 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multivalency in healable supramolecular polymers: the effect of supramolecular cross-link density on the mechanical properties and healing of non-covalent polymer networks†
Lewis R.
Hart
a,
James H.
Hunter
a,
Ngoc A.
Nguyen
b,
Josephine L.
Harries
c,
Barnaby W.
Greenland
d,
Michael E.
Mackay
be,
Howard M.
Colquhoun
*a and
Wayne
Hayes
*a
aDepartment of Chemistry, University of Reading, Whiteknights, Reading, RG6 6AD, UK. E-mail: w.c.hayes@reading.ac.uk; h.m.colquhoun@reading.ac.uk; Fax: +44 (0)118 378 6331; Tel: +44 (0)118 378 6491
bDepartment of Materials Science and Engineering, University of Delaware, Newark, Delaware 19716, USA
cDomino UK Ltd, Trafalgar Way, Bar Hill, Cambridge, CB23 8TU, UK
dThe Reading School of Pharmacy, University of Reading, Whiteknights, Reading, RG6 6AD, UK
eDepartment of Chemical and Biomolecular Engineering, University of Delaware, Newark, Delaware 19716, USA
First published on 13th March 2014
Abstract
Polymers with the ability to heal themselves could provide access to materials with extended lifetimes in a wide range of applications such as surface coatings, automotive components and aerospace composites. Here we describe the synthesis and characterisation of two novel, stimuli-responsive, supramolecular polymer blends based on π-electron-rich pyrenyl residues and π-electron-deficient, chain-folding aromatic diimides that interact through complementary π–π stacking interactions. Different degrees of supramolecular “cross-linking” were achieved by use of divalent or trivalent poly(ethylene glycol)-based polymers featuring pyrenyl end-groups, blended with a known diimide–ether copolymer. The mechanical properties of the resulting polymer blends revealed that higher degrees of supramolecular “cross-link density” yield materials with enhanced mechanical properties, such as increased tensile modulus, modulus of toughness, elasticity and yield point. After a number of break/heal cycles, these materials were found to retain the characteristics of the pristine polymer blend, and this new approach thus offers a simple route to mechanically robust yet healable materials.
Introduction
Polymers with the ability to repair themselves after damage1–8 could be used in a variety of applications to extend the lifetime of the material. Known examples of such materials demonstrate the ability to heal autonomically9–14 or upon the application of external stimuli such as heat,2,15–20 light21–25 or pressure.26–28 Healable polymeric systems can be achieved through a variety of approaches, including the use of encapsulated liquid monomers and microvascular networks,9–14 irreversible covalent bond formation,25 reversible covalent bond formation,18,23,29–32 and supramolecular33–43 polymer systems.44Within the field of supramolecular polymers, materials utilising hydrogen bonding,26,45–49 metal-ligand interactions,21,50–52 or π–π stacking interactions19,53–58 between electronically complementary moieties have all been shown to exhibit healable characteristics. These polymers are generally elastomeric in nature and require heating well above the Tg before healing can take place. Our previous work has focused on a two component polymer blend which is able to self-assemble through π–π stacking interactions between a π-electron-deficient chain-folding59–64 diimide residue and a π-electron-rich pyrenyl moiety (Scheme 1). Whilst the elastomeric materials produced did indeed demonstrate excellent healing characteristics, improving their mechanical properties would result in materials that could find use in more demanding applications. To address this problem, the polymer mid-blocks were first re-optimised19,53 to afford a more mechanically robust material. End-group modification56,64 was also studied through the incorporation of tweezer-type π-electron rich moieties with higher binding constants. This resulted in materials with greater mechanical strength and toughness, although longer times at higher temperatures were required for healing to be achieved. Finally, reinforced supramolecular polymer blends with cellulose nanowhiskers57 or gold nanoparticles58 have been developed, which also demonstrated improved mechanical properties.
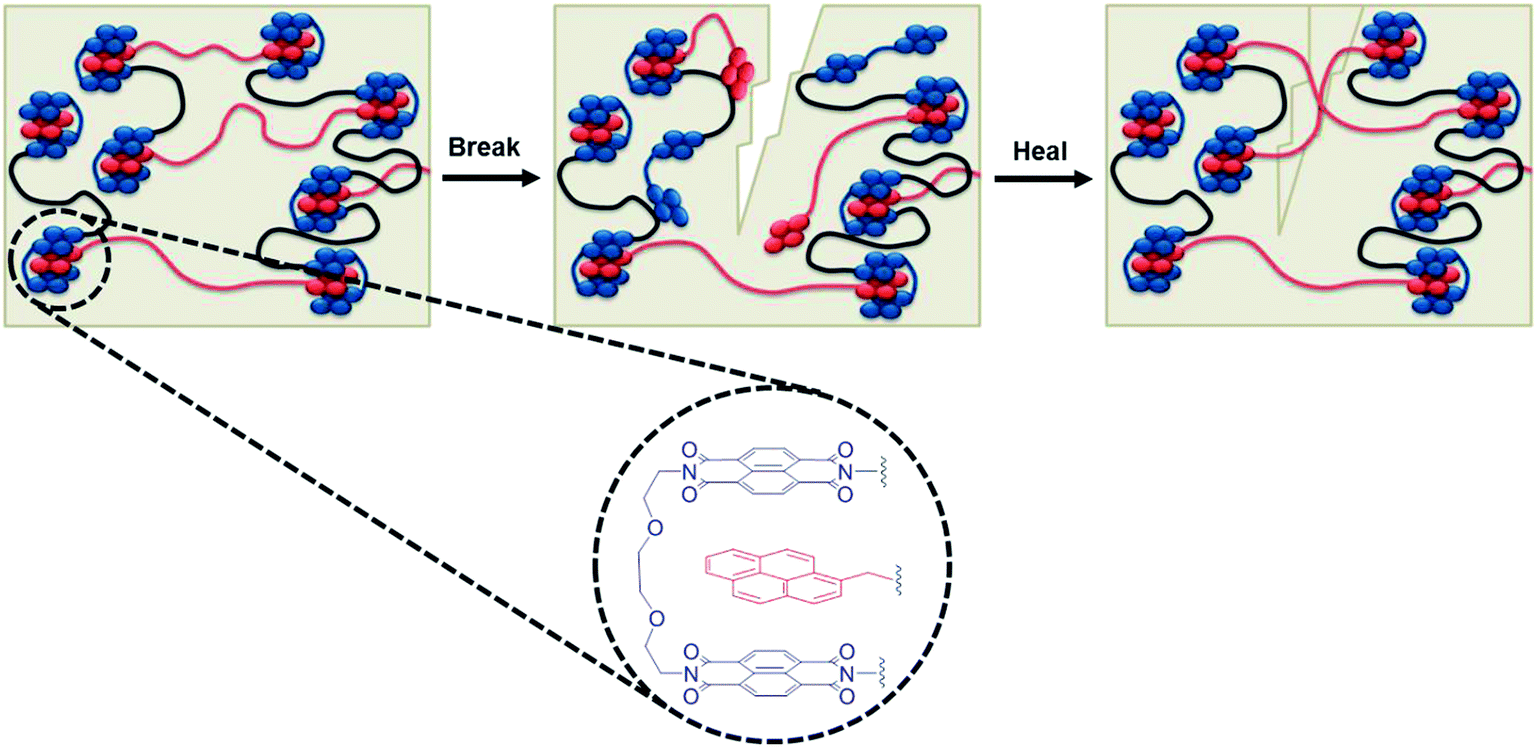 | ||
| Scheme 1 Schematic of a complementary π–π stacking polymer blend, illustrating the process of breaking the polymer matrix by disengagement of the weaker supramolecular interactions before applying an appropriate stimulus to facilitate healing. Insert shows the atomistic structure of the π–π stacked complex54 with π-electron-rich pyrenyl end-groups (red) and π-electron-deficient naphthalene diimide units (blue) linked by an appropriate spacer to form a chain-folding residue. | ||
In the present study we investigate the effect of increasing supramolecular valency65 as a means of enhancing the mechanical properties of such healable materials. Novel divalent and trivalent poly(ethylene glycol) (PEG) derived polymer with π-electron-rich pyrenyl end groups were readily synthesised. Blending these with a known multivalent π-electron-deficient chain-folding co-polymer gave ready access to polymer blends with differing degrees of supramolecular cross-link density. Rapid and reversible complexation was observed in the resulting supramolecular polymer blends, both in solution and in the solid state, and a clear relationship between supramolecular valency and mechanical properties has been established.
Results and discussion
To assess the effects of supramolecular valency, polymers with increasing numbers of pyrenyl end-capping residues (shown red in Scheme 2) were required. Blending these polymers with a multivalent chain-folding copolymer (blue/black/grey in Scheme 2) would allow access to supramolecular blends with differing supramolecular cross-link densities.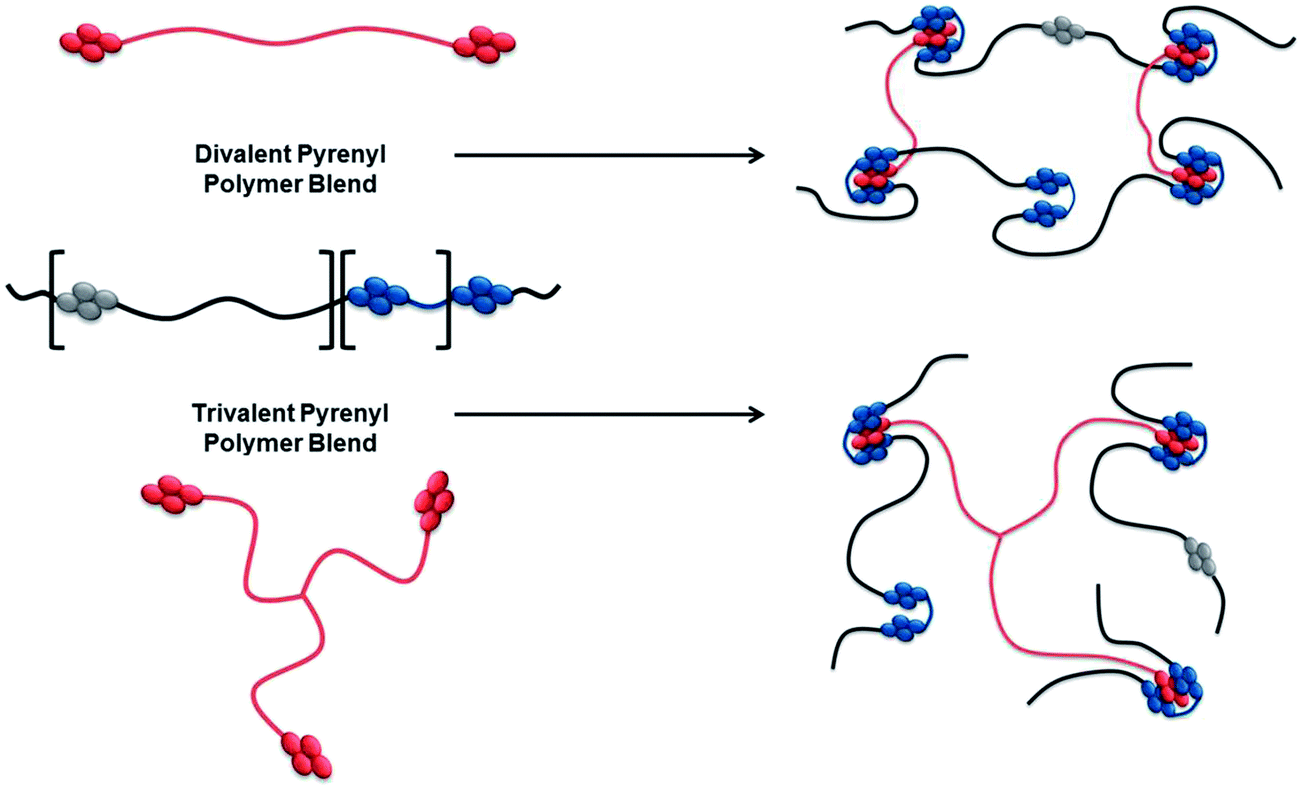 | ||
| Scheme 2 Schematic for supramolecular polymer blends in which species with different pyrenyl valencies (in red) complex with a chain-folding diimide (blue/black/grey). The naphthalene–diimide residues (grey) spaced by Jeffamine® D-400 residues do not form chain-folding sequences. | ||
Polymer synthesis and characterisation
To achieve polymer networks with varying degrees of supramolecular cross-link density, both divalent 1 (Scheme 3) and trivalent 2 (Scheme 4) polymers with π-electron-rich end-groups were designed and synthesised. A divalent polymer with pyrenyl end-groups (1) was synthesised by DCC-mediated coupling of pyrene butyric acid 3 with bis(3-aminopropyl) terminated poly(ethylene glycol) (Mn = 1500 g mol−1) 4 (Scheme 3). Analysis of 1 by 1H NMR spectroscopy showed no evidence of residual mono-end-capped product, whilst MALDI-TOF MS analysis showed a molecular ion corresponding to Mn at 2082.17 Da, with an average of 32 repeating ethylene glycol units. These data are in excellent agreement with a value of Mn (2082.18 Da) calculated for the divalent pyrenyl terminated polymer 1. For full spectroscopic analysis of the divalent polymer 1, see ESI Fig. 1–4.†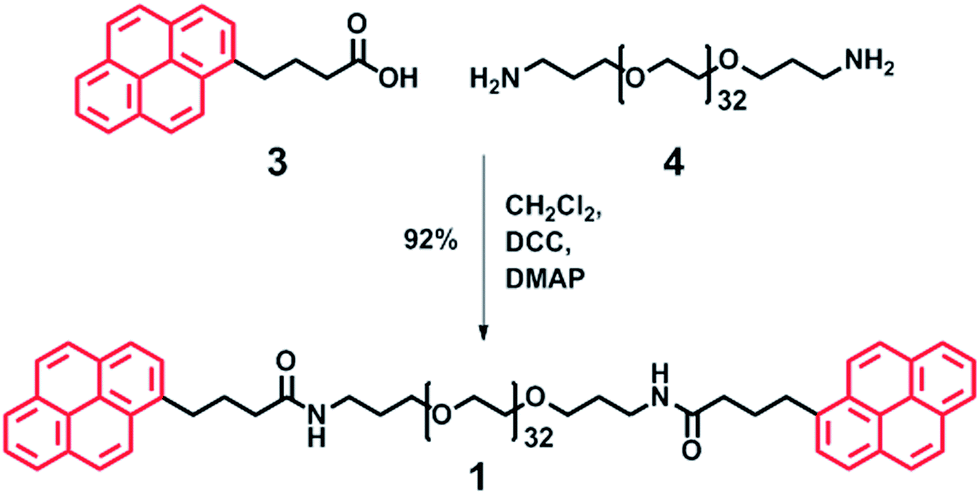 | ||
| Scheme 3 Synthesis of divalent pyrenyl-terminated poly(ethylene glycol) 1via DCC coupling. The pyrenyl moieties represent the π-electron-rich guests (Scheme 2) which are able to intercalate with a π-electron-deficient chain-folding motif. | ||
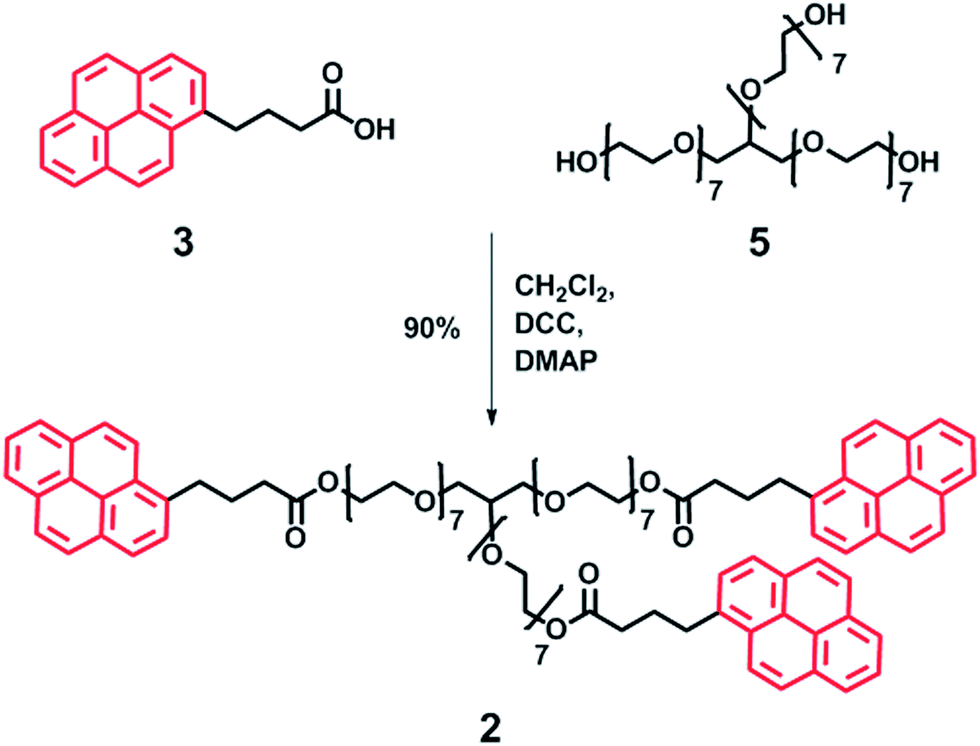 | ||
| Scheme 4 Synthesis of trivalent pyrenyl terminated poly(ethylene glycol) 2 by DCC mediated coupling. Red pyrenyl moieties represent the π-electron-rich end-groups. | ||
A similar synthetic approach was used to produce the trivalent polymer 2. Pyrene butyric acid 3 was coupled with glycerol ethoxylate (Mn = 1000 g mol−1) 5 using standard procedures (Scheme 4) in good yield (90%). Analysis by MALDI-TOF MS revealed a molecular ion at 2025.75 Da, in close agreement with the calculated molecular weight (2026.00 Da), whilst no residual mono or di-capped products were observed by 1H NMR spectroscopy (see ESI Fig. 5–8† for full spectroscopic analysis).
Analysis of polymers 1 and 2 by gel permeation chromatography (DMF containing 0.01 M LiBr as eluent) revealed values for Mn (1331 and 705 g mol−1, respectively) that were lower than expected for both the divalent and trivalent polymers. However, Gitsov and Fréchet66 have shown that solvation of poly(ethylene glycol)-based dendrimers having aromatic terminal units in polar solvents leads to encapsulation of the hydrophobic groups by the hydrophilic backbone, thus decreasing the radius of gyration. A comparable effect may account for the lower than expected values of Mn observed by GPC analysis of the current polymers. The effect of encapsulation is amplified in the trivalent polymer architecture, resulting in an even lower Mn as measured by GPC when compared to the divalent analogue.
As well as the pyrene-terminated π-electron-rich polymers, a π-electron-deficient polymer was required to form electronically-complementary polymer blends (Scheme 2). We have previously reported19 a chain-folding diimide copolymer 6 (Fig. 1) that shows healing characteristics when blended with π-electron-rich pyrenyl polymers. To allow detailed comparison with these earlier materials, this chain-folding diimide copolymer was also used in the present work (ESI Fig. 9–12), being obtained readily from commercially available materials as a tan powder (Mn = 2242 g mol−1, PDI = 1.87).19
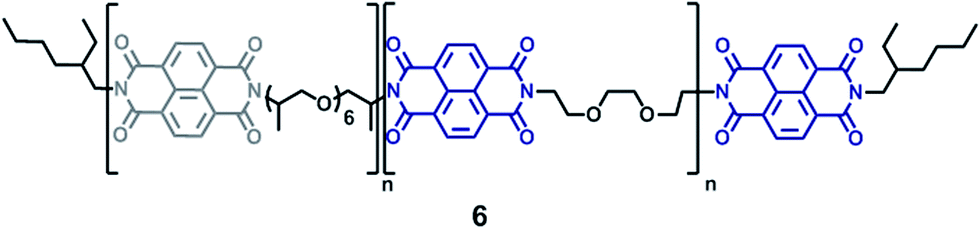 | ||
| Fig. 1 Structure of copolymer 6, with chain-folding naphthalene–diimide residues (blue) spaced by a linker derived from 2,2′-(ethylenedioxy)bis(ethylamine). The naphthalene–diimide residues (grey) spaced by Jeffamine® D-400 residues do not form chain-folding sequences. | ||
Solution complexation studies
Before analysing the mechanical properties of the supramolecular polymer blends ([1 + 6] and [2 + 6]) in the solid state, it was important to verify the presence of the π–π stacking interaction in solution. Blends were thus obtained by co-dissolving electronically complementary polymers (equimolar ratio with respect to binding motif equivalent weights) in chloroform. The resulting solutions were deep red in colour, whereas the diimide copolymer itself is a pale orange/brown and the pyrenyl-terminated polymers are essentially colourless (Fig. 2). | ||
| Fig. 2 Solutions of the divalent (1) and trivalent (2) pyrenyl end-capped polymer in chloroform, and their blends with the chain-folding diimide 6. The deep red colour of the blends arises from π–π stacking and charge-transfer between electronically complementary residues. | ||
The dramatic change in colour on mixing results from the presence of an absorption band at 535 nm in the visible spectrum (ESI Fig. 13 and 14†) as noted previously for related systems.54 Absorption at this wavelength is indicative of stacking-induced charge transfer between the π-electron-rich pyrenyl moiety (1 or 2) and the π-electron-deficient chain-folding diimide 6.54 Upon heating, the intensity of this absorption is visibly reduced, indicating a shift in the complexation equilibrium towards the unbound (“off”) state (ESI Fig. 15 and 16†). Conversely, when the solutions were cooled, an increase in the depth of colour was observed as a result of increased π–π stacking between the electronically complementary residues. In agreement with the increased supramolecular valency in blend [2 + 6], a ca. 50% increase in the absolute absorbance at 535 nm was noted for this system (1.20 a.u.) when compared to the divalent analogue [1 + 6] (0.85 a.u.).
On irradiating solutions of the pyrenyl terminated polymers (1 or 2) with a short wavelength UV source (310 nm), the solutions fluoresced visibly, with the typical blue emission expected from tethered pyrenyl end-groups. Fluorescence spectra revealed characteristic emission bands for the monomeric pyrenyl group at 379, 399, and 420 nm, respectively, as well as the excimer emission centred at 472 nm (ESI Fig. 17 and 18†). Formation of a pyrene excimer requires energy transfer between an electronically excited pyrenyl residue to a neighbouring pyrene moiety in its electronic ground state.67 This is seen more prominently in the spectrum of the trivalent polymer 2 than in that of divalent polymer 1, as consequence of the increased effective concentration of pyrenyl residues. When 1 or 2 is blended in an equimolar ratio (pyrene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :chain-fold motif) with poly(diimide) 6, both the monomer and excimer pyrenyl emissions are almost completely quenched, confirming complementary π–π stacking.
:chain-fold motif) with poly(diimide) 6, both the monomer and excimer pyrenyl emissions are almost completely quenched, confirming complementary π–π stacking.
As a further probe of the solution-state behaviour between electronically complementary π-electron-rich and π-electron-deficient polymers, 1H NMR spectra of the blends containing the divalent [1 + 6] and trivalent [2 + 6] polymer systems (equimolar with respect to binding motifs; 0.01 M in CDCl3–TFA, 9:1 v/v) were compared to the spectra obtained for the individual components (ESI Fig. 19 and 20†). Significant upfield shifts (>0.30 ppm) were observed in the aromatic proton resonances of both the π-electron-rich pyrenyl groups and π-electron-poor naphthalene diimide residues, as a result of mutual ring-current shielding between the intercalating pyrenyl end-groups and the chain-folding polydiimide. Additionally, in the blend, the aromatic (naphthalenyl) resonances of the co-polydiimide 6 are seen to split into two distinct populations, with chain-folding, binding sequences at 8.50–8.10 ppm, and non-binding sequences at ca. 8.60 ppm in which the naphthalene–diimide units are flanked by two Jeffamine® residues. The complex pattern of resonances observed for the “bound” naphthalenyl protons in the polymer blend arises from the range of different binding sequences within the random co-polymer backbone59,60,63 (i.e. doublets, triplets, etc.), whilst the broad nature of the proton resonances for both π-electron-rich and π-electron-deficient species in the bound state suggests that the system is approaching the slow exchange regime.
Variable temperature 1H NMR spectroscopy (CDCl3–TFA, 9:1 v/v) was carried out between 10 °C and 55 °C, to probe the association/dissociation of π–π stacking interactions in solution (ESI Fig. 21 and 22†). In both blends ([1 + 6] and [2 + 6]), resonances from the naphthalenyl chain-folding residues of 6 are seen to migrate downfield (ca. 0.25 ppm) with increasing temperature, in response to a shift in the equilibrium of the supramolecular interactions towards the unbound (“off”) state, represented also in the present system by the broad singlet at 8.75 ppm. The pyrenyl resonances also shift downfield (ca. 0.20 ppm), and regain resolution at elevated temperatures as a result of fast exchange between bound and unbound molecules.
Inherent viscosity measurements can give strong evidence for the formation of supramolecular polymers in solution.34 Divalent 1 and trivalent 2 polymers showed inherent viscosities (ηinh) of 0.15 and 0.11 dL g−1, respectively, whilst the chain-folding diimide 6 was found to have an inherent viscosity (ηinh) of 0.18 dL g−1, as shown in Fig. 3. Addition of the chain-folding polydiimide 6 to the divalent pyrenyl polymer 1, (equimolar ratio with respect to binding residues) revealed an increase in viscosity (ηinh = 0.20 dL g−1) as a consequence of the supramolecular interactions generating a more extended polymer network in solution. Importantly, a much greater increase in inherent viscosity was observed in the equimolar blend containing the trivalent pyrenyl polymer [2 + 6] (ηinh = 0.30 dL g−1), which can be attributed to formation of an even more highly extended supramolecular network being formed in solution as a result of the increased valency of the pyrenyl polymer 2. It was also noted that the viscosity of the blend containing the trivalent pyrenyl polymer [2 + 6] was 50% greater than that of the blend containing the divalent pyrenyl polymer [1 + 6]. This observation suggests strongly that the number of pyrenyl end-groups per polymer molecule is directly influencing the solution properties of the corresponding supramolecular blends.
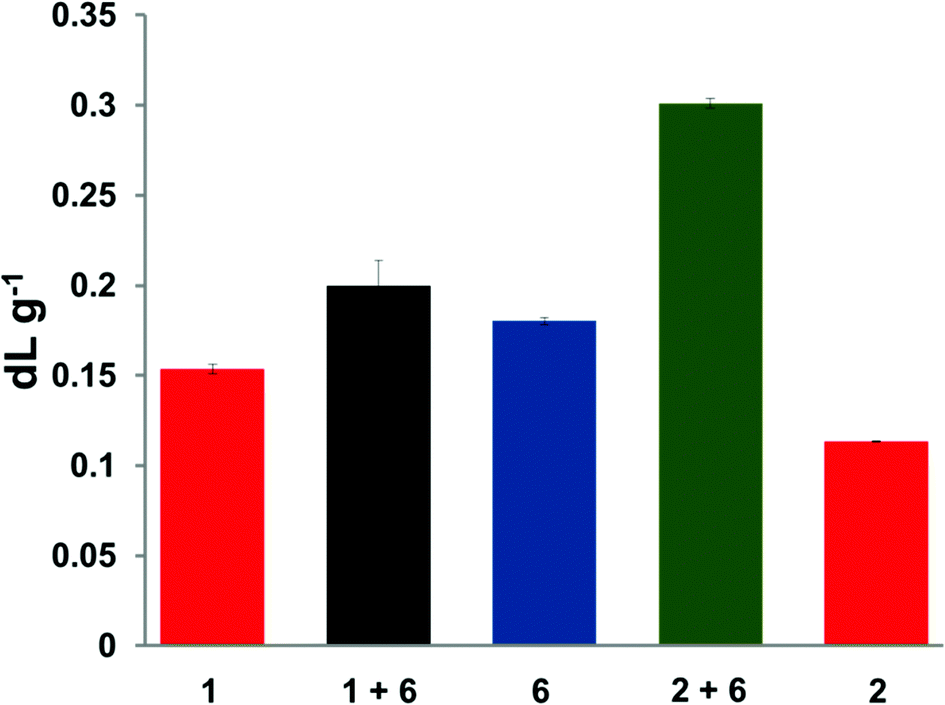 | ||
| Fig. 3 Inherent viscosities of individual polymers (1, 2 and 6) and the equimolar (with respect to binding motifs) polymer blends ([1 + 6] and [2 + 6]) at 10 mg mL−1 in chloroform–hexafluoroisopropanol (9:1, v/v). Viscosities are the mean of four repeat measurements in each case. | ||
Polymer films and solid state complexation studies
To investigate the formation of π–π stacked polymer complexes in the solid state, films were cast from 2,2,2-trichloroethanol and allowed to dry in an oven for 3 days, raising the temperature incrementally to 80 °C (Fig. 4). Films of the polydiimide 6 alone were extremely fragile, whilst both of the pyrenyl end-capped, PEG-based polymers 1 and 2 were viscous oils. However, films cast from solution blends of the divalent 1 or trivalent 2 pyrenyl end-capped polymer with the π-electron-deficient polydiimide 6 (1:1 with respect to binding motifs) were tough, deep-red elastomeric solids. The intense colour of these films is obviously indicative of the π–π stacking interaction between complementary π-electron-rich and π-electron-deficient polymers. The materials thus produced clearly demonstrate that complementary π–π stacking is not only retained in the solid state, but also leads to robust films from non-film-forming precursors. Previous studies19,53 on structurally related polymer utilising pyrenyl and chain-folding binding motifs demonstrated not only the requirement of electronically complementary binding motifs in producing homogenous polymer blends, but also healable polymeric materials. By way of a control experiment, the chain-folding polydiimide 6 was blended with the unfunctionalised PEG prepolymers (4 or 5) and films cast under analogous conditions to test their film-forming properties and healability (ESI Fig. 23†). The blend [5 + 6] resulted in an extremely brittle, non-pealable film similar in nature to 6, whilst the polymer blend of [4 + 6] produced a pealable, yet fragile film.
 | ||
| Fig. 4 Films cast from 2,2,2-trichloroethanol and dried at room temperature for 24 hours before drying in an oven at 40 °C for 24 hours and subsequently at 80 °C for 24 hours. | ||
To investigate the morphology of the supramolecular polymer blend, variable-temperature small-angle X-ray scattering (SAXS) analysis of the cast films was carried out. Samples were heated in 25 °C increments from ambient temperature to 200 °C and then back to room temperature (Fig. 5). At all temperatures in the range studied, both blends ([1 + 6] and [2 + 6]) showed a single scattering signal at 1/d = 0.005–0.020 Å−1 that is indicative of a phase-separated morphology with a domain spacing ca. 100 Å. While the domain spacing was essentially invariant within the temperature range studied, the X-ray contrast between the different domains increased with increasing temperature. This is demonstrated in the insert of Fig. 5, where a plot of the intensity of the scattering signal at 1/d = 0.01 Å−1 as a function of temperature shows the reversible nature of this relationship. Full variable temperature SAXS data is given in the ESI (Fig. 24 and 25†).
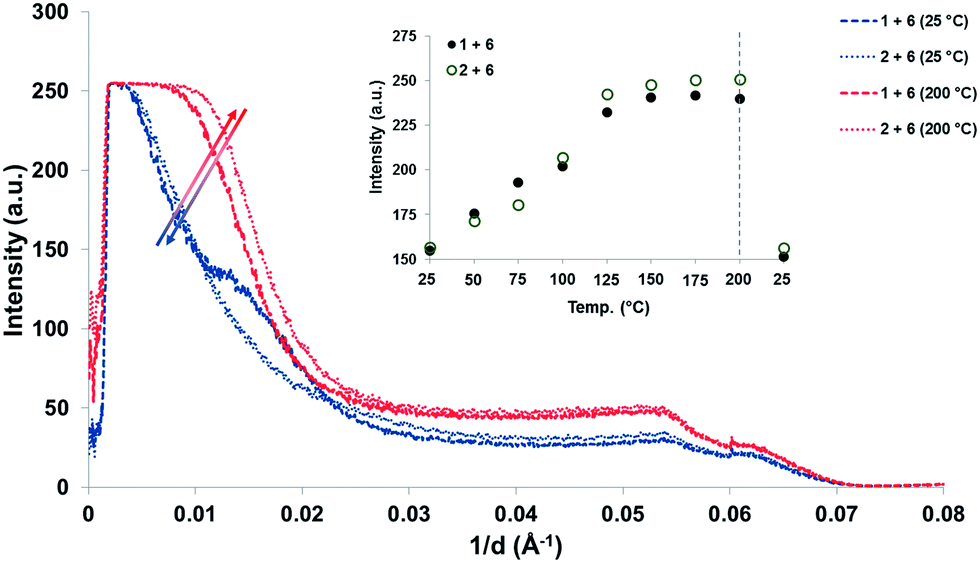 | ||
| Fig. 5 Small angle X-ray scattering from the divalent pyrenyl-polymer blend ([1 + 6]) and the blend containing the trivalent pyrenyl-polymer ([2 + 6]). Curves are shown for scattering at 25 °C and 200 °C, and the inset shows the incremental variation in scattering intensity at 1/d = 0.01 Å−1 over this temperature range. | ||
Healing studies
As a visual demonstration of the healing capabilities of the homogeneous polymer blends, a damaged film-sample (with a 75 μm wide cut) of the trivalent blend [2 + 6] was heated at 10 °C min−1 in an environmental scanning electron microscope (ESEM) from ambient temperature. As the temperature approached ca. 75 °C healing of the damaged area was seen to initiate. After further heating to 100 °C the material demonstrated almost complete healing and was found to be completely homogenous at 200 °C (Fig. 6). Original images can be found in the ESI (Fig. 26†). An analogous result was observed for the divalent blend [1 + 6] (ESI Fig. 27†) in which healing of the damaged area began to be evident at 50 °C, and again was fully healed on reaching 200 °C. In contrast, ESEM analysis of the control blend [4 + 6] revealed no spontaneous healing at elevated temperatures (ESI Fig. 28†). The broken sample was seen to expand at increased temperature, but no obvious healing was observed between the crack faces and indeed the fracture remained evident at 200 °C. | ||
| Fig. 6 False colour ESEM images of the healable polymer blend [2 + 6] containing a trivalent pyrenyl-terminated polymer. Micrographs were taken at (a) 25 °C, (b) 100 °C and (c) 200 °C. The images demonstrate both formation of a homogenous blend and the healabilty of the polymer at elevated temperatures. Scale bar represents 300 μm. | ||
To quantify healing efficiencies of these materials, a test-strip of film (0.2–0.3 mm in thickness and 4.0 mm in width) was elongated to break by tensometry over 3 test/break/heal/test cycles at 23 °C with a Hencky strain rate of 0.1 s−1 (Fig. 7).19,53 The pristine film of the divalent blend [1 + 6] exhibited a tensile modulus (E) of 29 MPa whilst the trivalent, pyrenyl–polymer containing blend [2 + 6] showed a much higher tensile modulus (49 MPa, an increase of ca. 70%). The substantial increase in modulus was attributed to the formation of a supramolecular network with a higher degree of cross-linking derived from additional π-electron-rich moieties within the trivalent pyrenyl-polymer 2. The healing efficiency relative to the pristine material was maintained at ca. 95% over three break/heal cycles for the divalent polymer blend [1 + 6] and at ca. 90% in the blend containing the trivalent pyrenyl polymer [2 + 6], as shown in Fig. 7.
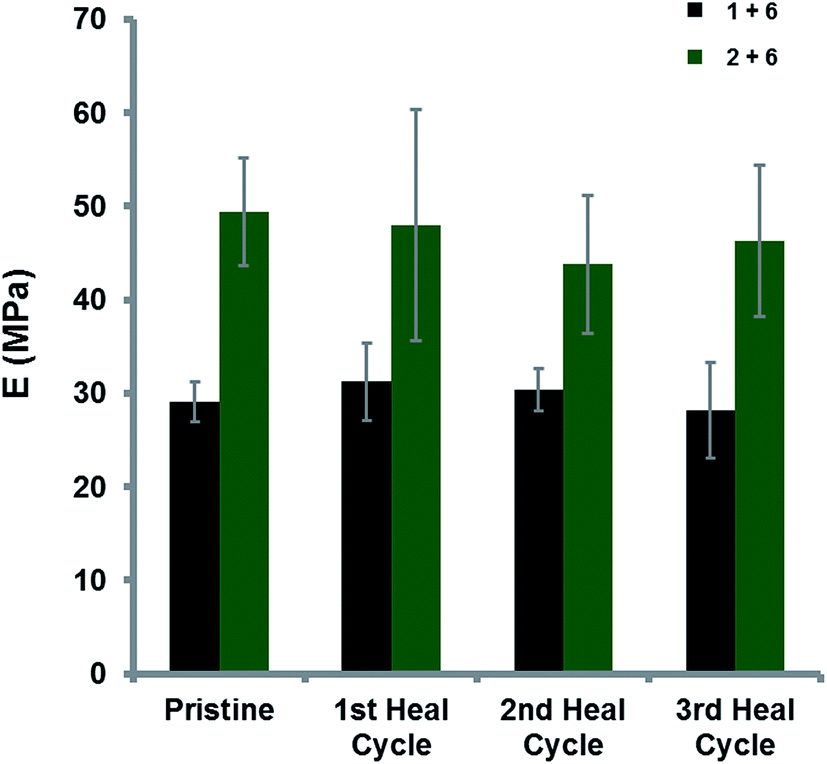 | ||
| Fig. 7 Tensile moduli of the blends containing the divalent pyrenyl-polymer [1 + 6] and trivalent pyrenyl-polymer [2 + 6], demonstrating the increased modulus of the pristine blend containing the trivalent pyrenyl polymer (49 MPa) when compared to the pristine divalent pyrenyl polymer blend (29 MPa) as a result of increased supramolecular cross-link density. This difference is maintained over three break/heal cycles. | ||
To quantify the increase in cross-linking within the pristine polymer blend [2 + 6] when compared to [1 + 6] the average molecular weight between cross-linking points (Mc) was calculated. Using the treatment of tensile module data described by Charlesby and Hancock68 (see eqn (1), ESI Fig. 29†) the relationship between Mc, tensile modulus and the density of the polymer blend allowed assessment of the cross-linking density in [1 + 6] and [2 + 6]. The divalent polymer blend [1 + 6] was shown to have a Mc of 313 g mol−1, whilst the trivalent blend [2 + 6] demonstrated a lower Mc (207 g mol−1), indicating a greater degree of supramolecular cross-linking points within the polymer network. The 50% decrease in Mc in the trivalent polymer blend again highlights the clear relationship between the valency of the polymer and the average molecular weight between cross-links.
The supramolecular polymer blend between the trivalent pyrenyl polymer 2 and the chain-folding polydiimide 6 demonstrated a significant increase in modulus of toughness when compared to the comparable blend containing the linear pyrenyl polymer analogue 1 (Fig. 8). The elastomeric nature of the blended supramolecular films allowed them to be stretched to 1.8 times ([1 + 6]) and 6.2 times ([2 + 6]) their original lengths, respectively, before breaking. These data corresponded to elongations of 80% and 520%, respectively, with corresponding modulus of toughness values of 1.45 MPa for [1 + 6] and 22.60 MPa for [2 + 6]. It was also observed that the yield point at 0.2% offset strain of the branched polymer blend [2 + 6], at 4.6 MPa, was approximately twice that of the divalent blended film [1 + 6] (2.4 MPa), again demonstrating the increase in strength of the material as the degree of supramolecular cross-linking is increased. However, over a number of break and heal cycles, the modulus of toughness was found to decrease for both materials (ESI Fig. 30 and 31†).
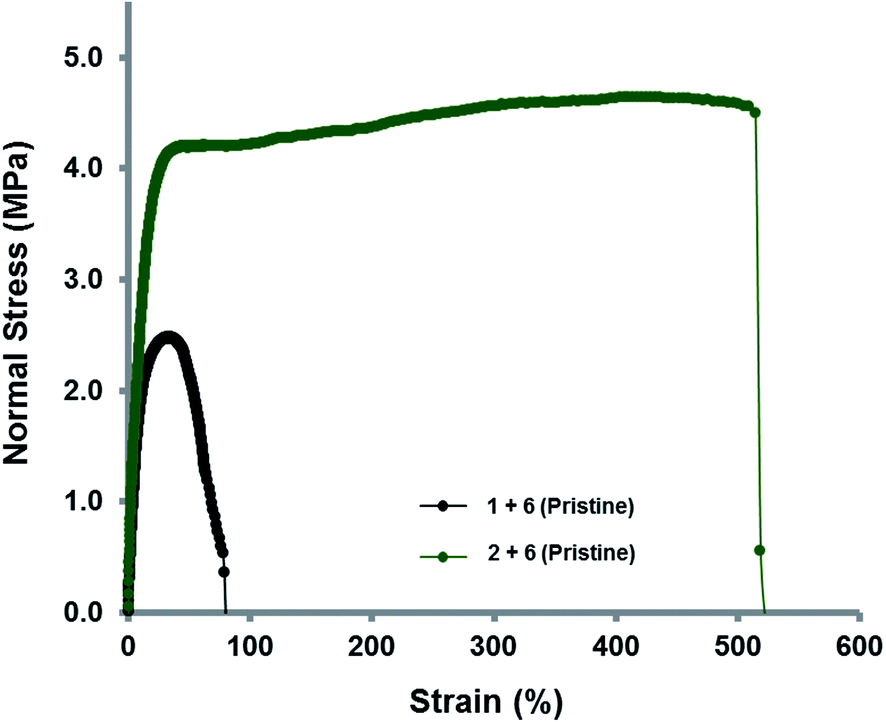 | ||
| Fig. 8 Stress–strain curves for supramolecular polymer blends of polydiimide 6 with the divalent pyrenyl-polymer 1 and trivalent pyrenyl-polymer 2. | ||
Conclusions
The properties of supramolecular polymer blends in which the two components associate through complementary aromatic π–π stacking interactions can be controlled by varying the valency of one of the components, leading to a clear relationship between the supramolecular “cross-link density” of the blend and its solution and solid state properties. Spectroscopic analyses of the divalent and trivalent polymer blends enabled the density of π–π stacking interactions within the supramolecular polymer network to be correlated with the change in valency. Robust, elastomeric films were successfully cast from these blends, and tensometric measurements showed a dramatic enhancement of both tensile modulus and toughness for the trivalent over the divalent blend, whilst maintaining comparable thermal healability.Experimental
Materials
Reagents and solvents were purchased from Sigma Aldrich and were used without further purification, with the exception of dichloromethane which was dried by distillation from calcium hydride under argon.Instrumentation
Proton NMR (400 MHz) and 13C NMR (100 MHz) spectra were obtained on a Bruker Nanobay 400 spectrometer using CDCl3 or CDCl3–trifluoroacetic acid (9:1 v/v) as solvent, with TMS as internal standard. Infrared (IR) spectroscopic analysis was carried out using a Perkin Elmer 100 FT-IR instrument with diamond-ATR sampling accessory and samples either as solids or oils. Ultraviolet-visible spectra were measured with a Varian Cary 300 spectrophotometer with heating attachment, using 1 cm2 quartz cuvettes, in the wavelength range 350–800 nm. Fluorescence spectroscopy was carried out in chloroform using a Varian Cary Eclipse fluorescence spectrophotometer and a 1 cm2 quartz cuvette, exciting at 345 nm and recording emissions in the wavelength range 350–700 nm. Matrix-assisted laser desorption–ionization time-of-flight mass spectra (MALDI-TOF MS) were obtained using a Bruker Daltonics Ultraflex 1 spectrometer operating in reflection mode. The instrument was calibrated using a standard peptide mixture (Bruker Daltonics, Calibration standard II), and 2,5-dihydroxybenzoic acid (DHB) was used as the matrix. A typical method of sample preparation follows: 3 μL of a solution of the analyte in acetonitrile (5 mg mL−1) was combined with 3 μL of the freshly prepared matrix (20 mg mL−1 in 1:1 acetonitrile–water with 1% TFA). An aliquot (1 μL) was then spotted onto a ground steel MALDI target plate and left to dry in air prior to analysis. Gel permeation chromatography (GPC) analysis was conducted using a Varian PL-GPC 220 chromatograph with poly(ethylene glycol) standards as calibrants. Samples for GPC were prepared in a solution of 0.01 M lithium bromide in N,N′-dimethylformamide, which was also used as the eluent. Inherent viscosities were measured in chloroform–hexafluoroisopropanol (9:1, v/v) on a Schott-Geräte CT-52 auto-viscometer using a size 03 Ubbelohde capillary in a thermostatted water bath at 25 °C. Differential scanning calorimetry (DSC) was carried out using a TA Instruments Q2000 calorimeter. Samples for DSC were heated to 110 °C to remove residual solvent, cooled to −90 °C, and then re-scanned from −90 to 250 °C. Small angle X-ray scattering (SAXS) data were collected using a Bruker Nanostar instrument with an Incoatec microfocus X-ray source operating at 45 kV. Scattering patterns were collected using a Vantec area detector (2048 × 2048 pixels) using silver behenate as a calibrant. Environmental Scanning Electron Microscope (ESEM) images were recorded with a FEI Quanta FEG 600 ESEM equipped with a hot stage. Dynamic mechanical analysis was carried out using a TA Instruments RSA III at 23 °C with a Hencky strain rate of 0.1 s−1. Samples were 0.20–0.3 mm in thickness, cut to dimensions of approximately 4 × 40 mm. Uniform films with a thickness from 0.2–0.3 mm were elongated to breaking, and the edges of the broken samples were then overlapped and pressed slightly on a pre-heated PTFE plate and healed in an oven at 100 °C for 30 minutes (divalent polymer blend) or 160 °C for 60 minutes (trivalent polymer blend). Broken samples were also left for specified times between 1 day and 1 week before testing their healing ability.
Preparation of pyrenyl end-capped divalent PEG (1)
Under an inert nitrogen atmosphere at 0 °C, poly(ethylene glycol) bis(3-amino propyl) 4 (1.50 g, 1.0 mmol) and 4-dimethylaminopyridine (0.05 g, 0.4 mmol) were mixed in dry dichloromethane (30 mL). Pyrene butyric acid 3 (0.69 g, 2.4 mmol) was added and stirred for 20 minutes at 0 °C, followed by dicyclohexylcarbodiimide (0.50 g, 2.4 mmol). The suspension was left to warm to room temperature and subsequently stirred for 18 hours before being cooled and filtered. The product was precipitated in diethyl ether (300 mL) in a dry ice/acetone bath and filtered whilst cold to yield 1 as an orange-brown oil (2.10 g, 92%); IR (ATR) v/cm−1 3319, 2927, 1636, 1552, 1410, 1234; 1H NMR (400 MHz/CDCl3) δ ppm: 8.30–7.86 (18H, m), 6.27 (2H, t, J = 6.35), 3.6–3.60 (148H, br), 3.39 (8H, q, J = 7.0 Hz), 2.28 (8H, m), 1.75 (4H, q, J = 6.0 Hz); 13C NMR (100 MHz/CDCl3) δ ppm: 172.6, 136.1, 131.4, 130.9, 129.9, 128.7, 127.5, 127.4, 127.3, 126.6, 125.8, 125.0, 124.9, 124.9, 124.8, 124.7, 123.5, 76.8, 70.5, 70.5, 70.4, 70.3, 70.1, 70.0, 69.1, 63.5, 53.5, 38.0, 36.1, 33.8, 32.8, 32.7, 28.9, 27.5, 26.8; (DMF/LiBr, 0.1 M) Mn = 1331 Da, Mw = 1485 Da, PDI = 1.12; MALDI-TOF MS (m/z) calc. = 2082.18 Da; found = 2082.17 Da ± 44n Da.Preparation of pyrenyl end-capped trivalent PEG (2)
Under an inert nitrogen atmosphere, glycerol ethoxylate 5 (3.47 g, 3.47 mmol) and 4-dimethylaminopyridine (1.27 g, 10.4 mmol) were dissolved in dry dichloromethane (100 mL) in an ice bath. Pyrene butyric acid 3 (3.6 g, 12.5 mmol) was added and the mixture was allowed to stir for 20 minutes. Dicyclohexylcarbodiimide (2.58 g, 12.5 mmol) was added whilst at 0 °C and the solution was allowed to warm to room temperature, before stirring for 18 hours. The solution was then cooled in ice, filtered and concentrated in vacuo. The concentrate was then precipitated into diethyl ether (350 mL) in a dry ice/acetone bath, and filtered whilst cold. The crude material was further purified by column chromatography (gradient chloroform–methanol 35:1, to 25:1). to yield 2 as a pure orange-brown oil (5.58 g, 90%); IR (ATR) v/cm−1 2871, 1732, 1102, 844; 1H NMR (400 MHz/CDCl3) δ ppm: 8.27–7.83 (27H, m), 4.24 (6H, t, J = 5.0 Hz), 3.67–3.48 (104H, br), 3.37 (6H, t, J = 7.5 Hz), 2.48 (6H, t, J = 7.0 Hz), 2.18 (6H, quin, J = 7.5 Hz); 13C NMR (100 MHz/CDCl3) δ ppm: 173.4, 135.7, 131.4, 130.9, 130.0, 128.7, 127.4, 126.7, 125.9, 125.1, 125.0, 124.9, 124.8, 124.7, 123.3, 78.4, 71.3, 70.8, 69.8, 69.2, 63.5, 33.8, 32.7, 26.8; GPC (DMF/LiBr, 0.1 M) Mn = 705 Da, Mw = 748 Da, PDI = 1.06; MALDI-TOF MS (m/z) calc. = 2026.00 Da, found = 2025.75 Da ± 44n Da.
Acknowledgements
We thank EPSRC and Domino Printing Sciences UK Ltd. for a studentship in support of LRH, and EPRSC for support of BWG (grants EP/G026203/1 and EP/D07434711). Funding for the healing studies at the University of Delaware was provided from the Department of Materials Science and Engineering, The College of Engineering, NIST, and the Center for Neutron Science through award 70NANOBIOH256. Spectroscopic and thermal data were acquired using equipment in the Chemical Analysis Facility (CAF) of the University of Reading. The authors are also grateful to the Electron Microscopy Laboratory (EMLab) of the University of Reading for providing access to the ESEM microscope. We also wish thank Mr N. Spencer of the Department of Chemistry, University of Reading for technical assistance with SAXS analysis.References
- E. B. Murphy and F. Wudl, Prog. Polym. Sci., 2010, 35, 223–251 CrossRef CAS.
- S. Burattini, B. W. Greenland, D. Chappell, H. M. Colquhoun and W. Hayes, Chem. Soc. Rev., 2010, 39, 1973–1985 RSC.
- S. D. Bergman and F. Wudl, J. Mater. Chem., 2008, 18, 41–62 RSC.
- R. P. Wool, Soft Matter, 2008, 4, 400–418 RSC.
- S. Burattini, H. M. Colquhoun, B. W. Greenland and W. Hayes, in Supramolecular Chemistry: Molecules to Nanomaterials, ed. J. W. Steed and P. A. Gale, John Wiley & Sons Ltd, Chichester, 2012 Search PubMed.
- W. Hayes and B. W. Greenland, Healable Polymer Systems, Royal Society of Chemistry Publishing, Cambridge, 1st edn, 2013 Search PubMed.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- D. Döhler, P. Michael and W. H. Binder, in Self-Healing Polymers: From Principles to Applications, ed. W. H. Binder, Wiley-VCH Verlag GmbH & Co, Weinheim, 2013 Search PubMed.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- S. H. Cho, H. M. Andersson, S. R. White, N. R. Sottos and P. V. Braun, Adv. Mater., 2006, 18, 997–1000 CrossRef CAS.
- M. W. Keller, S. R. White and N. R. Sottos, Adv. Funct. Mater., 2007, 17, 2399–2404 CrossRef CAS.
- K. Toohey, N. R. Sottos and J. Lewis, Nat. Mater., 2007, 6, 581–585 CrossRef CAS PubMed.
- M. M. Caruso, B. J. Blaiszik, S. R. White, N. R. Sottos and J. S. Moore, Adv. Funct. Mater., 2008, 18, 1898–1904 CrossRef CAS.
- K. S. Toohey, C. J. Hansen, J. A. Lewis, S. R. White and N. R. Sottos, Adv. Funct. Mater., 2009, 19, 1399–1405 CrossRef CAS.
- X. Chen, F. Wudl, A. K. Mal, H. Shen and S. R. Nutt, Macromolecules, 2003, 36, 1802–1807 CrossRef CAS.
- B. J. Adzima, C. J. Kloxin and C. N. Bowman, Adv. Mater., 2010, 22, 2784–2787 CrossRef CAS PubMed.
- Y.-L. Liu and C.-Y. Hsieh, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 905–913 CrossRef CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- S. Burattini, H. M. Colquhoun, J. D. Fox, D. Friedmann, B. W. Greenland, P. J. F. Harris, W. Hayes, M. E. Mackay and S. J. Rowan, Chem. Commun., 2009, 6717–6719 RSC.
- P. Zheng and T. J. McCarthy, J. Am. Chem. Soc., 2012, 134, 2024–2027 CrossRef CAS PubMed.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–338 CrossRef CAS PubMed.
- C.-M. Chung, Y.-S. Roh, S.-Y. Cho and J.-G. Kim, Chem. Mater., 2004, 16, 3982–3984 CrossRef CAS.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 1660–1663 CrossRef CAS PubMed.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444–2450 CrossRef CAS PubMed.
- B. Ghosh and M. Urban, Science, 2009, 323, 1458–1460 CrossRef CAS PubMed.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic, L. Leibler and C. Soulie, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- K. Haraguchi, K. Uyama and H. Tanimoto, Macromol. Rapid Commun., 2011, 32, 1253–1258 CrossRef CAS PubMed.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- M. Capelot, D. Montarnal, L. Leibler and F. Tournilhac, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- M. J. Barthel, T. Rudolph, A. Teichler, R. M. Paulus, J. Vitz, S. Hoeppener, M. D. Hager, F. H. Schacher and U. S. Schubert, Adv. Funct. Mater., 2013, 23, 4921–4932 CrossRef CAS.
- J. Kötteritzsch, S. Stumpf, S. Hoeppener, J. Vitz, M. D. Hager and U. S. Schubert, Macromol. Chem. Phys., 2013, 214, 1636–1649 CrossRef.
- J. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons Ltd, Chichester, 2nd edn, 2009, vol. 27 Search PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. Folmer, J. H. Hirschberg, R. F. Lange, J. K. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- J.-M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS PubMed.
- L. Brunsveld, B. J. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed.
- J.-M. Lehn, Polym. Int., 2002, 51, 825–839 CrossRef CAS.
- J. Emsley, Prog. Inorg. Chem., 1980, 9, 91–124 CAS.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- C. A. Hunter, K. R. Lawson, J. Perkins and C. J. Urch, J. Chem. Soc., Perkin Trans. 2, 2001, 651–669 RSC.
- C. G. Claessens and J. F. Stoddart, J. Phys. Org. Chem., 1997, 10, 254–272 CrossRef CAS.
- R. Quesada and P. A. Gale, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2005, 101, 148–170 RSC.
- X. Zhang and C. Wang, Chem. Soc. Rev., 2010, 40, 94–101 RSC.
- L. R. Hart, J. L. Harries, B. W. Greenland, H. M. Colquhoun and W. Hayes, Polym. Chem., 2013, 4, 4860–4870 RSC.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467–472 CrossRef CAS PubMed.
- R. P. Sijbesma and E. W. Meijer, Mater. Today, 2004, 7, 34–39 Search PubMed.
- A. W. Bosman, in Proceedings of the First International Conference on Self Healing Materials, 2007, pp. 1–6 Search PubMed.
- B. J. B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. Van Der Rijt and E. W. Meijer, Adv. Mater., 2000, 12, 874–878 CrossRef CAS.
- F. Herbst, S. Seiffert and W. H. Binder, Polym. Chem., 2012, 3, 3084–3092 RSC.
- G. L. Fiore, S. J. Rowan and C. Weder, Chim. Int. J. Chem., 2011, 65, 745 CrossRef CAS.
- S. Bode, L. Zedler, F. H. Schacher, B. Dietzek, M. Schmitt, J. Popp, M. D. Hager and U. S. Schubert, Adv. Mater., 2013, 25, 1634–1638 CrossRef CAS PubMed.
- S. Bode, R. K. Bose, S. Matthes, M. Ehrhardt, A. Seifert, F. H. Schacher, R. M. Paulus, S. Stumpf, B. Sandmann, J. Vitz, A. Winter, S. Hoeppener, S. J. Garcia, S. Spange, S. van der Zwaag, M. D. Hager and U. S. Schubert, Polym. Chem., 2013, 4, 4966–4973 RSC.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS PubMed.
- B. W. Greenland, S. Burattini, W. Hayes and H. M. Colquhoun, Tetrahedron, 2008, 64, 8346–8354 CrossRef CAS.
- S. Burattini, H. M. Colquhoun, B. W. Greenland and W. Hayes, Faraday Discuss., 2009, 143, 251–264 RSC.
- S. Burattini, B. W. Greenland, W. Hayes, M. E. Mackay, S. J. Rowan and H. M. Colquhoun, Chem. Mater., 2011, 23, 6–8 CrossRef CAS.
- J. Fox, J. J. Wie, B. W. Greenland, S. Burattini, W. Hayes, H. M. Colquhoun, M. E. Mackay and S. J. Rowan, J. Am. Chem. Soc., 2012, 134, 5362–5368 CrossRef CAS PubMed.
- R. Vaiyapuri, B. W. Greenland, H. M. Colquhoun, J. M. Elliott and W. Hayes, Polym. Chem., 2013, 4, 4902–4909 RSC.
- H. M. Colquhoun and Z. Zhu, Angew. Chem., Int. Ed., 2004, 43, 5040–5045 CrossRef CAS PubMed.
- H. M. Colquhoun, Z. Zhu, C. J. Cardin and Y. Gan, Chem. Commun., 2004, 2650–2652 RSC.
- H. M. Colquhoun, Z. Zhu, C. J. Cardin, Y. Gan and M. G. B. Drew, J. Am. Chem. Soc., 2007, 129, 16163–16174 CrossRef CAS PubMed.
- H. M. Colquhoun, Z. Zhu, C. J. Cardin, M. G. B. Drew and Y. Gan, Faraday Discuss., 2009, 143, 205–220 RSC.
- Z. Zhu, C. J. Cardin, Y. Gan and H. M. Colquhoun, Nat. Chem., 2010, 2, 653–660 CrossRef PubMed.
- B. W. Greenland, M. B. Bird, S. Burattini, R. Cramer, R. K. O'Reilly, J. P. Patterson, W. Hayes, C. J. Cardin and H. M. Colquhoun, Chem. Commun., 2013, 49, 454–456 RSC.
- A. Mulder, J. Huskens and D. N. Reinhoudt, Org. Biomol. Chem., 2004, 2, 3409–3424 CAS.
- I. Gitsov and J. M. J. Fréchet, J. Am. Chem. Soc., 1996, 118, 3785–3786 CrossRef CAS.
- F. Winnik, Chem. Rev., 1993, 93, 587–614 CrossRef CAS.
- A. Charlesby and N. H. Hancock, Proc. R. Soc. London, Ser. A, 1953, 218, 245–255 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4py00292j |
| This journal is © The Royal Society of Chemistry 2014 |