 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From particles to stabilizing blocks – polymerized ionic liquids in aqueous heterophase polymerization†
Ran
Yu
and
Klaus
Tauer
*
Max Planck Institute of Colloids and Interfaces, D-14424 Potsdam, Germany. E-mail: klaus.tauer@mpikg.mpg.de
First published on 16th May 2014
Abstract
Suspensions of polyvinylimidazolium salts with long alkyl chains (polymerized ionic liquids) are a new class of reactive electrostatic stabilizers for aqueous polymer dispersions with dual functions – as a reductant in a redox initiator system and as a colloidal stabilizer. Studying the properties of di- and triblock copolymer dispersions (with N-isopropyl acryl amide and/or hydrophobic monomers) made with these reactive suspensions reveals an interesting behavior which is described as the principle of relative hydrophilicity. Following this principle, the colloid chemical behavior of the polyvinylimidazolium salt block depends on the relative hydrophilicity in comparison with the other blocks.
1. Introduction
The search for new stabilizers in heterophase polymerization is an everlasting topic in research since the very first attempts to copy natural rubber in chemical reactors. The choice of an appropriate stabilization strategy is extremely crucial because it determines not only the stability of the latex but in addition also the morphology of the latex particles, the polymerization kinetics, and many application properties.1–3 A very recent development in this area is the application of polymerized ionic liquids (PIL) as stabilizers in aqueous heterophase polymerization.4–7 Particularly, it has been found that poly (3-n-tetradecyl-1-vinylimidazolium bromide) behaves extraordinarily and appears as a kind of ideal colloidal stabilizer for emulsion polymers because it changes from hydrophilic in the wet state to hydrophobic in the dry state. Consequently, the hydrophilicity of an emulsion polymer coating, expressed by means of the contact angle of a sessile water drop, made with the PIL stabilizer is significantly reduced compared with commonly used emulsifiers.8 These results support the hypothesis that the stabilizing PIL units are quite mobile and can adopt different conformations. The PIL with long alkyl chains might alternately be considered as a kind of polymerized self-assembled, self-organized structures (lyotropic mesophases; or surfactants), considered also as a subclass of polysoaps,9 which are key for membrane-based chemical and physical processes.10,11 Polysoaps have been a topic in colloid chemistry since the 1950s.12–14Ionic liquids are organic salts with quite low melting points (typically below 100 °C), enabling their use as solvents with very particular properties, such as high polarity and ionic conductivity with a very special effect on the kinetics of all kinds of chemical reactions.15
Ionic liquid monomers (ILM) containing a polymerizing double bond in their molecular structure resemble polymerizable surfactants or surface active monomers (surfmers).16 Like surfmers which are hydrophobically modified water-soluble monomers, ILMs show the typical behavior of self-associating monomers and polymers.17–21 Before, during, and after polymerization, interactions of the hydrophobic tails can cause quite strong association leading to gel formation even for copolymers where the content of hydrophobically modified units is only about 1 mol%.22 Interestingly, the solubility behavior of polymerized dimethylaminoethyl methacrylate salts quarternized with n-alkyl bromides (C8–C18)17 is very similar to that of the PIL used in this study. Both are insoluble in water but dissolve in polar organic solvents. Also, the formation of lamellar structures on substrates is observed for partially quarternized poly(styrene-b-2-vinylpyridine) copolymers.20 These few examples demonstrate the quite far-reaching similarity between PIL and polymerized surface active monomers. In fact, IL monomers, depending on the influence of the hydrophobic parts, are either ionic monomers or surfmers (IL-surfmers). Thus, in the family tree of polymers PILs can be spotted somewhere between polyelectrolytes and polysoaps.
The molecular architecture of the monomer, particularly the arrangement of different functional groups (ionic or hydrophilic, hydrophobic, and polymerizable double bond), determines the properties and the behavior of PIL as is generally the case in chemistry and also for polymerized amphiphiles.9
The imidazole group is known to be quite active in nucleophilic catalysis of carboxylic acid derivatives.23 Polymers bearing imidazole units have a history starting from the early 1960s as model compounds for synthetic enzymes based on the observation that the histidine group plays an active role in enzymatic hydrolysis.24–32 The first synthetic polymers of this kind mimicking enzymes were homo- and copolymers of 4-vinylimidazole25 prepared via free radical polymerization. The first polymerization of vinylimidazolium iodide and methylsulphate salts was described in the open scientific literature only in 1973.33 But already in the 1950s patents had been filed describing the polymerization of vinylimidazolium salts and possible uses of the (co)polymers.
Copolymers containing ionic liquid (IL) moieties (imidazolium cations) were described for the first time only in 2004.34,35 These were block copolymers with polystyrene made from poly(styrene-b-chloromethyl styrene) precursors. In a subsequent polymer analogous reaction, the chloromethyl styrene block was quaternized with 1-methylimidazole in the presence of NaBF4 to yield polystyrene-IL block copolymers. In the subsequent years more PIL-containing copolymers have been synthesized via various radical polymerization techniques. Amongst them are random copolymers and block copolymers synthesized via normal free radical and reversible addition-fragmentation transfer (RAFT) polymerization of N-2-thiazolylmethacrylamide and 2-(1-butylimidazolium-3-yl)ethyl methacrylate, respectively, as polymeric ligands in magnetic complexes with Ni2+ and Nd3+.36 Also, double hydrophilic block copolymers of 3-(1-ethylimidazolium-3-yl)propyl(meth)acrylamido bromide and acrylamide or methacrylic acid have been prepared via RAFT polymerization in methanol.37 Block copolymers with another type of IL group are described in ref. 20. Starting from poly(styrene-b-2-vinylpyridine) synthesized via anionic polymerization and subsequent quarternization of the 2-vinylpyridine units with Li-bis(trifluoromethylsulfonyl)imine the authors obtained polymeric IL-block copolymers with pyridinium cations with a tunable lamellae domain size in the bulk block copolymer.
All kinds of polymers containing IL-moieties are interesting materials because the counterion and solvent stimuli responsiveness of ILs add new properties to these hydrophilic or amphiphilic polymers and promise new materials for new applications. For instance, exchange of the bromide or chloride counterion with bulkier anions such as tetrafluoroborate, hexafluorophosphate, or bis(trifluoromethanesulfonyl) changes the properties of the IL from hydrophilic to hydrophobic. An interested reader can find instructive examples in ref. 37–45.
Temperature is an easily applicable trigger for property changes and hence, block copolymers with thermosensitive units became quite popular during the last decades. Thermosensitivity is a very typical and general property for solutions of all polymers.46 However, poly-N-isopropylacrylamide (PNIPAM) is the most prominent example because its lower critical solution temperature (LCST) of about 32 °C is in an easily accessible range and very attractive for biomedical applications.47,48 Therefore it is not surprising that nowadays block copolymers containing PIL and PNIPAM units as temperature responsive components are also in the focus of research to produce multi-responsive smart polymers.49–54 To the best of our knowledge the first reports regarding the synthesis of PIL–PNIPAM block copolymers appeared only in 2009. One procedure uses RAFT polymerization in organic solvents, starting with the PNIPAM block.49 The other procedures started from the PIL block with ceric ion redox radical generation in water.50
In this contribution, we describe the synthesis and characterization of block copolymer particles, with poly(vinylimidazolium) groups (particularly, poly (3-n-tetradecyl-1-vinylimidazolium bromide)) as the PIL block. The synthesis strategy takes advantage of the facile and robust ceric ion redox heterophase polymerization technique which has already frequently been applied for block copolymer production, cf.2,55–57 and references therein.
Aqueous poly (3-n-tetradecyl-1-vinylimidazolium bromide) dispersions, either crosslinked or not, with hydroxymethyl chain ends were used as reductants in the initiation process with ceric ion as an oxidant (cf.Scheme 1). Despite the fact that the PIL reductant is used as a suspension and that the final product is a suspension as well, the PIL particles do not serve as simple seed particles in the common sense. Here, we discuss the results regarding the colloidal and morphological characterization of diblock particles with N-isopropylacrylamide (NIPAM), butyl acrylate (BA), butyl methacrylate (BMA), and styrene (STY) as second monomer and triblock copolymer particles with NIPAM as second and BA, or BMA, or STY as third monomer blocks. In addition, we consider the thermo responsiveness of the copolymers containing PNIPAM blocks by means of dynamic light scattering and speed of sound data. The block copolymers under consideration consist of very different units with each of them possessing a very particular aggregation behavior under very specific conditions. Consequently, each block has a particular influence on the particles' morphology. The results convincingly reveal quite a high mobility of the hydrophilic blocks (PIL, PNIPAM), arranging themselves in order to minimize the free interfacial energy of the dispersion with substantial influence on the morphology of the particles. The properties of the hydrophobic block have a great influence on the morphology of the dried particles as observed on transmission electron microscopy grids.
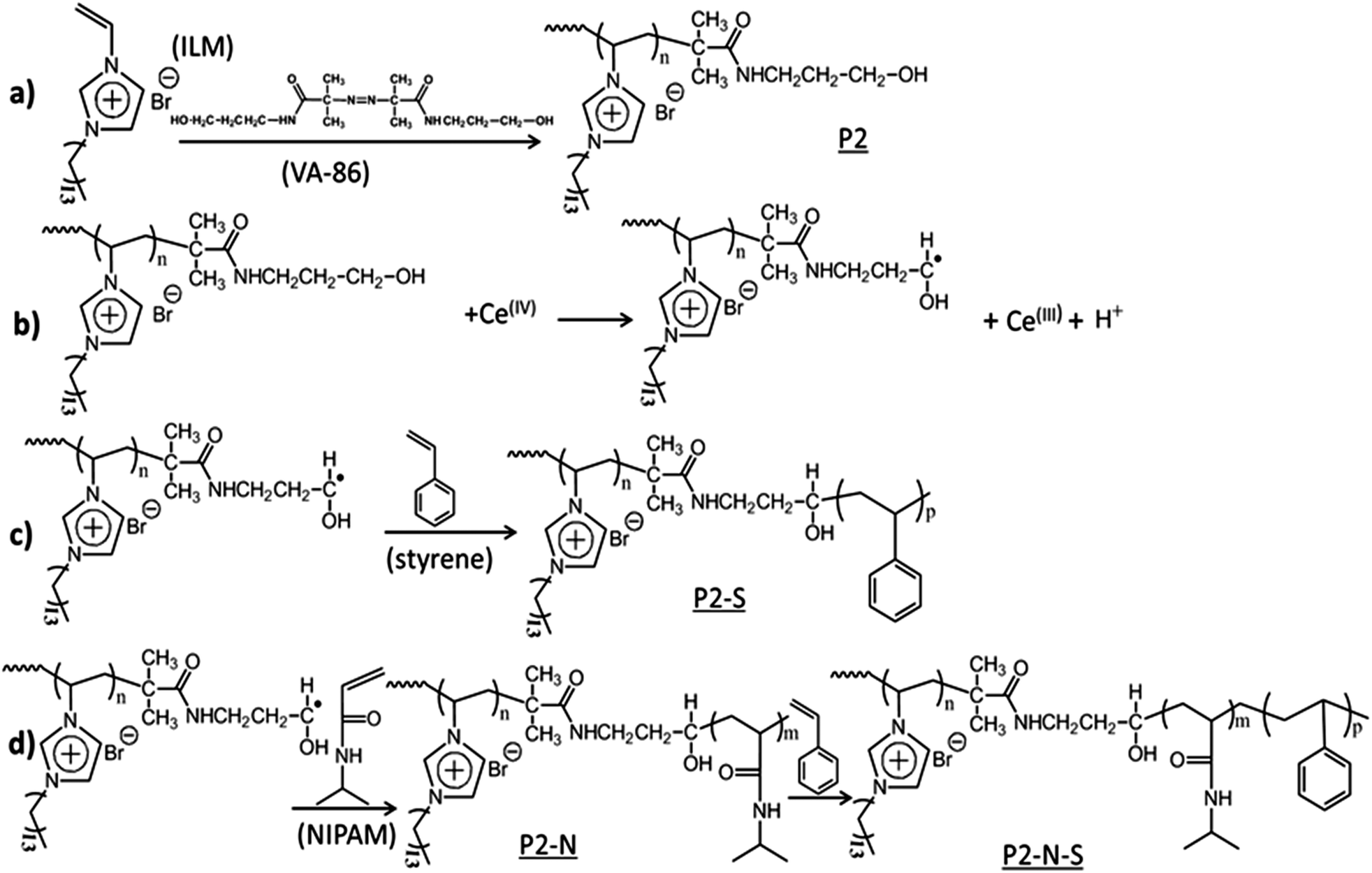 | ||
| Scheme 1 Illustration of the synthesis strategy leading to di- and triblock copolymer dispersions as applied in this study. (a) Synthesis of the PIL dispersion starting from the ionic liquid monomer (ILM), 3-n-tetradecyl-1-vinylimidazolium bromide, and VA-086, 2,2′-azobis[2-methyl-N-(2-hydroxyethyl)propionamide, leading to PIL with methylol end groups. (b) Redox initiation with ceric ion leading to radical generation at the methylol carbon atom. (c) Synthesis of diblock copolymer dispersions with styrene as the hydrophobic monomer, P2–S. (d) Synthesis of triblock copolymer dispersions with N-isopropylacrylamide (NIPAM) as the middle block and styrene as the middle block and styrene as the third block, P2–N–S third block, P2–N–S. | ||
2. Experimental section
2.1 Materials
Crosslinked poly(ionic liquid) (P1) and non-crosslinked poly(ionic liquid) (P2) are synthesized as described elsewhere58–60 using the following method: 15 g (0.04 mole) of 3-n-tetradecyl-1-vinylimidazolium bromide (ILM), 1.623 g (0.004 mole) of 1,4-butanediyl-3,3′-bis-1-vinylimidazolium dibromide as the crosslinker, 360 ml of water, 450 mg of 2,2′-azobis[2-methyl-N-(2-hydroxyethyl)propionamide] (VA-086) as the initiator, and polymerization temperature 70 °C. All the PIL samples were carefully dialyzed against deionized water for about 3 weeks until the conductivity of the dialysate reached a constant value.Two kinds of poly(ionic liquid) dispersions were used in our study, crosslinked (P1) and non-crosslinked samples (P2) with particle sizes 43.1 and 27.8 nm, respectively. Both of them were diluted to 1 wt% before being used in the experiments.
STY, BMA, and BA (all of 99% purity from Sigma-Aldrich) were distilled under reduced pressure to remove inhibitors. NIPAM (99% purity, Acros) was recrystallized from hexane. Ceric ammonium nitrate (CAN) from Sigma-Aldrich was used as received. Water was taken from a SG purification system (Hamburg, Germany) with a conductivity of 0.055 μS cm−1.
2.2 Synthesis
2.3 Characterization
During the polymerization samples were withdrawn and the solid content (SC) was determined with a HR 73 halogen moisture analyzer (Mettler Toledo, Giessen, Germany). The monomer conversion was calculated with the SC. The reaction products were dialysed against distilled water (dialysis tubing was made of regenerated cellulose, type: membra-cel MD44, cut-off 14 kilo-Dalton) and the water was replaced daily. The dialysis was continued until no more change in the conductivity of water in the dialysis bath was detected. This lasted on average for about three weeks. If not otherwise stated, only dialysed samples were characterized.Infrared spectroscopy was carried out on a Varian 1000 FT-IR spectrometer as ATR measurements.
The average hydrodynamic particle size is expressed as intensity weighed diameter (Di) determined with a Nicomp particle sizer (model 380, PSS, USA).
The zeta potential was measured with a Zetasizer Nano ZSP (Malvern) at room temperature.
The change in the speed of sound was measured with the ResoScan URT System (TF Instruments GmbH, Heidelberg, Germany) based on ultrasound resonator technology.
Shape and morphology of the particles were investigated by electron microscopy. Transmission electron microscopy (TEM) was performed with a Zeiss EM 912 Omega microscope operating at 120 kV according to standard procedures (suspension preparation on carbon coated copper grids).
2.4 Sample abbreviations
The following abbreviations for the block copolymer particles will be used throughout the manuscript: P1: crosslinked PIL; P2: non-crosslinked PIL; P1–N and P2–N: diblock copolymers of the corresponding PIL with PNIPAM; P1–BA and P2–BA: diblock copolymers of the corresponding PIL with poly(butyl acrylate); P1–BMA and P2–BMA: diblock copolymers of the corresponding PIL with poly(butyl methacrylate); P1–S and P2–S: diblock copolymers of the corresponding PIL with polystyrene; P1–N–BA and P2–N–BA: triblock copolymers of the corresponding diblock copolymers with poly(butyl acrylate); P1–N–BMA and P2–N–BMA: triblock copolymers of the corresponding diblock copolymers with poly(butyl methacrylate); P1–N–S and P2–N–S: triblock copolymers of the corresponding diblock copolymers with polystyrene.3. Results and discussion
3.1 Block copolymer formation
The polymerization strategy as outlined in Scheme 1 which has already been applied several times successfully for the synthesis of a variety of block copolymers relies on a heterogeneous intermediate generated by the collapse of the PNIPAM above the LCST. Starting with a hydrophilic precursor polymer bearing methylol end groups, the precipitated diblock generates an amphiphilic reaction environment enabling the uptake of both hydrophilic and hydrophobic monomers for further block building. This block copolymer formation strategy relies on the following facts. Firstly, the redox initiation is very fast and it lasts only a few minutes.55 Secondly, the radical termination probability is quite low as only polymeric radicals are involved.2 The whole process can be typically carried out at temperatures well above the LCST of PNIPAM, thus allowing fast polymerizations.For ‘common’ water soluble polymers such as poly(ethylene glycol), poly(styrene sulfonate), poly(diallyldimethylammonium chloride), temperature is not an issue and the final product is a stable thermoresponsive block copolymer latex.61 However, the situation changes when PILs are used as the reducing water soluble polymer to synthesize triblock copolymer latexes containing a hydrophobic polymer as the third block. In this case, a polymerization temperature above the LCST of PNIPAM causes breakdown of both the latex stability and the polymerization reaction after charging the hydrophobic monomer to the reaction mixture. Interestingly, when the polymerization is conducted at room temperature, colloidal stability is maintained throughout the entire reaction, however at the expense of the reaction time which will be extended accordingly. In contrast, diblock copolymer formation can be carried out at any temperature.
Scheme 1 shows only the synthetic pathway to non-crosslinked copolymers. The corresponding crosslinked samples were prepared starting with reaction (a) of Scheme 1 with ILM and 1,4-butanediyl-3,3′-bis-1-vinylimidazolium dibromide (10 mol% relative to ILM). Other hydrophobic monomers were butyl acrylate and butyl methacrylate resulting in P2–BA, P2–N–BA and P2–BMA, P2–N–BMA di- and triblock copolymers, respectively.
The data depicted in Fig. 1 show a conversion–time curve for the triblock copolymer formation process at room temperature with sequential NIPAM and STY addition starting with P1. The polymerization of NIPAM (diblock copolymer formation) takes only 3 h for a monomer conversion higher than 96%. The formation of the hydrophobic block takes much longer (about 38 h to reach a monomer conversion greater than 80%). This huge difference in the required polymerization time or polymerization rate reflects the different propagation rate constants between both the monomers.62,63
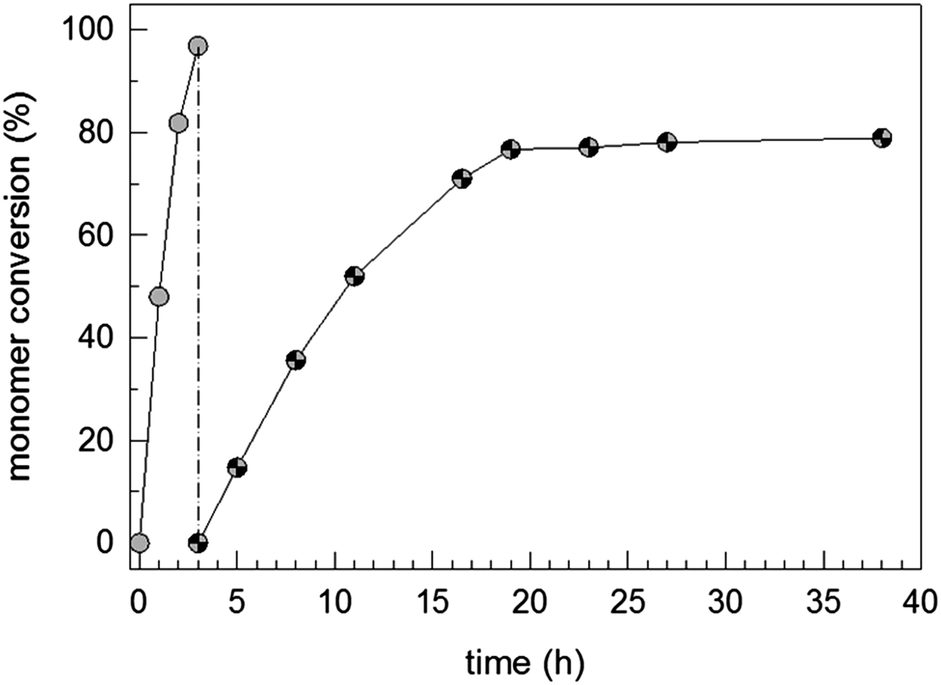 | ||
| Fig. 1 Monomer conversion–time curve for the synthesis of P1–N–S triblock copolymer dispersions; the grey and patterned symbols describe the NIPAM and styrene conversion, respectively. | ||
In the present study two different PIL dispersions have been used. P1 and P2 synthesized in the presence and absence of a cross-linker (1, 4-butanediyl-3,3′-bis-1-vinylimidazolium dibromide) as described in ref. 58–60. Both the crosslinked and non-crosslinked PIL form an aqueous dispersion with a positive zeta potential of about 50 and 60 mV and an average hydrodynamic particle size of about 43 and 28 nm, respectively. The inner morphology of these particles is very peculiar because the PIL molecules arrange themselves in circular lamellae58–60 consisting of consecutive non-polar/hydrophobic (alkyl chains) and polar/hydrophilic (ionic) layers. Interestingly, this kind of morphology has been reported for crosslinked and non-crosslinked PIL particles58,59 and observed only on cryo-TEM micrographs. The PIL particles on normal TEM micrographs in the dried state show different morphologies depending strongly on the concentration and maturation of the PIL dispersion. The conditions during drying, in addition to the local properties at the drying spot of the carbon coated copper TEM grid, are also of importance. Clearly, PIL particles behave differently compared to normal latex particles made of hydrophobic polymers.
The characterization of the block copolymer particles has been carried out in the dispersed and dried states and the results have been used for the characterization of the colloid chemical features and the block copolymer composition (to control the success of the applied polymerization strategy), respectively. For amphiphilic block copolymer particles under consideration it is important to bear in mind that the morphology as observed by electron microscopy in the dried state must not represent the dispersed state.64 Typically, hydrophilic polymer chains which stabilize the particles in the dispersed state, collapse in the dried state onto the hydrophobic core. Thus, sizing via dynamic light scattering and analysis of electron micrographs lead to different results.
All analytics have been carried out with carefully dialyzed samples. The composition of the copolymers was checked by FT-IR spectroscopy. The expected characteristic absorption bands of the different building blocks were clearly detected and the corresponding wavenumbers and assignments are summarized in Table 1.
| Sample | Functional group | Characteristic frequency (cm−1) | Bonds |
|---|---|---|---|
| P1, P2 | Imidazolium | 2920, 2850 and 1158 | C–H stretch, C–N stretch |
| P1–N, P2–N | Amide | 1636, 1527 and 1457 | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch, N–H bending, C–H bending O stretch, N–H bending, C–H bending |
| P1–BA, P2–BA | Ester | 1728 and 1150 | CO stretch, C–O–C stretch |
| P1–BMA, P2–BMA | Ester | 1728 and 1150 | CO stretch, C–O–C stretch |
| P1–S, P2–S | Aromatics | 3010, 750 and 699 | C–H stretch, o.o.p C–H ring |
| P1–N–BA, P2–N–BA | Amide ester | 1636, 1527, 1457, 1728 and 1150 | CO stretch, N–H bending, C–H bending, CO stretch, C–O–C stretch |
| P1–N–BMA, P2–N–BMA | Amide, ester | 1636, 1527, 1457, 1728 and 1150 | CO stretch, N–H bending, C–H bending CO stretch, C–O–C stretch |
| P1–N–S, P2–N–S | Amide, aromatics | 1636, 1527, 1457, 3010, 750 and 699 | CO stretch, N–H bending, C–H bending C–H stretch, o.o.p. C–H ring |
3.2 Zeta potential
Valuable information regarding the morphology of the particles in the dispersed state has been obtained from zeta potential measurements. The zeta potential is the electric potential in the diffuse electrical double layer surrounding the particles, measured at the slipping plane which is located at a certain distance from the geometrical particle surface because the particles tow a water layer which contains counter and co-ions while moving. So, the composition of the electrical double layer in the gap between the particles' surface and the slipping plane determines the potential. For the kind of particles considered here, this value is essentially governed by the concentration of imidazolium groups at the surface stretching out into the aqueous phase and the concentration of ions (counterions and co-ions) diffusing inside the gap up to the slipping plane. Model polystyrene particles prepared with cetyltrimethylammonium peroxodisulfate as a surfactant–initiator complex possess a positive zeta potential, proving that it is not the anionic polymer end group that determines the zeta potential but the surface active cationic counterion which is farther away from the surface. The inverse initiator–surfactant complex, that is cationic initiating radicals and anionic dodecyl sulfate as counterions, leads to PS particles with negative zeta potential. In this context, it is necessary to recall that for latex particles made surfactant-free with ionic initiators the charge of the polymer end group controls the sign of the zeta potential. These findings regarding the zeta potential-determining ions are important for evaluating the zeta potential data obtained for the PILs and block copolymer particles as discussed below. Fig. 2 shows clear differences in the dependence on the polymer/copolymer composition and, for the block copolymers, these data allow reasonable conclusion regarding the arrangement of the blocks. The most interesting behavior is observed for the diblock copolymers where the PIL diblocks with the hydrophobic partners possess a similar zeta potential like the pure PIL particles. In contrast, for the PIL-PNIPAM diblock particles (P1–N, P2–N) the zeta potential is an order of magnitude lower. However, adding a third hydrophobic block, the zeta potential slightly increases again to values of 15–20 mV. Notably, the increase for the triblocks made with the crosslinked PIL (P1) is higher compared with that for the non-crosslinked PIL (P2) as a starting material. These experimental data, particularly the data for the diblock copolymers, lead us to introduce the idea or the principle of relative hydrophilicity (PRH). The PRH means that a given hydrophilic polymer in combination with a more hydrophilic polymer behaves hydrophobic and vice versa. Accordingly and considering PIL and PNIPAM, the less hydrophilic PIL blocks form the core of these diblock copolymer particles. The particles are predominantly stabilized sterically by the PNIPAM blocks and consequently the zeta potential is drastically reduced compared with the PIL homopolymer particles. In contrast, in the diblock copolymer particles with more hydrophobic monomers the PIL blocks stretch into the aqueous phase and ensure the stability of the dispersion. The thermodynamic driving force behind the principle of relative hydrophilicity is the minimization of the free energy which always brings the more hydrophilic component in direct contact with water, thus lowering the excess interfacial free energy. For the triblock copolymers, the third hydrophobic block tries to avoid contact with water and moves towards the core of the particles. This causes drastic rearrangements of the particle morphology, bringing the PIL units again in closer contact with water as indicated by the moderate increase in the zeta potential. | ||
| Fig. 2 Correlation between the zeta potential (ζ) and polymer/block copolymer composition; the grey- and white-filled symbols represent the polymers with P1 and P2, respectively. | ||
3.3 Particle morphology
The properties of the PIL stabilizer and the PRH lead to the assumption that the morphology as observed with common TEM in the dried state might not reflect the particles' morphology in the dispersed state64 and might cause huge differences between the hydrodynamic average particle size and the average resulting from enumerating TEM micrographs. Additionally, the properties of the hydrophobic block, particularly the glass transition temperature (Tg), influence the appearance of the dried particles on the TEM grids.The TEM micrographs of the di- and triblock copolymer particles displayed in Fig. 3 and 4, respectively, clearly prove the enormous influence of the nature of the hydrophobic polymer block on the appearance of the polymer dispersions in both the wet state dispersed in water and the dried particles on the TEM grids. Mainly two important conclusions can be drawn. Firstly, the higher the glass transition temperature (Tg) of the hydrophobic block, the clearer the solid sphere morphology of both the di- and triblock copolymer particles. Tg of the hydrophobic blocks changes in the order PBA (−55 °C) < PBMA (20 °C) < PS (100 °C).65 Secondly, the differences between the morphology displayed by the diblock copolymer particles with crosslinked and non-crosslinked PIL are significantly larger than those observed for the triblock copolymer particles. This is especially obvious comparing the micrographs of P1–BMA and P2–BMA particles. The P1–BMA particles prepared with crosslinked PIL maintain their sphere morphology on TEM micrographs while film formation and microphase separation are observed for P2–BMA particles with non-crosslinked PIL. In general, the influence of crosslinking the PIL starting dispersion is less pronounced than one might expect for the 10 mol% crosslinker relative to the ILM. This indicates that the efficiency of the crosslinker is pretty much limited which is reasonable considering that a significant mismatch is observed in the molecular sizes of the ILM and the crosslinker. Fig. S1† details this behavior by means of additional TEM micrographs.
 | ||
| Fig. 3 TEM micrographs of the diblock copolymer dispersions and the starting PIL dispersions showing the typical structures observed when the dispersions were dried on carbon-coated copper grids; the photographic snapshots arranged on the sides of the figure illustrate the dispersions as obtained after dialysis. The bar indicates 200 and 500 nm for samples P1, P2, P1–S, P2–S and P1–BA, P2–BA, P1–BMA, P2–BMA, respectively. | ||
 | ||
| Fig. 4 TEM micrographs of the triblock copolymer dispersions and the starting PIL–NIPAM diblock dispersions showing the typical structures observed when the dispersions were dried on carbon coated copper grids; the photographic snapshots arranged on the sides of the figure illustrate the dispersions as obtained after dialysis. The bar indicates 200 and 500 nm for samples P1–N, P2–N, P1–N–S, P2–N–S, P1–N–BMA, P2–N–BMA and P1–N–BA, P2–N–BA, respectively. | ||
The combined evaluation of the corresponding average values as measured by DLS at room temperature (Di) and by enumerating TEM micrographs (D) is a way to quantify the only qualitative impression gained from the TEM micrographs (cf.Table 2 and Fig. 5).
| Sample | P1 | P1–S | P1–BA | P1–BMA | P2 | P2–S | P2–BA | P2–BMA |
|---|---|---|---|---|---|---|---|---|
| D i (nm) | 43.1 | 89.9 | 105.2 | 96.5 | 27.8 | 124.1 | 178.8 | 80.5 |
| ± SD (nm) | 19.1 | 41.3 | 51.8 | 42.4 | 9.8 | 65.8 | 74.8 | 41.6 |
| D (nm) | 47.9 | 75.2 | 103.9 | 213.8 | 127.0 | |||
| ± SD (nm) | 6.8 | 9.0 | 19.4 | 40.9 | 28.0 |
| Sample | P1–N | P1–N–S | P1–N–BA | P1–N–BMA | P2–N | P2–N–S | P2–N–BA | P2–N–BMA |
|---|---|---|---|---|---|---|---|---|
| D i (nm) | 116.4 | 281.9 | 486.3 | 359.9 | 155.3 | 287.8 | 381.1 | 324.0 |
| ± SD (nm) | 0.3 | 9.0 | 36.0 | 27.7 | 0.8 | 16.3 | 33.3 | 20.5 |
| D (nm) | 152.3 | 428.9 | 134.2 | 133.6 | 403.6 | 133.3 | ||
| ± SD (nm) | 15.8 | 87.1 | 19.9 | 10.7 | 63.0 | 12.1 |
 | ||
| Fig. 5 Comparison of hydrodynamic diameters from DLS (Di, grey spheres) and average diameters from TEM images (D, black spheres) of PILs and their corresponding block copolymer latexes at room temperature; DLS measurements and preparation of TEM grids at room temperature; lines are just guides to the eye. | ||
The comparison of the average diameters from DLS and TEM in Fig. 5 clearly supports the limitations regarding the applicability of TEM in the determination of the morphology of this kind of amphiphilic block copolymer particles in the dispersed state. The DLS data (grey spheres) decrease with increasing Tg of the hydrophobic block which is particularly clear for the triblock copolymers. The copolymers containing PBA as a hydrophobic block expectedly show a strong tendency towards film formation, thus leading to the largest particles on the TEM micrographs. For the triblock copolymers with PBMA and PS as the hydrophobic block the diameter from the TEM micrographs is much smaller than Di proving the collapse of the hydrophilic blocks in the dried state on the TEM grids. For the triblock copolymers the Di-values from DLS at room temperature are much higher than those of all other samples. This result is a consequence of the hydrophilicity of the PNIPAM-block at temperatures below its LCST.
3.4 Thermoresponsiveness
Both the morphology in the dispersed state and the hydrodynamic particle size of the particles containing PNIPAM in one or the other way more or less strongly depend on the temperature.66 The changes are initiated by the behavior of PNIPAM which is either hydrophilic or hydrophobic at temperature below or above the LCST, respectively. The temperature sensitivity of the polymers containing NIPAM-blocks was studied by means of DLS and the ultrasound resonator technology (URT). Both are complementary methods allowing the study of specific features of the polymer solution state, with dependence on temperature. URT technology evaluates the temperature dependence of the difference in the speed of sound (ΔU) between the polymer dispersion and water. U in fluids depends on the compressibility and density of the medium. At the lower critical solution temperature of PNIPAM, the polymer precipitates and water is released. The overall compressibility of the sample solution increases and hence, the speed of sound decreases compared with pure water. In essence, URT tracks quantitatively changes in the layer of water molecules surrounding the solutes.67,68 In contrast, DLS measures the change in the size of the dissolved/dispersed matter. Evaluating the DLS data of charged molecules or particles at different temperatures one must additionally consider the change of the electrostatic interactions. The higher the temperature, the shorter the Bjerrum and the larger the Debye length. Indeed, comparison experiments with electrostatically stabilized, surfactant-free polystyrene latexes show a slight increase of Di with increasing temperature which should be considered in the following discussion (data not shown). The data in Fig. S2,† prove an analogous behavior for the cationically charged PIL dispersions. Interestingly and regardless whether the particles are crosslinked or not, the temperature dependence (dDi/dT) for both dispersions is very similar. One can also now expect that the temperature sensitivity of P1–N and P2–N is qualitatively identical. The data summarized in Fig. 6a and 7a prove that this is in fact observed experimentally with both DLS and URT measurements. | ||
| Fig. 6 Correlation between Di and temperature for (a) P1–N, P2–N and (b) triblock copolymers with styrene; in graph (a) the grey and white symbols are P1–N and P2–N, respectively; in graph (b) the grey and white symbols are P1–N–S and P2–N–S, respectively. | ||
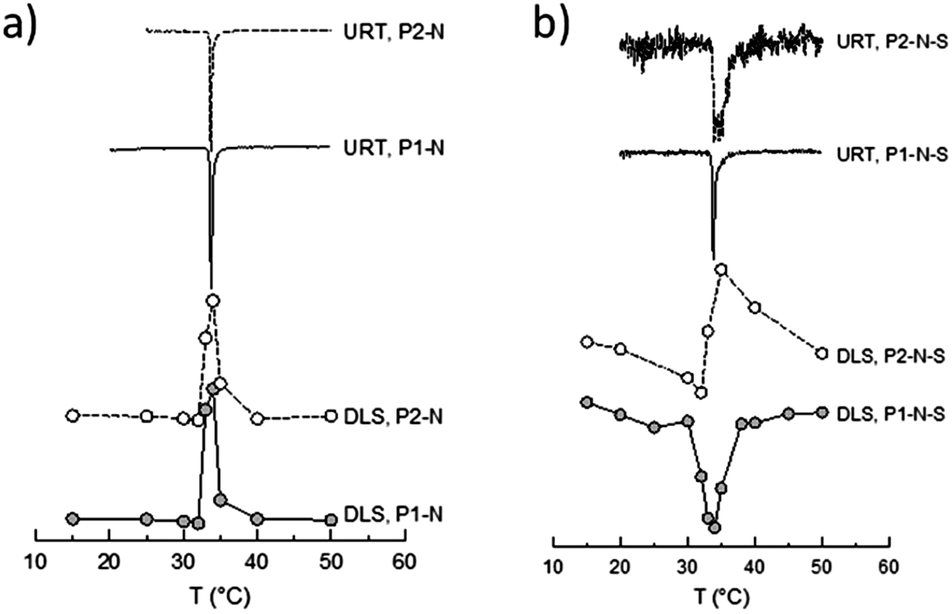 | ||
| Fig. 7 Correlation between the change in the temperature gradient of the hydrodynamic diameter (from DLS, dDi/dT, and the two lower curves) and of the speed of sound (from URT heating, dΔU/dT, and the two upper curves) with temperature for P1–N, P2–N diblock copolymer particles (graph a) and P1–N–S, P2–N–S triblock copolymer particles (graph b). | ||
The data in Fig. 6a show that the particle size increases for both samples sharply between 32 and 35 °C. The position of the extreme value in the temperature curve of the first derivative dDi/dT (cf. two lower curves of Fig. 7a and b) gives the LCST of the particular sample. Another way to determine the LCST is determining the position of the extreme value of the first derivative of the speed of sound (dΔU/dT) in dependence on temperature (upper two curves in Fig. 7a and b). For the samples P1–N and P2–N the maximum of dDi/dT and the minimum of dΔU/dT are positioned at the same temperature. This clearly proves that the sharp increase in temperature of the particle size in these samples is caused by the change in the solution behavior of the PNIPAM block. The increase of the hydrodynamic diameter above the LCST, which is much steeper than the temperature-caused changes of the electrostatic interactions of the pure PIL (cf. Fig. S2†) points towards aggregation processes of the particles. The aggregation is however limited and, at least at such low concentrations as used for the measurements, complete aggregation with macroscopic coagulum formation is prevented by a certain stabilization capability of the PIL chains. In contrast, the PNIPAM homopolymer leads to macroscopic coagulum formation (drastic increase in the hydrodynamic size) and block copolymers with styrene sulfonate or diallyldimethylammonium chloride change from transparent solutions to stable dispersions with a PNIPAM core and a stabilizing polyelectrolyte shell (decrease in the hydrodynamic size).61 Obviously at temperatures below the LCST, the PIL–PNIPAM particles are mainly stabilized by the PNIPAM chains and hence, they collapse after the PNIPAM chains turn hydrophobic. Generally, this interpretation is in accordance with the zeta-potential data (cf.Fig. 2).
Interestingly, for all triblock copolymers the DLS and URT data show a distinctly different behavior for the crosslinked (P1) and non-crosslinked (P2) PIL as exemplarily depicted in Fig. 6b and 7b for styrene as the hydrophobic block. The hydrodynamic diameter of the triblock particles with P1 decrease with increasing temperature and the dDi/dT curves show a clear minimum at the LCST. However, for the triblock copolymer particles with P2 (non-crosslinked) Di initially decreases with increasing temperature but in the range of the LCST Di changes the direction and increases to values larger than those at room temperature (cf.Fig. 6b). Such behavior is in accordance with limited aggregation as already discussed for the behavior of the PIL–PNIPAM diblock particles based on the DLS data. The limited coagulation finds in the URT data only indirect expression as the LCST (extreme value in the dΔU/dT curve) is unchanged and the width of the peak increases. The width of the base of the dΔU/dT curve measures the temperature range in which changes in the speed of sound or in the amount of released water take place. Thus, the increasing width as observed for the P2-triblock copolymers is expected for LCST-induced aggregation. A closer look at the base width of the peak in the dΔU/d –T plots for all samples containing PNIPAM blocks reveals interesting results (Fig. 8). In our understanding, this is a significant experimental observation because the width of the base is an expression of the sharpness of the LCST transition which is an important characteristic of thermoresponsive devices.
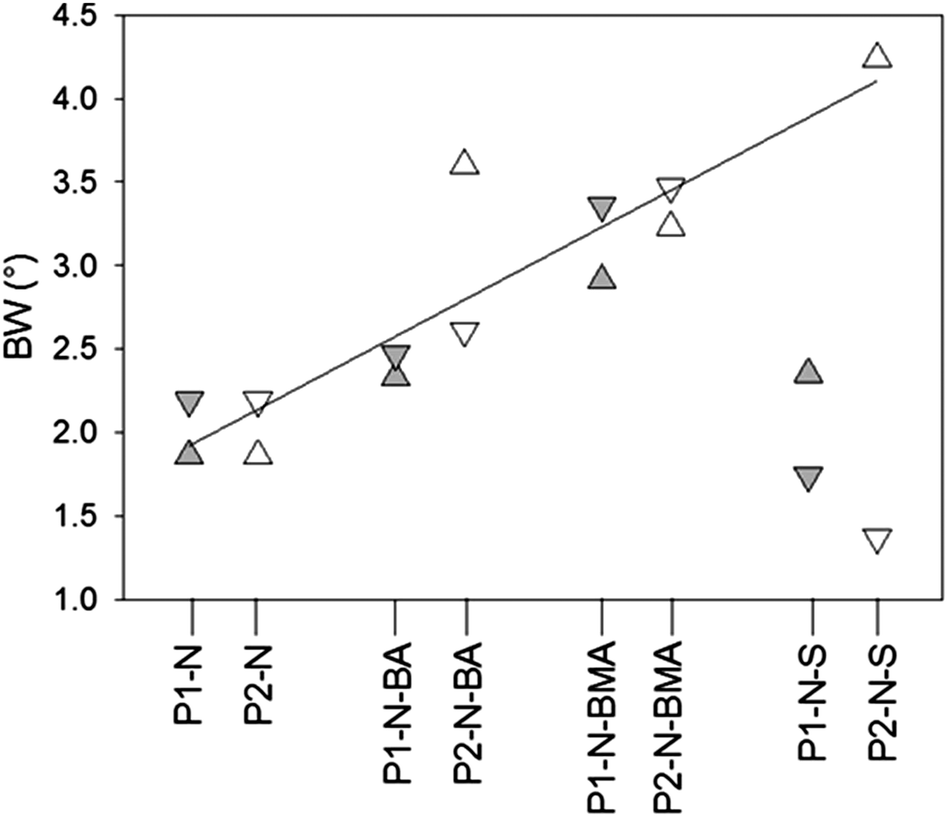 | ||
| Fig. 8 Width of the base of the dΔU/dT peaks (BW) for the various PNIPAM-containing block copolymer particles; grey and white symbols are samples prepared with crosslinked (P1) and non-crosslinked (P2) PIL, respectively, up and down triangles indicate heating and cooling cycles, respectively; the line is the linear regression of the corresponding data points (all data except for P1–N–S and cooling for P2–N–S). | ||
Quite surprisingly, the base width of the peak in the dΔU/dT–T curves strongly depends on the properties of the hydrophobic block and not so much on the property of the PIL block (crosslinked or non-crosslinked). With increasing Tg of the hydrophobic block, the base width increases except for heating and cooling of P1–N–S and cooling of P2–N–S. The behavior might be explained considering the temperature where the changes are observed (LCST) in relation to Tg of the hydrophobic block. For PBMA and PBA as hydrophobic blocks the LCST is much higher than Tg and hence, they are quite mobile and do not strongly influence the inflow and outflow of water molecules as well as the unfolding and folding of the PNIPAM block during cooling and heating, respectively. For PS as the hydrophobic block the situation is, however, different. The PS particles with crosslinked PIL (P1–N–S) are the hardest of the samples under consideration. In these particles, only the PNIPAM chains are mobile in the vicinity of the LCST. For the non-crosslinked PS particles (P2–N–S) the mobility of the PNIPAM is less influenced and hence, during heating the particles follow the trend as expressed by the regression line. To understand the behavior during cooling it is necessary to remember that the P2 particles form aggregates at temperatures above the LCST. Dissolution or unfolding of the collapsed PNIPAM chains requires disaggregation which, when compared with P2–N–BA and P2–N–BMA particles, is significantly hindered by the hard PS segments.
The data collected in Fig. 7 show that the LCST value determined by the URT heating curve and DLS measurement coincides quite nicely. In contrast to DLS, URT measurements allow the separate evaluation of the heating and cooling behavior and thus, the determination of two critical solution temperatures and a possible hysteresis. Typically, the critical temperature determined during heating (for precipitation) is greater than that during cooling (for dissolution). This difference, ΔTp−d, depends on the rate of the temperature change (dT/dt) and has a finite value even at dT/dt = 0.68 This underlines the conclusion that transient and equilibrium (isothermal) studies such as URT and DLS measurements, respectively, must not lead per se to identical results.69 For the samples in this study, the hysteresis, ΔTp−d, does not seem to significantly depend on the composition. ΔTp−d for all samples is 0.785 ± 0.23° in a similar range as observed for block copolymers of very different compositions and is mainly determined by the rate of the temperature change (300 mK min−1).57,68
4. Summary
The main experimental findings of our investigations are:(1) Within the class of reactive surfactants, PIL dispersions represent the first example proving that even polymer dispersions can act as a kind of reactive stabilizers for initiating aqueous heterophase polymerizations.
(2) Crosslinked and non-crosslinked PIL dispersions, P1 and P2, respectively, behave similarly in the polymerization reaction.
(3) The amphiphilic nature of the PIL chains enables them to adjust their behavior depending on the properties of the other block(s) to be either hydrophilic or hydrophobic.
(4) In diblock copolymer dispersions of PIL with a hydrophobic second block, the PIL is hydrophilic and stabilizes the particles.
(5) In triblock copolymer dispersions with a PNIPAM middle block, the PIL block is hydrophobic and the particles are stabilized by PNIPAM.
(6) The PNIPAM containing block copolymer dispersions show an LCST in the range typically observed for PNIPAM homopolymers.
(7) The sharpness of the LCST transition depends strongly on the composition of the block copolymer dispersions and is the highest for the triblock copolymer dispersions with polystyrene.
(8) Both the diblock copolymer PIL-PNIPAM and the triblock copolymers made with the non-crosslinked PIL dispersion undergo limited aggregation at temperatures higher than the LCST of PNIPAM.
(9) The morphology of the dried particles is governed mainly by the glass transition temperature of the hydrophobic block.
5. Conclusions
The experimental results show the quite easy applicability of suspensions of poly(3-n-tetradecyl-1-vinylimidazolium bromide), either crosslinked or not, in double function during the synthesis of polymer dispersions. On the one hand, when equipped with 2-hydroxyethylpropionamide end groups the PIL chains act as a reductant for ceric ions in an aqueous redox system thus leading to the formation of free initiating radicals at the methylol group carbon. On the other hand, the PIL molecules act as an electrostatic stabilizer during the heterophase polymerization of hydrophobic monomers. Within the class of reactive surfactants this kind of PIL dispersions represents the first example showing that polymer dispersions can act as a kind of reactive stabilizers for heterophase polymerization.There is no indication in the experimental results that the PIL particles act as simple seed particles. The stabilizing action of the PIL dispersion is rather unique and is characterized by quite a high mobility of the PIL chains. Zeta potential measurements clearly show that the PIL dispersion is amphiphilic in nature. This means that the PIL block is able to adjust its behavior depending on the properties of the other block(s) within the molecules following what we call the principle of relative hydrophilicity (PRH). Accordingly, the poly(ionic liquid) block is hydrophilic if connected to a less hydrophilic (more hydrophobic) block such as polystyrene but hydrophobic if attached to a less hydrophobic (more hydrophilic) block such as PNIPAM (at temperature below the LCST).
The behavior of the crosslinked and non-crosslinked PIL dispersions during the polymerization and with respect to the PRH is remarkably similar. This result indicates that the efficiency of the crosslinker is pretty much limited which is reasonable considering the layered structure of the PIL particles and the mismatch of the ILM and crosslinker size.
The introduction of a PNIPAM block gives to the corresponding di- and triblock particles reversible thermoresponsive properties. Interestingly, studying the temperature-dependent properties revealed an influence of the crosslinking of the PIL particles. The triblock particles made with non-crosslinked PIL lose their colloidal stability in the vicinity of the LCST and undergo limited aggregation which causes a net increase in the hydrodynamic particle size during the temperature increase. In contrast, the triblock copolymer particles originating from the crosslinked PIL shrink and swell during temperature increase and decrease relative to the LCST, respectively. Both kinds of diblock copolymer particles, with crosslinked and non-crosslinked PIL, show in accordance with the PRH, limited aggregation when the LCST is reached. The width of the LCST transition depends strongly on the composition of the block copolymer dispersions. It is the same for the diblocks with crosslinked and non-crosslinked PIL dispersions but strongly dependent on the nature of the hydrophobic block. The sharpness of the transition is the highest for the triblock copolymer dispersions with polystyrene as the hydrophobic block.
Acknowledgements
We thank Dr J. Y. Yuan for introducing us to the synthesis of PIL dispersions and Mrs Rona Pitschke and Mrs Heike Runge for the TEM investigations. R. Y. gratefully acknowledges a fellowship from the joint doctoral promotion program between the Max Planck Society and the Chinese Academy of Sciences.Notes and references
- H. F. Hernandez and K. Tauer, in Synthesis of Polymers, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, vol. 2, ch. 25, pp. 741–773 Search PubMed.
- K. Tauer, in Advanced Polymer Nanoparticles, ed. V. Mittal, CRC Press, Boca Raton, 2011, ch. 11, pp. 329–359 Search PubMed.
- K. Tauer, in Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Inc., 2002 Search PubMed.
- N. Rozik, M. Antonietti, J. Y. Yuan and K. Tauer, Macromol. Rapid Commun., 2013, 34, 665–671 CrossRef CAS PubMed.
- M. Antonietti, Y. F. Shen, T. Nakanishi, M. Manuelian, R. Campbell, L. Gwee, Y. A. Elabd, N. Tambe, R. Crombez and J. Texter, ACS Appl. Mater. Interfaces, 2010, 2, 649–653 CAS.
- D. England, N. Tambe and J. Texter, ACS Macro Lett., 2012, 1, 310–314 CrossRef CAS.
- C. Giordano, W. Yang, A. Lindemann, R. Crombez and J. Texter, Colloids Surf., A, 2011, 374, 84–87 CrossRef CAS PubMed.
- C. Wei, R. Yu and K. Tauer, Colloid Polym. Sci., 2014 DOI:10.1007/s00396-014-3210-3.
- H. Ringsdorf, B. Schlarb and J. Venzmer, Angew. Chem., Int. Ed., 1988, 27, 113–158 CrossRef.
- J. K. Thomas, Chem. Rev., 1980, 80, 283–299 CrossRef CAS.
- J. H. Fendler and P. Tundo, Acc. Chem. Res., 1984, 17, 3–8 CrossRef CAS.
- U. P. Strauss and E. G. Jackson, J. Polym. Sci., Part A: Polym. Chem., 1951, 6, 649–659 CAS.
- A. I. Medalia, H. H. Freedman and S. Sinha, J. Polym. Sci., Part A: Polym. Chem., 1959, 40, 15–33 CAS.
- P. Anton, P. Koberle and A. Laschewsky, Makromol. Chem., 1993, 194, 1–27 CrossRef CAS.
- M. Freemantle, in An Introduction to Ionic Liquids The Royal Society of Chemistry, Cambridge, UK, 2010, ch. 5, pp. 41–64 Search PubMed.
- A. Guyot and K. Tauer, Adv. Polym. Sci., 1994, 111, 43–65 CrossRef CAS.
- K. Nagai, Y. Ohishi, H. Inaba and S. Kudo, J. Polym. Sci. Pol. Chem., 1985, 23, 1221–1230 CrossRef CAS.
- V. V. Egorov and V. P. Zubov, Usp. Khim., 1987, 56, 2076–2097 CAS.
- J. Selb, S. Biggs, D. Renoux and F. Candau, Adv. Chem. Ser., 1996, 248, 251–278 CrossRef CAS PubMed.
- P. M. Carrasco, A. R. de Luzuriaga, M. Constantinou, P. Georgopanos, S. Rangou, A. Avgeropoulos, N. E. Zafeiropoulos, H. J. Grande, G. Cabanero, D. Mecerreyes and I. Garcia, Macromolecules, 2011, 44, 4936–4941 CrossRef CAS.
- J. M. Lu, F. Yan and J. Texter, Prog. Polym. Sci., 2009, 34, 431–448 CrossRef CAS PubMed.
- Y. Yang, D. Schulz and C. A. Steiner, Langmuir, 1999, 15, 4335–4343 CrossRef CAS.
- M. L. Bender, Chem. Rev., 1960, 60, 53–113 CrossRef CAS.
- E. Katchalski, G. D. Fasman, E. Simons, E. R. Blout, F. R. N. Gurd and W. L. Koltun, Arch. Biochem. Biophys., 1960, 88, 361–365 CrossRef CAS.
- C. G. Overberger, T. Stpierre, N. Vorchhei and S. Yaroslavsky, J. Am. Chem. Soc., 1963, 85, 3513–3515 CrossRef CAS.
- C. G. Overberger and N. Vorchhei, J. Am. Chem. Soc., 1963, 85, 951–955 CrossRef CAS.
- C. Overberg, T. Stpierre, N. Vorchhei, J. Lee and S. Yaroslav, J. Am. Chem. Soc., 1965, 87, 296–301 CrossRef.
- H. C. Kiefer, I. M. Klotz, I. S. Scarpa and W. I. Congdon, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 2155–2159 CrossRef CAS.
- T. Kunitake and Y. Okahata, Adv. Polym. Sci., 1976, 20, 161–227 CrossRef.
- M. R. Simmons and C. S. Patrickios, Macromolecules, 1998, 31, 9075–9077 CrossRef CAS.
- M. R. Simmons and C. S. Patrickios, J. Polym. Sci. Pol. Chem., 1999, 37, 1501–1512 CrossRef CAS.
- C. S. Patrickios and M. R. Simmons, Colloids Surf., A, 2000, 167, 61–72 CrossRef CAS.
- J. C. Salamone, S. C. Israel, P. Taylor and B. Snider, Polymer, 1973, 14, 639–644 CrossRef CAS.
- C. M. Stancik, A. R. Lavoie, P. A. Achurra, R. M. Waymouth and A. P. Gast, Langmuir, 2004, 20, 8975–8987 CrossRef CAS PubMed.
- C. M. Stancik, A. R. Lavoie, J. Schutz, P. A. Achurra, P. Lindner, A. P. Gast and R. M. Waymouth, Langmuir, 2004, 20, 596–605 CrossRef CAS.
- J. Yang, W. L. Sun, W. H. Lin and Z. Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5123–5132 CrossRef CAS.
- S. M. Yu, F. Yan, X. W. Zhang, J. B. You, P. Y. Wu, J. M. Lu, Q. F. Xu, X. W. Xia and G. L. Ma, Macromolecules, 2008, 41, 3389–3392 CrossRef CAS.
- F. Yan and J. Texter, Chem. Commun., 2006, 2696–2698 RSC.
- M. Yoshio, T. Kagata, K. Hoshino, T. Mukai, H. Ohno and T. Kato, J. Am. Chem. Soc., 2006, 128, 5570–5577 CrossRef CAS PubMed.
- D. Batra, S. Seifert, L. M. Varela, A. C. Y. Liu and M. A. Firestone, Adv. Funct. Mater., 2007, 17, 1279–1287 CrossRef CAS.
- F. Yan and J. Texter, Angew. Chem., Int. Ed., 2007, 46, 2440–2443 CrossRef CAS PubMed.
- T. Sekitani, Y. Noguchi, K. Hata, T. Fukushima, T. Aida and T. Someya, Science, 2008, 321, 1468–1472 CrossRef CAS PubMed.
- K. Vijayakrishna, S. K. Jewrajka, A. Ruiz, R. Marcilla, J. A. Pomposo, D. Mecerreyes, D. Taton and Y. Gnanou, Macromolecules, 2008, 41, 6299–6308 CrossRef CAS.
- M. Tokuda and H. Minami, J. Colloid Interface Sci., 2013, 398, 120–125 CrossRef CAS PubMed.
- J. Texter, Macromol. Rapid Commun., 2012, 33, 1996–2014 CrossRef CAS PubMed.
- B. A. Wolf, Adv. Polym. Sci., 2010, 238, 1–66 CrossRef.
- M. A. Ward and T. K. Georgiou, Polymers, 2011, 3, 1215–1242 CrossRef CAS PubMed.
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC.
- H. Mori, M. Yahagi and T. Endo, Macromolecules, 2009, 42, 8082–8092 CrossRef CAS.
- K. Tauer, N. Weber and J. Texter, Chem. Commun., 2009, 6065–6067 RSC.
- J. Y. Yuan, H. Schlaad, C. Giordano and M. Antonietti, Eur. Polym. J., 2011, 47, 772–781 CrossRef CAS PubMed.
- S. Soll, M. Antonietti and J. Y. Yuan, ACS Macro Lett., 2012, 1, 84–87 CrossRef CAS.
- E. Karjalainen, N. Chenna, P. Laurinmaki, S. J. Butcher and H. Tenhu, Polym. Chem., 2013, 4, 1014–1024 RSC.
- Y. J. Men, M. Drechsler and J. Y. Yuan, Macromol. Rapid Commun., 2013, 34, 1721–1727 CrossRef CAS PubMed.
- M. D. C. Topp, I. H. Leunen, P. J. Dijkstra, K. Tauer, C. Schellenberg and J. Feijen, Macromolecules, 2000, 33, 4986–4988 CrossRef CAS.
- K. Tauer and V. Khrenov, Macromol. Symp., 2002, 179, 27–52 CrossRef CAS.
- R. Yu, J. Hartmann and K. Tauer, Macromol. Rapid Commun., 2013, 34, 1629–1634 CrossRef CAS PubMed.
- M. Koebe, M. Drechsler, J. Weber and J. Y. Yuan, Macromol. Rapid Commun., 2012, 33, 646–651 CrossRef CAS PubMed.
- J. Y. Yuan and M. Antonietti, Macromolecules, 2011, 44, 744–750 CrossRef CAS.
- J. Y. Yuan, S. Soll, M. Drechsler, A. H. E. Muller and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 17556–17559 CrossRef CAS PubMed.
- K. Tauer, M. Mukhamedjanova, C. Holtze, P. Nazaran and J. Lee, Macromol. Symp., 2007, 248, 227–238 CrossRef CAS.
- F. Ganachaud, R. Balic, M. J. Monteiro and R. G. Gilbert, Macromolecules, 2000, 33, 8589–8596 CrossRef CAS.
- J. M. Asua, S. Beuermann, M. Buback, P. Castignolles, B. Charleux, R. G. Gilbert, R. A. Hutchinson, J. R. Leiza, A. N. Nikitin, J. P. Vairon and A. M. van Herk, Macromol. Chem. Phys., 2004, 205, 2151–2160 CrossRef CAS.
- N. Weber, B. Tiersch, M. M. Unterlass, A. Heilig and K. Tauer, Macromol. Rapid Commun., 2011, 32, 1925–1929 CrossRef CAS PubMed.
- J. Snuparek, Prog. Org. Coat., 1996, 29, 225–233 CrossRef CAS.
- R. H. Pelton and P. Chibante, Colloids Surf., 1986, 20, 247–256 CrossRef CAS.
- K. Van Durme, L. Delellio, E. Kudryashov, V. Buckin and B. Van Mele, J. Polym. Sci. Pol. Phys., 2005, 43, 1283–1295 CrossRef CAS.
- K. Tauer, D. Gau, S. Schulze and H. Hernandez, Polymer, 2008, 49, 5452–5457 CrossRef CAS PubMed.
- N. V. Grinberg, A. S. Dubovik, V. Y. Grinberg, D. V. Kuznetsov, E. E. Makhaeva, A. Y. Grosberg and T. Tanaka, Macromolecules, 1999, 32, 1471–1475 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4py00458b |
| This journal is © The Royal Society of Chemistry 2014 |