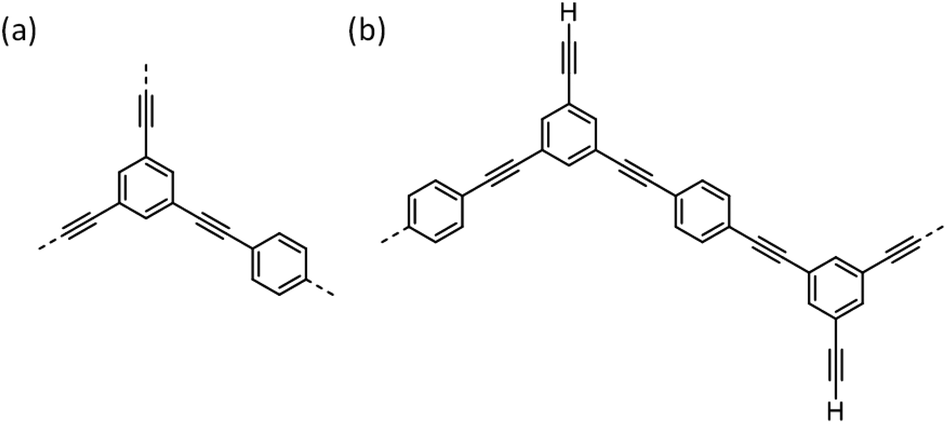 | ||
| Fig. 3 (a) Idealised network structures for CMP-1; (b) conceptual linear structure with terminal alkyne groups, taking into account the monomer stoichiometry. | ||
| Reaction time (min) | Deconvoluted peak area | Ratio 91.5:82.4 ppm |
Ratio (131.9 + 123.9):91.5 ppm |
|||
|---|---|---|---|---|---|---|
| 131.9 ppm | 123.9 ppm | 91.5 ppm | 82.4 ppm | |||
| a Predicted values based on structure shown in Fig. 3a. b Predicted values based on structure shown in Fig. 3b. | ||||||
| Predicteda | 1:0 |
1:0.40 |
||||
| Predictedb | 1:0.2 |
1:0.42 |
||||
| 60 | 4.20 | 3.60 | 2.16 | 0.86 | 1:0.40 |
1:0.28 |
| 120 | 3.39 | 3.56 | 2.14 | 0.80 | 1:0.37 |
1:0.30 |
| 300 | 3.80 | 3.07 | 2.34 | 0.11 | 1:0.05 |
1:0.34 |
| 1080 | 3.02 | 3.22 | 2.33 | 0.03 | 1:0.01 |
1:0.37 |
As expected, the peak area corresponding to alkyne end groups (82.4 ppm) decreases with increasing reaction time, and the polymerised, internal alkyne peak area (91.5 ppm) increases (Table 3). Hence, the ratio of internal alkyne to terminal end group alkynes increases significantly, from 1:0.40 at 60 minutes to 1:0.01 at 1080 minutes. The ratio of aromatic to polymerised, internal alkynes changes from 1:0.28 at 60 minutes to 1:0.37 at 1080 minutes, becoming closer to the ideal ratio of 0.40 with increasing reaction time. This observation suggests that the number of –C6H4– linkages increases with increasing reaction time. This is contrary to the stoichiometric expectation that we should observe a significant number of terminal alkyne end groups at the end of the reaction. In fact, the NMR spectrum at the end of the reaction looks extremely similar to those obtained for related CMPs where a stoichiometric balance of alkyne and halogen reactive groups was used.24,29,40
It therefore appears that Sonogashira–Hagihara cross-coupling is not the only reaction occurring here. It has been shown previously that alkyne–alkyne homocoupling can be used to prepare microporous networks.39,41 The solid-state NMR spectrum of these materials also shows a peak at ca. 136 ppm,39,41 which has been ascribed to alkene bonds, resulting from the formation of head-to-tail 1,3-disubstituted enynes.39 The reaction conditions used for the homocoupling are very similar to those used for Sonogashira–Hagihara coupling, so it is likely that both of these reactions are occurring simultaneously. We therefore hypothesise that alkyne homocoupling may be beneficial from the perspective of microporosity via the generation of a more highly cross-linked network in the late stages of the reaction.
These data show that insoluble, precipitated oligomers or polymers are formed at short reaction times. The relatively low yield of solid intermediates at this stage in the reaction seems to reflect particle size, as opposed to absolute yield, with the filtration failing to isolate all of the solid products. The isolated materials at short reaction times contain more unreacted end groups than at later stages, implying that heterogeneous coupling reactions occur in the solvent-swollen nascent network after the initial phase separation.
The nitrogen adsorption–desorption isotherms at 77 K for four polymers isolated at different reaction times are shown in Fig. 4. The polymers isolated at early reaction times show a low uptake of nitrogen. The isotherms are Type I, albeit with a very modest micropore step, with some Type IV character. At high relative pressure (P/P0 > 0.9) the isotherm steepens as a result of nitrogen adsorption in large mesopores and interparticulate spaces. H3/4 hysteresis is observed upon desorption. These data suggest that porosity in the polymers at early reaction times arises mainly from inter-particulate adsorption, rather than from micropores.
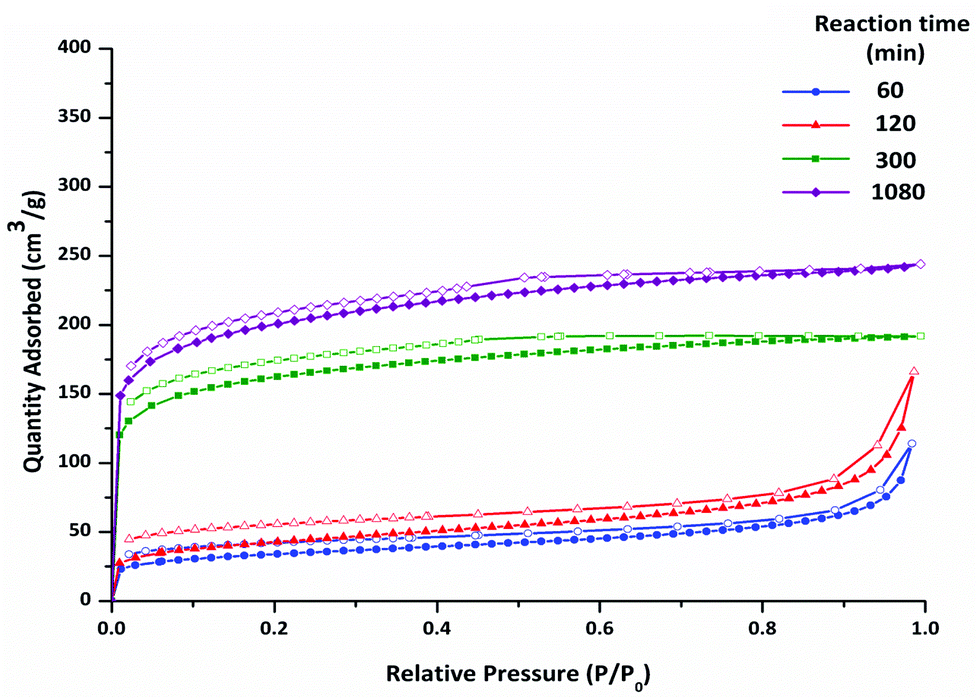 | ||
| Fig. 4 Nitrogen adsorption–desorption isotherms at 77 K for CMP-1 materials synthesised at varying reaction times (no offset). Adsorption (filled symbols), desorption (hollow symbols). | ||
At longer reaction times the networks display Type I isotherms, with slight hysteresis upon desorption. For these networks, a significantly larger uptake of gas is observed at low pressure (P/P0 < 0.1), compared with materials collected at 60 and 120 minutes. The gas uptake plateaus at high relative pressure (P/P0 > 0.9). These isotherms indicate microporous polymers, and they are similar to those observed for CMPs described previously.11,12 There is a significant difference in the gas sorption data for the networks isolated at 120 minutes and at 300 minutes, even though these materials are very similar on the basis of elemental analysis and ssNMR data. It is possible, therefore, that these differences in sorption arise from changes in the meso-structure, or that even small changes to the degree of crosslinking, which are difficult to assay by ssNMR, lead to substantial changes in microporosity.
We have previously used the ratio of the pore volume calculated at low relative pressure to the pore volume calculated at high relative pressure, V0.1/VTot, to estimate the level of microporosity in CMP networks.11,12 Non-porous materials display V0.1/VTot ratios that are close to zero. V0.1/VTot ratios closer to 1 indicate highly microporous materials. As shown Table 4, V0.1/VTot ratios of less than 0.21 were found for the polymers collected before 300 minutes. Such values suggest that the majority of gas sorption for these materials arises from interparticulate mesoporosity or macroporosity. Mesoporosity is also common for polymers where agglomerated structures are formed during liquid–liquid phase separation.42 A V0.1/VTot ratio of 0.77 was found for the polymer collected at 300 minutes, with the polymer formed after 1080 minutes having a V0.1/VTot ratio of 0.66. These values are similar to those reported previously for other CMP-1 networks.9,11,12,28
| Reaction time (min) | SABETa (m2 g−1) | V 0.1 (cm3 g−1) | V Tot (cm3 g−1) | V 0.1/VTot |
|---|---|---|---|---|
| a Based on an isotherm pressure range of 0.06–0.12. b Pore volume at P/P0 = 0.1. c Total pore volume at P/P0 = 0.98. Data collected at 77 K using N2 as the sorbate. | ||||
| 60 | 123 | 0.04 | 0.18 | 0.22 |
| 120 | 153 | 0.06 | 0.29 | 0.21 |
| 300 | 755 | 0.23 | 0.30 | 0.77 |
| 1080 | 733 | 0.23 | 0.38 | 0.61 |
Non-local density functional theory (NL-DFT) was used to calculate pore sizes and this provides further confirmation of the pore size distributions implied by the gas sorption isotherms. NL-DFT was chosen as it can be used to calculate pores over a wide range of sizes (ultramicropore to macropores).43 NL-DFT has also been used for pore size determination of CMPs and similar materials,11,12,31,37,39,44,45 allowing a comparison between the materials in this work with those that are published elsewhere. Pore size distribution curves are shown in Fig. 5. No data below 10 Å are shown on the NL-DFT plots because there is a lack of experimental points at low pressure. For reaction products collected before 300 minutes, pore sizes of 300–450 Å were suggested by the model. These pore sizes fall within the large mesopore range. Polymers collected after 300 minutes display pore sizes within the micropore range at ca. 20 Å. The pore sizes of materials collected after 300 minutes are similar to those reported for other CMP-1 networks9,11,12 although it should be noted that there are slight apparent differences in pore size distributions which can be explained by the difference in partial pressures over which the data were collected (see above).
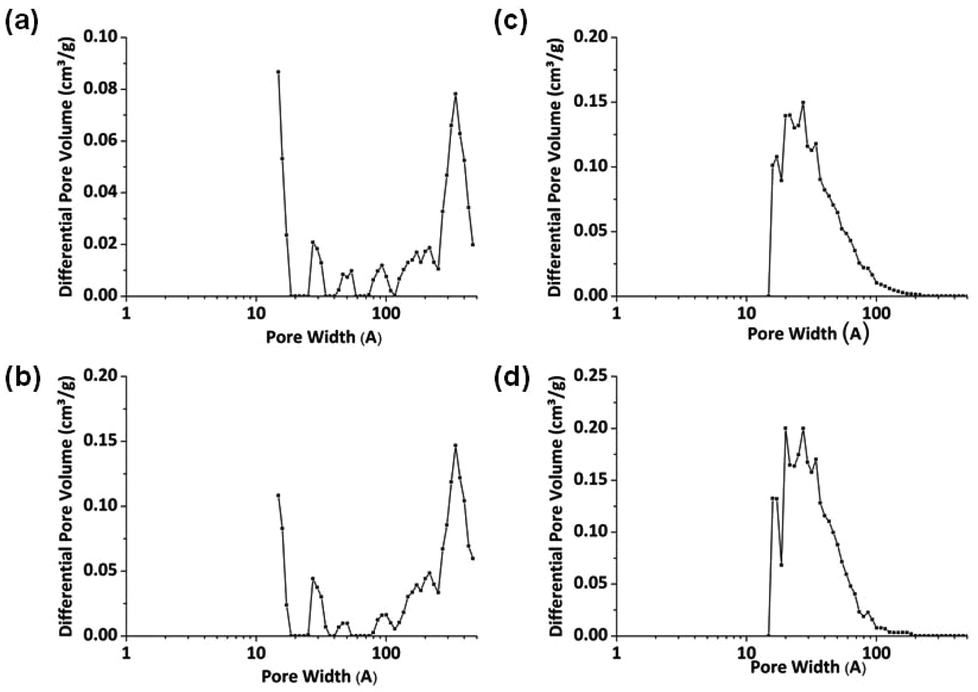 | ||
| Fig. 5 NL-DFT pore size distributions of CMP-1 intermediates synthesised at varying reaction times: (a) 60 min, (b) 120 min, (c) 300 min, and (d) 1080 min. The NL-DFT model for slit pores was used in the calculations. | ||
We further investigated the polymers by scanning electron microscopy (SEM). For CMP networks, a strong link exists between polymer morphology and gas sorption properties.12 Networks prepared in toluene were generally mesoporous and showed the presence of smooth, spherical morphologies by SEM. When synthesised in DMF, the same networks were significantly more microporous and SEM showed that these networks consisted of larger fused masses with rough surfaces. We concluded that the change in morphology was a consequence of different solubility of reaction intermediates in the solvent. Indeed, spherical morphologies have been reported for aromatic polymers that undergo liquid–liquid phase separation (i.e., premature precipitation of oligomers) during their synthesis. Spherical structures are also observed just after the gel point for rigid polymers such as polyamides.46 In this work, polymers collected at 60 and 120 minutes exhibit interparticulate mesoporosity and consist of fused-spherical morphologies with smooth surfaces (Fig. 6). These morphologies are similar to the CMP networks prepared in toluene.12 However, the polymers collected at 300 and 1080 minutes display larger particles with rough surfaces (Fig. 6). These results are akin to the previously reported CMP-1 synthesised in DMF.12 Hence, the differences in porosity between the polymers isolated at 120 minutes and 300 minutes seems to correlate well with the differences in morphology, as evidenced by SEM, as opposed to the chemical composition. It is not clear, however, whether these changes in morphology affect the microporosity in the materials or whether, perhaps more likely, that these morphology changes introduce effects in the mesopore region that are overlaid with changes to the micropore volume arising from crosslinking at the molecular level.
 | ||
| Fig. 6 SEM images of CMP-1 materials collected at reaction times of: (1) 60 min, (2) 120 min, (3) 300 min, and (4) 1080 min. Scale bars are 2 μm (left images) and 20 μm (right images). | ||
Aharoni and Edwards have discussed in detail the formation of networks from rigid polymers.47 A morphological change from spherical structures to a three-dimensional ensemble of polyhedra is expected when, at the gel point, residual monomers and oligomers are pushed into the interstitial volumes of the fractal polymers, which results in gelation. Hence, spherical structures become deformed as further monomer attachment occurs in the interstitial regions. However, this suggests that the size distribution of the resulting polymer structures should correspond to the distribution of the spherical precursors. Here, we observe instead an apparent evolution in size of the aggregates. Hence, from the combination of the microscopy and sorption data, we postulate the following mechanism. During the initial stages of the reaction (0–30 min), all starting materials are soluble. Short oligomers start to form, as soluble structures or as sufficiently small precipitates or microgels such that they are not easily removed by filtration. Once the oligomers reach a specific molecular weight, gelation of the reaction mixture occurs (∼40 min) and phase separation leads to the formation of insoluble spherical particles, as seen by SEM. At times shortly after gelation (40–120 min), there is insufficient crosslinking in the network to generate much permanent microporosity. However, further homo- and hetero-polymerisation takes place between catalyst-activated end groups in the oligomer-rich droplets (>300 min), and this results in a higher degree of intra-particle cross-linking, and also reaction between particles, thus generating a CMP network that remains microporous after desolvation.
Conclusions
The mechanism of network formation for CMP-1 has been investigated in detail. Polymers collected before 120 minutes are composed of spherical particles exhibiting interparticulate mesoporosity, while materials collected after this time consist of fused particles exhibiting microporosity in the particles themselves. Based upon the change in textural properties, a reaction mechanism for the formation of CMP-1 networks is suggested. The proposed mechanism involves formation of oligomers in solution that react to give clusters. These clusters then precipitate from solution and continue to react in the solid-state by cross-linking, ultimately leading to the formation of extended CMP-1 networks (Scheme 2). The initially precipitated material exhibits a low degree or microporosity and significant inter-particulate mesoporosity; true microporous materials are only formed at longer times upon fusion of the clusters and further crosslinking within the particles.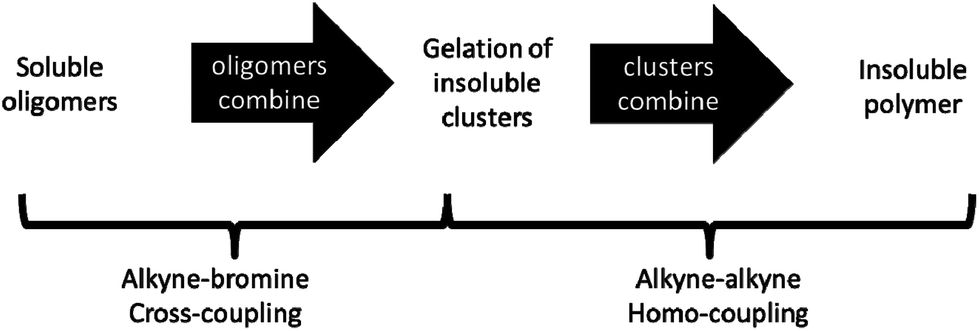 | ||
| Scheme 2 Proposed reaction mechanism for the formation of CMP networks. | ||
This mechanism was validated by chemical and structural analyses. Particular attention was paid to the identification and quantification of end groups. All materials contain low concentrations of alkyne end groups, as evidenced by FTIR and solid-state NMR. Considering the reaction stoichiometry, this suggests that homo-coupling between alkynes occurs in addition to the primary Sonogashira–Hagihara coupling reaction. The exact mechanism and homocoupled product are unclear, but this process may lead to the formation of alkenic bonds. It is intuitive that an excess of the bromine monomer would be detrimental to porosity formation, since bromine–bromine homocoupling is less likely to occur under these reaction conditions. However, the precise reason for the maximization of surface area with excess alkyne monomer is still unclear. In principle, alkyne–bromine cross-coupling can also occur in the phase separated state. As such, an equimolar ratio of alkynes and bromines should still favour extended network formation. However, experiments show, consistently, that lower surface areas are attained with equimolar monomer ratios.9,12,28 Since this reaction involves precipitation of intermediate species, the factors that influence the solubility of these species are likely to affect the final polymer network morphology. It is possible, therefore, that excess alkyne end groups influence the early-stage phase separation process in a way that maximizes porosity.
A key question is whether these side reactions are driven by our choice of reaction stoichiometry. We note here that the presence of a broad shoulder can also be observed at approximately 137 ppm in the work of others where a stoichiometric balance of alkyne and halogenated monomers is used.29,40 Hence, we suspect that these side reactions might be inherent to the system, and present to some degree whether or not an excess of alkyne is used.
To conclude, while these polymerisation reactions are superficially simple, it should be understood that factors such as concentration, solvent, temperature, and monomer structure will affect the network formation in complex, interrelated ways. Hence, optimising the network properties and pore structure requires control over these experimental conditions. This suggests that a single, generic set of reaction conditions is unlikely to be optimal for all possible monomer combinations.
Acknowledgements
We thank the EPSRC for funding (EP/F057865/1 and EP/H000925). We thank the EPSRC and E.ON for funding (EP/C511794/1) through the E.ON−EPSRC strategic call on CCS. A.I.C. is a Royal Society Wolfson Merit Award holder.Notes and references
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS PubMed.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS.
- F. Svec, J. Germain and J. M. J. Frechet, Small, 2009, 5, 1098–1111 CrossRef PubMed.
- T. A. Makal, J. R. Li, W. G. Lu and H. C. Zhou, Chem. Soc. Rev., 2012, 41, 7761–7779 RSC.
- R. Dawson, A. I. Cooper and D. J. Adams, Polym. Int., 2013, 62, 345–352 CrossRef CAS.
- Y. H. Xu, S. B. Jin, H. Xu, A. Nagai and D. L. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- D. C. Wu, F. Xu, B. Sun, R. W. Fu, H. K. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959–4015 CrossRef CAS PubMed.
- Q. Liu, Z. Tang, M. Wu and Z. Zhou, Polym. Int., 2014, 63, 381–392 CrossRef CAS.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- R. Dawson, A. Laybourn, R. Clowes, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Macromolecules, 2009, 42, 8809–8816 CrossRef CAS.
- R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Macromolecules, 2010, 43, 8524–8530 CrossRef CAS.
- R. Dawson, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1173–1177 RSC.
- R. Dawson, E. Stöckel, J. R. Holst, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2011, 4, 4239–4245 CAS.
- J.-X. Jiang, C. Wang, A. Laybourn, T. Hasell, R. Clowes, Y. Z. Khimyak, J. Xiao, S. J. Higgins, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2011, 50, 1072–1075 CrossRef CAS PubMed.
- Q. Chen, M. Luo, T. Wang, J.-X. Wang, D. Zhou, Y. Han, C.-S. Zhang, C.-G. Yan and B.-H. Han, Macromolecules, 2011, 44, 5573–5577 CrossRef CAS.
- K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Angew. Chem., Int. Ed., 2013, 52, 1432–1436 CrossRef CAS PubMed.
- P. Zhang, Z. Weng, J. Guo and C. Wang, Chem. Mater., 2011, 23, 5243–5249 CrossRef CAS.
- S. Fischer, A. Schimanowitz, R. Dawson, I. Senkovska, S. Kaskel and A. Thomas, J. Mater. Chem. A, 2014, 2, 11825–11829 CAS.
- W. Z. Yuan, R. Hu, J. W. Y. Lam, N. Xie, C. K. W. Jim and B. Z. Tang, Chem. – Eur. J., 2012, 18, 2847–2856 CrossRef CAS PubMed.
- J. Chun, J. H. Park, J. Kim, S. M. Lee, H. J. Kim and S. U. Son, Chem. Mater., 2012, 24, 3458–3463 CrossRef CAS.
- B. Kiskan and J. Weber, ACS Macro Lett., 2011, 1, 37–40 CrossRef.
- J. L. Novotney and W. R. Dichtel, ACS Macro Lett., 2013, 2, 423–426 CrossRef CAS.
- Z. J. Yan, H. Ren, H. P. Ma, R. R. Yuan, Y. Yuan, X. Q. Zou, F. X. Sun and G. S. Zhu, Microporous Mesoporous Mater., 2013, 173, 92–98 CrossRef CAS PubMed.
- X. Han, L. Li, Z. Huang, J. Liu and Q. Zheng, Chin. J. Chem., 2013, 31, 617–623 CrossRef CAS.
- Q. Liu, Z. Tang, M. Wu and Z. Zhou, Polym. Int., 2014, 63, 381–392 CrossRef CAS.
- T. Ben, H. Ren, S. Q. Ma, D. P. Cao, J. H. Lan, X. F. Jing, W. C. Wang, J. Xu, F. Deng, J. M. Simmons, S. L. Qiu and G. S. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef CAS PubMed.
- D. Z. Tan, W. J. Fan, W. N. Xiong, H. X. Sun, Y. Q. Cheng, X. Y. Liu, C. G. Meng, A. Li and W. Q. Deng, Macromol. Chem. Phys., 2012, 213, 1435–1440 CrossRef CAS.
- A. E. Bennett, C. M. Rienstra, M. Auger, K. V. Lakshmi and R. G. Griffin, J. Chem. Phys., 1995, 103, 6951–6958 CrossRef CAS PubMed.
- N. Kang, J. H. Park, K. C. Ko, J. Chun, E. Kim, H. W. Shin, S. M. Lee, H. J. Kim, T. K. Ahn, J. Y. Lee and S. U. Son, Angew. Chem., Int. Ed., 2013, 52, 6228–6232 CrossRef CAS PubMed.
- S. Yuan, B. Dorney, D. White, S. Kirklin, P. Zapol, L. Yu and D.-J. Liu, Chem. Commun., 2010, 46, 4547–4549 RSC.
- M. G. Schwab, B. Fassbender, H. W. Spiess, A. Thomas, X. L. Feng and K. Mullen, J. Am. Chem. Soc., 2009, 131, 7216–7217 CrossRef CAS PubMed.
- M. G. Schwab, M. Hamburger, X. L. Feng, J. Shu, H. W. Spiess, X. C. Wang, M. Antonietti and K. Mullen, Chem. Commun., 2010, 46, 8932–8934 RSC.
- D. Yuan, W. Lu, D. Zhao and H.-C. Zhou, Adv. Mater., 2011, 23, 3723–3725 CrossRef CAS PubMed.
- A. Wilke and J. Weber, J. Mater. Chem., 2011, 21, 5226–5229 RSC.
- H. Lee, H. W. Park and J. Y. Chang, Macromol. Res., 2013, 21, 1274–1280 CrossRef CAS PubMed.
- E. Stöckel, X. F. Wu, A. Trewin, C. D. Wood, R. Clowes, N. L. Campbell, J. T. A. Jones, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Chem. Commun., 2009, 212–214 RSC.
- J. X. Jiang, F. Su, H. Niu, C. D. Wood, N. L. Campbell, Y. Z. Khimyak and A. I. Cooper, Chem. Commun., 2008, 486–488 RSC.
- D. Z. Tan, W. N. Xiong, H. X. Sun, Z. Zhang, W. Ma, C. G. Meng, W. J. Fan and A. Li, Microporous Mesoporous Mater., 2013, 176, 25–30 CrossRef CAS PubMed.
- D. Z. Tan, W. J. Fan, W. N. Xiong, H. X. Sun, A. Li, W. Q. Deng and C. G. Meng, Eur. Polym. J., 2012, 48, 705–711 CrossRef CAS PubMed.
- K. Kimura, S. Kohama and S. Yamazaki, Polym. J., 2006, 38, 1005–1022 CrossRef CAS.
- J. Landers, G. Y. Gor and A. V. Neimark, Colloids Surf., A, 2013, 437, 3–32 CrossRef CAS PubMed.
- A. Laybourn, R. Dawson, R. Clowes, J. A. Iggo, A. I. Cooper, Y. Z. Khimyak and D. J. Adams, Polym. Chem., 2012, 3, 533–537 RSC.
- A. Bhunia, V. Vasylyeva and C. Janiak, Chem. Commun., 2013, 49, 3961–3963 RSC.
- S. M. Aharoni, N. S. Murthy, K. Zero and S. F. Edwards, Macromolecules, 1990, 23, 2533–2549 CrossRef CAS.
- S. M. Aharoni and S. F. Edwards, Adv. Polym. Sci., 1994, 118, 1–231 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Further NMR data and hypothetic polymer networks. See DOI: 10.1039/c4py00647j |
| This journal is © The Royal Society of Chemistry 2014 |