 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The synthesis of microporous polymers using Tröger's base formation
Mariolino
Carta
a,
Richard
Malpass-Evans
b,
Matthew
Croad
b,
Yulia
Rogan
b,
Michael
Lee
a,
Ian
Rose
a and
Neil B.
McKeown
*a
aEaStCHEM and School of Chemistry, University of Edinburgh, West Mains Road, Edinburgh EH9 3JJ, Scotland, UK. E-mail: neil.mckeown@ed.ac.uk
bSchool of Chemistry, Cardiff University, Cardiff, CF10 2AT, UK
First published on 20th June 2014
Abstract
A step-growth polymerisation based on the formation of Tröger's base, performed by simple reaction of a suitable aromatic diamine monomer with dimethoxymethane in trifluoroacetic acid, provides polymers of high average molecular mass. The properties of the resulting polymers can be tailored by the choice of monomer. In particular, the Tröger's base polymerisation is highly suited to the preparation of soluble polymers of intrinsic microporosity (PIMs) due to the resulting fused-ring TB linking group, which is both highly rigid and prohibits conformational freedom.
Introduction
Leo Baekeland's phenol–formaldehyde resin, patented in 1907 and later sold in huge quantities as Bakelite, initiated the age of synthetic polymers.1 Bakelite's rapid commercial success encouraged investigation of the similar acid mediated reaction between aniline and formaldehyde (Fig. 1a) and resulted in a polymer originally sold as Dilectene or Cibanit.2–4 These aniline–formaldehyde resins proved useful as insulating materials for electronic equipment and for making composite building materials. In contrast, the analogous reaction between 4-methylaniline (p-toluidine) and formaldehyde, first studied by Julius Tröger in 1887,5 yields a non-polymeric product that was later identified as the bridged bicyclic diamine, 2,8-dimethyl-6H,12H-5,11-methanodibenzo[b,f][1,5]diazocine, now known as Tröger's base (TB; Fig. 1b).6 TB and its derivatives are of continuing interest as molecular building blocks for supramolecular assemblies, enantiomeric separations and as scaffolds for molecular replication.7–12 In addition, TB-containing polymers have been prepared by linking together preformed TB monomers using well-established polymerisation reactions.13,14 However, despite its long history, the TB-forming reaction has only very recently been used to make polymers (e.g. PIM–EA–TB; Fig. 1c).15–18 This oversight is surprising given the continued use of formaldehyde-based aromatic electrophilic substitution reactions for the preparation of commodity polymers and the ready availability of suitable aromatic diamine monomers.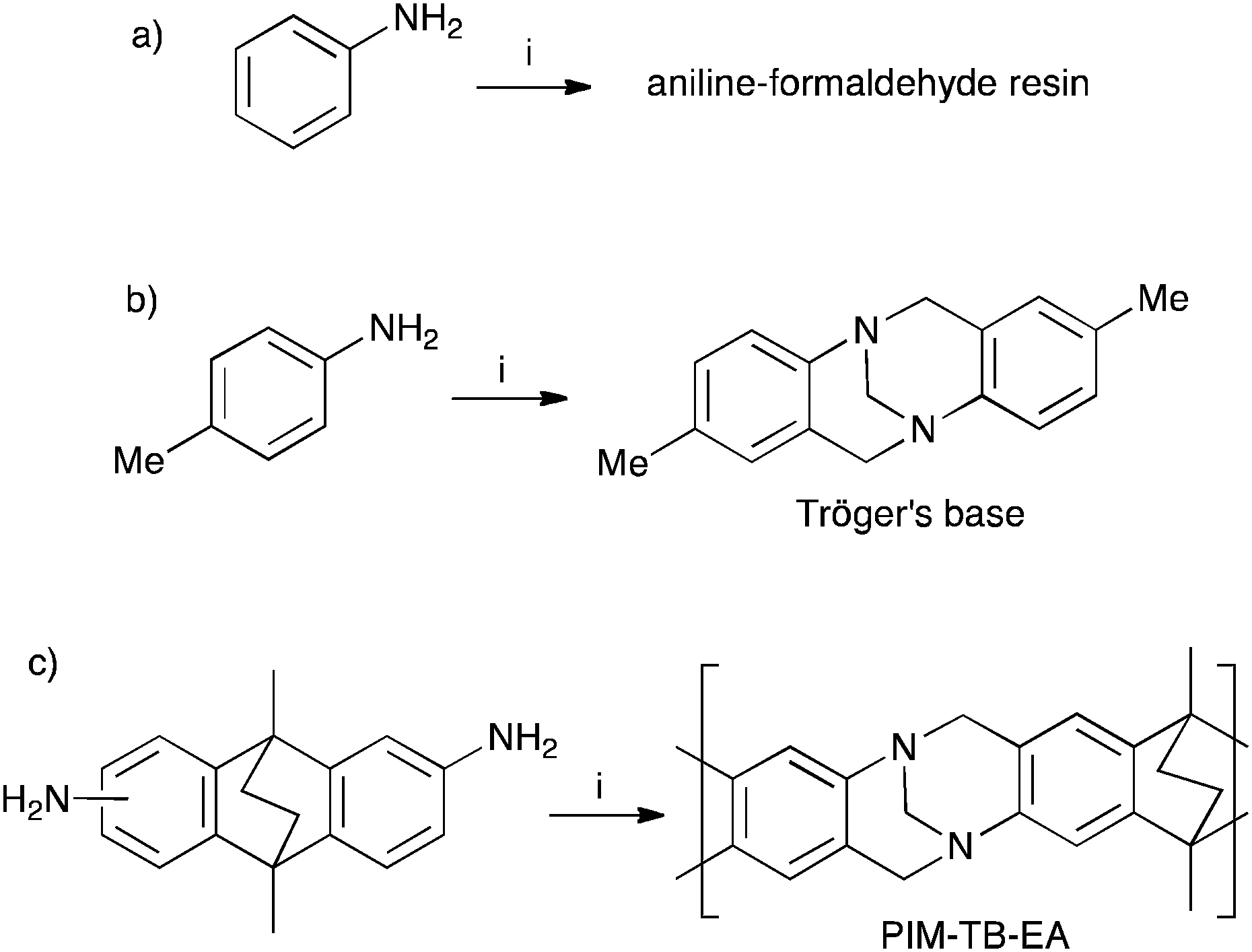 | ||
| Fig. 1 The synthesis of (a) aniline–formaldehyde resin; (b) Tröger's base and (c) PIM–EA–TB. In each case the reaction is mediated by a strong acid with formaldehyde or formaldehyde proxy (e.g. dimethoxymethane) as reagent. | ||
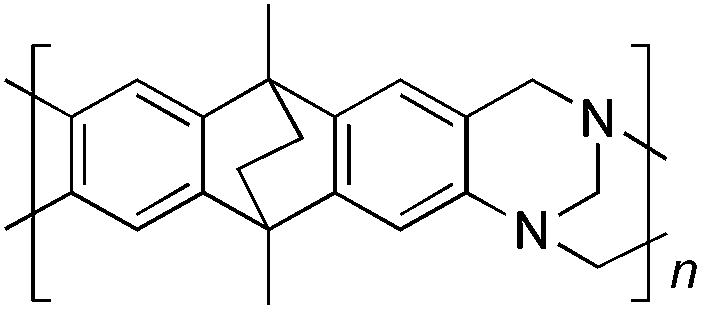
As part of an on-going programme to develop polymers of intrinsic microporosity (PIMs)19–21 for use in gas separation membranes,16,17,22–35 as adsorbents,36–38 for heterogeneous catalysis39 and as materials for sensors,40,41 we are interested in polymerisation reactions that can provide highly rigid and contorted polymers. In addition, the TB unit is an attractive component for PIMs as it can act as a strong base and therefore has potential for catalysis, CO2 adsorption, metal-binding etc. Here we assess the potential of TB formation for making polymers with particular emphasis on generating intrinsic microporosity.19–21
Experimental
Materials and methods
Commercially available reagents were used without further purification. 1H NMR spectra were recorded in the solvent stated using an Avance Bruker DPX 400 (400 MHz) or DPX 500 (500 MHz) instruments, with 13C NMR spectra recorded at 100 MHz or 125 MHz respectively. Matrix Assisted Laser Desorption Ionisation (MALDI) mass spectroscopic analysis was performed with a Waters MALDI Micro MX spectrometer. GPC analysis was carried out on a Viscotek GPCmax VE2001 with RI (VE3580) detector, using chloroform as eluent (columns: KF-805L SHODEX) at ambient temperature and flow rate of 1 ml min−1 calibrated with a series of polystyrene standards with molecular mass up to 9.4 × 105 g mol−1 with a narrow polydispersity. Low-temperature (77 K) N2 adsorption/desorption measurements of PIM powders were made using a Coulter SA3100. Samples were degassed for 800 min at 120 °C under high vacuum prior to analysis. Thermogravimetric analysis (TGA) was performed using a Thermal Analysis SDT Q600 at a heating rate of 10 °C min−1 from room temperature to 1000 °C.General procedure for TB polymerisation
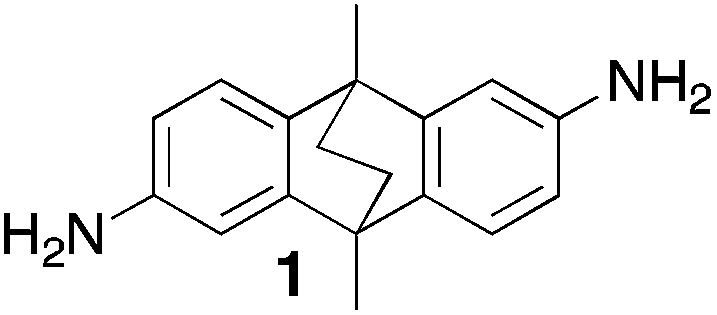
The aromatic diamine (1 mol eq.) was dissolved in dimethoxymethane (5 mol eq.) and the solution cooled to 0 °C. Trifluoroacetic acid (120 mol eq.) was added drop-wise over 0.5 h and the mixture was stirred at room temperature until the solution became viscous. The reaction mixture was then poured into vigorously stirred aqueous ammonium hydroxide solution and left for 2 h. The resulting solid was collected by filtration, washed with water and then acetone. The solid was dissolved in chloroform and methanol added to precipitate the polymer. This process was repeated twice. Finally the polymer was dried in a vacuum oven.From Monomer 1, 2,6(7)-diamino-9,10-dimethyl-9,10-dihydro-9,10-ethanoanthracene;16 yield = 82%; 1H NMR (500 MHz, CDCl3) δ ppm = 7.28 (br, m, 4H), 4.57 (br, s, 2H), 4.05 (br, s, 4H), 1.81 (br, m, 6H), 1.65 (br, m, 4H); 13C NMR (100 MHz, solid state) δ ppm 145.8, 142.3, 124.4, 117.3, 67.4, 59.2, 41.1, 36.6, 18.0; GPC Mn = 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700, Mw = 156000 g mol−1; elemental analysis: calculated C, 84.25; H, 6.40; N, 9.36 found C, 80.19; H, 6.43; N, 8.81%. BET surface area = 1028 m2 g−1; total pore volume = 0.75 cm3 g−1 at (p/po = 0.98); TGA: 4% loss of mass between 20 and 50 °C, mass loss due to thermal degradation commences at ∼260 °C with a 10% loss of mass below 400 °C.
700, Mw = 156000 g mol−1; elemental analysis: calculated C, 84.25; H, 6.40; N, 9.36 found C, 80.19; H, 6.43; N, 8.81%. BET surface area = 1028 m2 g−1; total pore volume = 0.75 cm3 g−1 at (p/po = 0.98); TGA: 4% loss of mass between 20 and 50 °C, mass loss due to thermal degradation commences at ∼260 °C with a 10% loss of mass below 400 °C.
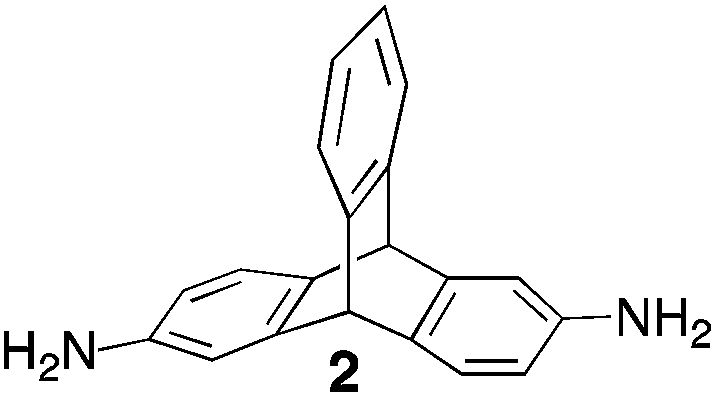
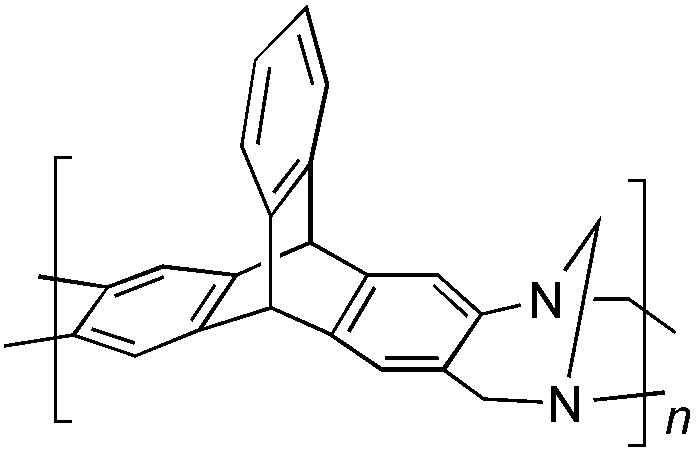
From Monomer 2, 2,6(7)-diaminotriptycene;17 yield = 79%; 1H NMR (500 MHz, CDCl3) δ ppm = 3.89 (br s, 2H), 4.41 (br s, 2H), 4.80 (br s, 2H), 5.07 (br s, 2H), 6.97 (br m, 8H); 13C NMR (solid state, 100 MHz) δ ppm = 53.7, 58.4, 67.0, 124.2, 145.0; GPC = Mn = 21500, Mw = 50700; elemental analysis: calculated C, 86.22; H, 5.03; N, 8.74 found C, 76.63; H, 4.15; N, 7.69%. BET surface area = 870 m2 g−1, total pore volume = 0.51 cm3 g−1 at (p/po = 0.98); TGA: 3% loss of mass between 20 and 50 °C, mass loss due to thermal degradation commences at ∼400 °C with a 30% loss of mass below 1000 °C.
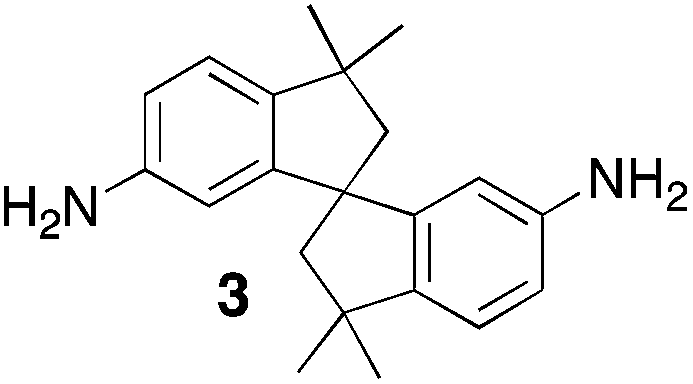
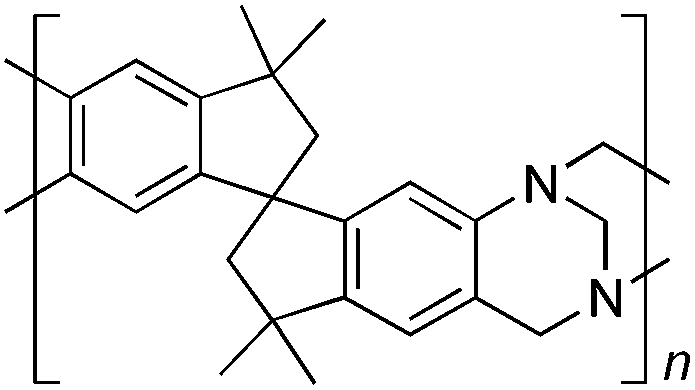
From Monomer 3, 5(6),5′(6′)-diamino-3,3,3′,3′-tetramethyl-1,1′-spirobisindane;16 yield = 73%; 1H NMR (500 MHz, CDCl3) δ ppm = 1.32 (br s, 12H), 2.24 (br s, 4H), 4.22 (br s, 4H); 4.66 (br s, 2H), 6.72 (br m, 4H); 13C NMR (100 MHz, solid state) δ ppm 148.4, 127.0, 120.1, 67.0, 57.7, 43.1, 31.0; GPC = Mn = 96000, Mw = 360000; elemental analysis: calculated C, 84.42; H, 7.28; N, 8.20 found C, 84.31; H, 7.46; N, 8.09%. BET surface area = 745 m2 g−1, total pore volume = 0.54 cm3 g−1 at (p/po = 0.98); TGA: 3% loss of mass between 20 and 50 °C, mass loss due to thermal degradation commences at ∼435 °C with a 33% loss of mass below 1000 °C.
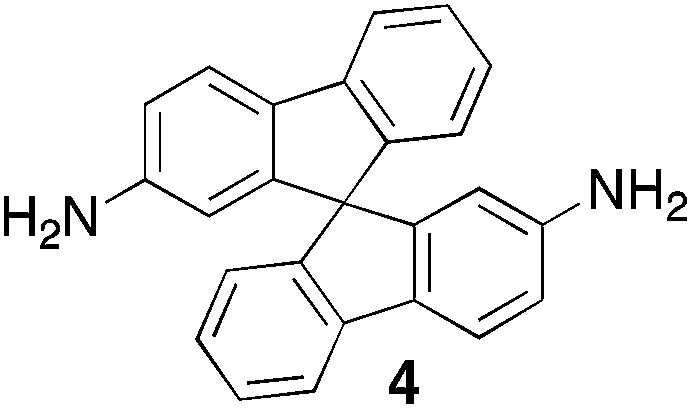
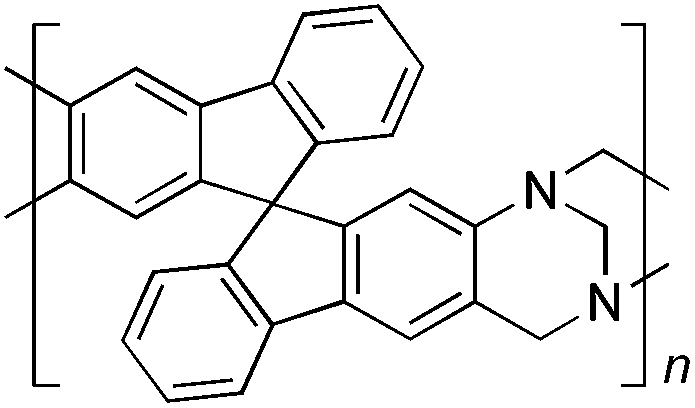
From Monomer 4, 9,9′-spirobisfluorene-2,2′-diamine;42 yield = 90%; 1H NMR (400 MHz, CDCl3) δ ppm = d 7.60 (br m, 4H), 7.30 (br m, 2H), 6.96 (br m, 2H), 6.43 (br m, 4H), 4.62(br s, 2H), 4.25(br s, 2H), 3.82 (br s, 2H); 13C NMR (100 MHz, solid state) δ ppm 148.6, 141.7, 127.5, 119.1, 66.2, 59.9, 54.1; GPC = Mn = 15000, Mw = 30000; elemental analysis: calculated C, 88.16; H, 4.49; N, 7.34 found C, 80.62; H, 4.34; N, 6.79%. BET surface area = 566 m2 g−1, total pore volume = 0.37 cm3 g−1 at (p/po = 0.98); TGA: 3% loss of mass between 20 and 100 °C, mass loss due to thermal degradation commences at ∼270 °C.
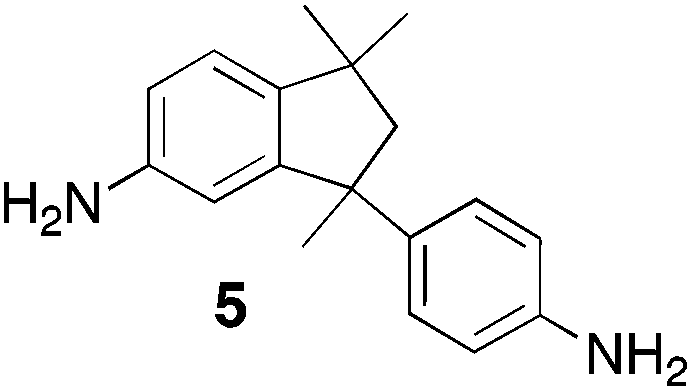
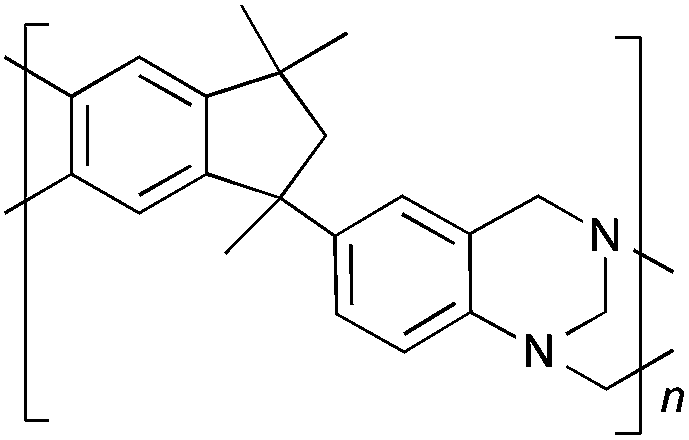
From Monomer 5, 5(6)-amino-1,1,3-trimethyl-3-(4-aminophenyl)indan;43 yield = 63%; 1H NMR (500 MHz, CDCl3) δ ppm = 6.60 (br, m, 5H), 4.44 (br, m, 2H), 4.10 (br, m, 4H), 2.10 (br, s, 1H), 1.86 (br, s, 1H), 1.34 (br, s, 3H), 1.05 (br, s, 3H), 0.81 (br, s, 3H); 13C NMR (100 MHz, solid state) δ ppm 147.5, 126.8, 67.2, 60.0, 50.3, 42.6, 30.9; GPC = Mn = 36000, Mw = 45000; elemental analysis: calculated C, 83.68; H, 7.02; N, 9.29 found C, 83.45; H, 6.91; N, 9.24%. BET surface area = 535 m2 g−1, total pore volume = 0.61 cm3 g−1 at (p/po = 0.98); TGA: 4% loss of mass between 20 and 50 °C, mass loss due to thermal degradation commences at ∼350 °C with a 45% loss of mass below 1000 °C.
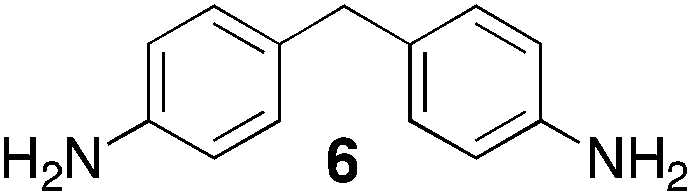
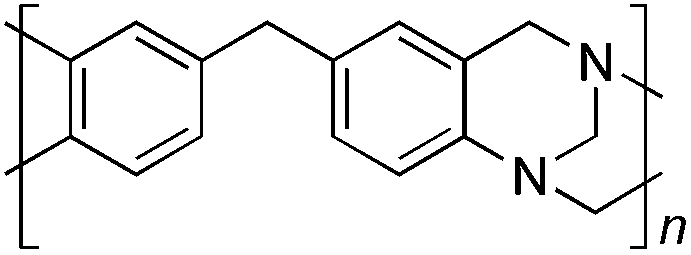
From Monomer 6, 4,4′-diaminophenylmethane; yield = 95%; insoluble solid. 13C NMR (100 MHz, solid state) δ ppm 146.7, 137.6, 128.0, 66.5, 59.2, 41.0; elemental analysis: calculated C, 82.37; H, 5.62; N, 12.01 found C, 76.93; H, 6.40; N, 11.91% (note low values for carbon likely to be due to solvent or water trapped in insoluble network).
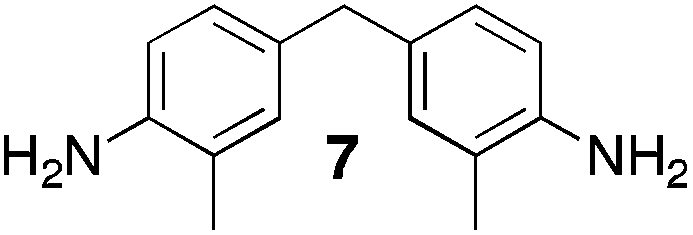
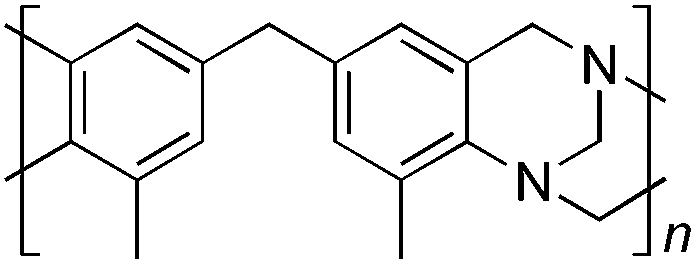
From Monomer 7, 4,4′-diamino-3,3′-dimethylphenylmethane; yield = 54%; 1H NMR (500 MHz, CDCl3) δ ppm = 6.81 (br, s, 2H), 6.58 (br, s, 2H), 4.54 (br, m, 2H), 4.29 (br, s, 2H), 3.95 (br, m, 2H), 3.67 (br, s, 2H), 2.37 (br, s, 6H); 13C NMR (100 MHz, solid state) δ ppm 144.7, 137.0, 132.6, 128.2, 68.2, 55.6, 40.6, 16.8; GPC = Mn = 49500, Mw = 94500; elemental analysis: calculated C, 82.72; H, 6.56; N, 10.72 found C, 82.56; H, 6.45; N, 10.59%. BET surface area = 38 m2 g−1, total pore volume = 0.14 cm3 g−1 at (p/po = 0.98); TGA: 1% loss of mass between 20 and 50 °C, mass loss due to thermal degradation commences at ∼350 °C with a 45% loss of mass below 1000 °C.
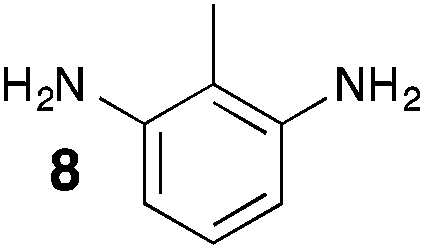
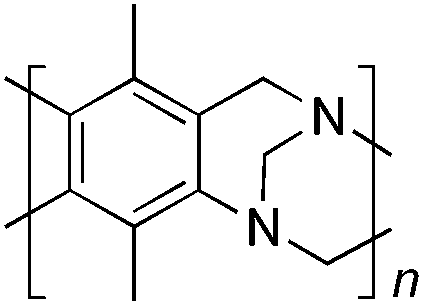
From Monomer 8, 2-methyl-1,3-benzenediamine; yield = 91%; 1H NMR (400 MHz, CDCl3) δ ppm = 6.30 (br s, 1H), 4.11 (br m, 6H), 2.35 (br s, 3H); 13C NMR (100 MHz, solid state) δ ppm 146.0, 124.4, 67.6, 55.5, 44.6, 11.8; GPC = Mn = 3800, Mw = 8100; elemental analysis: calculated C, 76.42; H, 5.77; N, 17.82 found C, 65.72; H, 4.83; N, 14.64%. BET surface area = 504 m2 g−1, total pore volume = 0.37 cm3 g−1 at (p/po = 0.98); TGA: 5% loss of mass below 150 °C, mass loss due to thermal degradation commences at ∼380 °C.
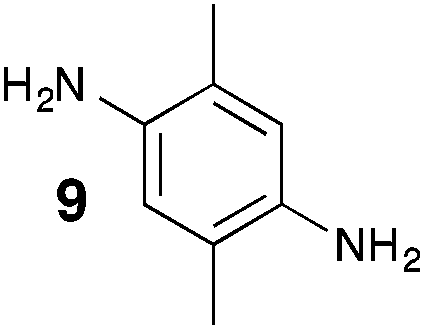
From Monomer 9, 2,5-dimethyl-1,4-phenylenediamine; yield = 95%; 1H NMR (400 MHz, CDCl3) δ ppm = 4.31 (br m, 2H), 4.09 (br m, 2H), 3.79 (br m, 2H), 2.15 (br s, 3H), 1.79 (br s, 3H); 13C NMR (100 MHz, solid state) δ ppm 142.8, 126.5, 66.3, 54.7, 11.7; GPC = Mn = 6500, Mw = 10500; elemental analysis: calculated C, 77.16; H, 6.48; N, 16.36 found C, 69.21; H, 6.98; N, 13.03%. BET surface area = 677 m2 g−1, total pore volume = 0.49 cm3 g−1 at (p/po = 0.98); TGA: 5% loss of mass below 150 °C, mass loss due to thermal degradation commences at ∼350 °C.
Results and discussions
Aromatic diamines are commonly employed as monomers for polyamide, polyimide and polyurea preparation. Hence many thousands of monomers with potential for TB polymerisation are commercially available or can be prepared readily. Diamines 1–4, based on ethanoanthracene,16 triptycene,17 1,1-spirobisindane16 and 9,9-spirobifluorene,42 respectively, were selected for investigation because they all contain fused ring or spirocyclic structures and hence have the potential to generate intrinsic microporosity.19 Diamine monomer 5, based on 1,3,3-trimethyl-1-phenylindane,43 was chosen due to its use as a monomer for making commercial polyimides (e.g. Matrimid) that are exploited for many different applications including the fabrication of gas separation membranes.44 Monomers 6 and 7, based on diphenylmethane, and the diaminobenzene monomers 8–9 are commercially available.A meticulous study by Didier et al.12 showed that TB and its simple derivatives can be prepared from suitable anilines in excellent isolated yields (>95%) from the reaction with a convenient source of formaldehyde such as paraformaldehyde or dimethoxymethane in trifluoroacetic acid (TFA), which acts as both acid catalyst and solvent. Hence, these reaction conditions were used as the initial basis for the step-growth polymerisations of aromatic diamine monomers 1–9 (Table 1).
| Monomer | Polymer | Yield | M n × 10−3(g mol−1) | M w × 10−3(g mol−1) | n | SABET(m2 g−1) | V p(ml g−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
|
|
82 | 41 | 156 | 133 | 1028 | 0.75 | 16 |
|
|
79 | 22 | 51 | 66 | 899 | 0.51 | 17 |
|
|
73 | 96 | 360 | 281 | 745 | 0.54 | 16 |
|
|
90 | 15 | 30 | 39 | 566 | 0.37 | — |
|
|
63 | 36 | 45 | 120 | 535 | 0.61 | — |
|
|
95 | n.a | n.a | n.a | n.a | n.a | — |
|
|
54 | 50 | 95 | 175 | 38 | 0.14 | — |
|
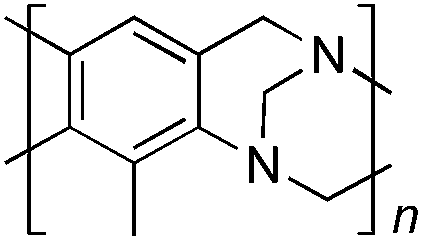
|
91 | 3.8 | 8.1 | 24 | 504 | 0.37 | — |
|
|
95 | 6.5 | 10.5 | 32 | 677 | 0.49 | — |
In general, the TB polymerisations proved remarkably easy to perform by simply mixing the monomer and dimethoxymethane in TFA at 0 °C and leaving under ambient conditions until the solution becomes too viscous to stir using a conventional magnetic stirring bar. Preliminary investigations showed that dimethoxymethane produces polymers with higher average molecular mass than paraformaldehyde and that an excess of dimethoxymethane (i.e. molar ratio of aromatic diamine to dimethoxymethane of approximately 1:5) also gave better results. The lack of the requirement for an exact stoichiometric balance between the two monomers is an unusual but helpful feature of this step-growth polymerisation. Work-up involves the precipitation of the resulting polymer from the reaction mixture by pouring into water or aqueous ammonia solution followed by collecting the solid polymer by simple filtration. Purification (i.e. removal of oligomers) is achieved by reprecipitation of the polymer from chloroform solution into methanol or hexane.
With the exception of monomer 6, polymers were obtained from all monomers 1–9 that were wholly soluble in chloroform allowing for confirmation of the expected structure by solution 1H NMR and an estimation of their average molecular mass by Gel Permeation Chromatography (GPC), calibrated to polystyrene standards (Table 1). Polymers with promisingly high average molecular masses were obtained. In addition, a 13C solid state NMR spectrum consistent with the ideal structure was obtained for each polymer. For monomers 4 and 5, which both possess additional sites adjacent to the amine groups activated towards electrophilic substitution, a high concentration of monomer or a prolonged reaction time resulted in insoluble gel formation presumably due to cross-linking via further reaction involving the excess dimethoxymethane. Gel formation is less problematic for monomers 1–3 probably because of steric hindrance provided by the central hydrocarbon framework blocking further electrophilic substitution. Cross-linking is also likely to be responsible for the failure to obtain a soluble polymer from monomer 6, whereas gel formation is not observed for the similar monomer 7 for which the methyl substituents block one of the activated sites adjacent to the amine group. It should be noted that the cross-linked polymer from monomer 6, itself prepared from an excess of aniline and formaldehyde, is likely to be structurally similar to aniline–formaldehyde resin (AFR). Conversely, the possibility of the formation of TB components within AFR is overlooked.
Matrix Assisted Laser Desorption Ionisation Mass Spectrometry (MALDI-MS) analysis of the relatively low molar mass material removed from the polymers during purification by reprecipitation confirms the structure of the expected repeat unit obtained from TB formation. For monomers 5 and 7, in which the two aniline units possess conformational freedom relative to one another, these MALDI spectra are dominated by the full series of cyclic oligomers (e.g.Fig. 2). In contrast, MALDI-MS indicated the formation of only a small proportion of possible cyclic products from monomers 1–4, within which the relative conformation of the aniline units is locked due to a fused ring structure, and from monomers 8–9, for which the amines share a common benzene ring. For example, the MALDI-MS spectrum of the small amount of material removed during reprecipitation of PIM–EA–TB, prepared from monomer 1, showed a significant peaks only for the cyclic oligomer composed of three (m/z = 901) repeat units (Fig. 2). This cyclic oligomer is composed of six alternating TB and ethanoanthracene components, that both contain “bite” angles of ∼120 °C, and is therefore unstrained. Unfortunately, this cyclic oligomer could not be isolated from the other low molar mass by-products of the polymerisations. The small yield of such by-products from monomers with locked conformations of the amine groups relative to one another (i.e.1–4, 8 and 9) suggests that cyclic oligomers form under strict kinetic control and that TB formation is not reversible under the conditions used for the polymerisation. This is consistent with previous conclusions on the non-reversibility of TB formation in strong acids such as TFA based on isomerisation studies of oligomers.45 For the monomers with fixed conformations the polymer yields after purification by reprecipitation are high (79–90%), whereas for monomers 5 and 7, for which the aniline units have rotational freedom with respects to each other, the yields are lower (54–63%), presumably due to the greater proportion of small cyclic oligomers which are subsequently lost during reprecipitation.
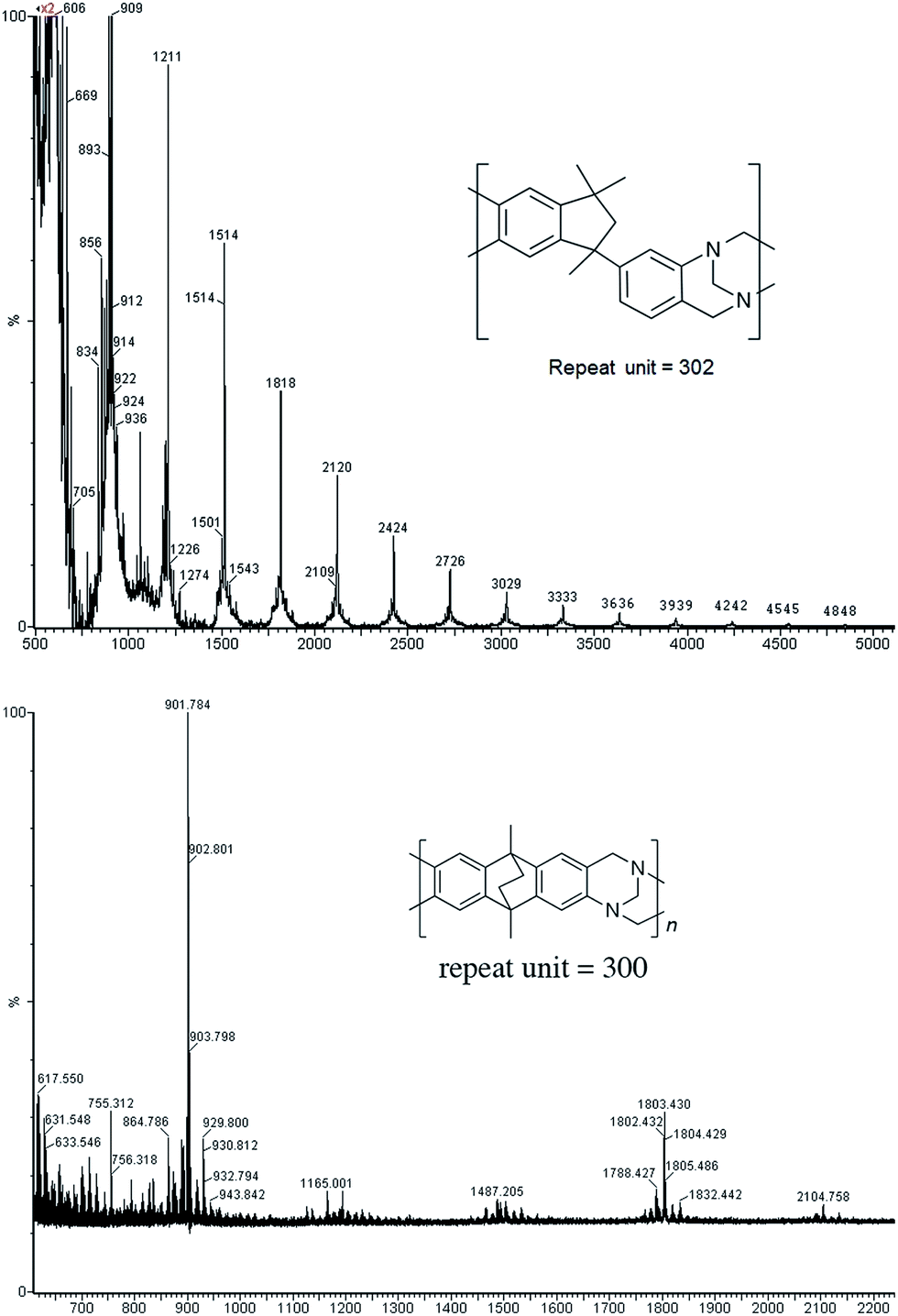 | ||
| Fig. 2 MALDI-MS (Top) of oligomers from the TB polymerisation of monomer 5 showing the full family of cyclics and (bottom) from monomer 1 showing a significant peak only for the cyclic based on three repeat units. | ||
Properties
Thermal Gravimetric Analysis (TGA) indicates that the TB polymers are stable up to temperatures of 350 °C with the exception of the ethanoanthracene-based polymer from monomer 1 which loses mass (∼10%), corresponding to loss of ethene via a retro Diels–Alder commencing above 250 °C. Polymers from monomers 1, 2, 4, 8 and 9 provided exceptionally high char yield (60–80%) and in each case the elemental analysis is low in carbon possible to incomplete combustion. The polymers do not degrade on prolonged contact with hot water or warm dilute aqueous acids or bases (70 °C). As noted, with the exception of the product from monomer 6, which appears cross-linked, the TB polymers are all freely soluble in chloroform but insoluble in most other organic solvents (e.g. acetone, methanol, hexane, THF, toluene, DMF). Polymers prepared with sufficient average molecular mass, such as those from monomers 1–3, 5 and 7, form robust self-standing films by simple casting from chloroform solution. Further optimisation of reaction conditions involving monomers 4, 8 and 9 to provide TB polymers with higher average molecular mass is required to yield film-forming materials.All TB polymers whose ideal structure consists entirely of fused rings demonstrate significant intrinsic microporosity with apparent BET surface areas within the range of 550–1028 m2 g−1 as calculated from nitrogen adsorption isotherms of powdered samples collected at 77 K. The apparent surface area for the PIM–EA–TB prepared from the ethanoanthracene-based monomer 1 (1028 m2 g−1) is of particular note as it exceeds all other non-network PIMs reported to date. In addition, it appears that for the polymers derived from simple diamino-benzene monomers 7–9 the V-shaped TB unit itself acts as an efficient site of contortion for the generation of intrinsic microporosity. As expected, the TB polymer from monomer 6, which contains a flexible methylene unit about which there is conformational freedom, does not demonstrate intrinsic microporosity. However, the TB polymer from monomer 5 has a respectable BET surface area of 535 m2 g−1, therefore, it appears that rotation about the single C–C bonds within its 1,3,3-trimethyl-1-phenylindane component must be sufficiently hindered to suppress efficient polymer packing via conformation rearrangement.
Conclusions
Despite its long history and wide-spread exploitation in synthetic chemistry, only recently has TB formation been employed directly as a polymerisation reaction. This reaction can provide polymers of high average molecular mass by simple mixing of suitable aromatic diamine monomers and dimethoxymethane in TFA solvent under ambient conditions. Film-forming polymers can be prepared from a number of monomers and the choice of monomer facilitates control over the properties of the resulting polymer. In particular, the TB polymerisation reaction is suitable for the preparation of polymers of intrinsic microporosity (PIMs) due to the resulting fused-ring TB linking group, which is both highly rigid and prohibits conformation freedom. Recent studies have demonstrated that microporous polymers prepared by this polymerisation reaction have potential for membrane gas separations,16,17,46,47 CO2 adsorption,18,48 catalysis49 and electrochemical membranes.50–52Acknowledgements
We thank the EPSRC for funding through grants EP/G01244X and EP/G062129/1 and the EPSRC National Solid State NMR Service (Durham University).Notes and references
- D. Crespy, M. Bozonnet and M. Meier, Angew. Chem., Int. Ed., 2008, 47, 3322–3328 CrossRef CAS PubMed.
- R. Goldschmidt, Helv. Phys. Acta, 1939, 12, 305 CAS.
- S. A. Rulon, Mod. Plast., 1942, 19, 72–74 CAS.
- J. A. Brydson, Plastic Materials, Butterworth–Heinemann, Oxford, 1999 Search PubMed.
- J. Tröger, J. Prakt. Chem., 1887, 36, 227 CrossRef.
- M. A. Spielman, J. Am. Chem. Soc., 1935, 57, 583–584 CrossRef CAS.
- O. V. Runarsson, J. Artacho and K. Warnmark, Eur. J. Org. Chem., 2012, 7015–7041 CrossRef CAS.
- B. Dolensky, M. Havlik and V. Kral, Chem. Soc. Rev., 2012, 41, 3839–3858 RSC.
- C. X. Yuan, Q. A. Xin, H. J. Liu, L. Wang, M. H. Jiang and X. T. Tao, Sci. China: Chem., 2011, 54, 587–595 CrossRef CAS.
- T. Weilandt, U. Kiehne, J. Bunzen, G. Schnakenburg and A. Lutzen, Chem.–Eur. J., 2010, 16, 2418–2426 CrossRef CAS PubMed.
- S. Sergeyev, Helv. Chim. Acta, 2009, 92, 415–444 CrossRef CAS.
- D. Didier, B. Tylleman, N. Lambert, C. Velde, F. Blockhuys, A. Collas and S. Sergeyev, Tetrahedron, 2008, 64, 6252–6262 CrossRef CAS PubMed.
- A. Abdolmaleki, S. Heshmat-Azad and M. Kheradmand-fard, J. Appl. Polym. Sci., 2011, 122, 282–288 CrossRef CAS.
- X. Du, Y. L. Sun, B. E. Tan, Q. F. Teng, X. J. Yao, C. Y. Su and W. Wang, Chem. Commun., 2010, 46, 970–972 RSC.
- N. B. McKeown, M. Carta and M. Croad, UK Patent Application, WO2012035327, 2010.
- M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Science, 2013, 339, 303–307 CrossRef CAS PubMed.
- M. Carta, M. Croad, R. Malpass-Evans, J. C. Jansen, P. Bernardo, C. J. Gabriel, K. Friess, M. Lanč and N. B. McKeown, Adv. Mater., 2014, 26, 3526–3531 CrossRef CAS PubMed.
- X. Zhu, D.-T. Chi-Linh, C. R. Murdock, K. M. Nelson, C. Tian, S. Brown, S. M. Mahurin, D. M. Jenkins, J. Hu, B. Zhao, H. Liu and S. Dai, ACS Macro Lett., 2013, 2, 660–663 CrossRef CAS.
- N. B. McKeown and P. M. Budd, Macromolecules, 2010, 43, 5163–5176 CrossRef CAS.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230–231 RSC.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- P. M. Budd, N. B. McKeown, B. S. Ghanem, K. J. Msayib, D. Fritsch, L. Starannikova, N. Belov, O. Sanfirova, Y. P. Yampol'skii and V. Shantarovich, J. Membr. Sci., 2008, 325, 851–860 CrossRef CAS PubMed.
- Y. Rogan, L. Starannikova, V. Ryzhikh, Y. Yampolskii, P. Bernardo, F. Bazzarelli, J. C. Jansen and N. B. McKeown, Polym. Chem., 2013, 4, 3813–3820 RSC.
- C. G. Bezzu, M. Carta, A. Tonkins, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Adv. Mater., 2012, 24, 5930–5933 CrossRef CAS PubMed.
- R. Short, M. Carta, C. G. Bezzu, D. Fritsch, B. M. Kariuki and N. B. McKeown, Chem. Commun., 2011, 47, 6822–6824 RSC.
- D. Fritsch, G. Bengtson, M. Carta and N. B. McKeown, Macromol. Chem. Phys., 2011, 212, 1137–1146 CrossRef CAS.
- P. M. Budd and N. B. McKeown, Polym. Chem., 2010, 1, 63–68 RSC.
- B. S. Ghanem, N. B. McKeown, P. M. Budd, N. M. Al-Harbi, D. Fritsch, K. Heinrich, L. Starannikova, A. Tokarev and Y. Yampolskii, Macromolecules, 2009, 42, 7881–7888 CrossRef CAS.
- B. S. Ghanem, N. B. McKeown, P. M. Budd, J. D. Selbie and D. Fritsch, Adv. Mater., 2008, 20, 2766–2771 CrossRef CAS.
- B. S. Ghanem, N. B. McKeown, P. M. Budd and D. Fritsch, Macromolecules, 2008, 41, 1640–1646 CrossRef CAS.
- M. Carta, K. J. Msayib, P. M. Budd and N. B. McKeown, Org. Lett., 2008, 10, 2641–2643 CrossRef CAS PubMed.
- N. Du, H. B. Park, G. P. Robertson, M. M. Dal-Cin, T. Visser, L. Scoles and M. D. Guiver, Nat. Mater., 2011, 10, 372–375 CrossRef CAS PubMed.
- N. Y. Du, G. P. Robertson, I. Pinnau and M. D. Guiver, Macromolecules, 2010, 43, 8580–8587 CrossRef CAS.
- S. Thomas, I. Pinnau, N. Y. Du and M. D. Guiver, J. Membr. Sci., 2009, 338, 1–4 CrossRef CAS PubMed.
- S. Thomas, I. Pinnau, N. Y. Du and M. D. Guiver, J. Membr. Sci., 2009, 333, 125–131 CrossRef CAS PubMed.
- A. V. Maffei, P. M. Budd and N. B. McKeown, Langmuir, 2006, 22, 4225–4229 CrossRef CAS PubMed.
- N. B. McKeown, B. Ghanem, K. J. Msayib, P. M. Budd, C. E. Tattershall, K. Mahmood, S. Tan, D. Book, H. W. Langmi and A. Walton, Angew. Chem., Int. Ed., 2006, 45, 1804–1807 CrossRef CAS PubMed.
- P. M. Budd, B. Ghanem, K. Msayib, N. B. McKeown and C. Tattershall, J. Mater. Chem., 2003, 13, 2721–2726 RSC.
- H. J. Mackintosh, P. M. Budd and N. B. McKeown, J. Mater. Chem., 2008, 18, 573–578 RSC.
- Y. Wang, N. B. McKeown, K. J. Msayib, G. A. Turnbull and I. D. W. Samuel, Sensors, 2011, 11, 2478–2487 CrossRef CAS PubMed.
- N. A. Rakow, M. S. Wendland, J. E. Trend, R. J. Poirier, D. M. Paolucci, S. P. Maki, C. S. Lyons and M. J. Swierczek, Langmuir, 2010, 26, 3767–3770 CrossRef CAS PubMed.
- J. Weber, O. Su, M. Antonietti and A. Thomas, Macromol. Rapid Commun., 2007, 28, 1871–1876 CrossRef CAS.
- I. V. Farr, D. Kratzner, T. E. Glass, D. Dunson, Q. Ji and J. E. McGrath, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2840–2854 CrossRef CAS.
- W. F. Yong, F. Y. Li, Y. C. Xiao, P. Li, K. P. Pramoda, Y. W. Tong and T. S. Chung, J. Membr. Sci., 2012, 407, 47–57 CrossRef PubMed.
- B. Dolensky, J. Kessler, M. Jakubek, M. Havlik, J. Cejka, J. Novotna and V. Kral, Tetrahedron Lett., 2013, 54, 308–311 CrossRef CAS PubMed.
- M. Carta, M. Croad, J. C. Jansen, P. Bernardo, G. Clarizia and N. B. McKeown, Polym. Chem., 2014 10.1039/c4py00607k.
- Y. Zhuang, J. G. Seong, Y. S. Do, H. J. Jo, Z. Cui, J. Lee, Y. M. Lee and M. D. Guiver, Macromolecules, 2014, 47, 3254–3262 CrossRef CAS.
- A. Del Regno, A. Gonciaruk, L. Leay, M. Carta, M. Croad, R. Malpass-Evans, N. B. McKeown and F. R. Siperstein, Ind. Eng. Chem. Res., 2013, 52, 16939–16950 CrossRef CAS.
- M. Carta, M. Croad, K. Bugler, K. J. Msayib and N. B. McKeown, Polym. Chem., 2014 10.1039/c4py00608a..
- F. Xia, M. Pan, S. Mu, R. Malpass-Evans, M. Carta, N. B. McKeown, G. A. Attard, A. Brew, D. J. Morgan and F. Marken, Electrochim. Acta, 2014, 128, 3–9 CrossRef CAS PubMed.
- Y. Rong, R. Malpass-Evans, M. Carta, N. B. McKeown, G. A. Attard and F. Marken, Electroanalysis, 2014, 26, 904–909 CrossRef CAS.
- Y. Rong, R. Malpass-Evans, M. Carta, N. B. McKeown, G. A. Attard and F. Marken, Electrochem. Commun., 2014 DOI:10.1016/j.elecom.2014.06.005.
| This journal is © The Royal Society of Chemistry 2014 |