 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploiting end group functionalization for the design of antifouling bioactive brushes†
A. R.
Kuzmyn
a,
A.
de los Santos Pereira
a,
O.
Pop-Georgievski
a,
M.
Bruns
b,
E.
Brynda
a and
C.
Rodriguez-Emmenegger
*a
aInstitute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic, v.v.i., Heyrovsky sq. 2, 162 06 Prague, Czech Republic. E-mail: rodriguez@imc.cas.cz
bInstitute for Applied Materials and Karlsruhe Nano Micro Facility (KNMF), Karlsruhe Institute of Technology (KIT), 76344 Eggenstein-Leopoldshafen, Germany
First published on 21st March 2014
Abstract
Biologically active surfaces are essential in many applications in the fields of biosensing, bioimplants, and tissue engineering. However, the introduction of bioactive motifs without impairment of their ability to resist non-specific interactions with biological media remains a challenge. Herein, we present a straightforward, facile strategy for the creation of bioactive surfaces based on the end-group biofunctionalization of state-of-the-art polymer brushes via an ultra-fast Diels–Alder “click” reaction. Surface-initiated atom transfer radical polymerization is employed to grow antifouling polymers preserving the end groups. These groups are then further converted to a reactive cyclopentadienyl moiety and exploited for the immobilization of biomolecules on the topmost layer of the brush. The minimal chemical modification of the antifouling polymer brush accounts for the full preservation of the fouling resistance of the surface even after biofunctionalization, which is critical for the aforementioned applications.
Introduction
Biologically active surfaces are crucial for in vitro and in vivo applications, such as biomedical devices, label-free biosensing and tissue engineering.1 Bioactive surfaces refer to surfaces with immobilized bioactive molecules aimed specifically at promoting or supporting particular biological functions.2 Simultaneously, they must be able to prevent any unwanted adsorption or deposition of material from the media in which they operate. Complex biological media, in particular blood plasma and bodily fluids, cause non-specific adsorption on virtually any artificial surface with which they are contacted. This phenomenon, which is referred to as fouling, leads to the impairment of performance and the loss of function of biomedical devices and biosensors as well as any surface-based material in contact with biological fluids.2,3 Critical examples of the detrimental effects of fouling encompass the non-specific protein adsorption on bioimplants, which triggers foreign body response leading to rejection, a high non-specific signal in biosensors, which precludes label-free measurements in biological matrices, and the reduced blood circulation time of nanocarriers.2,4–7Therefore, the creation of bioactive surfaces requires the use of antifouling layers capable of preventing non-specific adsorption from biological media. Simultaneously, a means of attaching bioactive molecules to the surface must be provided without impairing the antifouling properties.2 Several different strategies for the design and preparation of antifouling surfaces have been applied, namely functional self-assembled monolayers (SAMs),8–10 “grafted-to” polymer layers,9,11–14 and polymer brushes.15–20 The much acclaimed antifouling SAMs have been widely used for the introduction of functional groups to surfaces.3,21 In particular mixed SAMs containing oligo(ethylene glycol)-terminated alkanethiolates are able to decrease adsorption from model solutions of single proteins.22,23 The direct end-grafting of poly(ethylene glycol) to a surface has also been exploited with moderate success for reducing fouling.6,8 Nevertheless, both of these approaches fail when contacted with complex biological matrices such as undiluted blood plasma and serum.9,24 The advances in controlled radical polymerization reactions have permitted the tailoring of surfaces, enabling access to unmatched antifouling properties,18 the precise control of cell adhesion,17,25 highly stable nanocapsules even in complex biological media,7 hierarchically structured polymer coatings,26 and modification of non-reactive substrates.27,28 Even though atom transfer radical polymerization (ATRP) has been the most widely employed method for the growth of antifouling polymers,9 approaches based on radical addition–fragmentation chain transfer (RAFT)16 and single electron transfer living radical polymerization reactions (SET-LRP)29 have also been introduced.
However, even with all of these remarkable advances, the biofunctionalization of antifouling polymer brushes still poses serious challenges as it generally affects their structure impairing their resistance to fouling.30 Due to their chemical inertness it is generally necessary to properly activate or modify them prior to the attachment of bioactive molecules.2 There are three main strategies for the postpolymerization biofunctionalization of polymer brushes: side chain modification, diblock copolymer brushes with only the upper block being activated, and chain end modification.31 Typically, the first two methods of biofunctionalization have been carried out by activating functional groups of the side chains, present in a very high amount along all or part of the length of the polymer chain, to activate esters or carbonates.4,28,30 These activated groups are then capable of reacting with groups available on biomolecules, leading to large quantities of immobilized molecules. However, the introduction of a large number of activated groups results in irreversible changes in the structure of the brush due to the changes in the hydrodynamic properties of the polymer chains, crosslinking of lateral chains, and steric hindrance leading to concomitant impairment of the antifouling properties.2,30 Furthermore, the activity of the biomolecule may be severely impaired upon immobilization as parts of it may penetrate into the polymer brush or the formation of too many new bonds damages its structure. Until now, these problems preclude the application of bioactive surfaces as platforms for the creation of biosensors.30
In contrast, chain end modification is a more promising alternative, since it would only minimally alter the brush itself. On the other hand, this comparatively little explored approach poses serious challenges on the chemical protocols applicable for effective biofunctionalization. For the immobilization reaction to occur, the biomacromolecule needs to approach the surface and remain there for a sufficient time until it collides with an active end-group. However, the exposure to end-groups of antifouling polymer brushes can be very limited as biomacromolecules are repelled and the end-groups are mobile and can be buried in a thermal blob of the brush.32,33 Thus, for a reaction to be suitable for the end-group biofunctionalization of polymer brushes, it must be ultra-rapid and proceed under mild conditions that, as highlighted before, do not impair the structure and properties of the antifouling layer. Additionally, it should be bioorthogonal, modular, and not require a catalyst or create unwanted products. Classical approaches of biofunctionalization of the end-group of antifouling polymer brushes, such as active ester or amidation, usually failed due to the harsh reaction conditions, slow reaction rate, and chemical instability of the reactive intermediates.34 Less commonly used techniques of biofunctionalization proceed faster under milder conditions but generally require catalysis by transition metals or irradiation.35 Several new ligation protocols have been introduced for the conjugation of biomolecules and polymers and the functionalization of SAMs. Various techniques such as the Cu-catalyzed alkyne–azide cycloaddition,36,37 the thiol–ene click reaction,38–40 and the strain-promoted alkyne–azide cycloaddition41 have been demonstrated for functionalization of surfaces. However, the application of such reactions for the biofunctionalization of antifouling polymer brushes has not been possible probably due to the higher complexity of these systems. Thus, there is a great need for a new, efficient, simple method which will allow the conjugation of bioactive molecules onto antifouling layers based on polymer brushes while simultaneously preserving the functions of both.
Previously, (hetero) Diels–Alder ((H)DA) reactions were utilized for clicking polymers and the conjugation of low molecular weight polymers and peptides to surfaces.42–50
It has been shown that a judicious selection of the diene allows these reactions to be efficiently performed using water as the reaction medium with even higher reaction rates43,51 and enhancement of the selectivity.52 Recently, a new type of ultra-fast DA “click” chemistry protocol was introduced by the group of Barner-Kowollik.53–56 It takes place between cyclopentadiene (Cp) moieties and reactive dienophiles, such as maleimide.53–55 In particular, when the dienophile is capable of accepting hydrogen bonds, the reaction rate is enhanced. Thus, the ultra-fast maleimide–cyclopentadiene cycloaddition shows several distinct advantages over previously reported ligation protocols, as it proceeds in drastically shorter reaction times at room temperature, in water-based media, and does not require a metal catalyst or irradiation. These features make the maleimide–cyclopentadiene “click” reaction a prime candidate for the biofunctionalization of antifouling polymer brushes.
In order to tackle the severe challenges involved in the end-group biofunctionalization of antifouling polymer brushes we exploited the ultra-fast DA “click” reaction between Cp and maleimide. The bromine terminus of brushes of poly[oligo(ethylene glycol) methyl ether methacrylate], grown by surface-initiated atom transfer radical polymerization (SI-ATRP), was substituted by Cp utilizing nickelocene (NiCp2). The highly reactive Cp units were utilized for the immobilization of a maleimide-conjugated protein. This constitutes the first example of such application of this reaction. In sharp contrast to other procedures previously presented, we demonstrate the full preservation of the original antifouling properties of the polymer brush after functionalization while achieving a similar level of immobilization. This brings the ultimate goal of achieving a simple, modular platform for the creation of bioactive surfaces within reach.
Materials and methods
Materials
All chemical reagents were used without further purification. Oligo(ethylene glycol) methyl ether methacrylate (Mn = 300 g mol−1, MeOEGMA), CuBr (99.999%), CuBr2 (99.999%), 2,2′-dipyridyl (BiPy), 2-bromoisobutyryl bromide, 11-mercapto-1-undecanol, α-maleimide-ω-methoxy-(polyethylene glycol) (Mn = 5000 g mol−1), α-hydroxy-ω-methoxy-poly(ethylene glycol) (Mn = 5000 g mol−1), bis(cyclopentadienyl) nickel(II) (nickelocene, NiCp2), triphenylphosphine (99%), and sodium iodide were purchased from Sigma-Aldrich. ATRP initiator ω-mercaptoundecyl bromoisobutyrate was synthesized by reacting α-bromoisobutyryl bromide with 11-mercapto-1-undecanol according to the method published previously.57 Blood plasma was kindly provided by the Institute of Hematology and Blood Transfusion, Prague, Czech Republic. Maleimide-conjugated bovine serum albumin (BSA-M) and bovine serum albumin (BSA) were purchased from Sigma-Aldrich, and anti-albumin rabbit polyclonal antibody from ThermoScientific.Preparation of the self-assembled monolayers of the ATRP initiator
The substrates used were gold-coated silicon wafers for ellipsometry and X-ray Photoelectron Spectroscopy (XPS) or gold-coated glass chips for other measurements. The gold-coated substrates were rinsed with ethanol and deionized water, dried with nitrogen, cleaned in a UV–ozone cleaner (Jelight) for 20 min, and immediately immersed in a 1 mM ethanolic solution of ω-mercaptoundecyl bromoisobutyrate (SAM-Br) overnight at room temperature.Preparation of bromine-ended MeOEGMA polymer brushes by SI-ATRP
Polymerization was carried out according to our modified procedure published previously.26 A solution of MeOEGMA (13.65 g, 45.5 mmol) in water (12 mL) was degassed by bubbling Ar for 1 h, separately from the solid compounds, CuBr2 (24.3 mg, 0.18 mmol), CuBr (37 mg 0.9 mmol) and 2,2-dipyridyl (435 mg, 2.4 mmol), and methanol (12 mL). The methanol and the monomer solutions were transferred under Ar-protection to the flask containing the degassed solid components and the polymerization mixture was stirred under an inert atmosphere until full dissolution. The polymerization solution was transferred under an Ar atmosphere to the degassed reactors containing the initiator-SAM-coated substrates. The polymerization was allowed to proceed at 30 °C and was stopped by removing the substrates and rinsing them with copious amounts of ethanol and water. The chips were stored in deionized water.Synthesis of Cp-ended poly(MeOEGMA) brushes and SAM
The procedure was carried out according to the modified method published previously.53 In a glove-box, poly(MeOEGMA)-Br brushes with different thicknesses and SAM-Br were individually placed in sealable reactors to each of which a solution containing NiCp2 (94.4 mg, 0.5 mmol), triphenylphosphine (65.5 mg, 0.25 mmol), and sodium iodide (112.4 mg, 0.75 mmol) in dry THF (5 mL) was added. The reactors were subsequently sealed, removed from the glove-box and gently agitated for 14 h. After this time the reactors were opened and the substrates were removed and rinsed with copious amounts of dry THF, methanol, and water.Surface plasmon resonance (SPR)
A custom-built SPR instrument (Institute of Photonics and Electronics, Academy of Sciences of the Czech Republic, Prague) based on the Kretschmann geometry of the attenuated total reflection method and spectral interrogation of the SPR conditions was used.8 The tested solutions were driven using a peristaltic pump through four independent channels of a flow cell in which the SPR responses were simultaneously measured as shifts in the resonant wavelength, λres. The sensor response (Δλres) was obtained as the difference between the baselines in phosphate buffered saline (PBS) before and after the injection of the tested samples. The sensor response was calibrated to the mass deposited at the surface of bound molecules. According to a calibration made by FTIR-GASR, a shift Δλres = 1 nm corresponds to a change in the deposited protein mass of 150 pg mm−2.6,9Immobilization of bovine serum albumin
The specific immobilization of BSA-M onto the Cp-groups at the end of the polymer brush was monitored in real time via SPR spectroscopy. After obtaining a stable resonant wavelength, λres, in PBS (pH 7.4), a solution of 1 mg mL−1 of BSA-M in PBS was injected over the surface for 45 min with a flow rate of 5 μL min−1. As a control, on an independent channel, a solution of 5 mg mL−1 BSA in PBS was flowed simultaneously. Subsequently, the flow rate was increased to 25 μL min−1 and unbound proteins were washed with PBS for 30 min. A solution of anti-albumin rabbit polyclonal antibody (anti-BSA) in PBS (100 μg mL−1) was injected over the surface for 15 min at a pump flow rate of 25 μL min−1.Fouling measurements
Non-specific adsorption of proteins was monitored in real time by SPR spectroscopy. In a typical experiment, PBS was driven through the flow cell using a peristaltic pump until a stable baseline was achieved. After obtaining a stable resonant wavelength, λres, in PBS (pH 7.4), undiluted blood plasma was injected over the surface for 15 min after which it was replaced with PBS. The sensor response is the difference in the resonance wavelength of the surface plasmons in PBS, before and after contact of the surface with blood plasma.X-ray photoelectron spectroscopy (XPS)
XPS measurements were performed using a K-Alpha XPS spectrometer (ThermoFisher Scientific, East Grinstead, UK). All the samples were analyzed using a microfocused, monochromated Al Kα X-ray source (400 μm spot size). The kinetic energy of the electrons was measured using a 180° hemispherical energy analyzer operated in the constant analyzer energy mode (CAE) at 50 eV pass energy for elemental spectra. Data acquisition and processing using the Thermo Advantage software is described elsewhere.58 The spectra were fitted with one or more Voigt profiles (binding energy uncertainty: ±0.2 eV). The analyzer transmission function, Scofield sensitivity factors,59 and effective attenuation lengths (EALs) for photoelectrons were applied for quantification. EALs were calculated using the standard TPP-2M formalism.60 All spectra were referenced to the C1s peak of hydrocarbons at 285.0 eV binding energy controlled by means of the well-known photoelectron peaks of metallic Cu, Ag, and Au.Time-of-flight secondary ion mass spectrometry (ToF-SIMS)
ToF-SIMS was performed with a TOF.SIMS 5 instrument (ION-TOF GmbH, Münster, Germany) equipped with a Bi cluster liquid metal primary ion source and a non-linear time of flight analyzer. The Bi source was operated in ‘bunched’ mode providing 0.7 ns Bi1+ ion pulses at 25 keV energy and a lateral resolution of approximately 4 μm. The short pulse length allowed for high mass resolution to analyze the complex mass spectra of the immobilized organic layers. Negative polarity spectra were calibrated on the C−, C2−, and C3− peaks. Primary ion doses were kept below 1011 ions cm−2 (static SIMS limit).Results and discussion
The strategy employed for the creation of biofunctionalized antifouling surfaces consists of: synthesis of antifouling polymer brushes by SI-ATRP, introduction of Cp groups to the brushes, and coupling of a maleimide-conjugated protein to the surface (Scheme 1). The macromonomer chosen for the fabrication of the brushes was MeOEGMA due to the excellent resistance to fouling and non-specific cell adhesion that can be achieved.8,17,24 The use of SI-ATRP allows the preservation of the living chain-end groups after polymerization, which can be exploited for subsequent postpolymerization modification to highly reactive Cp-moeities. Finally, these groups were utilized for the ultra-fast Diels–Alder “click” reaction with BSA-M as a model biomolecule (Scheme 1). | ||
| Scheme 1 Schematic graphical representation of the DA “click” reaction for the end-group conjugation of biomolecules on a modified antifouling surface. | ||
Synthesis, modification and characterization of polymer brushes
Polymer brushes were grown from a SAM of initiator ω-mercaptoundecyl bromoisobutyrate on a gold surface (Scheme 2). The polymerization procedure was optimized using a catalyst system based on copper bromide with copper(II) bromide as the added deactivator, without the use of halogen exchange to chloride, which is typically employed to facilitate the control of ATRP.26,61 Importantly, this enabled the Cp-substitution to proceed faster, as the polymer chains are capped with bromine, a better leaving group than chlorine. No sacrificial initiator was added in solution, as this would lead to additional termination events which must be avoided.62 The polymerization kinetics were followed by measuring the dry thickness of the polymer after different reaction times by ellipsometry. As evidenced by the linear increase of thickness with time, the SI-ATRP of poly(MeOEGMA) brushes proceeded in a well-controlled fashion (ESI†). This indicates that termination reactions were minimized, assuring a maximum amount of bromine end-groups after the polymerization, which are necessary for the effective introduction of Cp and further maleimide–cyclopentadiene “click” reaction. The chemical composition of the poly(MeOEGMA) brushes was confirmed by XPS, FTIR-GASR, and ToF-SIMS. XPS analysis of the C1s spectra of poly(MeOEGMA)-Br brushes shows a strong increase of the peaks assigned to [C–O] bonds at 286.4 eV, stemming from oligo(ethylene glycol) side chains of the brush and a decrease of the signal corresponding to [C–H, C–C] at 285.0 eV (Fig. 1a).17 The presence of strong bands at 1800 cm−1 (ester carbonyl), 1145 cm−1 (C–O–C stretching modes) and others in the FTIR-GASR spectrum (Fig. S3 in ESI†) further confirms successful polymerization. ToF-SIMS was utilized to assess the presence of the bromine end-groups, critical for the subsequent transformations (Fig. 1b). ToF-SIMS is a commonly utilized highly sensitive technique allowing access to the precise chemical composition of the topmost layer of ultra-thin films.63 In particular, the inherent isotopic pattern of bromine was used to unambiguously confirm its presence as an end-group in polymer brushes (Fig. 1). The morphology was studied by AFM, confirming that all surfaces were homogeneous and without pin-holes, and the water contact angles indicate a hydrophilic surface (ESI†). A smooth topography is usually associated with a better control in polymerization as well as a higher resistance to protein fouling and cell adhesion.64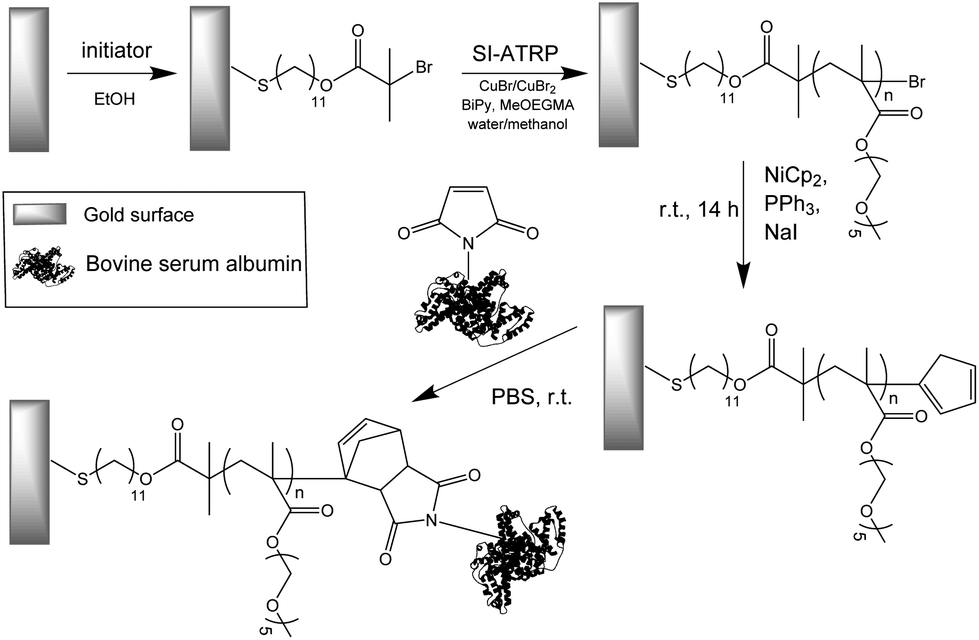 | ||
| Scheme 2 Synthetic route for biofunctionalization with maleimide-conjugated bovine serum albumin of antifouling poly(MeOEGMA) brushes grown via SI-ATRP from an initiator SAM and further transformation of bromine-end groups of the brushes to Cp. | ||
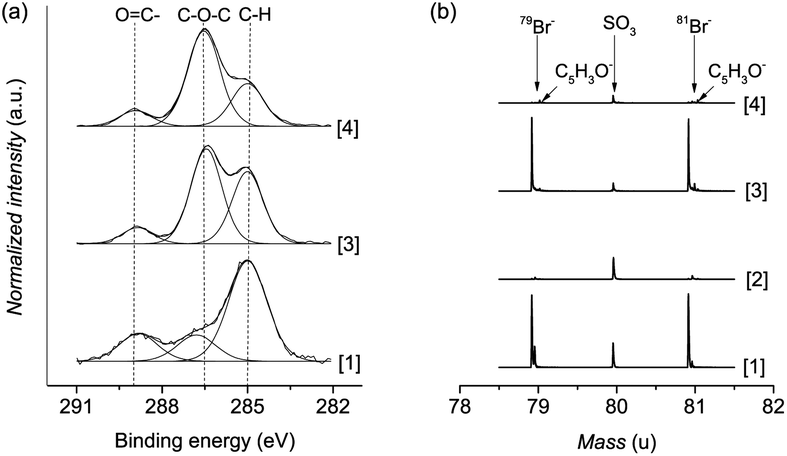 | ||
| Fig. 1 Results of high-resolution XPS spectra of the C1s region (a) and ToF-SIMS (b); [1] SAM of ω-mercaptoundecyl bromoisobutyrate (SAM-Br), [2] cyclopentadiene-capped SAM (SAM-Cp), [3] bromine-ended poly(MeOEGMA) brushes (poly(MeOEGMA)-Br), and [4] cyclopentadiene-ended poly(MeOEGMA) brushes (poly(MeOEGMA)-Cp). | ||
Transformation of the terminal group of polymer brushes with thicknesses of 10 nm, 20 nm, and 30 nm to the cyclopentadienyl moiety was carried out employing NiCp2 as the substituting agent by using a modified procedure reported previously.53,65 The rather covalent nature of the Ni–Cp bond makes it a milder reagent than the previously reported and much more ionic NaCp.66 Thus, a procedure based on NiCp2 is tolerant to functional groups such as esters and allows circumventing the various ill-desired side reactions encountered by the use of NaCp.65,66 The reaction was performed by immersing the corresponding surfaces in a solution of NiCp2, triphenylphosphine and sodium iodide in dry tetrahydrofuran for 14 hours (Scheme 2). It is worth noting that the addition of triphenylphosphine and sodium iodide as halogen metathesis reagents has been shown to enhance the alkylation reaction.67,68 Considering the challenges to detect end-groups in polymer brushes due to their low concentration and mobility, the Cp-substitution of the bromine groups was additionally performed directly on initiator-SAM-coated gold substrates. The FTIR-GASR spectra of SAM-Cp and poly(MeOEGMA)-Cp (ESI†) showed distinctive spectral features characteristic for the Cp distal end-group. The spectrum of SAM-Cp was characterized by a weak and broad band at about 1600 cm−1 arising from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C modes and a family of bands at 1570, 1310, 1257 and 1160 cm−1 characteristic for the C–H bending vibrations of the cyclopentadienyl ring.69,70 The spectrum of poly(MeOEGMA)-Cp is dominated by the strong contributions of the oligo(ethylene glycol) methyl ether side chains and the aliphatic main chain which overlap the contributions of the Cp distal end, making its identification impossible. However, in the region of 1650–1550 cm−1, which is free from any vibrations characteristic for the polymer brushes, the presence of Cp is evidenced as a weakly pronounced band at about 1600 cm−1. The success of the transformation was further corroborated by monitoring the disappearance of bromine in ToF-SIMS (Fig. 1). Comparison of the advancing water contact angles reveals a significant change after Cp-substitution, probably associated with a rearrangement of the hydrophobic end-groups in the topmost layer of the polymer brush. Conversely, only negligible differences were observed in the receding water contact angle, a more representative parameter of the conditions to which the brushes will be exposed, i.e. hydrated brushes. The unaltered wettability displayed suggests that the poly(MeOEGMA)-Cp and poly(MeOEGMA)-Br surfaces have similar surface energies, and a concomitant similar resistance to protein fouling. In addition, XPS (Fig. 1) and FTIR-GASR spectra confirmed that the chemical structure of the brushes was preserved after the modification. AFM and ellipsometry analyses further confirm that there were no changes in the homogeneity or thickness of the polymer brushes after the transformation (ESI†). The presence of residual quantities of Ni could pose severe disadvantages for various bioapplications. Importantly, no appreciable amount of Ni could be detected. The XPS survey spectrum of samples after the substitution of end-groups with Cp shows no peaks corresponding to Ni (ESI†). This was further confirmed by ToF-SIMS.
C modes and a family of bands at 1570, 1310, 1257 and 1160 cm−1 characteristic for the C–H bending vibrations of the cyclopentadienyl ring.69,70 The spectrum of poly(MeOEGMA)-Cp is dominated by the strong contributions of the oligo(ethylene glycol) methyl ether side chains and the aliphatic main chain which overlap the contributions of the Cp distal end, making its identification impossible. However, in the region of 1650–1550 cm−1, which is free from any vibrations characteristic for the polymer brushes, the presence of Cp is evidenced as a weakly pronounced band at about 1600 cm−1. The success of the transformation was further corroborated by monitoring the disappearance of bromine in ToF-SIMS (Fig. 1). Comparison of the advancing water contact angles reveals a significant change after Cp-substitution, probably associated with a rearrangement of the hydrophobic end-groups in the topmost layer of the polymer brush. Conversely, only negligible differences were observed in the receding water contact angle, a more representative parameter of the conditions to which the brushes will be exposed, i.e. hydrated brushes. The unaltered wettability displayed suggests that the poly(MeOEGMA)-Cp and poly(MeOEGMA)-Br surfaces have similar surface energies, and a concomitant similar resistance to protein fouling. In addition, XPS (Fig. 1) and FTIR-GASR spectra confirmed that the chemical structure of the brushes was preserved after the modification. AFM and ellipsometry analyses further confirm that there were no changes in the homogeneity or thickness of the polymer brushes after the transformation (ESI†). The presence of residual quantities of Ni could pose severe disadvantages for various bioapplications. Importantly, no appreciable amount of Ni could be detected. The XPS survey spectrum of samples after the substitution of end-groups with Cp shows no peaks corresponding to Ni (ESI†). This was further confirmed by ToF-SIMS.
It is worth noting that this surface modification can be easily extended to a vast variety of surfaces by the use of substrate-independent approaches previously reported provided the materials are compatible with the solvent required for the metathesis reaction.27,28
Biofunctionalization of poly(MeOEGMA)-Cp polymer brushes
To demonstrate the power of the novel ligation protocol for the design of bioactive surfaces we “clicked” a medium-sized protein – bovine serum albumin, BSA in phosphate buffered saline (PBS). This protein has a Mw of 65 kDa and an isoelectric point of 4.7, thus being an excellent model for bioactive compounds or bioreceptors. A 1 mg mL−1 solution of BSA-M in PBS buffer was injected over the previously prepared poly(MeOEGMA)-Cp brushes and the SPR spectroscopy (Fig. 2(a)) response was recorded. The extremely high sensitivity (3.0 pg mm−2) of the SPR allows access to the kinetics of immobilization of BSA-M onto the surface of poly(MeOEGMA)-Cp in real time. Fig. 2 shows the strikingly fast biofunctionalization of the brushes. Remarkably, contact times of less than 1 h resulted in levels of functionalization of 67% (838 pg mm−2), 25% (314 pg mm−2) and 12% (154 pg mm−2) of a monolayer of BSA8 for poly(MeOEGMA)-Cp brushes of 10, 20, and 30 nm thickness. The decreasing amount of BSA-M that can be immobilized with increasing thickness of the brushes can be explained by the increase of the mobility of the polymer chains with thickness, making the polymer end-groups more available for conjugation on thinner brushes.71 Immobilization of proteins can lead to impairment of their biological function. To assess whether the immobilized proteins retain their structures a solution of anti-BSA, capable of binding only to the preserved and accessible epitopes of BSA, was flowed over the surface (Fig. 2). The molar ratio of anti-BSA (Mw 150 kDa) to the attached BSA (Mw 65 kDa) increases as the overall amount of BSA decreases. This indicates that BSA becomes more available for recognition as its density decreases, suggesting that the immobilization occurs exclusively on the surface of the brush. | ||
| Fig. 2 Immobilization of BSA-M on poly(MeOEGMA)-Cp surfaces with three different thicknesses: 10 nm [1], 20 nm [2], and 30 nm [3]. Reference sensogram of adsorption from non-modified BSA on a 10 nm thick MeOEGMA-Cp surface [4]. Presented squares in the histogram below show the molar ratio between BSA and anti-BSA. | ||
Further confirmation of the presence and reactivity of the Cp-moieties on the surface was provided on a Cp-modified SAM, due to the greater ease of characterization by XPS and FTIR-GASR after clicking. However, since the clicking of proteins on a SAM would also result in non-specific adsorption, maleimide-conjugated poly(ethylene glycol) was used instead. The spectra show the specific attachment onto the surface of the Cp-capped SAM (ESI†).
Arguably, specificity and orthogonality are of central importance when selecting a biofunctionalization technique.39 The high specificity of the maleimide–Cp conjugation was evidenced by injecting non-modified BSA on poly(MeOEGMA)-Cp (Fig. 2) and by injecting BSA-M on poly(MeOEGMA)-Br (ESI†). SPR sensograms showed no immobilization or adsorption of BSA or BSA-M, as well as no attachment of anti-BSA confirming the high specificity of the maleimide–Cp bioconjugation (Fig. 2 and ESI†). Importantly, the prepared surfaces of poly(MeOEGMA)-Cp brushes showed high stability, preserving the reactivity towards bioconjugation with BSA-M even after 6 months of storage in deionized water (ESI†). The high stability of reactive poly(MeOEGMA)-Cp is of great importance when real applications are considered.
As referred to before, the design of antifouling biointerfaces not only requires high immobilization of bioreceptors but also an excellent resistance to fouling from complex biological media. Thus, the prepared polymer brushes were challenged by contacting the surfaces with undiluted human blood plasma (BP) and the fouling was assessed by SPR. Poly(MeOEGMA)-Br surfaces, before any modification, were able to reduce the fouling from BP by 97% (Fig. 3, (1), (4) and (7)) for the three thicknesses tested. The resistance of poly(MeOEGMA) brushes to fouling remained unchanged after modification with Cp, i.e. 160, 150, and 81 pg mm−2 for 10, 20 and 30 nm thick brushes (Fig. 3). This implies that the structure and conformation of the polymer brushes is fully preserved during this step, in agreement with the physico-chemical data presented above. Remarkably, while previous reports of similar antifouling polymer brushes resulted in a 4-fold increase in BP fouling after biofunctionalization,30 the resistance to BP remained unchanged in the current approach (Fig. 3 and ESI†). Moreover, the minute fouling observed for the poly(MeOEGMA)-Cp-BSA-anti-BSA was even lower than that observed for the much acclaimed ultra-low fouling poly(carboxybetaine)s after attachment of bioreceptors (ca. 190 pg cm−2).30,72
 | ||
| Fig. 3 Fouling from undiluted human blood plasma on bare gold (Au), poly(MeOEGMA)-Br of thickness 10 nm (1), 20 nm (4), 30 nm (7), poly(MeOEGMA)-Cp of thickness 10 nm (2), 20 nm (5), 30 nm (8), poly(MeOEGMA)-Cp after immobilization of BSA-M and capture of anti-BSA 10 nm (3), 20 nm (6), and 30 nm (9). (a) SPR sensogram, (b) irreversible fouling from BP obtained from SPR measurements. | ||
Thus, the strategy introduced herein is not only modular, but also very mild on the brush itself, fully preserving the antifouling characteristics of the original brush. Compared to other surface modifications where the properties of the brush were impaired after immobilization, the current approach paves the way for the creation of bioactive antifouling surfaces and biosensors which would be able to recognize analytes in different biological media, in particular undiluted blood plasma. In addition, due to the extremely fast reaction rate, it can be envisioned that this technique can be exploited for the patterning of proteins on surfaces by micro-contact printing, avoiding the problems of current approaches such as lateral diffusion or droplet evaporation.
Conclusions
In summary, we presented an efficient room-temperature conjugation strategy for the design of end-group-functionalized antifouling polymer brushes. The novel approach for the creation of bioactive surfaces is based on the Diels–Alder “click” reaction of maleimide-functionalized biomolecules with cyclopentadiene. The mild conditions utilized for the transformation of the polymer end-groups to cyclopentadiene ensured the preservation of the excellent antifouling properties of the original brush, as opposed to other commonly employed biofunctionalization techniques, which rely on extensive chemical modification of the polymer. Further conjugation with maleimide-conjugated BSA proceeded rapidly and was confirmed with a secondary antibody, without impairment of the fouling resistance. We envision that the strategy presented herein can be extended to applications such as biosensing, tissue engineering, and patterning of DNA and proteins arrays.Acknowledgements
This research was supported by the Grant Agency of the Czech Republic (GACR) under contract numbers: P205-12-1702 and P108-11-1857, by the project “BIOCEV – Biotechnology and Biomedicine Centre of the Academy of Sciences and Charles University” (CZ.1.05/1.1.00/02.0109), from the European Regional Development Fund, and by the Karlsruhe Nano Micro Facility (KNMF), a Helmholtz Research Infrastructure at KIT.Notes and references
- D. F. Williams, Biomaterials, 2009, 30, 5897–5909 CrossRef CAS PubMed
.
- Q. Yu, Y. Zhang, H. Wang, J. Brash and H. Chen, Acta Biomater., 2011, 7, 1550–1557 CrossRef CAS PubMed
- J. Homola, Chem. Rev., 2008, 108, 462–493 CrossRef CAS PubMed
- C. Rodriguez-Emmenegger, O. A. Avramenko, E. Brynda, J. Skvor and A. Bologna Alles, Biosens. Bioelectron., 2011, 26, 4545–4551 CrossRef CAS PubMed
- C. Blaszykowski, S. Sheikh and M. Thompson, Chem. Soc. Rev., 2012, 41, 5599–5612 RSC
- T. Riedel, Z. Riedelova-Reicheltova, P. Majek, C. Rodriguez-Emmenegger, M. Houska, J. E. Dyr and E. Brynda, Langmuir, 2013, 29, 3388–3397 CrossRef CAS PubMed
- C. Rodriguez-Emmenegger, A. Jäger, E. Jäger, P. Stepanek, A. B. Alles, S. S. Guterres, A. R. Pohlmann and E. Brynda, Colloids Surf., B, 2011, 83, 376–381 CrossRef CAS PubMed
- C. Rodriguez-Emmenegger, M. Houska, A. B. Alles and E. Brynda, Macromol. Biosci., 2012, 12, 1413–1422 CrossRef CAS PubMed
- C. Rodriguez-Emmenegger, E. Brynda, T. Riedel, Z. Sedlakova, M. Houska and A. B. Alles, Langmuir, 2009, 25, 6328–6333 CrossRef CAS PubMed
- K. Feldman, G. Hahner, N. D. Spencer, P. Harder and M. Grunze, J. Am. Chem. Soc., 1999, 121, 10134–10141 CrossRef CAS
- O. Pop-Georgievski, S. Popelka, M. Houska, D. Chvostova, V. Proks and F. Rypacek, Biomacromolecules, 2011, 12, 3232–3242 CrossRef CAS PubMed
- O. Pop-Georgievski, D. Verreault, M. O. Diesner, V. Proks, S. Heissler, F. Rypáček and P. Koelsch, Langmuir, 2012, 28, 14273–14283 CrossRef CAS PubMed
- S. I. Jeon and J. D. Andrade, J. Colloid Interface Sci., 1991, 142, 159–166 CrossRef CAS
- S. I. Jeon, J. H. Lee, J. D. Andrade and P. G. De Gennes, J. Colloid Interface Sci., 1991, 142, 149–158 CrossRef CAS
- G. Gunkel and W. T. Huck, J. Am. Chem. Soc., 2013, 135(18), 7047–7052 CrossRef CAS PubMed
- M. Zamfir, C. Rodriguez-Emmenegger, S. Bauer, L. Barner, A. Rosenhahn and C. Barner-Kowollik, J. Mater. Chem. B, 2013, 1, 6027–6034 RSC
- C. Rodriguez-Emmenegger, C. M. Preuss, B. Yameen, O. Pop-Georgievski, M. Bachmann, J. O. Mueller, M. Bruns, A. S. Goldmann, M. Bastmeyer and C. Barner-Kowollik, Adv. Mater., 2013, 25, 6123–6127 CrossRef CAS PubMed
- C. Rodriguez-Emmenegger, E. Brynda, T. Riedel, M. Houska, V. Šubr, A. Bologna Alles, E. Hasan, J. E. Gautrot and W. T. S. Huck, Macromol. Rapid Commun., 2011, 32, 952–957 CrossRef CAS PubMed
- S. Jiang and Z. Cao, Adv. Mater., 2010, 22, 920–932 CrossRef CAS PubMed
- H. W. Ma, M. Wells, T. P. Beebe and A. Chilkoti, Adv. Funct. Mater., 2006, 16, 640–648 CrossRef CAS
- H. Vaisocherová, K. Mrkvová, M. Piliarik, P. Jinoch, M. Šteinbachová and J. Homola, Biosens. Bioelectron., 2007, 22, 1020–1026 CrossRef PubMed
- K. Prime and G. Whitesides, Science, 1991, 252, 1164–1167 CrossRef CAS
- L. Li, S. Chen, J. Zheng, B. D. Ratner and S. Jiang, J. Phys. Chem. B, 2005, 109, 2934–2941 CrossRef CAS PubMed
- A. de los Santos Pereira, C. Rodriguez-Emmenegger, F. Surman, T. Riedel, A. Bologna and E. Brynda, RSC Adv., 2014, 4, 2318–2321 RSC
- A. Hucknall, S. Rangarajan and A. Chilkoti, Adv. Mater., 2009, 21, 2441–2446 CrossRef CAS
- C. Rodriguez-Emmenegger, E. Hasan, O. Pop-Georgievski, M. Houska, E. Brynda and A. Bologna Alles, Macromol. Biosci., 2011, 12, 525–532 CrossRef
- O. Pop-Georgievski, C. Rodriguez-Emmenegger, A. de los Santos Pereira, V. Proks, E. Brynda and F. Rypacek, J. Mater. Chem. B, 2013, 1, 2859–2867 RSC
- C. Rodriguez-Emmenegger, O. Kylian, M. Houska, E. Brynda, A. Artemenko, J. Kousal, A. Bologna Alles and H. Biederman, Biomacromolecules, 2011, 12, 1058–1066 CrossRef CAS PubMed
- U. Edlund, C. Rodriguez-Emmenegger, E. Brynda and A. C. Albersson, Polym. Chem., 2012, 3, 2920–2927 RSC
- H. Vaisocherova, V. Sevcu, P. Adam, B. Spackova, K. Hegnerova, A. de los Santos Pereira, C. Rodriguez-Emmenegger, T. Riedel, M. Houska, E. Brynda and J. Homola, Biosens. Bioelectron., 2014, 51, 150–157 CrossRef CAS PubMed
- R. Barbey, L. Lavanant, D. Paripovic, N. Schuwer, C. Sugnaux, S. Tugulu and H. A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS PubMed
- W. J. Brittain and S. Minko, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3505–3512 CrossRef CAS
- P. Gong, T. Wu, J. Genzer and I. Szleifer, Macromolecules, 2007, 40, 8765–8773 CrossRef CAS
- A. T. Nguyen, J. Baggerman, J. M. Paulusse, H. Zuilhof and C. J. van Rijn, Langmuir, 2012, 28, 604–610 CrossRef CAS PubMed
- O. Michel and B. J. Ravoo, Langmuir, 2008, 24, 12116–12118 CrossRef CAS PubMed
- X. Deng, C. Friedmann and J. Lahann, Angew. Chem., Int. Ed., 2011, 50, 6522–6526 CrossRef CAS PubMed
- F. Bally, K. Cheng, H. Nandivada, X. Deng, A. M. Ross, A. Panades and J. Lahann, ACS Appl. Mater. Interfaces, 2013, 5, 9262–9268 CAS
- P.-C. Lin, D. Weinrich and H. Waldmann, Macromol. Chem. Phys., 2010, 211, 136–144 CrossRef CAS
- Y.-X. Chen, G. Triola and H. Waldmann, Acc. Chem. Res., 2011, 44, 762–773 CrossRef CAS PubMed
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS
- A. Kuzmin, A. Poloukhtine, M. A. Wolfert and V. V. Popik, Bioconjugate Chem., 2010, 21, 2076–2085 CrossRef CAS PubMed
- M. Glassner, K. K. Oehlenschlaeger, T. Gruendling and C. Barner-Kowollik, Macromolecules, 2011, 44, 4681–4689 CrossRef CAS
- M. Glassner, G. Delaittre, M. Kaupp, J. P. Blinco and C. Barner-Kowollik, J. Am. Chem. Soc., 2012, 134, 7274–7277 CrossRef CAS PubMed
- M. Glassner, K. K. Oehlenschlaeger, A. Welle, M. Bruns and C. Barner-Kowollik, Chem. Commun., 2013, 49, 633–635 RSC
- K. K. Oehlenschlaeger, J. O. Mueller, N. B. Heine, M. Glassner, N. K. Guimard, G. Delaittre, F. G. Schmidt and C. Barner-Kowollik, Angew. Chem., Int. Ed. Engl., 2013, 52, 762–766 CrossRef CAS PubMed
- B. Yameen, C. Rodriguez-Emmenegger, I. Ahmed, C. M. Preuss, C. J. Durr, N. Zydziak, V. Trouillet, L. Fruk and C. Barner-Kowollik, Chem. Commun., 2013, 49, 6734–6736 RSC
- T. Tischer, T. K. Claus, M. Bruns, V. Trouillet, K. Linkert, C. Rodriguez-Emmenegger, A. S. Goldmann, S. Perrier, H. G. Borner and C. Barner-Kowollik, Biomacromolecules, 2013, 14, 4340–4350 CrossRef CAS PubMed
- T. Pauloehrl, A. J. Inglis and C. Barner-Kowollik, Adv. Mater., 2010, 22, 2788–2791 CrossRef CAS PubMed
- A. D. de Araújo, J. M. Palomo, J. Cramer, M. Köhn, H. Schröder, R. Wacker, C. Niemeyer, K. Alexandrov and H. Waldmann, Angew. Chem., Int. Ed., 2006, 45, 296–301 CrossRef PubMed
- C. M. Preuss, T. Tischer, C. Rodriguez-Emmenegger, M. M. Zieger, M. Bruns, A. S. Goldmann and C. Barner-Kowollik, J. Mater. Chem. B, 2014, 2, 36–40 RSC
- S. Otto and J. B. F. N. Engberts, Pure Appl. Chem., 2000, 72, 1365–1372 CrossRef CAS
- T. Rispens and J. B. F. N. Engberts, J. Phys. Org. Chem., 2005, 18, 725–736 CrossRef CAS
- J. P. Blinco, V. Trouillet, M. Bruns, P. Gerstel, H. Gliemann and C. Barner-Kowollik, Adv. Mater., 2011, 23, 4435–4439 CrossRef CAS PubMed
- C. M. Preuss, A. S. Goldmann, V. Trouillet, A. Walther and C. Barner-Kowollik, Macromol. Rapid Commun., 2013, 34, 640–644 CrossRef CAS PubMed
- A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2009, 48, 2411–2414 CrossRef CAS PubMed
- B. Yameen, C. Rodriguez-Emmenegger, C. M. Preuss, O. Pop-Georgievski, E. Verveniotis, V. Trouillet, B. Rezek and C. Barner-Kowollik, Chem. Commun., 2013, 49, 8623–8625 RSC
- D. M. Jones, A. A. Brown and W. T. S. Huck, Langmuir, 2002, 18, 1265–1269 CrossRef CAS
- K. L. Parry, A. G. Shard, R. D. Short, R. G. White, J. D. Whittle and A. Wright, Surf. Interface Anal., 2006, 38, 1497–1504 CrossRef CAS
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1994, 21, 165–176 CrossRef CAS
- C. H. Peng, J. Kong, F. Seeliger and K. Matyjaszewski, Macromolecules, 2011, 44, 7546–7557 CrossRef CAS
- D. Zhou, X. Gao, W.-j. Wang and S. Zhu, Macromolecules, 2012, 45, 1198–1207 CrossRef CAS
- C.-Y. Lee, G. M. Harbers, D. W. Grainger, L. J. Gamble and D. G. Castner, J. Am. Chem. Soc., 2007, 129, 9429–9438 CrossRef CAS PubMed
- X. Shi, Y. Wang, D. Li, L. Yuan, F. Zhou, Y. Wang, B. Song, Z. Wu, H. Chen and J. L. Brash, Langmuir, 2012, 28, 17011–17018 CrossRef CAS PubMed
- A. J. Inglis, T. Paulohrl and C. Barner-Kowollik, Macromolecules, 2010, 43, 33–36 CrossRef CAS
- K. A. Rufanov, N. B. Kazennova, A. V. Churakov, D. A. Lemenovskii and L. G. Kuz'mina, J. Organomet. Chem., 1995, 485, 173–178 CrossRef CAS
- N. E. Leadbeater, Tetrahedron Lett., 2002, 43, 691–693 CrossRef CAS
- R. P. Hughes and H. A. Trujillo, Organometallics, 1996, 15, 286–294 CrossRef CAS
- S. F. Tayyari, S. Laleh, M. Zahedi-Tabrizi and M. Vakili, J. Mol. Struct., 2013, 1038, 177–187 CrossRef CAS PubMed
- S. F. Tayyari, S. Laleh, M. Zahedi-Tabrizi and M. Vakili, J. Mol. Struct., 2013, 1036, 151–160 CrossRef CAS PubMed
- K. Binder and A. Milchev, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1515–1555 CrossRef CAS
- N. D. Brault, C. Gao, H. Xue, M. Piliarik, J. Homola, S. Jiang and Q. Yu, Biosens. Bioelectron., 2010, 25, 2276–2282 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental methods, ellipsometry, water contact angle, AFM, FTIR-GASR, Br 3d XPS spectra, Cp-substitution of the bromine end-group of SAMs and immobilization of α-maleimide-ω-methoxy-(polyethylene glycol) on the resulting SAM-Cp, additional SPR sensograms of blank samples, and the preservation of the reactivity of the Cp moieties. See DOI: 10.1039/c4py00281d |
| This journal is © The Royal Society of Chemistry 2014 |