 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amphiphilic block copolymers featuring a reversible hetero Diels-Alder linkage†
Marcel
Langer
ab,
Josef
Brandt
c,
Albena
Lederer
c,
Anja S.
Goldmann
ab,
Felix H.
Schacher
*d and
Christopher
Barner-Kowollik
*ab
aSoft Matter Synthesis Laboratory, Institut für Biologische Grenzflächen, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: christopher.barner-kowollik@kit.edu
bPreparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128 Karlsruhe, Germany
cLeibniz Institut für Polymerforschung, Dresden e.V., Hohe Straße 6, 01069 Dresden, GermanyTechnische Universität Dresden, 01062 Dresden, Germany
dInstitut für Organische Chemie und Makromolekulare Chemie and Jena Center for Soft Matter, Friedrich Schiller Universität Jena, 07743 Jena, Germany. E-mail: felix.schacher@uni-jena.de
First published on 3rd June 2014
Abstract
The present article reports the preparation of a novel class of switchable amphiphilic diblock copolymers with a temperature switchable linkage. Reversible addition fragmentation chain transfer (RAFT) polymerization was used to synthesize the individual blocks: for the preparation of the non-polar block, i.e. poly(isoprene-co-styrene) (P(I-co-S)) (9200 g mol−1 ≤ Mn ≤ 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, 1.22 ≤ Đ ≤ 1.36), a chain transfer agent (CTA, 3-((2-bromo-2-methylpropanoyl)oxy)propyl 2-(((dodecylthio)carbonothioyl)thio)-2-methylpropanoate) carrying a bromine group was employed, ready for subsequent cyclopentadienyl (Cp) transformation. For the preparation of the polar block, triethylene glycol methyl ether acrylate (TEGA) was polymerized (6600 g mol−1 ≤ Mn ≤ 35000 g mol−1, 1.12 ≤ Đ ≤ 1.30) using a RAFT agent carrying a phosphoryl Z-group, which is able to undergo hetero Diels-Alder (HDA) ligation with Cp moieties. Both building blocks were conjugated at ambient temperature in the presence of ZnCl2 as catalyst yielding the amphiphilic block copolymer P(I-co-S)-b-PTEGA (16000 g mol−1 ≤ Mn ≤ 68000 g mol−1, 1.15 ≤ Đ ≤ 1.32). To investigate the bonding/debonding capability of the HDA linkage, high temperature nuclear magnetic resonance (HT-NMR) spectroscopy, high temperature dynamic light scattering (HT-DLS) and high temperature size exclusion chromatography (HT-SEC) were carried out, evidencing that efficiently switchable amphiphilic block copolymers were generated (>4 cycles).
000 g mol−1, 1.22 ≤ Đ ≤ 1.36), a chain transfer agent (CTA, 3-((2-bromo-2-methylpropanoyl)oxy)propyl 2-(((dodecylthio)carbonothioyl)thio)-2-methylpropanoate) carrying a bromine group was employed, ready for subsequent cyclopentadienyl (Cp) transformation. For the preparation of the polar block, triethylene glycol methyl ether acrylate (TEGA) was polymerized (6600 g mol−1 ≤ Mn ≤ 35000 g mol−1, 1.12 ≤ Đ ≤ 1.30) using a RAFT agent carrying a phosphoryl Z-group, which is able to undergo hetero Diels-Alder (HDA) ligation with Cp moieties. Both building blocks were conjugated at ambient temperature in the presence of ZnCl2 as catalyst yielding the amphiphilic block copolymer P(I-co-S)-b-PTEGA (16000 g mol−1 ≤ Mn ≤ 68000 g mol−1, 1.15 ≤ Đ ≤ 1.32). To investigate the bonding/debonding capability of the HDA linkage, high temperature nuclear magnetic resonance (HT-NMR) spectroscopy, high temperature dynamic light scattering (HT-DLS) and high temperature size exclusion chromatography (HT-SEC) were carried out, evidencing that efficiently switchable amphiphilic block copolymers were generated (>4 cycles).
Introduction
Amphiphilic block copolymers are composed of at least one hydrophilic and one hydrophobic block, which are typically covalently bonded. Such macromolecules find many applications, e.g. in drug delivery systems,1,2 membrane filtration and separation systems3 and electronic materials,4 based on their ability to phase separate in solution as well as in the solid state. In general, many different architectures can be realized, e.g. linear, star shaped or comb like block copolymers.5 In general, unlike polymer segments are not miscible but the covalent linkage present in block copolymers prevents macroscopic phase separation. Thus, block copolymers undergo microphase separation on the length scale of the constituting building blocks.6,7 Linear AB diblock copolymers are the simplest block copolymers and have been studied extensively. It was found that depending on the volume fraction of the blocks, the materials form spherical, cylindrical, gyroid, or lamellar nanostructures.8 Block copolymers with a more complex architecture (e.g. ABC triblock terpolymers) can form even more sophisticated nanostructures, including perforated lamellae9 and helices.10,11 In light of lithography applications, block copolymers with one block which can be selectively removed are highly interesting.12 Since 1988,13 various strategies have developed, e.g. degradation via UV radiation,14 chemical etching,15 or ozonolysis.16 However, either relatively harsh reaction conditions are required or the method is limited to specific materials. These drawbacks can be avoided by the introduction of an efficiently cleavable junction between adjacent blocks and the use of distinct differences in solubility (in case of amphiphilic block copolymers).17,18 However, amphiphilic block copolymers featuring a temperature-reversible linkage have not been reported so far.For the synthesis of block copolymers, two general approaches are possible: either sequential polymerization of several monomers or modular ligation of pre-synthesized polymeric building blocks can be targeted.19 Both preparation methods require living polymerization techniques and high end group fidelity, or at least chain growth polymerization techniques featuring living characteristics. Living anionic polymerization is the most powerful technique in terms of polymerization control for the synthesis of polymers with defined molecular weight, architectures and narrow molecular weight distributions.20 However, substantial restrictions apply to anionic polymerization such as a limited monomer choice associated with the required demanding experimental conditions.21 In comparison, controlled/living radical polymerization (CLRP) techniques are simpler to implement and have less limitations regarding the monomer choice.22Via such approaches, good control over molecular weight, architecture, and the molecular weight distribution is obtained. The most common CLRP techniques are nitroxide mediated polymerization (NMP),23,24 atom transfer radical polymerization (ATRP),25–27 and reversible addition chain transfer (RAFT)28,29 polymerization. In order to achieve the successful modular conjugation of polymers, reactions adhering to the strict click criteria30 are required. Barner-Kowollik and co-workers demonstrated that under certain conditions the hetero Diels-Alder (HDA) reaction between a cyclopentadienyl (Cp) moiety and a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S double bond of RAFT agents with an electron withdrawing Z group, fulfills these criteria.31 More recently, the same group demonstrated that the HDA linkage can undergo efficient bonding and debonding behavior via a mild temperature gradient.32
S double bond of RAFT agents with an electron withdrawing Z group, fulfills these criteria.31 More recently, the same group demonstrated that the HDA linkage can undergo efficient bonding and debonding behavior via a mild temperature gradient.32
Herein we present the modular synthesis of amphiphilic diblock copolymers via HDA ligation chemistry with the aim of creating reversibly cleavable block junctions. These might be promising candidates for the design of nanostructured materials.12 As hydrophobic block, a copolymer constituted of styrene and isoprene was chosen as the residual vinyl groups of isoprene in the side chain can be used for subsequent crosslinking. Thus, the mechanical stability and the persistence against solvents of the generated material can be increased. As hydrophilic segment, a polymer with an acrylate backbone is required, since the HDA–RAFT agent is not able to undergo HDA conjugations when a methacrylate monomer is employed in the RAFT polymerization (however, the reverse functions by using a Cp-terminal poly(methacrylate) and a RAFT terminal poly(acrylate)). Moreover, RAFT agents may be sensitive towards amide and amine moieties. Therefore, poly(triethylene glycol methyl ether acrylate) (PTEGA) is employed and both building blocks are combined using HDA chemistry. The reversible HDA linkage enables the cleavage of the resulting amphiphilic block copolymer, potentially also after microphase separation and crosslinking. Thus, this will open up the way to a novel class of nanostructured materials with highly reactive Cp moieties on the surface. We investigate the reversible linkage between both blocks via high temperature nuclear magnetic resonance (HT-NMR) spectroscopy, high temperature dynamic light scattering (HT-DLS), and high temperature size exclusion chromatography (HT-SEC).
The experimental data, including the used materials, synthesis procedures and characterization methods, can be found in the ESI.†
Results and discussion
In order to obtain amphiphilic block copolymers with a reversible hetero Diels-Alder (HDA) linkage, the synthetic strategy depicted in Scheme 1 was followed. For the preparation of the polar building block polymer, triethylene glycol methyl ether acrylate (TEGA)35 was polymerized using a HDA capable RAFT agent.36 The non-polar building block was synthesized via RAFT polymerization with a bromine carrying CTA (DMP-Br). After the polymerization, the bromine end group was substituted with a Cp moiety. Subsequently, both building blocks were ligated via a HDA reaction to form the amphiphilic block copolymer P(S-co-I)-b-PTEGA.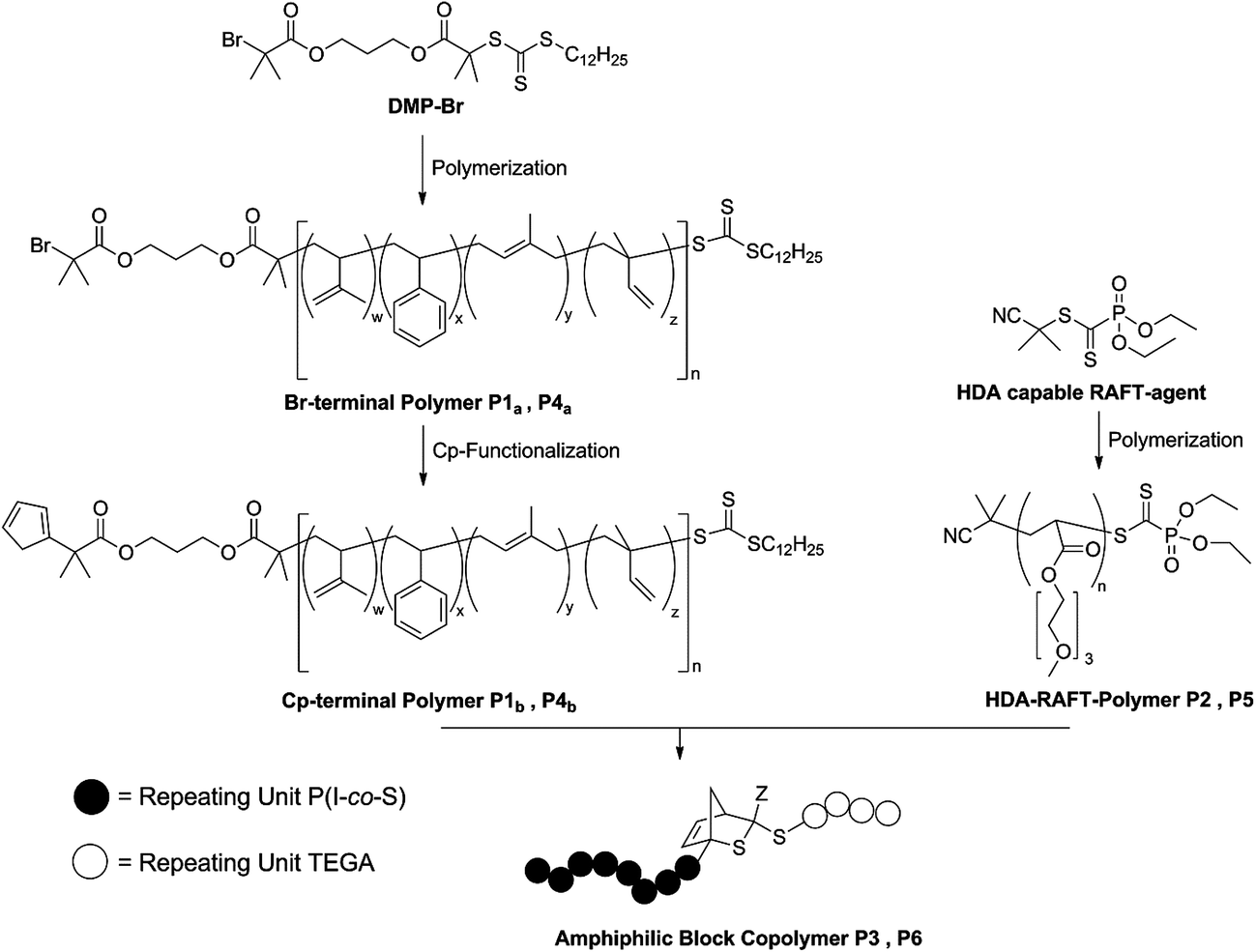 | ||
| Scheme 1 Synthetic strategy for the preparation of amphiphilic P(S-co-I)-b-PTEGA block copolymers with a reversible hetero Diels-Alder linkage. | ||
RAFT agent design
The newly developed RAFT agent DMP-Br is based on the well-known CTA DMP.37,38 It is based on a trithiocarbonate with a tertiary R-group, able to control the polymerizations of styrene and isoprene.39,40 The facile preparation via a Steglich esterification involves 3-hydroxypropyl-2-bromo-2-methylpropanoate33 to introduce a bromine functionality at the R-group. The bromine substituted R-group can act as an initiator for atom transfer radical polymerizations (ATRP)34 and can further be substituted with a Cp moiety.41 Thus, complex block copolymers can be prepared by simultaneous RAFT and ATRP.42 The molecule was characterized via NMR (1H and 13C, see Fig. S1 and S2†) and ESI-MS (see Fig. S3†).RAFT polymerizations
For the modular block copolymer formation, high end group fidelity of both building blocks is essential. Among the controlled/living radical polymerization techniques, RAFT provides not only high end group fidelity, but concomitantly benefits from higher radical concentrations, compared to nitroxide mediated polymerization (NMP) and atom transfer radical polymerizations (ATRP).43 Thus, higher polymerization rates and shorter reaction times are accessible. To verify the control afforded by the RAFT process leading to polymers with high end group fidelity, kinetic studies were carried out for the copolymerization of styrene and isoprene using CTA-1 (see Fig. S4, S5 and Table S1†) and the polymerization of TEGA using CTA-2 (see Fig. S6, S7 and Table S2†). The linear increase of the molar mass with good agreement of Mn,theo and Mn,exp and low Đ values evidences the control of the polymerizations. For each building block (non-polar side with P(S-co-I) and polar with PTEGA) two molecular weights have been generated, one lower (P(S-co-I): P1, Mn = 9200 g mol−1; PTEGA: P2, Mn = 6600 g mol−1) and one higher example (P(S-co-I): P4, Mn = 50000 g mol−1; PTEGA: P5, Mn = 35000 g mol−1). Due to smaller repeating unit to end group ratio and better solubility features, P1 and P2 were subjected to a detailed NMR analysis and DLS experiments, whereas the large differences in solubility of P4 and P5 were used for macroscopic separation experiments (will be discussed later).
Cp-transformation
In general, two approaches for the substitution of bromine with Cp exist. The use of sodium cyclopentadienyl as source for nucleophilic Cp entails a high reactivity of the Cp-anion towards functional groups such as ester moieties.44,45 Since two ester moieties are present in the polymer, the NaCp based approach for the Cp transformation is not suitable. Nickelocene (NiCp2) as nucleophilic source, on the other hand, is chosen as a mild and effective transformation that tolerates a wide range of functional groups, including ester moieties.46 Since it is known that Cp groups undergo dimerization,47 the comparison of the SEC traces before and after the functionalization of the polymers P1 and P4 is essential (see Fig. S8† for polymer P1 and Fig. S9† for polymer P4). The SEC traces of both polymers, P1 and P4, do not show any side products due to dimerization. The successful substitution of bromine with Cp can be verified by the resonances appearing between 6.5 ppm and 6.1 ppm associated with the vinyl protons of the Cp moiety in the 1H-NMR spectra of polymer P1b (see Fig. 2). Full conversion – and therefore close to quantitative Cp transformation – was assessed by the successful conjugation of the building blocks P1b and P2 verified via SEC.Block copolymer formation
All conjugation reactions were conducted in ethyl acetate at ambient temperature in the presence of ZnCl2 as catalyst. It is not mandatory to use a catalyst for the polymer conjugation (refer to the HT-SEC analysis section). However, more than seven times higher reaction rates are found, especially for larger building blocks. The total concentration of polymers was kept at 50 mg mL−1 for the preparation of P3 and P6. The SEC traces for the generated block copolymers and the corresponding building blocks are presented in Fig. 1.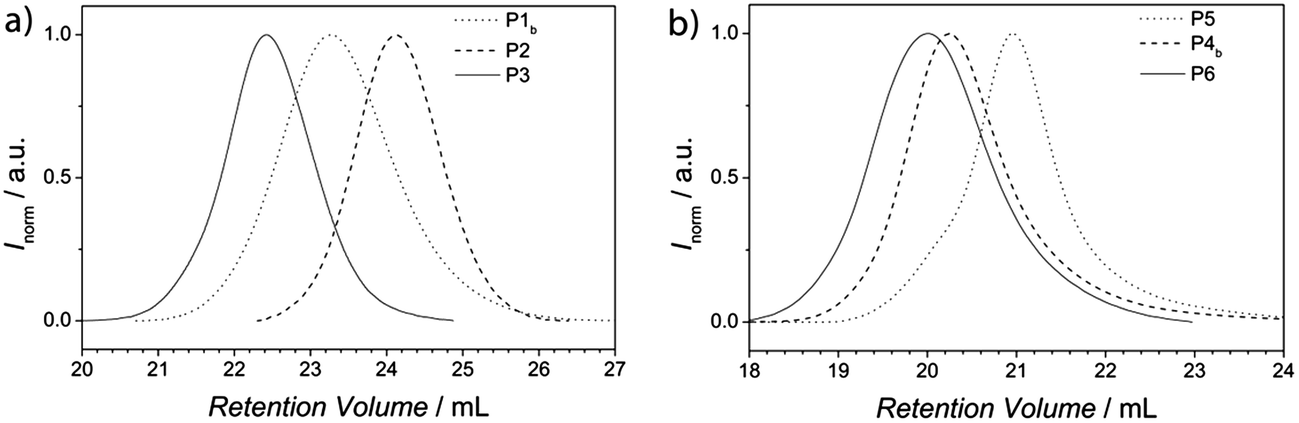 | ||
| Fig. 1 (a) SEC traces of the polar building block PTEGA (P2, dashed line, Mn = 6600 g mol−1 (PMMA calibration), Đ = 1.12), the non-polar building block P(S-co-I) (P1b, dotted line, Mn = 9200 g mol−1 (PS calibration), Đ = 1.22) and the resulting block copolymer P(S-co-I)-b-PTEGA (P3, solid line, Mn = 16000 g mol−1 (PS calibration), Đ = 1.15). (b) SEC traces of the polar building block PTEGA (P5, dotted line, Mn = 35000 g mol−1 (PMMA calibration), Đ = 1.30), the unpolar building block P(S-co-I) (P4b, dashed line, Mn = 50000 g mol−1 (PS calibration), Đ = 1.36) and the resulting block copolymer P(S-co-I)-b-PTEGA (P6, solid line, Mn = 68000 g mol−1 (PS calibration), Đ = 1.32). | ||
As expected, the resulting block copolymer P3 shows a significant shift to lower retention volume, compared to the building blocks P1b and P2. Moreover, the Đ value of P3 (Đ = 1.15) is in between the values of the building blocks P1b and P2 (P1b: Đ = 1.22 and P2: Đ = 1.12). The low Đ value and the shift to a lower retention volume indicate the successful conjugation and thus, close to quantitative Cp-transformation of the building block P1.48,49 For the block copolymer with higher molecular weight (P6), the shift to lower retention volumes, compared to its building blocks P4b and P5, is less pronounced than for polymer P3. This observation is associated with the fact that high molecular weight polymers have a small retention volume and therefore have a larger effect on the resulting molecular weight shift after ligation compared to polymers with lower molecular weights. Thus, the shift of the block copolymer compared to its building blocks is in the expected range. Furthermore, the Đ value of P6 (Đ = 1.32) is in between the values of the building blocks P4b and P5 (P4b: Đ = 1.36 and P5: Đ = 1.30). Again, low Đ values and the shift to lower retention volumes indicate a successful and quantitative conjugation. In addition, P1, P2 and P3 were analyzed via1H-NMR spectroscopy (see Fig. S10†). The spectrum of the block copolymer P3 is the sum of the resonances from the individual building block polymers P1b and P2. The molar ratio of PTEGA and P(S-co-I) in the resulting block copolymer can be determined by comparison of the integrals from the resonances of the individual building blocks. A table of the calculated ratios from both block copolymers and the content of isoprene is shown in the ESI (see Table S3†). Inspection of the 1H-NMR spectra of the polymers P1b and P3 in the region of the vinyl moiety (6.7 ppm–5.2 ppm, see Fig. 2) clearly indicates the strong decrease of the resonances of the Cp moiety from polymer P1b after the HDA reaction. Consequently, the block copolymer P3 reveals a new resonance, corresponding to a proton of the HDA reaction product. The two resonances at 6.16 ppm and 5.72 ppm of the Cp moiety and the HDA reaction product are essential for following the bonding and debonding of the hetero Diels-Alder linkage by HT-NMR analysis.
 | ||
| Fig. 2 1H-NMR spectra of the relevant region (500 MHz, CDCl3, ambient temperature) of polymer P1 before (P1a) the Cp-transformation, after (P1b) the Cp-transformation and of the block copolymer P3. Comparing the spectra of P1a and P1b, the resonance of the cyclopentadienyl moiety can be identified (left dashed line). After the block copolymer formation, the Cp-associated resonance disappears and an additional resonance associated with the DA reaction product in a different region is visible (P3, right dashed line). An enlargement of the relevant peaks employed for HT-NMR (recorded in toluene-d6/DMSO-d6) is depicted for the 25 °C sample in Fig. 3. | ||
HT-NMR analysis
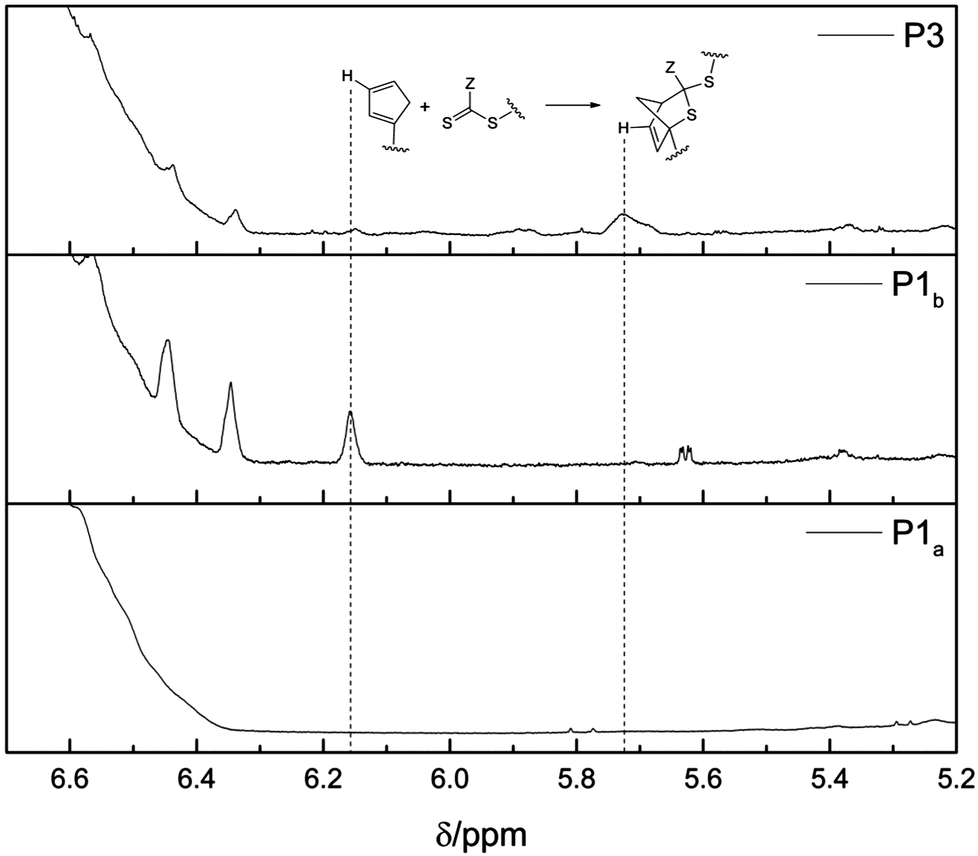
A (H)DA reaction is at all times in an equilibrium with its retro (H)DA reaction. At ambient temperature, the equilibrium of the HDA reaction of P1b with P2 is completely shifted towards the HDA reaction product, i.e. the block copolymer P3. However, by increasing the temperature the retro HDA reaction is favored and the equilibrium shifts towards the reactants P1b and P2. HT-NMR analysis allows for the determination of the temperature when the equilibrium is completely shifted towards the reactants P1b and P2. Thus, 1H-NMR spectra of the block copolymer P3 were recorded while increasing the temperature from 25 °C to 85 °C (see Fig. 3). To obtain information of the position of the equilibrium between the block copolymer P3 and the reactants P1b and P2, the region of the resonance of the Cp proton Ha (6.2–6.0 ppm) and the region from the resonance of the proton from the HDA reaction product Hb (5.85–5.6 ppm) were inspected. To ensure equilibrium conditions for each temperature step, successive NMR spectra have been recorded until the actual spectrum is identical with the one recorded before. From 45 °C to 85 °C, the intensity of the resonance of the Cp moiety is increasing continuously. Simultaneously, the intensity of the resonance of the HDA reaction product is decreasing. At 85 °C, the resonance of the proton of the hetero Diels-Alder product completely disappears, indicating complete debonding. The complete disappearance of the peaks at 5.85–5.6 ppm and concurrent appearance of the resonance peak at 6.2–6.0 ppm indicate that the equilibrium is completely shifted towards the building blocks P1b and P2. The shift of the signals towards a lower field is due to the increasing temperature. The NMR experiment was performed in a mixture of toluene-d8 and DMSO-d6 (1:1). The solvent mixture was used to ensure a moderate polarity to keep both building blocks in solution after debonding. In addition, a high boiling point was preferred because the cleavage temperature is relatively high. Having determined the cleavage temperature, further HT-NMR experiments have been performed in CDCl3 in an NMR pressure tube in the presence of ZnCl2 as catalyst. Note that the different employed NMR solvents slightly affect the resonances appearance and position.
 | ||
| Fig. 3
1H-NMR spectra (400 MHz, toluene-d8–DMSO-d6, 1:1) of the block copolymer P3 at variable temperatures. The resonances of the Cp moiety are depicted on the left hand side (6.2–6.0 ppm, Ha) and the resonances associated with the DA reaction product are depicted on the right hand side (5.85–5.6 ppm, Hb). As the temperature increases, the intensity of the Cp resonance (left side) increases, whereas the intensity of the DA reaction product resonance decreases (right side). Above 85 °C the building blocks are completely separated. | ||
To evidence that the bonding and debonding of the building blocks P1b and P2 is reversible with temperature, NMR spectra of the block copolymer P3 were conducted and cycled 4 times between 25 °C and 90 °C (see Fig. 4). Again, the regions of the resonance from the Cp proton (6.1–6.0 ppm) and of the proton of the HDA reaction product (5.85–5.6 ppm) were investigated.
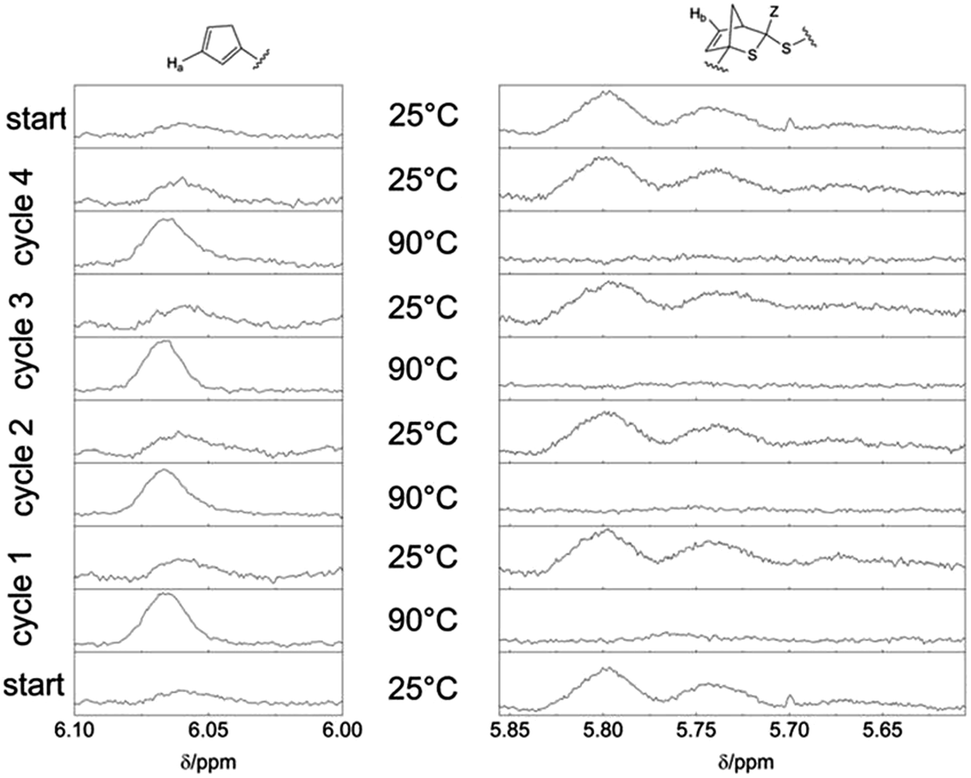 | ||
| Fig. 4 1H-NMR spectra (400 MHz, CDCl3) of the block copolymer P3 at alternating temperatures in four heating/cooling cycles between 25 °C and 90 °C. The resonances of the cyclopentadienyl moiety are depicted on the left side (6.1–6.0 ppm, Ha) and the resonances associated with the DA reaction product are shown on the right side (5.85–5.6 ppm, Hb). After the cleavage at 90 °C, the block copolymer P3 is reformed at 25 °C. For better comparison, the starting spectrum is depicted at the bottom and on top of the figure. Note that the forward ligation is not 100% quantitative, due to the Diels-Alder reaction being an equilibrium reaction and some limited debonded material remains at ambient temperature. | ||
The bonding/debonding behavior can be observed over all 4 cycling processes. For every cycle, the resonance of the Cp moiety has full intensity at 90 °C, whereas in the region of the HDA reaction product only the baseline can be detected, revealing complete transformation into the two separate building blocks. Concomitantly, the resonance of the Cp moiety disappears for each cycle at 25 °C with highest intensity of the resonance of the HDA reaction product Hb. For the cleavage of the block copolymer at 90 °C an equilibrium time of approximately 30 min was observed. Complete rebonding was achieved after 24 h at 25 °C. The difference in reaction time, compared to the synthesis of P3 (16 h), can be explained by the absence of stirring of the polymer solution in the NMR tube and thus the bonding is only diffusion controlled, leading to longer reaction time for the block copolymer formation. In CDCl3 no shift of the signals towards a lower field at elevated temperatures could be observed.
HT-DLS analysis
To further underpin the results of the HT-NMR experiments, HT-DLS analysis of the block copolymer P3 has been carried out. Since no pressure stable DLS cuvettes were available, the choice of solvents was limited by their boiling points and scattering properties as well as their ability to dissolve both individual building blocks. P3 was dissolved in a mixture of N,N-dimethylacetamide (DMAc, 80 vol%) and toluene (20 vol%) in the presence of ZnCl2 as catalyst revealing the best possible conditions for the HT-DLS measurements. Representative examples of autocorrelation functions and the resulting size distributions can be found in Fig. S11 and S12,† respectively. For the block copolymer, unimers with a radius of close to 2.5 nm are detected at 30 °C (refer to Fig. 5). When the sample is heated to 90 °C, the detected radius decreases to values below 2.0 nm, hinting at the block copolymer being cleaved into the individual building blocks, which form significantly smaller unimers. As soon as the temperature is decreased again to 30 °C, the block copolymer – and hence the corresponding unimers – start to reform and the radius increases again continuously with time up to the initially recorded average size (2.4 nm).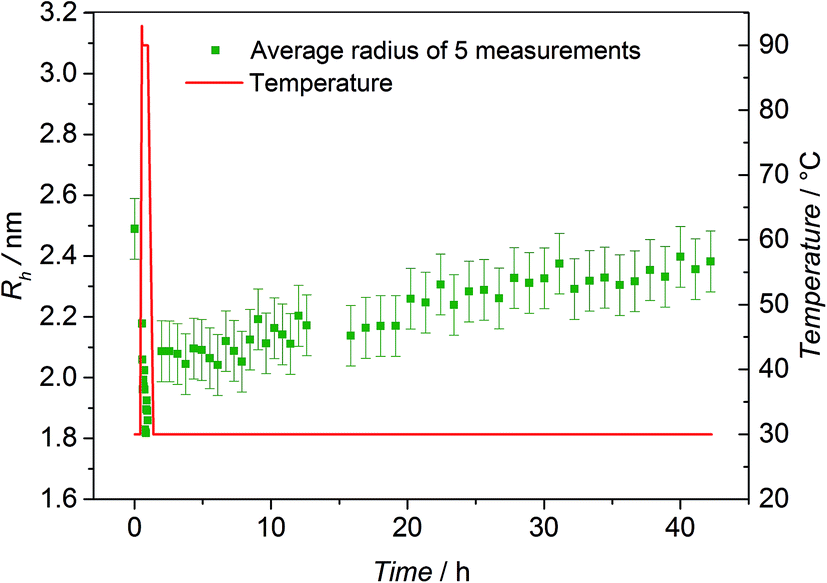 | ||
| Fig. 5 HT-DLS experiment of block copolymer P3 in DMAc in the presence of ZnCl2 as catalyst. The green squares depict the average radius of 5 subsequently measured values in the specified time interval. The experimental standard deviation is close to 0.1 nm. Due to the fast and large change of the actual radius when the sample is heated at 90 °C, no average radius is calculated for that temperature. The red solid line displays the temperature evolution. | ||
To examine the reversible bonding and debonding behavior of the HDA linkage, 4 temperature cycles between 30 °C and 90 °C were performed (refer to Fig. 6). At 90 °C, the system was allowed to equilibrate for 30 min until no further changes in Rh could be detected. Similarly, at 30 °C, the detected radius was measured after the equilibrium was established. For every temperature switch the expected change of the detected radius can be observed.
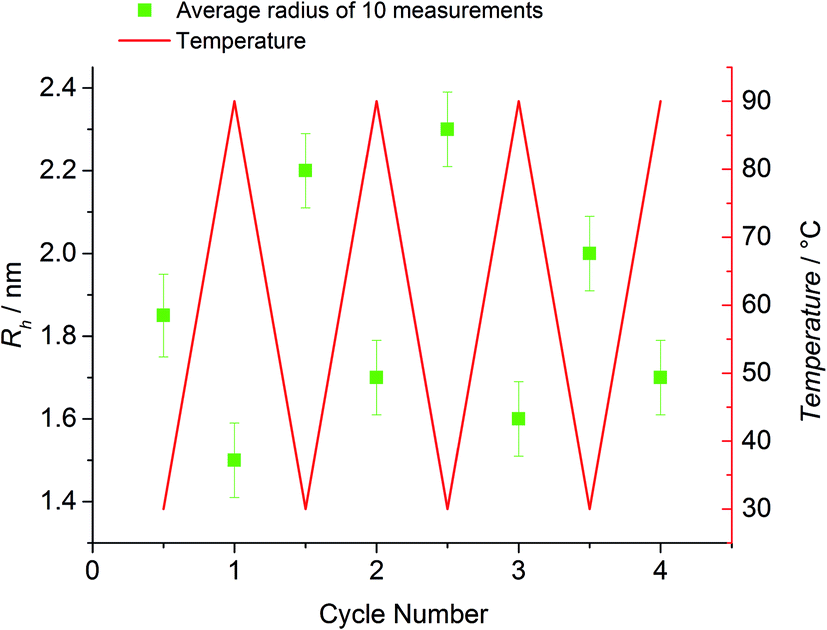 | ||
| Fig. 6 HT-DLS cycles of block copolymer P3 in DMAc (80 vol%) and toluene (20 vol%) in the presence of ZnCl2 as catalyst. The green squares represent the average radius of 10 subsequently measured values with an experimental standard deviation of 0.1 nm. The red solid line displays the temperature evolution. | ||
In contrast, the building block (P1b) as reference exhibits a radius of approximately 1.6 nm and no significant change with varying temperature could be observed. However, the respective radii for each cycle at 90 °C and at 30 °C vary more than expected (90 °C: 1.5–1.7 nm; 30 °C: 1.9–2.3 nm). A possible explanation is associated with the change of the actual ratio of the employed solvent mixture during the measurement. Since sealed cuvettes are not commercially available and thus could not be used, a certain amount of solvent evaporates constantly during the experiment (boiling point DMAc: 165 °C, boiling point toluene: 110 °C). Thus, continually solvent had to be added, possibly affecting the experiment and the data evaluation. The solubility of the polymers is influenced by the composition of the solvent mixture. A change in the ratio of toluene to DMAc can possibly lead to small aggregates that affect the measured Rh averages. In addition, the data evaluation depends on the solvent composition since the calculated Rh is indirectly proportional to the solution viscosity. In summary, the results of the HT-DLS experiments clearly underpin the results of the HT-NMR analysis.
HT-SEC analysis
To further support the results of the HT-NMR and HT-DLS analysis, cycled HT-SEC experiments were performed. A solution of the block copolymer P3 in TCB was prepared (5 mg mL−1) and placed into the autosampler (set to 90 °C). The sample was measured immediately in order to monitor the bonded state (see Fig. 7, solid black line, 0 min). However, it must be noted that at this point 100% bonding cannot be monitored because the sample is subjected to 90 °C for approximately 7–10 minutes during the analysis and, therefore, debonding of the hetero Diels-Alder linkage partially takes place before the differential refractive index (dRI) detector is reached. Thus, three distinct distributions can be observed, i.e. the two building blocks and the block copolymer. The unpolar building block shows a positive dRI signal at a retention time of close to 7.8 min (peak maximum), whereas the polar block shows a large negative dRI signal at approximately 8.6 min retention time (peak maximum). The block copolymer reveals a small negative signal at a retention time of close to 8.1 min. The calculated distributions are depicted as dashed lines. The assignments are based on SEC measurements of the particular building blocks. For validation, the sum of the three calculated distributions is compared with the experimental data, showing good agreement.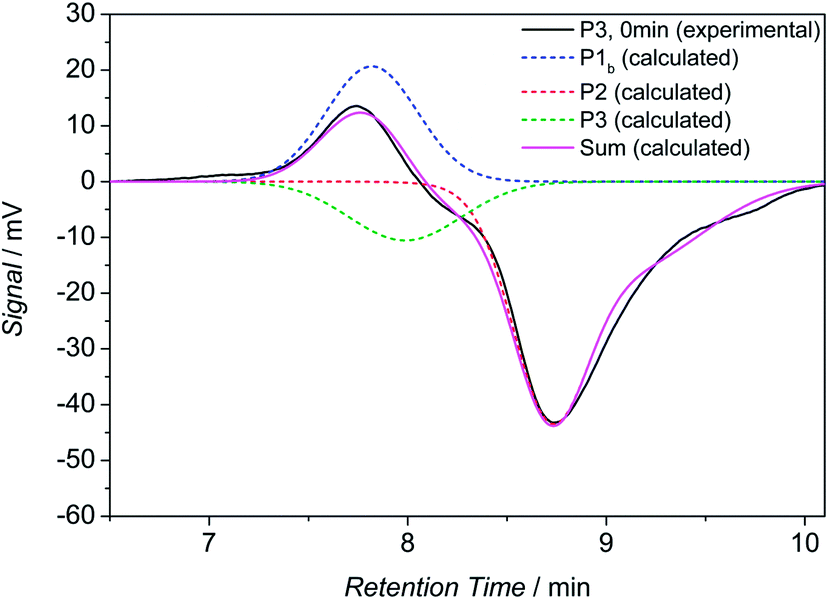 | ||
| Fig. 7 SEC trace of P3 in TCB at 90 °C. The black solid line represents the measurement immediately after the sample was placed into the autosampler (0 min), the dashed lines indicate the mathematically fitted fractions of the polar block (P2), the non-polar block (P1b) and the block copolymer (P3). The pink solid line represents the sum of all theoretical peaks. | ||
Next, the sample was allowed to equilibrate for 30 minutes at 90 °C where the retro HDA reaction proceeds completely. Subsequently, a new chromatogram was recorded, representing the debonded state (refer to Fig. 8, black solid line, 30 min) with maximum intensity (respectively in positive and negative direction) of the building blocks. Again, the calculated distributions are depicted in dashed lines. The distribution of the block copolymer disappears completely.
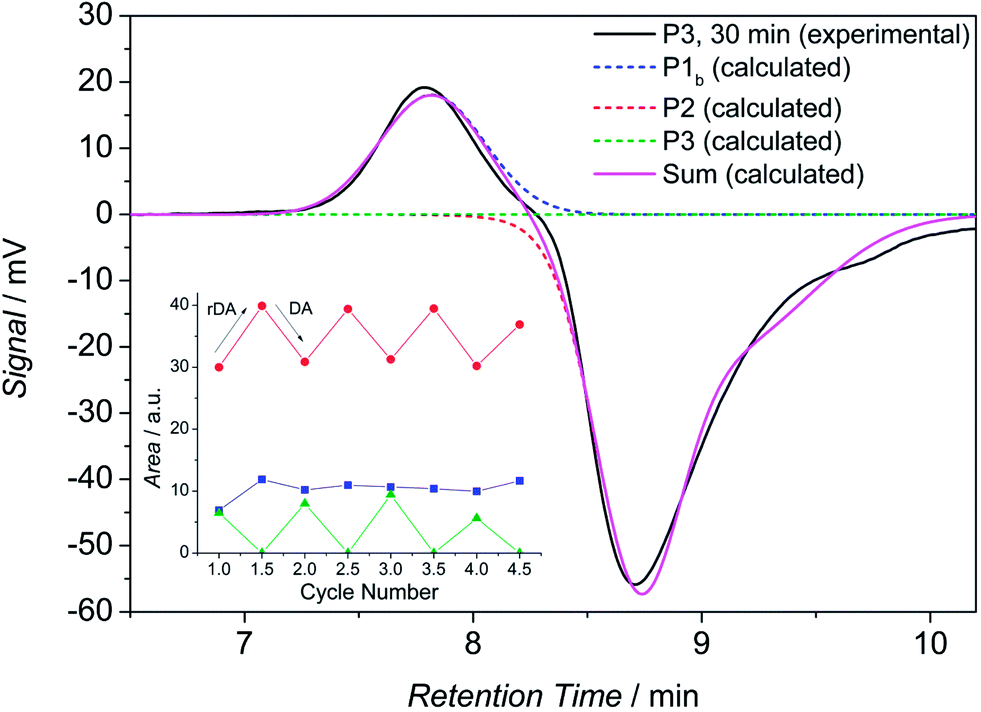 | ||
| Fig. 8 SEC trace of P3 in TCB at 90 °C. The black solid line represents the measurement 30 min after the sample was placed into the autosampler, the dashed lines indicate the mathematically fitted fractions of the polar block (P2), the non-polar block (P1b) and the block copolymer (P3). The pink solid line represents the sum of all theoretical peaks. The inset diagram shows the evolution of the peak areas of the three distributions, alternating for every bonding/debonding cycle. | ||
Next, the sample was removed from the autosampler and kept at ambient temperature for 5 days in which the HDA conjugation of the building blocks takes place (without the presence of any catalyst). This procedure was repeated 4 times, all SEC traces can be found in Fig. S13.† The signal intensities have been evaluated in detail by deconvolution of the chromatograms to determine the peak areas. The results are displayed in the inset diagram in Fig. 8. The trend of the data clearly demonstrates the expected effects: the peak area and thus the concentration of both building blocks increases during the retro DA reaction and it decreases during the DA reaction. As expected, the observation for the block copolymer is inversed (the intensity of P1b is minor because of the low dn/dc value and thus the changes in peak area are less pronounced). The separation of the specific polymers on the SEC column is not purely entropically driven but also due to enthalpic interactions (adsorption of polar polymers on the column material at high temperatures with TCB as eluent was already observed in other experiments). Thus, the retention time of the amphiphilic block copolymer is in between the retention times of the constituting building blocks. A standard calibration for calculating molar masses is not feasible since enthalpic interactions are occurring. However, the constant difference of the RI signal intensities of the building blocks between the measurements after 0 min and 30 min for the respective cycles confirms again the temperature dependent cleavage of the HDA linkage, here without the presence of any catalyst.
Macroscopic cleavage
To corroborate the results of the aforementioned bonding and debonding studies of the amphiphilic block copolymer, the thermo-responsiveness and the opposite polarities of the building blocks have been harnessed for a macroscopic separation study. First, an aqueous dispersion of the block copolymer P6 was generated. Therefore, P6 was dissolved in THF and subsequently added to water. Next, the THF was removed under reduced pressure. The dispersion was heated at 90 °C for 30 min and then cooled to ambient temperature (see Fig. 9). At 90 °C the amphiphilic block copolymer P6 is debonded to yield the building blocks P4b* and P5*. P4b* is not soluble in water and thus remains as precipitate. In contrast, P5* is soluble in water, yet has a lower critical solution temperature (LCST) at 70 °C. Above the LCST, P5 does not precipitate as a solid, but as a sticky liquid. Hence, at 90 °C the polymers P4b* and P5* aggregate and form a large macroscopic agglomerate. When the mixture is cooled to ambient temperature P5* dissolves in water, whereas P4b* remains insoluble and forms an aqueous dispersion. Finally, P4b* and P5* (one dissolved in water, the other as dispersion) have been separated via centrifugation (for a separation via filtration, the particles of the dispersion have been too small in size). | ||
| Fig. 9 Images of the macroscopic cleavage of the block copolymer P6, into the building block polymers P4b* and P5*. (a) Block copolymer dispersion in water at ambient temperature. (b) At 90 °C the building blocks are separated. Due to the LCST of P5, the building block polymers P4b* and P5* aggregate and form a macroscopic agglomerate (red arrow). (c) Cooled to ambient temperature, P5* dissolves in water, whereas P4b* forms an aqueous dispersion. | ||
The obtained polymers P4b* and P5* were analyzed via SEC and compared with the SEC traces of the original building blocks P4b and P5 and the block copolymer (see Fig. 10). For P4b* the SEC trace is in good agreement with the SEC trace of P4b. However, the SEC trace of P5* shows a shoulder in the retention volume region of P4b. A small amount of P4b* particles were probably not fully separated from the solution of P5*. These findings again support the temperature-dependent studies via HT-NMR and HT-DLS discussed earlier.
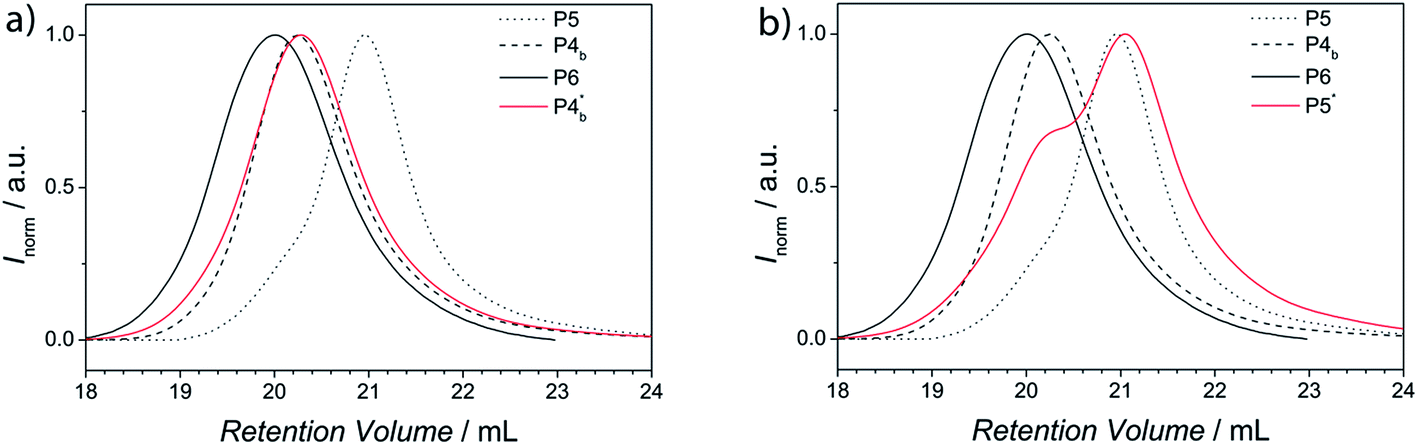 | ||
| Fig. 10 SEC traces of the building blocks after macroscopic separation (a) non-polar block P(S-co-I), P4b*, solid red line; (b) polar block PTEGA, P5*, solid red line), in comparison to the initial building blocks (P4b and P5) and the block copolymer (P6). | ||
Conclusions
An in-depth study on a novel amphiphilic bonding/debonding on demand system, i.e. the amphiphilic block copolymer P(S-co-I)-b-PTEGA featuring a temperature switchable linkage, is presented. Reversible addition fragmentation chain transfer (RAFT) polymerization was used to synthesize the constituting building blocks. Therefore, a novel trithiocarbonate RAFT agent was synthesized carrying a bromine moiety at the tertiary R-group for subsequent Cp transformation of the end group to afford P(S-co-I). Subsequently, a HDA capable, phosphorous RAFT agent was used for the controlled polymerization of triethylene glycol methyl ether acrylate. The building blocks were ligated by a hetero Diels-Alder reaction at ambient temperature in the presence of ZnCl2 as catalyst to prepare the amphiphilic block copolymer. The bonding/debonding capability of P(S-co-I)-b-PTEGA was evidenced by high temperature nuclear magnetic resonance (HT-NMR) spectroscopy, high temperature dynamic light scattering (HT-DLS) and high temperature size exclusion chromatography (HT-SEC). The current study shows that the concept of reversible HDA linkages in block copolymers can be expanded to amphiphilic systems. The linkage can be repeatedly temperature cycled with high molecular precision, thus paving the way for the use of the herein introduced synthetic platform technology for the preparation of nanostructured and, perspectively, porous materials.Abbreviations
| AIBN | 2,2′-Azobis(2-methylpropionitrile) |
| ATRP | Atom transfer radical polymerization |
| CLRP | Controlled/living radical polymerization |
| Cp | Cyclopentadienyl |
| CTA | Chain transfer agent |
| DCC | N,N′-dicyclohexylcarbodiimide |
| DCM | Dichloromethane |
| DMAc | Dimethylacetamide |
| DMAP | 4-(Dimethylamino)pyridine |
| DMP-Br | 3-((2-Bromo-2-methylpropanoyl)oxy)propyl 2-(((dodecylthio)carbonothioyl)thio)-2-methylpropanoate |
| DMSO | Dimethylsulfoxide |
| dRI | Differential refractive index |
| ESI-MS | Electrospray ionization-mass spectrometry |
| HDA | Hetero Diels-Alder |
| HT-DLS | High temperature dynamic light scattering |
| HT-NMR | High temperature nuclear magnetic resonance |
| HT-SEC | High temperature size exclusion chromatography |
| LCST | Lower critical solution temperature |
| MHS | Mark–Houwink–Sakurada |
| NMP | Nitroxide mediated polymerization |
| PI | Polyisoprene |
| PMMA | Polymethylmethacrylate |
| PS | Polystyrene |
| PTEGA | Polytriethyleneglycol methylether acrylate |
| RAFT | Reversible addition–fragmentation chain transfer |
| TCB | 1,2,4-Trichlorobenzene |
| THF | Tetrahydrofuran |
| VAm-110 | 2,2′-Azobis(N-butyl-2-methylpropionamide) |
Acknowledgements
F. H. S. and C. B.-K. acknowledge funding from the German ministry for education and research (BMBF) in the framework of the Biotechnology 2020+ initiative (031A119A and 031A119B). C. B.-K. acknowledges continued support from the Karlsruhe Institute of Technology (KIT) in the context of the Helmholtz BIF program supporting the Soft Matter Synthesis (SML) platform. Dr Leonie Barner is thanked for managing the SML platform. In addition, F. H. S. is grateful to the Verband der Chemischen Industrie (VCI) for a fellowship. The authors thank Christian Pietsch and Christoph Hörenz for helpful discussions.References
- M. L. Adams, A. Lavasanifar and G. S. Kwon, J. Pharm. Sci., 2003, 92, 1343–1355 CrossRef CAS PubMed.
- G. Gaucher, M.-H. Dufresne, V. P. Sant, N. Kang, D. Maysinger and J.-C. Leroux, J. Controlled Release, 2005, 109, 169–188 CrossRef CAS PubMed.
- F. H. Schacher, P. A. Rupar and I. Manners, Angew. Chem., Int. Ed., 2012, 51, 7898–7921 CrossRef CAS PubMed.
- H.-C. Kim, S.-M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146–177 CrossRef CAS PubMed.
- A. W. York, S. E. Kirkland and C. L. McCormick, Adv. Drug Delivery Rev., 2008, 60, 1018–1036 CrossRef CAS PubMed.
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS PubMed.
- K. Koo, H. Ahn, S.-W. Kim, D. Y. Ryu and T. P. Russell, Soft Matter, 2013, 9, 9059–9071 RSC.
- N. A. Lynda, A. J. Meulerb and M. A. Hillmyer, Prog. Polym. Sci., 2008, 33, 875–893 CrossRef PubMed.
- F. H. Schacher, H. Sugimori, H. Song, H. Jinnai and A. H. E. Müller, Macromolecules, 2012, 45, 7956–7963 CrossRef CAS.
- H. Jinnai, T. Kaneko, K. Matsunaga, C. Abetz and V. Abetz, Soft Matter, 2009, 5, 2042–2046 RSC.
- F. H. Schacher, T. Rudolph, M. Drechsler and A. H. E. Müller, Nanoscale, 2011, 3, 288–297 RSC.
- C. J. Hawker and T. P. Russell, MRS Bull., 2005, 30, 952–966 CrossRef CAS.
- J. S. Lee, A. Hirao and S. Nakahama, Macromolecules, 1988, 21, 274–276 CrossRef CAS.
- J. Bang, S. H. Kim, E. Drockenmuller, M. J. Misner, T. P. Russell and C. J. Hawker, J. Am. Chem. Soc., 2006, 128, 7622–7629 CrossRef CAS PubMed.
- J. M. Leiston-Belanger, T. P. Russell, E. Drockenmuller and C. J. Hawker, Macromolecules, 2005, 38, 7676–7683 CrossRef CAS.
- P. Mansky, C. K. Harrison, P. M. Chaikin, R. A. Register and N. Yao, Appl. Phys. Lett., 1996, 68, 2586–2588 CrossRef CAS PubMed.
- J. T. Goldbach, T. P. Russell and J. Penelle, Macromolecules, 2002, 35, 4271–4276 CrossRef CAS.
- H. Zhao, W. Gu, E. Sterner, T. P. Russell, E. B. Coughlin and P. Theato, Macromolecules, 2011, 44, 6433–6440 CrossRef CAS.
- N. Hadjichristidis, M. Pitsikalis and H. Iatrou, Adv. Polym. Sci., 2005, 189, 1–124 CrossRef CAS.
- K. Hong, D. Uhrig and J. W. Mays, Curr. Opin. Solid State Mater. Sci., 1999, 4, 531–538 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, S. Pispas and M. Pitsikalis, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3211–3234 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS PubMed.
- G. Moad and E. Rizzardo, Macromolecules, 1995, 28, 8722–8728 CrossRef CAS.
- D. A. Shipp, Polym. Rev., 2011, 51, 99–103 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; (b) C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS PubMed.
- M. Glassner, G. Delaittre, M. Kaupp, J. P. Blinco and C. Barner-Kowollik, J. Am. Chem. Soc., 2012, 134, 7274–7277 CrossRef CAS PubMed.
- N. K. Guimard, J. Ho, J. Brandt, C. Y. Lin, M. Namazian, J. O. Mueller, K. K. Oehlenschlager, S. Hilf, A. Lederer, F. G. Schmidt, M. Coote and C. Barner-Kowollik, Chem. Sci., 2013, 4, 2752–2759 RSC.
- C. N. Urbani, A. B. Craig, M. R. Whittaker and M. J. Monteiro, Macromolecules, 2008, 41, 1057–1060 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- F. Hua, X. Jiang, D. Li and B. Zhao, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2454–2467 CrossRef CAS.
- A. Alberti, M. Benaglia, M. Laus and K. Sparnacci, J. Org. Chem., 2002, 67, 7911–7914 CrossRef CAS PubMed.
- X. Yin, A. S. Hoffman and P. S. Stayton, Biomacromolecules, 2006, 7, 1381–1385 CrossRef CAS PubMed.
- C. W. Scales, A. J. Convertine and C. L. McCormick, Biomacromolecules, 2006, 7, 1389–1392 CrossRef CAS PubMed.
- S. R. Gondi, A. P. Vogt and B. S. Sumerlin, Macromolecules, 2007, 40, 474–481 CrossRef CAS.
- D. S. Germack and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4100–4108 CrossRef CAS.
- C. J. Dürr, L. Hlalele, A. Kaiser, S. Brandau and C. Barner-Kowollik, Macromolecules, 2013, 46, 49–62 CrossRef.
- R. Nicolay, Y. Kwak and K. Matyjaszewski, Macromolecules, 2008, 41, 4585–4596 CrossRef CAS.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- M. D. Rausch, W. P. Hart and D. W. Macomber, J. Macromol. Sci., Chem., 1981, A16, 243–250 CrossRef CAS.
- M. T. Blankenbuehler and J. P. J. Selegue, Organomet. Chem., 2002, 642, 268–274 CrossRef CAS.
- A. J. Inglis, T. Pauloehrl and C. Barner-Kowollik, Macromolecules, 2010, 43, 33–36 CrossRef CAS.
- D. J. am Ende, D. C. Whritenour and J. W. Coe, Org. Process Res. Dev., 2007, 11, 1141–1146 CrossRef CAS.
- C. Barner-Kowollik, Macromol. Rapid Commun., 2009, 30, 1625–1631 CrossRef CAS PubMed.
- C. M. Preuss and C. Barner-Kowollik, Macromol. Theory Simul., 2011, 20, 700–708 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The experimental part, including the used materials, synthetic procedures and used characterization methods, 1H-, 13C-NMR and ESI-MS spectra of the new RAFT agent, kinetic studies of the polymerization and SEC traces of the polymers before and after Cp-transformation, 1H-NMR spectra of the building blocks and the resulting block copolymer, a table of the calculated ratios from both block copolymers and the content of isoprene and all measured HT-SEC traces. See DOI: 10.1039/c4py00644e |
| This journal is © The Royal Society of Chemistry 2014 |