DOI:
10.1039/C4PY00620H
(Paper)
Polym. Chem., 2014,
5, 6039-6050
Controlled synthesis of β-sheet polymers based on side-chain amyloidogenic short peptide segments via RAFT polymerization†
Received
4th May 2014
, Accepted 1st July 2014
First published on 1st July 2014
Abstract
A strategy is documented that exploits the controlled synthesis of side-chain peptide based stimuli-responsive hybrid macromolecules inspired by amyloid based β-sheet forming peptides. Well-defined side-chain peptidic polymers have been synthesized via the reversible addition–fragmentation chain transfer (RAFT) polymerization technique using a representative methacrylate monomer with a short peptide segment (Leu-Val-Phe) corresponding to the amyloid β-peptide Aβ1–42. Furthermore, the monomethoxy poly(ethylene glycol) (mPEG) macro-chain transfer agent is employed for RAFT polymerization of this peptidic monomer to prepare peptide based amphiphilic nano-carriers, a promising reservoir for hydrophobic Nile red dye or doxorubicin based anticancer drug molecules. These hybrid materials can easily produce primary amino groups in terminals of side-chains that can be protonated or deprotonated by changing the pH of the aqueous medium, thus providing “smart” pH-responsiveness, cationic characteristics and complexation capabilities with DNA to form polyplexes. Circular dichroism (CD) spectroscopy demonstrates the formation of a secondary structure from these peptidic polymers and they adopted the β-sheet conformation, which were stable against heat in the experimental temperature range and solvent polarity. Solid-state FT-IR spectroscopy has been performed to understand the formation of β-sheets as well as to distinguish between their parallel and antiparallel propensity. In vitro cytotoxicity testing indicates the biocompatible nature of these peptidic materials, and confocal laser scanning microscopy (CLSM) images revealed cellular uptake of their drug/dye loaded micelles, which potentially offers them as a suitable candidate with dual bioapplications for drug delivery as well as gene transfer.
Introduction
In recent years, there has been growing interest in the development of synthetic strategies for the creation of multifunctional materials by taking inspiration from nature.1–3 The palette of natural peptides or proteins offers an impressive scope for the design of novel structures and functionalities.4 Bioactive peptides control many physiological processes and illustrate several valuable characteristics for human health including antifungal, antimicrobial, antiviral, and antitumor activities.5 Peptides with unique and exciting biological functions such as cell penetrating and tumor homing have been documented and are being utilized in a wide range of bioapplications.6,7 Peptide incorporation promotes synthetic polymers in terms of providing much higher complexity due to the possibility of peptides to self-assemble into high-order hierarchical nanostructures,8–10 and also can encode their structural information in the polymeric nanomaterials.11 On the other hand, polymer components can affect the reactivity of peptides by altering their self-association properties, and also provide additional functionalities for developing highly multifunctional architectures.12,13 Therefore, these hybrid biomaterials have found numerous diverse applications such as drug delivery systems,14,15 bioactive hydrogels,16 structured nanomaterials,17etc.18–21
Ring-opening metathesis polymerization (ROMP) has been successfully used for the synthesis of homopolymers and copolymers substituted with the biologically active oligopeptides. Significant enhancements towards biological activity were observed with polymers substituted with peptides than the free peptide due to multivalent interactions provided by the polymer scaffold.22 A water soluble polymer containing a cell adhesive Arg-Gly-Asp-Ser sequence in the side chain was synthesized by conventional radical polymerization.23 van Hest and co-workers polymerized a VPGVG (V = valine, P = proline, and G = glycine) peptide based monomer via the atom transfer radical polymerization (ATRP) technique to prepare homopolymers and poly(ethylene glycol) (PEG) containing triblock copolymers.24 To introduce the β-sheet functionality into a synthetic polymer, van Hest's group synthesized an Ala-Gly-Ala-Gly tetrapeptide side chain monomer based on known β-sheet-forming sequences, and polymerized by the ATRP technique.25 Adams et al. reported the inter- and intra-chain β-sheet motif structuring in aqueous solution by the peptidic component of the PEG-b-poly(side-chain peptide) block copolymers prepared by ATRP.11 The aggregation behaviour of stiff polymer–peptide nanofibers, self-assembled from well-defined PEG–peptide conjugates, was described, where the peptide domain formed β-sheet structures.26 Hamley et al. reported that conjugation of PEG in the PEG hybrid block copolymers containing amphiphilic β-strand peptide sequences stabilized the adopted secondary structure of the peptide and also reduced their sensitivity towards pH variations as compared to the native peptide sequence.27 Oligopeptide-based chain transfer agents have been synthesized and subsequently utilized for the polymerization of n-butyl acrylate, where the chirality of the peptide segment has not been affected by the polymerization procedures.28 Although side-chain peptide based monomers have been polymerized via a variety of techniques, to the best of our knowledge, there are very limited reports,10,13,29 in which reversible addition–fragmentation chain transfer (RAFT) polymerization has been used. Those studies mainly demonstrated the utility of RAFT towards controlled polymerization of bioinspired materials. To investigate higher order structure formation by side-chain tripeptidic polymers, in this study we have synthesized a C-terminus modified tripeptidic vinyl monomer and polymerized it in a controlled fashion via the RAFT technique to prepare pH-responsive side-chain tripeptide containing homopolymers and the corresponding PEGylated block copolymers. We have adopted the short peptide fragment Leu-Val-Phe (LVF) from the central hydrophobic cluster (CHC) of the amyloid β-peptide, Aβ1–42, which is critical for fibril formation in Alzheimer's disease,30,31 because its derived polymers can have possible potential inhibition of fibril formation by Aβ to treat Alzheimer's disease.32 Moreover, the acetylated derivatives of this tripeptide containing a C-terminal aldehyde (Ac-LVF-CHO) have been shown to act as potent HIV protease inhibitors and showed increased activity against cathepsin D and pepsin.33,34 Since self-assembly of the peptide into β-sheet structures has been a topic of significant interest recently,35 formation of β-sheet like secondary structures from these peptidic polymers was studied in detail.
Experimental section
Materials
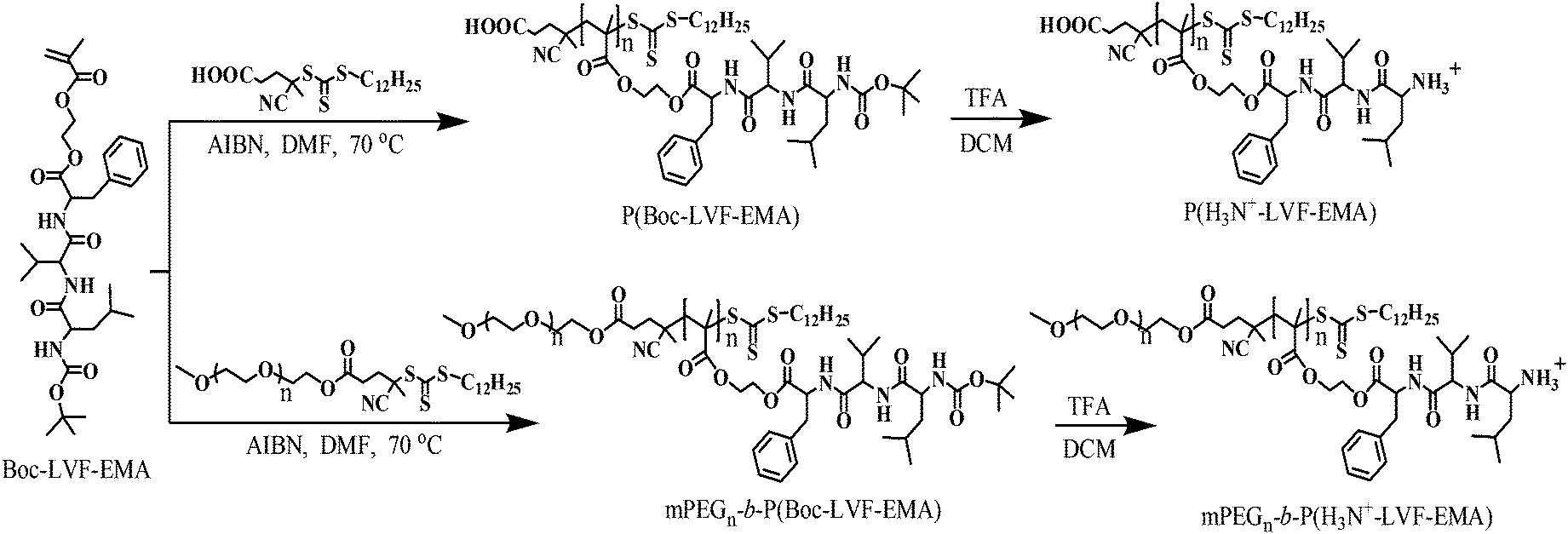
Commercial source and purity of 4-dimethylaminopyridine (DMAP), 1-hydroxybenzotriazole hydrate (HOBt hydrate), trifluoroacetic acid (TFA), Boc-L-leucine (Boc-L-Leu-OH), anhydrous N,N-dimethylformamide (DMF), dicyclohexylcarbodiimide (DCC), 2-hydroxyethyl methacrylate (HEMA), 2,2′-azobisisobutyronitrile (AIBN), CDCl3, D2O and DMSO-d6 are reported elsewhere.10 The L-valine methyl ester hydrochloride (HCl·H2N-Val-OMe, 99%), L-phenylalanine methyl ester hydrochloride (HCl·H2N-Phe-OMe, 98%), pyrene (98%), Nile red, doxorubicin hydrochloride (Dox·HCl), and monomethoxy poly(ethylene glycol) with molecular weights of 2 kDa (mPEG2k) and 5 kDa (mPEG5k) were purchased from Sigma. The Dox·HCl was deprotonated according to the method reported in the literature.36 The solvents such as hexanes, acetone, ethyl acetate, tetrahydrofuran (THF), and dichloromethane (DCM) were purified by standard procedures. The mPEG based macro chain transfer agents (mPEG2k-CTA and mPEG5k-CTA) were prepared from the 4-cyano-4-(dodecylsulfanylthiocarbonyl)sulfanylpentanoic acid (CDP) based chain transfer agent (CTA) according to our previous report.10 Synthesis of the side-chain Boc-protected tripeptide, containing the methacrylate monomer Boc-Leu-Val-Phe-oxyethyl methacrylate (Boc-LVF-EMA), has been described in the ESI.†
Instrumentation
The molecular weights and molecular weight distributions were determined by gel permeation chromatography (GPC) in THF relative to narrow molecular weight poly(methyl methacrylate) (PMMA) standards at 35 °C and 1.0 mL min−1 flow rate. The system is equipped with a Waters 515 HPLC pump, a 2414 refractive index (RI) detector and two 300 × 7.5 mm Polargel-M columns from Agilent Technologies. 1H and 13C NMR spectroscopy was carried out on a Bruker AVANCEIII 500 MHz spectrometer operating at 500 and 125 MHz, respectively. Positive mode electrospray ionization mass spectroscopy (ESI-MS), FT-IR studies, fluorescence spectroscopy, UV-vis analysis, particle size and zeta potential (ξ) measurements were carried out as reported elsewhere.10 Experimental details for atomic force microscopy (AFM) and field emission scanning electron microscopy (FE-SEM) studies are provided in the ESI.† Circular dichroism (CD) spectroscopic measurements were performed on a JASCO J-185 CD spectrometer. For the cytotoxicity test, the MCF-7 cell line was grown in Eagle's minimal essential medium (EMEM, HiMedia) containing 10% fetal bovine serum (HiMedia) and penicillin–streptomycin (HiMedia) solution. Formazan crystals produced from 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were dissolved in a MTT solubilization solution (In Vitro Toxicology Assay Kit MTT based, Sigma-Aldrich). For microscopic imaging, MCF-7 cells were fixed with 4% paraformaldehyde (Merck) and cells were permeabilized with Triton-X-100 (Sigma). Then, the cells were subsequently stained with 4′,6-diamidino-2-phenylindole (DAPI, USB Corporation).
RAFT polymerization of Boc-LVF-EMA
Typically, Boc-LVF-EMA (0.30 g, 0.51 mmol), CDP (8.21 mg, 0.204 μmol), AIBN (0.334 mg, 2.04 μmol; 0.1 mL solution of 33.4 mg AIBN in 10 mL DMF), DMF (0.5 mL) and a magnetic stir bar were taken in a 20 mL septum sealed vial. The vial was purged with dry N2 for 20 min and placed in a preheated reaction block at 70 °C. Around 0.1 mL reaction mixture was removed periodically using a N2 purged syringe to determine the number average molecular weight (Mn,GPC) and molecular weight distribution (PDI) by GPC, and monomer conversion by 1H NMR spectroscopy by comparing the integration of the monomer vinyl protons with the DMF protons at 8.02 ppm. Polymerization reaction was quenched by cooling the vial on an ice-water bath and exposing the solution to air, then it is diluted with acetone and precipitated into cold hexanes. The polymer, P(Boc-LVF-EMA), was reprecipitated four times from acetone–hexanes and dried under high vacuum at room temperature to a constant weight.
Synthesis of block copolymers from mPEG-based macro-CTA
A typical block copolymerization procedure is described as follows: Boc-LVF-EMA (0.2 g, 0.34 mmol), mPEG5k-CTA (5.400 g mol−1, 36.6 mg, 6.78 μmol), AIBN (0.111 mg, 0.678 μmol; 0.1 mL solution of 11.1 mg AIBN in 10 mL DMF), DMF (0.5 mL) and a magnetic stir bar were sealed in a 20 mL vial. After N2 purging (20 min), the vial was placed in a preheated reaction block at 70 °C. Samples were removed periodically using N2 purged syringes and analyzed by NMR spectroscopy for the monomer conversion data. After a predetermined time, the vial was cooled in an ice-water bath, and diluted with acetone as necessary. The polymer was precipitated into cold hexanes and allowed to stand, and then the solvent was decanted off. Finally, the block copolymer, mPEG5k-b-P(Boc-LVF-EMA), was reprecipitated four times from acetone–hexanes and dried under high vacuum at room temperature.
Nile red/Dox loaded micelle preparation
Nile red or Dox-loaded micelles were prepared by a dialysis method following the method reported elsewhere.37 Typically, the block copolymer (20 mg) and Dox/Nile red (10 mg) were dissolved in 2 mL of DMSO. Under gentle stirring, the solution was added drop-wise to 10 mL of DI water. After stirring for 2 h at room temperature, the solution was transferred to a 6–8 kDa molecular weight cut off (MWCO) dialysis bag (Spectra/Pro Membrane) and dialyzed for 48 h to remove the organic solvents and free Dox/Nile red. During the dialysis water was changed in every 2 to 6 h. The micellar solution was filtered through a 0.45 μm syringe filter and freeze-dried.
Determination of the critical aggregation concentration (CAC)10
To a volumetric flask, a predetermined amount of pyrene in acetone solution was added and acetone was then evaporated completely. Polymer micellar solutions of different concentrations were added to pyrene in the flask and left to equilibrate with pyrene overnight; the pyrene concentration in the final solution was 6.0 × 10−7 mol L−1. Fluorescence spectra of as prepared solutions were obtained with the excitation wavelength set at 339 nm and the ratios of pyrene probe emission intensities at 392 and 373 nm (I392/I373) were plotted as a function of the logarithm of polymer concentrations (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) C). The CAC was obtained from the intersection of two tangent plots of the intensity ratio I392/I373versus logC.
C). The CAC was obtained from the intersection of two tangent plots of the intensity ratio I392/I373versus logC.
Deprotection of Boc-protecting groups from polymers
Typically, P(Boc-LVF-EMA) (50.0 mg) was taken in 1.0 mL DCM and 0.5 mL TFA was added drop-wise in an ice-water bath. Then, the reaction mixture was stirred at room temperature for 2 h. The resulting Boc-deprotected polymer, P(H3N+-LVF-EMA), was isolated by precipitation from diethyl ether, and dried under high vacuum at room temperature.
Agarose gel retardation assay
The Boc-deprotected polymer was dissolved in phosphate buffered saline (PBS) solutions (pH 4.8) at various concentrations and then mixed with 0.1 μg of plasmid DNA (pEGFP-C1, 4.7 kbp) to a volume of 10 μL. The complexes were vortexed and incubated for 30 min at room temperature. After adding 1 μL of loading buffer, each complex solution was loaded onto 1% (w/v) agarose gel containing 1× tris-acetate-EDTA (TAE) buffer solution. Electrophoresis was carried out with a current of 80 V for 100 min. Naked plasmid DNA (pDNA) diluted with the same buffer without the polymer was used as the control. DNA was illuminated with ethidium bromide and was visualized with a UV lamp, using a gel documentation system.
In vitro cytotoxicity test
The MTT reduction assay was used for investigating the cytotoxicity of prepared polymers.38 MCF-7 cells were seeded at a density of 4 × 104 cells per well in a 96 well plate and incubated for 24 h to reach ∼80% confluency. Cells were treated with polymers having different concentrations (20 μg mL−1 to 200 μg mL−1) and incubated for 24 h. A set of untreated control cells was also incubated for the same time period. The untreated control and polymer treated samples were measured in triplicates. To each well, 10% culture volume MTT solution was added and incubated for 4 h. After incubation, the cell culture medium was disposed off, and MTT solubilization solution was added equally to the culture media. After 10 min of incubation, the absorbance at 570 nm was measured. For fluorescence microscopy, two polymer concentrations with 20 μg mL−1 and the untreated control, MCF-7 cells were seeded at a density of 2 × 104 cells per well in a 96 well plate and incubated for 24 h. After incubation, these cells were fixed with 4% paraformaldehyde and then stained with DAPI. Finally, it was used for microscopy (BD Pathway855) at 20× magnification. The filter set used for DAPI imaging was the excitation filter 380/10 nm, the epifluorescence dichroic filter 400DCLP, emission at 430LP with the exposure time of 6 milliseconds. Images were acquired using the BD AttoVision™ software.
Cellular uptake studies
The MCF-7 cells treated with polymeric micelles (loaded with Dox or Nile red) at various concentrations were seeded at a density of 2 × 104 cells per well in a 96 well plate and incubated for 24 h. Then, these cells were fixed with 4% paraformaldehyde and stained with DAPI and images were taken using the BD AttoVision software. The excitation filter was 548/20 and emission collection was performed using 570LP filters.
Results and discussion
Monomer synthesis
The synthesis of the amyloidogenic short peptide derived monomer is summarized in Scheme S1.† Firstly, tripeptide Boc-LVF-OH was prepared by solution-phase synthesis (Fig. S1–S7†) and was coupled with HEMA in the presence of DCC and DMAP to generate the side-chain peptide containing the methacrylate monomer, Boc-LVF-EMA. This as synthesized monomer was purified by silica gel based column chromatography and the structure was fully characterized by NMR (1H and 13C), FT-IR and mass spectroscopy measurements. In the 1H NMR spectrum of Boc-LVF-EMA (Fig. 1A), vinylic protons were clearly observed as two distinct peaks at δ 6.11 and 5.59 ppm. Characteristic resonance signals of tripeptidic segments in Boc-LVF-EMA were also clearly seen at 7.29–7.07 (phenyl group protons), 6.79–6.60 (amide protons of phenylalanine and valine), 5.0 (amide proton of Boc-leucine), 3.17–3.00 (benzyl protons), 1.42 (methyl protons of Boc group), and 0.94–0.77 (dimethyl peaks of leucine and valine). In the 13C NMR spectrum of the Boc-LVF-EMA monomer, the signals corresponding to vinyl and methyl carbons of the Boc-group are observed at 126.3 and 28.2 ppm, respectively. The other carbon signals of the monomer are also clearly assigned in Fig. S8.† The structure of Boc-LVF-EMA was further confirmed by ESI-MS study, where the observed molecular mass [M + Na]+ = 612.15 m/z matched nicely with the theoretical [M + Na]+ = 612.34 m/z (Fig. S9†). The FT-IR spectrum of Boc-LVF-EMA also evidenced the formation of the tripeptidic monomer (Fig. S10A†), where the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (ester linkage) was confirmed by the appearance of two strong peaks at 1753 and 1728 cm−1 due to the stretching vibrations of carbonyl groups. An absorption band at 1162 cm−1 was observed due to the stretching vibration of C–O. The characteristic absorption bands of the amide group of Boc-leucine can be attributed to the peaks at 3300 and 1521 cm−1.
O (ester linkage) was confirmed by the appearance of two strong peaks at 1753 and 1728 cm−1 due to the stretching vibrations of carbonyl groups. An absorption band at 1162 cm−1 was observed due to the stretching vibration of C–O. The characteristic absorption bands of the amide group of Boc-leucine can be attributed to the peaks at 3300 and 1521 cm−1.
 |
| | Fig. 1
1H NMR spectra of (A) monomer Boc-LVF-EMA, (B) homopolymer P(Boc-LVF-EMA), and (C) corresponding Boc-deprotected homopolymer P(H3N+-LVF-EMA). | |
RAFT polymerization
Recently, we demonstrated that the CDP based RAFT agent effectively polymerized side-chain amino acid39 or dipeptide10 based methacrylate monomers and produced narrow-disperse polymers with high conversion and good molecular weight control. Therefore, RAFT polymerization of the tripeptidic monomer Boc-LVF-EMA was carried out under similar conditions at 70 °C in DMF using AIBN as the initiator and CDP or mPEGn-CTA as RAFT agents for preparing peptidic side-chain homo- and block co-polymers. Firstly, the controlled character for the RAFT polymerization of Boc-LVF-EMA was examined using CDP as CTA for achieving well-defined homopolymers. As depicted in Fig. 2A and 2B, linear pseudo-first-order kinetics plots and a linear increment of Mn with monomer conversion were observed, indicating the absence of side reactions such as chain transfer and termination. However, an induction period of approximately 10–20 min was observed, which could generally originate from a small amount of residual oxygen and/or other trace impurities in the polymerization system.40,41 The obvious increment in the kinetic rate was observed by increasing the concentration of AIBN from 0.1 to 0.2 equivalent (Fig. 2A). The unimodal RI traces in both homopolymerization compositions shifted smoothly toward the lower elution volume with increasing monomer conversion and no high molecular weight species was observed from bimolecular termination (Fig. 2C and 2D). The polymers exhibited narrow PDI (<1.4) and the number average molecular weight (Mn,GPC) determined from GPC matches reasonably with the number average theoretical molecular weight (Mn,theo) throughout the conversion ranges (Table 1). A range of polymerization conditions was used to investigate the effect of reaction stoichiometry on polymerization control and, as expected, polymers of higher molecular weight were achieved with the increasing ratio of [Boc-LVF-EMA]/[CDP] (Fig. S11A†).
 |
| | Fig. 2 Pseudo-first-order kinetics plots for the RAFT polymerization of Boc-LVF-EMA (A), corresponding Mnversus monomer conversion plots (B), and GPC traces as a function of elution volume (C and D). ([M]/[CTA]/[I] = [Boc-LVF-EMA]/[CDP]/[AIBN]). | |
Table 1 Characterization results for P(Boc-LVF-EMA) homopolymers and mPEG-b-P(Boc-LVF-EMA) block copolymers
| Polymer |
[M]/[CTA]/[I] |
Time (min) |
Conv.a (%) |
M
n,GPCb (g mol−1) |
PDIb |
M
n,theoc (g mol−1) |
M
n,NMRd (g mol−1) |
|
Calculated by 1H NMR spectroscopy.
Measured by GPC.
The theoretical molecular weight (Mn,theo) = ([Boc-LVF-EMA]0/[CTA]0 × molecular weight (MW) of Boc-LVF-EMA × conversion) + (MW of CTA).
Determined by 1H NMR study.
CTA = CDP.
CTA = mPEG-macro CTA.
Not determined.
|
| P(Boc-LVF-EMA) |
15/1/0.1e |
210 |
53 |
6400 |
1.13 |
5100 |
7760 |
| P(Boc-LVF-EMA) |
25/1/0.1e |
200 |
55 |
8400 |
1.20 |
8820 |
9630 |
| P(Boc-LVF-EMA) |
25/1/0.2e |
130 |
74 |
9700 |
1.17 |
11780 |
11580 |
| P(Boc-LVF-EMA) |
50/1/0.1e |
240 |
63 |
15800 |
1.39 |
18980 |
NDg |
| mPEG2k-b-P(Boc-LVF-EMA) |
50/1/0.1f |
240 |
62 |
17500 |
1.44 |
20680 |
— |
| mPEG5k-b-P(Boc-LVF-EMA) |
50/1/0.1f |
270 |
59 |
15500 |
1.28 |
22500 |
— |
In the next stage, to prepare amphiphilic peptidic side-chain block copolymers, we performed RAFT polymerization of the Boc-LVF-EMA monomer using PEGylated macro-CTA, mPEG2k-CTA and mPEG5k-CTA, in DMF at 70 °C (Table 1). As depicted in Fig. S11B,† GPC chromatograms shifted toward the higher molecular weight region after the block copolymerization, suggesting that the chain extension was effective although little unreacted macro-CTA remained in the block copolymer samples. Similar observations have already been reported using mPEG-macro CTA.42 The Mn,GPC values determined for the block copolymers were somewhat lower than the Mn,theo values determined based on conversion, probably due to the GPC calibration with respect to PMMA standards. The hydrodynamic volume of PMMA may be different from these block copolymers.43 Nevertheless, these results suggest that PEGylated block copolymers with a side-chain peptide containing segment can be successfully synthesized. All polymers are characterized by 1H NMR spectroscopy. In the case of P(Boc-LVF-EMA), typical resonance signals for the different protons in the repeating units of the polymers are clearly assigned in the spectrum (Fig. 1B). Comparison of the integration areas from the terminal –CH2–CH2– protons (from the HOOC–CH2–CH2–C(CN)(CH3)– chain end) at 2.3–2.6 ppm and the chiral proton of phenylalanine at 4.7–5.0 ppm of the repeating unit (for –OCO–CH–NH–) allowed calculation of the number-average molecular weight (Mn,NMR) by NMR spectroscopy. Good agreement between the Mn,GPC, Mn,theo and Mn,NMR suggests controlled RAFT polymerization (Table 1). For mPEGn-b-P(Boc-LVF-EMA) block copolymers, 1H NMR signals from both the blocks are observed (Fig. 3A).
 |
| | Fig. 3
1H NMR spectra of mPEG2k-b-P(Boc-LVF-EMA) in CDCl3 (A) and in D2O (B), and mPEG2k-b-P(H3N+-LVF-EMA) in D2O (C). | |
Deprotection of Boc-groups from the side-chain peptide moiety
To obtain the primary amino group in the side chain terminal of each peptidic unit in the homo- and block copolymers, Boc-deprotection was carried out using TFA–DCM (1:1 v/v) at room temperature (Scheme 1).44 Successful deprotection of Boc-groups was confirmed by the disappearance of Boc-proton signals at about 1.4 ppm in the 1H NMR spectrum of the Boc-deprotected homopolymer (Fig. 1C) and block copolymer (Fig. 3C). In addition, Boc-deprotection was further confirmed by FT-IR spectroscopy, by the disappearance of the absorption peak at 1521 cm−1 attributed to the N–H (amide II band) due to the conversion of the Boc-group into the –NH3+ functionality (Fig. S10 and S12†). The appearance of free primary amino groups in those pendant peptide terminals transformed these hydrophobic homopolymers and amphiphilic block copolymers into the hydrophilic homopolymer P(H3N+-LVF-EMA) and the double hydrophilic block copolymer mPEGn-b-P(H3N+-LVF-EMA), respectively. These terminal free primary amino groups carrying peptidic polymers may find potential utility in a wide range of post-polymerization modification reactions to modulate their properties for various applications, which we will study separately in future.
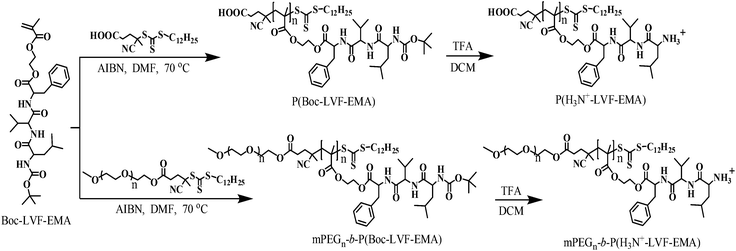 |
| | Scheme 1 Synthesis of tripeptide monomer derived homopolymers and block-copolymers by RAFT polymerization, followed by deprotection of Boc groups. | |
Recently, Mori et al. have reviewed a variety of amino acid based poly[(meth)acrylamide] chiral polymers with side-chain terminal free carboxylic acid groups, which showed pH responsiveness due to the ionization/deionization of carboxyl groups at different pH.13 Herein, the terminal primary amino groups of the peptidic moiety can be reversibly protonated/deprotonated by altering the pH of their aqueous solution.45 Hence, pH induced stimuli responsiveness of the synthesized polymers was analyzed from the turbidity measurement at 500 nm by UV-vis spectroscopy as a function of pH at 27 °C (Fig. 4A).46 Aqueous solutions (0.1 wt%) of P(H3N+-LVF-EMA), mPEG2k-b-P(H3N+-LVF-EMA) and mPEG5k-b-P(H3N+-LVF-EMA) showed phase transitions at pH 5.8, 6.0 and 6.2, respectively. Below this transition pH value the corresponding polymers have hydrophilic character due to protonation of pendant amino groups and are soluble in aqueous medium, while above this pH they undergo phase transition to the hydrophobic nature due to deprotonation of amino groups and achieve the inter/intra-molecular aggregated state (Fig. S13†). This phenomenon of pH induced phase transition has been further illustrated by the size distribution plot for a typical polymer mPEG2k-b-P(H3N+-LVF-EMA) (Fig. S14†). Interestingly, these primary amine containing polymers were retaining their pH based stimuli responsive behavior in PBS buffer solution also (Fig. S15†).
 |
| | Fig. 4 (A) Effect of pH on the transmittance of 0.1 wt% aqueous solution of P(H3N+-LVF-EMA) at 500 nm, and (B) CAC determination for the amphiphilic mPEG2k-b-P(Boc-LVF-EMA) block copolymer. Inset in (B) shows variation in emission intensity (E. I.) of the encapsulated pyrene dye inside polymer aggregates. | |
Self-assembly of polymers
The 1H NMR spectrum of the block copolymer mPEG2k-b-P(Boc-LVF-EMA) in CDCl3 (Fig. 3A) exhibited all NMR resonances from both of the block segments. While the corresponding 1H NMR spectrum in D2O displayed peaks corresponding to the hydrophilic mPEG block, and complete disappearance of Boc-LVF-EMA resonances was observed, engendered by possible suppressed molecular activity of the aggregated hydrophobic P(Boc-LVF-EMA) core encircled by the hydrophilic mPEG block (Fig. 3B).47 This phenomenon strongly exhibits formation of a stable higher-order morphology having a highly viscous inner core.48 The self-aggregation behaviour of amphiphilic mPEGn-b-P(Boc-LVF-EMA) block copolymers in aqueous medium was investigated by fluorescence spectroscopy. The critical aggregation concentration (CAC) was determined by employing pyrene as an extrinsic probe.10 The relative emission intensities of the pyrene probe at 392 and 373 nm (I392/I373) were indicative of the polarity of its microenvironment. The ratios of fluorescence intensities I392/I373 (λex = 339 nm) were plotted against the concentration logarithm of the block copolymer (Fig. 4B). The CAC values were determined from the calculated polymer concentration value at which the relative fluorescence intensity ratio began to change. The observed CAC values of mPEG2k-b-P(Boc-LVF-EMA) and mPEG5k-b-P(Boc-LVF-EMA) were 3.0 and 3.2 μg mL−1, respectively. The lower CAC value of the former amphiphilic polymer can be attributed to its longer hydrophobic amino acid block length.
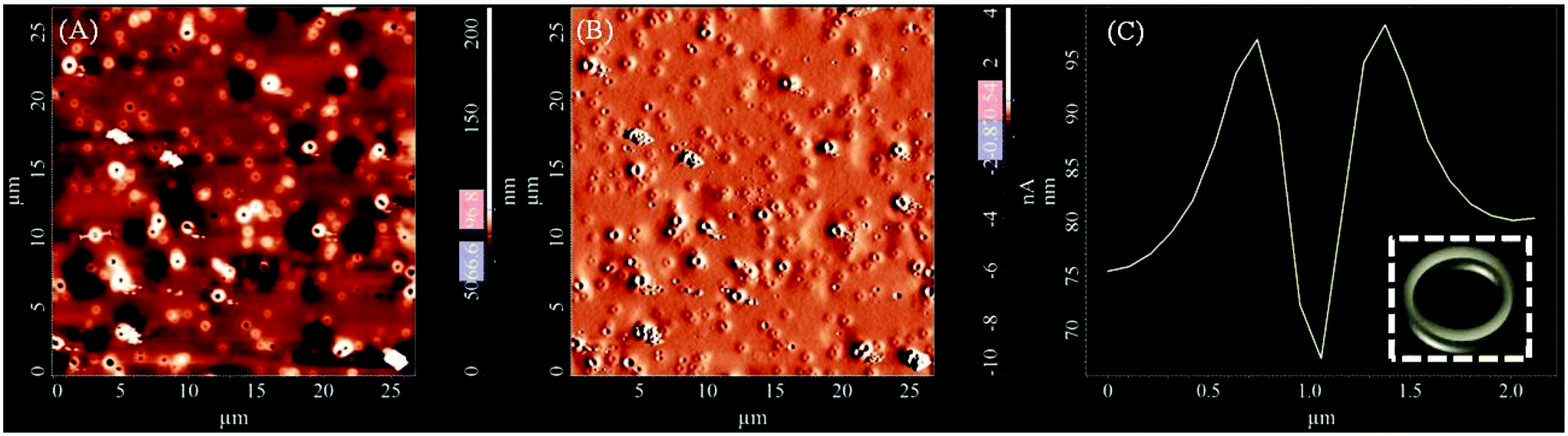
As illustrated by FE-SEM and AFM studies, the hydrophobic homopolymer P(Boc-LVF-EMA) self-assembled to form spherical morphology in polar methanol solution with an average diameter of about 450 nm (Fig. S16†). However, the corresponding Boc-deprotected homopolymer P(H3N+-LVF-EMA) assembled to form toroid like morphology in methanol solution with an average diameter of 1.7 μm (Fig. 5 and Fig. S17†).49 The correlation of these AFM results with its CD result (vide infra) suggests the presence of secondary structuring in the inside region of these nanostructures.11 Interestingly, the investigation by FE-SEM of the drop casting of the aqueous solution of the primary amine group carrying homopolymer P(H3N+-LVF-EMA) over silicon wafer displayed micellar aggregated nanostructures of 327 nm diameter, correlated with the dynamic light scattering (DLS) data that exhibited the size distribution of 340 nm (Fig. S18†). This phenomenon was engendered by the presence of the hydrophobic segment having the methacrylate backbone, alkyl groups (in leucine and valine) and the benzyl group (in phenylalanine) in each peptidic repeating unit as well as the presence of hydrophilic primary amine terminals and the polar segment of the oxyethyl peptidic backbone of each monomer unit.50 Additionally, the presence of amide and amine groups in each of the repeating unit can further induce secondary interactions in solution creating supramolecular assemblies.
 |
| | Fig. 5 AFM height (A), magnitude (B) image of homopolymer P(H3N+-LVF-EMA), (C) height profile plot (line marked in A) (prepared from 1 mg mL−1 methanol solution) (inset in C: toroidal structure). | |
Chiroptical properties
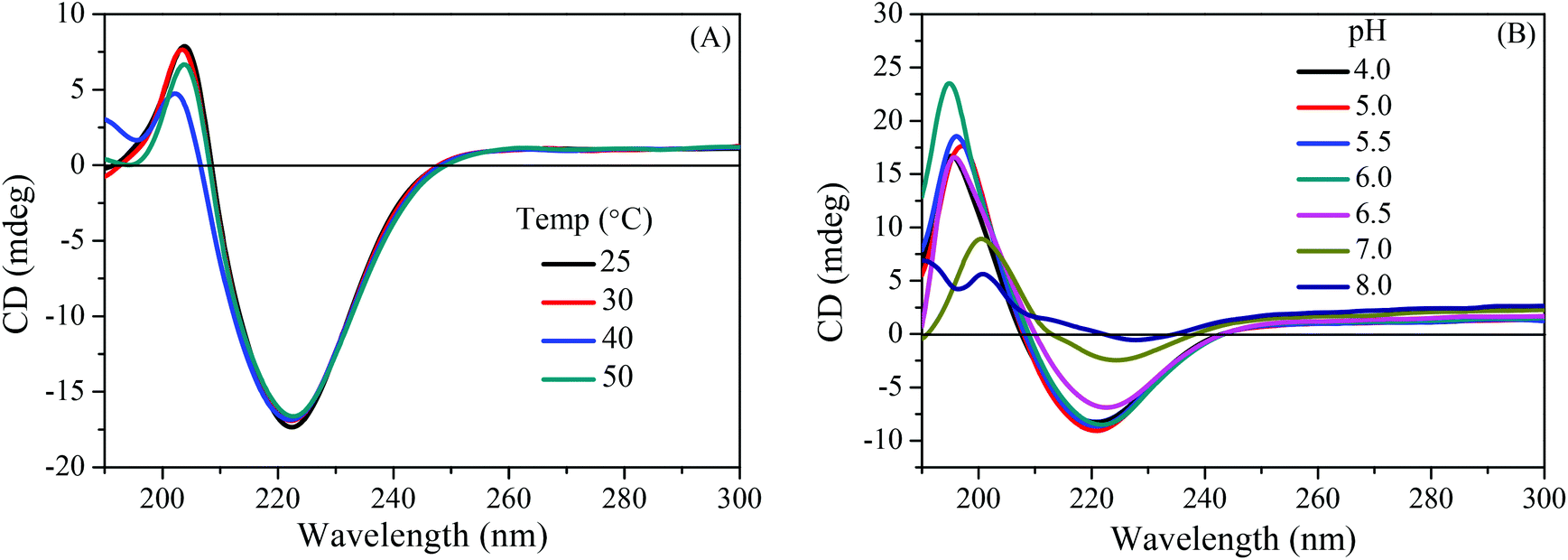
The as synthesized polymers can have supramolecular chirality originating from the self-association of their tripeptidic constituents during the process of self-assembly of polymers in solution. Therefore, CD spectroscopy was performed to assess the ability of these peptidic polymers to adopt secondary structures in solution. As compared to the tripeptide Boc-LVF-OMe, the homopolymer P(Boc-LVF-EMA) in dilute methanol solution exhibited a strong negative minimum at 222 nm with a very weak positive signal at 202 nm (Fig. 6A), demonstrating the alteration in conformation by transforming the tripeptide moiety into its derived homopolymer. The pattern of the peaks for P(Boc-LVF-EMA) in the CD spectrum is similar to that generally found (but not by exact positions of peaks) for the β-sheet secondary structure (positive maximum at 197 nm and negative minimum at 218 nm) of proteins.51 However, the particular spectral trend confirmed that the self-assembled structure of P(Boc-LVF-EMA) was mostly governed by β-pleated sheet arrangement.11,52–54 Furthermore, we employed solid-state FT-IR spectroscopy to understand the formation of β-sheets as well as to distinguish between its parallel and antiparallel propensity. Typically, the FT-IR spectrum of the P(Boc-LVF-EMA) homopolymer depicted the existence of the amide I band at 1692 cm−1 which can be assigned to antiparallel β-sheet conformations (Fig. 6B).4,17,55 Additionally, the appearance of a peak in the amide II region at 1521 cm−1 and in the amide V region at 700 cm−1 along with the absence of the band at 620 cm−1 (corresponds to the α-helix conformation) further supported the existence of β-sheet characteristics of the polymer.56,57 For the corresponding Boc-deprotected polymer P(H3N+-LVF-EMA) a negative CD signal at 223 nm persists with the strengthening of peak maxima at 202 nm, suggesting that the β-sheet conformation persists even after the terminal Boc-group deprotection (Fig. 6A). In the case of the Boc-protected PEGylated peptidic block copolymer mPEG2k-b-P(Boc-LVF-EMA), a strong clear negative minimum at 222 nm was observed along with a positive peak at 201 nm (Fig. 7), whereas after Boc-deprotection, mPEG2k-b-P(H3N+-LVF-EMA) shows the corresponding positive maximum shifted to 205 nm with the persistence of a negative peak at 223 nm. In the case of the mPEG5k-CTA derived block copolymer, we observed similar CD patterns both before and after Boc group deprotection (Fig. S19A†). These results demonstrated that these side-chain tripeptidic polymers exhibit specific β-sheet secondary structures in solution (Fig. S20†), which are independent of the mPEG block length and also of the molecular weight of the peptidic block segment. To understand the effect of temperature on side-chain tripeptidic polymer's conformation in solution, CD spectra are recorded for the Boc-protected homopolymer P(Boc-LVF-EMA) in methanol solution (Fig. 8A) and the Boc-deprotected block copolymer mPEG2k-b-P(H3N+-LVF-EMA) in aqueous media (Fig. S19B†) at different temperatures. Apparently, these polymers did not show substantial conformation changes upon temperature increase, suggesting that the adopted β-sheet conformation was thermodynamically stable in the experimental temperature range. To study the effect of solution pH on the chiroptical properties of the deprotected polymers, we analyzed aqueous solutions of mPEG2k-b-P(H3N+-LVF-EMA) at different pH by CD spectroscopy. As illustrated in Fig. 8B, almost similar CD spectra having superimposable negative minima were observed at pH lower than 6.0, suggesting the persistence of its β-sheet conformation up to pH 6.0. Note that we previously showed the hydrophilic to hydrophobic phase transition pH for mPEG2k-b-P(H3N+-LVF-EMA) as 6.0. Subsequent increment in pH above this responsive pH point showed continuous disruption of its adopted β-sheet conformation and complete disorder at a high pH value of 8.0. Moreover, we also investigated the effect of the solvent on the acquired conformation of side-chain tripeptidic polymers. The CD spectrum of mPEG2k-b-P(H3N+-LVF-EMA) in methanol (Fig. 7) is comparable to its spectrum in aqueous media (Fig. 8B), exhibiting the persistence of the β-sheet conformation and further indicates that the interaction of the polymer with the solvent is not apparently strong enough to disrupt the H-bonding found in the folded structure. The solvent polarity-independent conformational properties of the polymers were further illustrated in Fig. S21† by recording the CD spectra in solvents of different polarity. Since external addition of urea can generally denature the H-bonded secondary structure of proteins,58 we further investigated the effect of urea on the acquired conformation of the side-chain tripeptidic polymers. The CD spectral pattern remained unchanged with the persistence of negative maxima at 222 nm even up to a very high concentration of urea (Fig. S22†). However, we observed little decrement in the intensity of the 222 nm peak, due to the decreased concentration of the polymer after the addition of urea from the stock solution. Such a high stability can be attributed to the presence of strong H-bonding in the β-sheet conformations.37
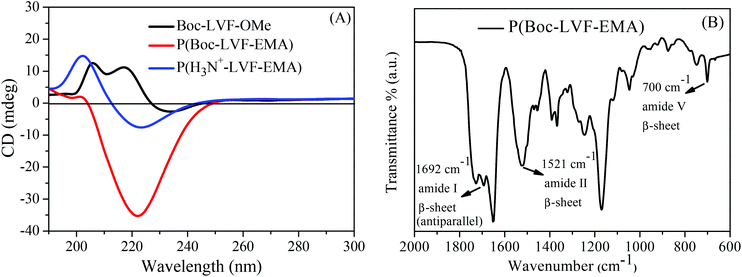 |
| | Fig. 6 CD spectra in methanol of (A) Boc-LVF-OMe tripeptide, Boc protected and deprotected homopolymer, and (B) solid state FT-IR spectra of P(Boc-LVF-EMA) polymer. | |
 |
| | Fig. 7 CD spectra of mPEG2k-CTA derived Boc-protected and deprotected block copolymers in methanol solution. | |
 |
| | Fig. 8 CD spectra of (A) the homopolymer P(Boc-LVF-EMA) in methanol at different temperatures, and (B) the block copolymer mPEG2k-b-P(H3N+-LVF-EMA) in an aqueous solution at various pH. | |
Characterization of polymer/DNA complexes
The surface charges (ξ) of the Boc-deprotected peptidic homo- and block copolymers in deionized (DI) water (1.0 mg mL−1) at pH 4.8 (below the transition pH) were quantified by zeta potential measurements. The ξ values of P(H3N+-LVF-EMA), mPEG2k-b-P(H3N+-LVF-EMA) and mPEG5k-b-P(H3N+-LVF-EMA) were found to be +13.2, +10.4 and +11.6 mV, respectively. The positive ξ values were attributed to the presence of ammonium (–NH3+) pendants at acidic pH, indicating the cationic nature of the deprotected polymers. To investigate the DNA binding ability of the side-chain peptide containing homo- and block co-polymers, different polymer/DNA weight ratios ranging from 0.1 to 10 were mixed to obtain polyplexes. Their ionic interaction, electrolytic stabilities and electrophoretic mobility were studied by gel electrophoresis.59,60 In the gel lower migration of DNA indicates the more stability of the ionic polyplex, which also denotes a strong interaction between the polymer and DNA. In the case of P(H3N+-LVF-EMA), it was observed that the polyplex at a weight ratio from 0.1 to 0.6 displayed migration of pDNA across the gel similar to that observed for the free pDNA but with a decrement in the intensity (Fig. 9A). With further increments in the weight ratio, no migration of the pDNA confirmed the formation of stable polyplexes at higher ratios of polymer/DNA and all pDNA molecules were involved in the formation of complexes with the peptidic homopolymer, suggesting their possible gene transfer applications. For the mPEG5k-b-P(H3N+-LVF-EMA) block copolymer, we observed similar formation of stable polyplexes, but the corresponding complete complexation was achieved at a relatively higher polymer/DNA weight ratio (Fig. 9B). Here, relatively more amount of cationic polymer was required to achieve complete DNA complexation due to the steric hindrance offered by the PEG chains.61 The achieved ratio for complete complexation of pDNA by these peptidic polymers depends on the peptide block length and the presence of the mPEG block would influence and tend to coat the bundling of pDNA with peptide side chains.62
 |
| | Fig. 9 Electrophoretic mobility of pDNA in the polyplexes formed by P(H3N+-LVF-EMA) homopolymer (A), and mPEG5k-b-P(H3N+-LVF-EMA) block copolymer (B) at different polymer/DNA weight ratios; pDNA without a polymer was used as a control. (C) Cytotoxicity of polymers on MCF-7 cells; P(H3N+-LVF-EMA) (blue color), mPEG2k-b-P(H3N+-LVF-EMA) (red color), and mPEG5k-b-P(H3N+-LVF-EMA) (green color). (D) Fluorescence microscopy images for the uptake of mPEG2k-b-P(Boc-LVF-EMA)-Nile red in MCF-7 cells treated at different concentrations for 24 h. | |
Cytotoxicity and cellular uptake studies
As a drug delivery system, the cytotoxicity assessment of the carrier is a key index for its future biomedical applications. Thus, the biocompatibility of these peptidic side-chain polymers in vitro was evaluated by the MTT assay against MCF-7 (human breast cancer) cell lines. Fig. 9C shows negligible cytotoxicity profiles of the polymers at concentrations up to 200 μg mL−1 following long-term incubation for 24 h. Confocal microscopy study of peptidic block copolymer treated MCF-7 cells revealed no obvious change in the nuclear structure with retained morphological integrity of the cells as compared to the control (Fig. S23A†).
Lastly, side-chain peptide containing polymers compatible for cell encapsulation were studied. Cellular uptake of drug (Dox) or dye (Nile red) loaded PEGylated peptidic block copolymer based micelles (Fig. S24†) was further determined by confocal laser scanning microscopy (CLSM). Fig. 9D shows dose dependent cellular uptake of the hydrophobic Nile red dye loaded mPEG2k-b-P(Boc-LVF-EMA) micelle in MCF-7 cells after incubation for 24 h. Diffused accumulation of dye loaded micelles in the intracellular matrix as well as in the nucleus suggests their potential application as a biomarker for pharmaceutical applications.63 Furthermore, the fluorescence microscopy images of MCF-7 cells treated with doxorubicin based anticancer drug loaded mPEG5k-b-P(Boc-LVF-EMA) micelles further confirmed their dose dependent cellular internalization, which suggests their possible application for cancer treatment (Fig. S23B†).
Conclusions
In summary, we have successfully demonstrated a simple and efficient approach to synthesize side-chain peptide containing polymers by the RAFT technique in a controlled fashion. Using mPEG-macro CTA, we have further prepared tripeptide based amphiphilic hybrid biomaterials mPEGn-b-P(Boc-LVF-EMA) and the cellular uptake studies of their dye or drug encapsulated micelles confirmed their promising bioapplications for cancer detection or treatment. Boc-group deprotection from the side-chain peptide units engendered terminal primary amino group pendants with pH responsiveness and cationic properties, which were able to form strong complexes with pDNA. These peptidic polymers exhibited the biocompatible nature as confirmed by the MTT assay against MCF-7 cells. CD studies of these peptidic polymers in solution illustrated the formation of strong β-sheet motifs which retained even after the Boc-group deprotection. The antiparallel β-sheet motif was independent of solvent polarity, PEGylation of homopolymers, the block length of PEG or peptidic segments in the block copolymer, as well as temperature. However, the β-sheet conformation was retained only up to the transition pH point, and continuous disruption was observed as the pH was further increased. Finally, it can be proclaimed that the strategy employed for preparing well-defined peptidic hybrid synthetic macromolecules with cationic, biocompatible and pH-responsive properties may provide new opportunities and fundamental guidelines to design promising ‘smart’ nano-carriers with excellent performance for both drug and gene delivery systems in biomedicine.64 We are currently studying highly ordered hierarchical organization from these amphiphilic block copolymers in detail and application of these macromolecules towards inhibition of amyloid-β 40 (Aβ40) peptide aggregation, involved in Alzheimer's disease.
Acknowledgements
The financial support for this work by the Department of Science and Technology (DST), New Delhi, India [project no. SR/S1/OC-51/2010] is gratefully acknowledged. We thank Dr. Sankar Maiti (Department of Biological Sciences, IISER-Kolkata) for the electrophoresis study.
Notes and references
- M. Motornov, Y. Roiter, I. Tokarev and S. Minko, Prog. Polym. Sci., 2010, 35, 174–211 CrossRef CAS PubMed.
- M. A. Gauthier and H.-A. Klok, Chem. Commun., 2008, 2591–2611 RSC.
- R. K. O'Reilly, Polym. Int., 2010, 59, 568–573 Search PubMed.
- J. Hentschel and H. G. Börner, J. Am. Chem. Soc., 2006, 128, 14142–14149 CrossRef CAS PubMed.
- G. L. Bidwell, Ther. Delivery, 2012, 3, 609–621 CrossRef CAS.
- S. Lim, W. J. Kim, Y. H. Kim and J. M. Choi, Mol. Cells, 2012, 34, 577–582 CrossRef CAS PubMed.
- J. Hamzah, V. R. Kotamraju, J. W. Seo, L. Agemy, V. Fogal, L. M. Mahakian, D. Peters, L. Roth, M. K. J. Gagnon, K. W. Ferrara and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7154–7159 CrossRef CAS PubMed.
- I. W. Hamley, Angew. Chem., Int. Ed., 2007, 46, 8128–8147 CrossRef CAS PubMed.
- J. Nicolas, G. Mantovani and D. M. Haddleton, Macromol. Rapid Commun., 2007, 28, 1083–1111 CrossRef CAS.
- S. Kumar, R. Acharya, U. Chatterji and P. De, J. Mater. Chem. B, 2013, 1, 946–957 RSC.
- D. J. Adams, D. Atkins, A. I. Cooper, S. Furzeland, A. Trewin and I. Young, Biomacromolecules, 2008, 9, 2997–3003 CrossRef CAS PubMed.
- H.-A. Klok, Macromolecules, 2009, 42, 7990–8000 CrossRef CAS.
- H. Mori and T. Endo, Macromol. Rapid Commun., 2012, 33, 1090–1107 CrossRef CAS PubMed.
- K. Osada and K. Kataoka, Adv. Polym. Sci., 2006, 202, 113–153 CrossRef CAS.
- S. Kumar, R. Acharya, U. Chatterji and P. De, Langmuir, 2013, 29, 15375–15385 CrossRef CAS PubMed.
- M. P. Lutolf, G. P. Raeber, A. H. Zisch, N. Tirelli and J. A. Hubbell, Adv. Mater., 2003, 15, 888–892 CrossRef CAS.
- A. Rösler, H.-A. Klok, I. W. Hamley, V. Castelletto and O. O. Mykhaylyk, Biomacromolecules, 2003, 4, 859–863 CrossRef PubMed.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS PubMed.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- J.-F. Lutz and H. G. Börner, Prog. Polym. Sci., 2008, 33, 1–39 CrossRef CAS PubMed.
- S. Kumar and P. De, Polymer, 2014, 55, 824–832 CrossRef CAS PubMed.
- H. D. Maynard, S. Y. Okada and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 1275–1279 CrossRef CAS PubMed.
- H. Komazawa, I. Saiki, Y. Igarashi, I. Azuma, M. Kojima, A. Orikasa, M. Ono and I. Itoh, J. Bioact. Compat. Polym., 1993, 8, 258–274 CrossRef CAS PubMed.
- L. Ayres, M. R. J. Vos, P. J. H. M. Adams, I. O. Shklyarevskiy and J. C. M. van Hest, Macromolecules, 2003, 36, 5967–5973 CrossRef CAS.
- L. Ayres, H. H. M. Adams, D. W. P. M. Lowik and J. C. M. van Hest, Biomacromolecules, 2005, 6, 825–831 CrossRef CAS PubMed.
- H. G. Börner, B. M. Smarsly, J. Hentschel, A. Rank, R. Schubert, Y. Geng, D. E. Discher, T. Hellweg and A. Brandt, Macromolecules, 2008, 41, 1430–1437 CrossRef.
- I. W. Hamley, I. A. Ansari and V. Castelletto, Biomacromolecules, 2005, 6, 1310–1315 CrossRef CAS PubMed.
- M. G. J. ten Cate, H. Rettig, K. Bernhardt and H. G. Börner, Macromolecules, 2005, 38, 10643–10649 CrossRef CAS.
- F. Fernández-Trillo, A. Duréault, J. P. M. Bayley, J. C. M. van Hest, J. C. Thies, T. Michon, R. Weberskirch and N. R. Cameron, Macromolecules, 2007, 40, 6094–6099 CrossRef.
- B. Urbanc, L. Cruz, S. Yun, S. V. Buldyrev, G. Bitan, D. B. Teplow and H. E. Stanley, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17345–17350 CrossRef CAS PubMed.
- D. Haldar and A. Banerjee, Int. J. Pept. Res. Ther., 2006, 12, 341–348 CrossRef CAS.
- H. Skaat, R. Chen, I. Grinberg and S. Margel, Biomacromolecules, 2012, 13, 2662–2670 CrossRef CAS PubMed.
- E. Sarubbi, P. F. Seneci, M. R. Angelastro, N. P. Peet, M. Denaro and K. Islam, FEBS Lett., 1993, 319, 253–256 CrossRef CAS.
- R. L. Johnson, J. Med. Chem., 1982, 25, 605–610 CrossRef CAS.
- N. R. Lee, C. J. Bowerman and B. L. Nilsson, Biomacromolecules, 2013, 14, 3267–3277 CrossRef CAS PubMed.
- K. Kataoka, T. Matsumoto, M. Yokoyama, T. Okano, Y. Sakurai, S. Fukushima, K. Okamoto and G. S. Kwon, J. Controlled Release, 2000, 64, 143–153 CrossRef CAS.
- A. Das and S. Ghosh, Macromolecules, 2013, 46, 3939–3949 CrossRef CAS.
- S. G. Roy, R. Acharya, U. Chatterji and P. De, Polym. Chem., 2013, 4, 1141–1152 RSC.
- S. Kumar, S. G. Roy and P. De, Polym. Chem., 2012, 3, 1239–1248 RSC.
- P. De, S. R. Gondi and B. S. Sumerlin, Biomacromolecules, 2008, 9, 1064–1070 CrossRef CAS PubMed.
- P. De, S. R. Gondi, D. Roy and B. S. Sumerlin, Macromolecules, 2009, 42, 5614–5621 CrossRef CAS.
- J. W. Bartels, S. I. Cauèt, P. L. Billings, L. Y. Lin, J. Zhu, C. Fidge, D. J. Pochan and K. L. Wooley, Macromolecules, 2010, 43, 7128–7138 CrossRef CAS PubMed.
- H. Z. Cao, W. Zhang, J. Zhu, X. R. Chen, Z. P. Cheng, J. H. Wu and X. L. Zhu, eXPRESS Polym. Lett., 2008, 2, 589–601 CrossRef CAS.
- K. Bauri, S. Pant, S. G. Roy and P. De, Polym. Chem., 2013, 4, 4052–4060 RSC.
- K. Bauri, P. De, P. N. Shah, R. Li and R. Faust, Macromolecules, 2013, 46, 5861–5870 CrossRef CAS.
- K. Yamamoto, T. Serizawa, Y. Muraoka and M. Akashi, Macromolecules, 2001, 34, 8014–8020 CrossRef CAS.
- C. Kim, S. C. Lee, I. C. Kwon, H. Chung and S. Y. Jeong, Macromolecules, 2002, 35, 193–200 CrossRef CAS.
- M. Nakayama and T. Okano, Biomacromolecules, 2005, 6, 2320–2327 CrossRef CAS PubMed.
- S. H. Kim, F. Nederberg, L. Zhang, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Nano Lett., 2008, 8, 294–301 CrossRef CAS PubMed.
- E. N. Savariar, S. V. Aathimanikandan and S. Thayumanavan, J. Am. Chem. Soc., 2006, 128, 16224–16230 CrossRef CAS PubMed.
- Z.-X. Lu, K.-F. Fok, B. W. Erickson and T. E. Hugli, J. Biol. Chem., 1984, 259, 7367–7370 CAS.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS PubMed.
- J. Hwang and T. J. Deming, Biomacromolecules, 2001, 2, 17–21 CrossRef CAS PubMed.
- M. Higuchi, T. Inoue, H. Miyoshi and M. Kawaguchi, Langmuir, 2005, 21, 11462–11467 CrossRef CAS PubMed.
- T. Miyazawa and E. R. Blout, J. Am. Chem. Soc., 1961, 83, 712–719 CrossRef CAS.
- A. Panitch, K. Matsuki, E. J. Cantor, S. J. Cooper, E. D. T. Atkins, M. J. Fournier, T. L. Mason and D. A. Tirrell, Macromolecules, 1997, 30, 42–49 CrossRef CAS.
- T. Miyazawa, Y. Masuda and K. Fukushima, J. Polym. Sci., 1962, 62, S62–S64 CrossRef CAS.
- L. B. Sagle, Y. Zhang, V. A. Litosh, X. Chen, Y. Cho and P. S. Cremer, J. Am. Chem. Soc., 2009, 131, 9304–9310 CrossRef CAS PubMed.
- X. Liu, J. W. Yang, A. D. Miller, E. A. Nack and D. M. Lynn, Macromolecules, 2005, 38, 7907–7914 CrossRef CAS.
- J.-L. Zhu, H. Cheng, Y. Jin, S.-X. Cheng, X.-Z. Zhang and R.-X. Zhuo, J. Mater. Chem., 2008, 18, 4433–4441 RSC.
- Y. Ping, D. Wu, J. N. Kumar, W. Cheng, C. L. Lay and Y. Liu, Biomacromolecules, 2013, 14, 2083–2094 CrossRef CAS PubMed.
- E. Froehlich, J. S. Mandeville, C. M. Weinert, L. Kreplak and H. A. Tajmir-Riahi, Biomacromolecules, 2011, 12, 511–517 CrossRef CAS PubMed.
- J. Yan, Z. Ye, M. Chen, Z. Liu, Y. Xiao, Y. Zhang, Y. Zhou, W. Tan and M. Lang, Biomacromolecules, 2011, 12, 2562–2572 CrossRef CAS PubMed.
- A. J. Convertine, C. Diab, M. Prieve, A. Paschal, A. S. Hoffman, P. H. Johnson and P. S. Stayton, Biomacromolecules, 2010, 11, 2904–2911 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, ESI-MS, FT-IR, 1H and 13C NMR spectra of various compounds and polymers, GPC and CD spectra of polymers, AFM and SEM images of homopolymers, schematic illustration, CLSM images of block copolymers and dye/drug-loaded micelles and DLS data for the pH response. See DOI: 10.1039/c4py00620h |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a