 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aggregation induced emission and amplified explosive detection of tetraphenylethylene-substituted polycarbazoles
Wenyue
Dong
a,
Teng
Fei
b,
Alex
Palma-Cando
a and
Ullrich
Scherf
*a
aBergische Universität Wuppertal, Macromolecular Chemistry Group (buwmakro) and Institute for Polymer Technology, Gauss-Str. 20, D-42097 Wuppertal, Germany. E-mail: Scherf@uni-wuppertal.de
bState Key Laboratory on Integrated Optoelectronics, College of Electronic Science and Engineering, Jilin University, Changchun 130012, PR China
First published on 12th March 2014
Abstract
Novel conjugated polymers based on 3,6-carbazole repeat units were synthesized by nickel-catalyzed Yamamoto coupling under microwave heating. The resulting poly(3,6-carbazole)s contain tetraphenylethylene (TPE) units in their side chains. The resultant polymers show aggregation induced emission (AIE) behavior. Hereby, the photoluminescence (PL) intensity of PCzTPE0.5 in 90% water–THF is 35 times higher than that in pure THF, connected to the introduction of TPE side chains. The ability of polymer PCzTPE0.5 for explosive sensing was also studied. A maximum Stern–Volmer quenching constant of 1.26 × 106 M−1 was observed for PL quenching of PCzTPE0.5 aggregates by trinitrobenzene (TNB). A solid state paper strip test based on PCzTPE0.5 and PCzTPE also demonstrates effective PL quenching towards both TNB vapor and solution.
Introduction
Most “conventional” organic luminophores exhibit high photoluminescence (PL) efficiency in solution but weaker PL efficiency in the solid state (e.g. in the films). The effect is called aggregation caused quenching (ACQ)1–3 and is often detrimental for practical applications, e.g. in sensor devices. To solve this problem, several chemical, physical or engineering approaches4–11 have been developed, but with limited success, because one has to fight against an intrinsic process – the energetically favored formation of chromophore aggregates in the solid state. In 2001, Tang et al. first described a novel phenomenon opposite to the ACQ effect: propeller-shaped molecules such as hexaphenylsilole (HPS) and tetraphenylethylene (TPE) emit strongly in the solid state but are weakly emissive in solution. The phenomenon is called aggregation induced emission (AIE).12–14 Thanks to the AIE effect, the design of efficient solid emitters was possible. The main reason for the AIE behavior is the restriction of intramolecular rotation (RIR) of the peripheral phenyl rotors in the aggregated state as has been concluded both from experimental and theoretical studies.15–19The detection of explosives, such as nitroaromatic compounds or peroxides, becomes increasingly important in modern society, due to the concerns on global security and environmental protection.20–23 Many methods have been developed for explosive detection, such as gas chromatography,24 mass spectrometry,25,26 surface enhanced Raman spectroscopy,27 ion mobility spectrometry,28,29 electrochemical sensing,30 PL spectroscopy,31–34 and others. Among them, PL sensors based on conjugated polymers35 have been widely tested because of their simplicity and high sensitivity. Trinitroaromatic compounds containing three electron-withdrawing nitro groups are potent electron acceptors. The working principle of PL sensors for nitroaromatic compounds is based on a photo-induced electron transfer from donor (conjugated polymer) to acceptor (nitroaromatic compound) thus resulting in PL quenching. Recently, AIE luminogens have been used in PL sensors for the detection of nitroaromatics36–42 due to their high solid state fluorescence quantum yields. Moreover, their twisted structure creates an increased number of 3D exciton diffusion channels, thus enhancing the quenching efficiency.
In this study, we prepared novel AIE-active conjugated polymers and investigated their ability for sensing nitroaromatic compounds. Two carbazole-based polymers (PCzTPE and PCzTPE0.5) with TPE side chain units were successfully synthesized. The 3,6-carbazole repeat units43–45 were chosen for constructing the polycarbazole backbone since their electron-donor character should be beneficial for the interaction with electron-poor trinitroaromatics. The incorporation of TPE into the side chains guarantees AIE activity, without strongly affecting the electronic properties of the polycarbazole backbone. Based on this design principle we expected a high sensitivity for the detection of nitroaromatic compounds. Our novel polymers are the first polymeric AIE materials with the AIE active groups in the side chain.
Experimental
Characterization of materials
NMR spectra were recorded on a Bruker AVANCE 400 or AVANCE III 600. 1H and 13C NMR spectra were recorded with tetramethylsilane (TMS) as an internal standard. Gel permeation chromatography (GPC) measurements were carried out on a PSS/Agilent SECurity GPC System equipped with polystyrene gel columns using chloroform as an eluent. APLI (Atmospheric Pressure Laser Ionization) measurements were carried out on a Bruker Daltronik Bremen with micrOTOF. UV-visible absorption spectra were recorded on a Jasco V-670 spectrometer and PL spectra on a Varian CARY Eclipse F2500. Elemental analyses were performed on a Vario EL II (CHNS) instrument. Thermal gravimetric analysis (TGA) was undertaken on a TGA/DSC1 STAR System (Mettler Toledo) at a heating rate of 10 °C min−1 and an argon flow rate of 50 mL min−1. Differential scanning calorimetry (DSC) was performed on a DSC1 STAR System (Mettler Toledo) at a heating rate of 10 °C min−1 under argon. The PL quantum efficiencies of polymer films were measured with an integrating sphere. Cyclic voltammetry (CV) measurements of the polymer films were performed on a standard three-electrode electrochemical cell attached to a VersaSTAT 4 electrochemical workstation in dichloromethane for polymers and acetonitrile for trinitrobenzene (TNB) with 0.1 M tetrabutylammonium perchlorate as a supporting electrolyte at a scan rate of 0.1 V s−1 for polymers and 0.2 V s−1 for TNB. The potentials were measured against an Ag/AgNO3 reference electrode (0.1 M AgNO3 in acetonitrile/0.6 V vs. NHE). The onset potentials were determined from the intersection of two tangents drawn at the rising current and background current of the cyclic voltammogram.Synthesis
All reagents were obtained from commercial suppliers and were used without further purification. All reactions were carried out under an argon atmosphere using standard and Schlenk techniques. The solvents used were of commercial p.a. quality.The crude alcohol was dissolved in about 250 mL of toluene containing p-toluenesulphonic acid (2.0 g, 10.5 mmol) in a 500 mL flask, and the mixture was refluxed overnight. The toluene layer was washed with 10% aqueous NaHCO3 solution and dried over MgSO4 and evaporated to afford the crude tetraphenylethylene derivative. The crude product was purified by recrystallization from the mixture of dichloromethane and methanol to give the target compound as a white solid in 71% yield (10.0 g). 1H NMR (400 MHz, C2D2Cl4) δ 7.15–7.07 (m, 9H), 7.06–6.95 (m, 8H), 6.80 (t, J = 8.8 Hz, 2H). 13C NMR (C2D2Cl4, 100 MHz) δ (ppm) 160.24, 143.73, 143.69, 143.62, 141.41, 140.01, 139.97, 139.94, 133.23, 133.15, 131.51, 128.06, 128.00, 127.95, 126.76, 126.69, 114.94, 114.73.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300, and Mw/Mn 2.10 (GPC, PS calibration).
400, and Mw/Mn 2.54 (GPC, PS calibration).
300, and Mw/Mn 2.10 (GPC, PS calibration).
400, and Mw/Mn 2.54 (GPC, PS calibration).
Results and discussion
Synthesis and characterization
The synthesis routes to the monomers and polymers are depicted in Scheme 1. Monofluoro-TPE 1 was obtained by treating 4-fluorobenzophenone with diphenylmethyl lithium followed by acid-catalyzed dehydration.46 The N-carbazolyl-TPE derivative 2 was synthesized by catalyst-free N-arylation in a direct nucleophilic substitution of 1 as nonactivated fluorobenzene with 3,6-dibromocarbazole.47 3,6-Dibromo-9-octylcarbazole 3 was synthesized according to a reported procedure.48 All monomers were fully characterized prior to polymerization. Homopolymer PCzTPE and random copolymer PCzTPE0.5 were synthesized from monomers 2 and 3 by Yamamoto-type coupling using Ni(COD)2 as a coupling reagent in a mixture of THF, COD and Bpy under microwave (MW) heating.49 Following these protocols we could obtain the target conjugated polymers in short reaction times. The chemical structures of the obtained polymers were confirmed by NMR spectroscopy, GPC, thermal analysis and optical spectroscopy.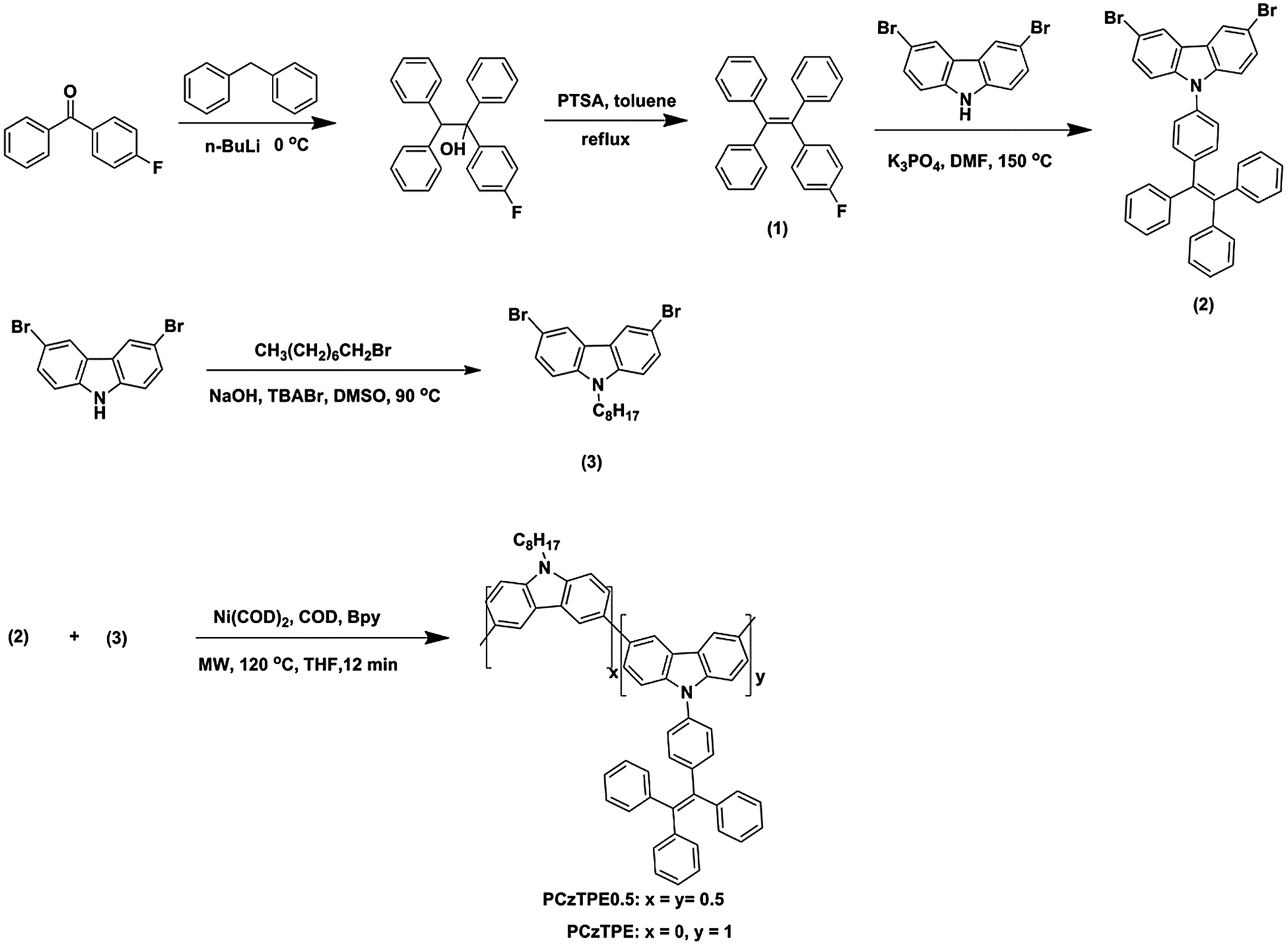 | ||
| Scheme 1 Synthetic procedures for monomer and polymer synthesis. | ||
Thermal properties
The thermal properties of PCzTPE0.5 and PCzTPE were investigated by TGA and DSC. Both polymers exhibit high thermal stability with 5% weight loss occurring at 490 and 430 °C, respectively. In DSC analysis we could not record glass transitions (Tg) up to 300 °C. High thermal stability is important for practical application in solid state sensors.Photophysical properties
Fig. 1 shows the absorption and PL spectra of PCzTPE and PCzTPE0.5 in THF solution and solid state films. The absorption spectra of PCzTPE and PCzTPE0.5 are very similar, with solid state peak maxima at 314 and 310 nm, respectively. In the PL spectra both polymers exhibit green fluorescence peaking around 495 nm, both in solution and as thin films. This behavior is attributed to the incorporation of the TPE units – they effectively suppress π-stacking in the condensed phase due to the presence of the propeller-shaped TPE side chain. The photoluminescence quantum yields (PLQYs) of PCzTPE and PCzTPE0.5 in diluted THF solution, estimated by using quinine sulfate as the standard, have been determined as 1.1% and 0.8%, respectively. The PLQYs distinctly increase to 20% and 21%, respectively, in solid state films, 18- and 26-fold higher when compared to THF solutions. Evidently, the transition into the condensed state dramatically enhances the PL of the polymers. The AIE properties will be discussed in detail in the following paragraph.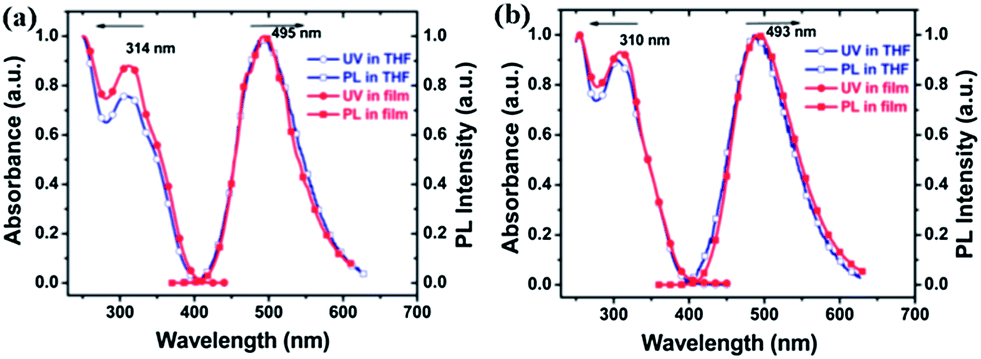 | ||
| Fig. 1 Absorption and PL spectra (excitation wavelength: 320 nm) of (a) PCzTPE and (b) PCzTPE0.5 in dilute THF solution (10−5 M) and as spin-coated films recorded at room temperature. | ||
To further investigate the AIE effect with the polymers PCzTPE and PCzTPE0.5, a series of PL spectra in THF–water mixtures with increasing water fraction were recorded. Fig. 2a and c show PL spectra of the polymers in such water–THF mixtures. The PL intensity increases progressively with increasing water fraction for both polymers. Hereby, polymer PCzTPE0.5 showed a more pronounced AIE effect when compared to the PCzTPE copolymer. For PCzTPE the PL intensity is 11 times higher for a water content of 80% when compared to pure THF (Fig. 2b), and for PCzTPE0.5 35 times higher for a water content of 90% (Fig. 2d). As water is a non-solvent for PCzTPE and PCzTPE0.5, both polymers are assumed to form solid-state aggregates in the THF–water mixtures with a high water content, thus exhibiting aggregation-induced PL enhancement: they are AIE active. Caused by the high rotational freedom of the TPE side-chain moieties in solution, a high internal conversion rate results in weak emission. Within the aggregated (solid) state, however, the rotation of phenyl rings of the TPE units is strongly restricted, thus blocking non-radiative deactivation channels and leading to the AIE effect. Poly(N-octyl-3,6-carbazole) without TPE side groups that was generated as a reference did not show any AIE activity.
 | ||
| Fig. 2 PL spectra of (a) PCzTPE and (c) PCzTPE0.5 in THF–water mixtures with different water contents; increase of PL intensity with increasing water fraction for (b) PCzTPE and (d) PCzTPE0.5, respectively (polymer concentration: 10−5 M; excitation wavelength: 320 nm); (e) fluorescence images of PCzTPE0.5 in THF–water mixtures with different water contents. | ||
Explosive detection
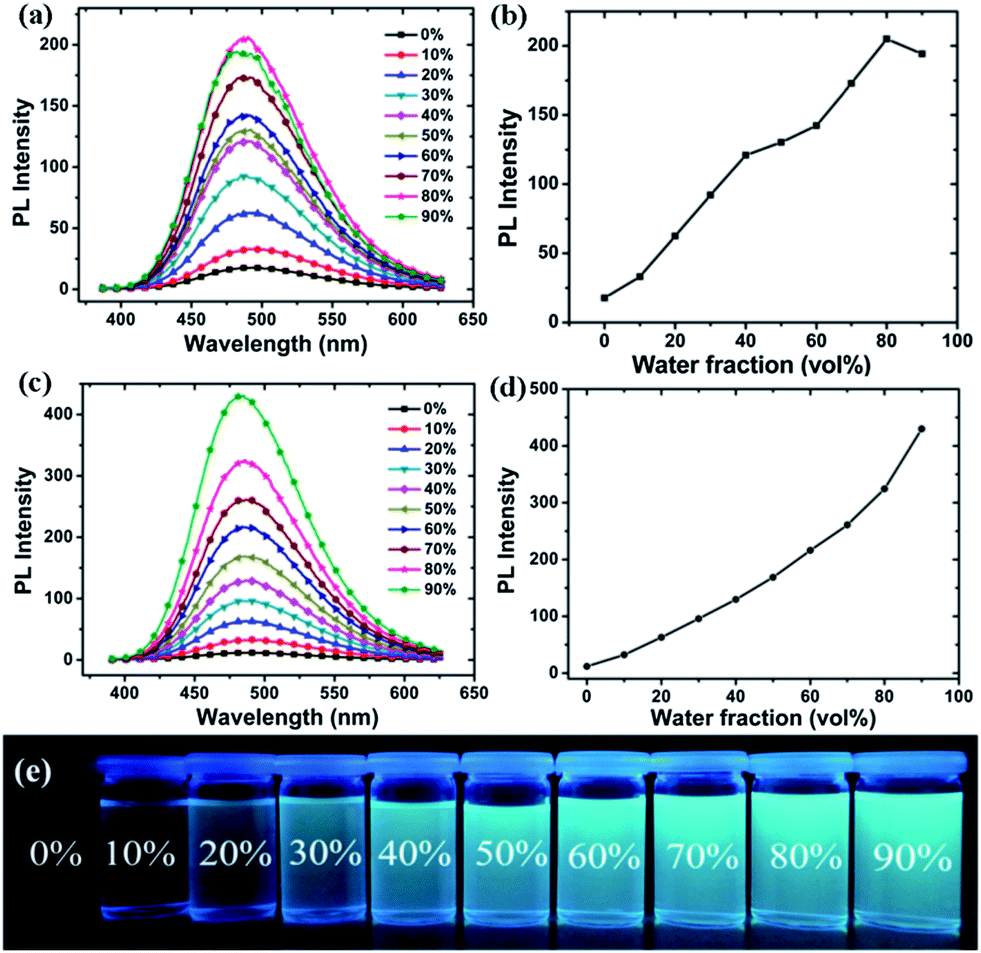
PCzTPE and PCzTPE0.5 should show good electron-donor ability due to their electron-rich polycarbazole backbones. Moreover, their twisted 3D-structure should create effective pathways for interchain exciton diffusion leading to amplified PL quenching properties. Based on the above presented results we started our investigations with aggregated PCzTPE0.5 in THF–water 1:9 (polymer concentration 10 μM). 1,3,5-Trinitrobenzene (TNB) was chosen as a prototypical nitroaromatic analyte. As shown in Fig. 3a, the PL intensity of PCzTPE0.5 in THF–water 1:9 decreases progressively during addition of TNB, without changing the PL peak position, suggesting that different emissive species are not formed. The onset of PL quenching is found for addition of 50 nM TNB, low enough for the detection of submillimolar TNB concentrations. For a TNB concentration of 58 μM, the PL of the dispersed polymer nanoaggregates is fully quenched. The quenching response was analyzed by fitting the data with the Stern–Volmer equation, as depicted in Fig. 3b. For TNB concentrations below 21 μM, the Stern–Volmer plot is linear with a quenching constant of 2.14 × 105 M−1. For a higher analyte concentration, the curve bends upward, thus demonstrating an amplified quenching.50 The quenching constant reaches ca. 1.26 × 106 M−1 between TNB concentrations of 43 μM and 58 μM. This amplified quenching is attributed to the twisted 3D topology of the polymer chains in the nanoaggregates, leading to the formation of an increased number of quenching sites that can interact with TNB molecules and/or to an improved exciton diffusion to quenching sites.51
 | ||
| Fig. 3 (a) PL spectra of PCzTPE0.5 in THF–water 1:9 containing different amounts of TNB. Polymer concentration: 10−5 M; λex: 320 nm. (b) Stern–Volmer plots of PL intensity of PCzTPE0.5 I0/I − 1 versus TNB concentration (I = PL intensity, I0 = PL intensity at a TNB concentration of 0 M). | ||
Fig. 4a shows that there is no spectral overlap between the absorption spectrum of TNB and the PL spectrum of PCzTPE0.5 (as a prerequisite for Förster-type energy transfer) thus indicating that the main quenching mechanism for TNB addition should be an excited state charge transfer between the excited state of the host and the ground state of the TNB quencher. The occurrence of charge transfer was further confirmed by cyclic voltammetry (Fig. 4b). The HOMO (highest occupied molecular orbital) level of PCzTPE0.5 was estimated to be ca. −5.1 eV, and the LUMO (lowest unoccupied molecular orbital) level of TNB to be ca. −3.1 eV. Considering an optical bandgap (Eg) of 3.2 eV for PCzTPE0.5 from the onset of its UV/vis absorption band, the LUMO level of PCzTPE0.5 is calculated to be ca. −1.9 eV. Therefore, the LUMO energy (−1.9 eV) of PCzTPE0.5 allows for an excited state electron transfer to the lower-lying LUMO level of TNB with a LUMO–LUMO offset of ca. 1.2 eV.52
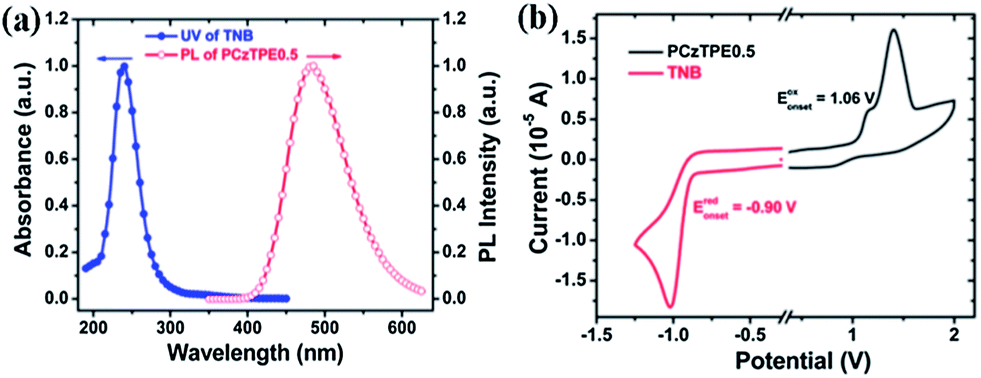 | ||
| Fig. 4 (a) Normalized absorption spectrum of TNB and the PL spectrum of PCzTPE0.5 nanoaggregates in THF–water 1:9. (b) CV plots of PCzTPE0.5 in dichloromethane (oxidative scan) and TNB in acetonitrile (reductive scan), for the experimental conditions please see the Characterization of materials chapter. | ||
For practical explosive detection, the availability of solid state sensor devices is of primary importance. Towards this goal, we prepared test strips by dip-coating Whatman filter paper into solutions of PCzTPE and PCzTPE0.5 in THF (10−4 M) followed by drying the strips in an air stream. First, for vapor-mode tests, we placed the fluorescent paper strips on top of a glass vial containing solid TNB for 5 min at room temperature. In this way, a circular area of the strip was exposed to TNB vapor. Within the exposed area the PL of the polymers was obviously distinctly quenched (Fig. 5A). Second, for solution tests, the test strips containing both polymers were dipped into pure THF (as reference) and a solution of TNB in THF (10−4 M). As shown in Fig. 5B, the fluorescence of the strips was quenched completely after contact with the TNB solution for both polymers. The reference strips dipped into pure THF did not show significant PL quenching thus demonstrating that the majority of the polymers remain adsorbed at the test strips. These first promising results demonstrate the potential of our new polycarbazole-type polymers PCzTPE0.5 and PCzTPE for the fabrication of solid state sensors for nitroaromatic explosives with sufficient sensitivity.
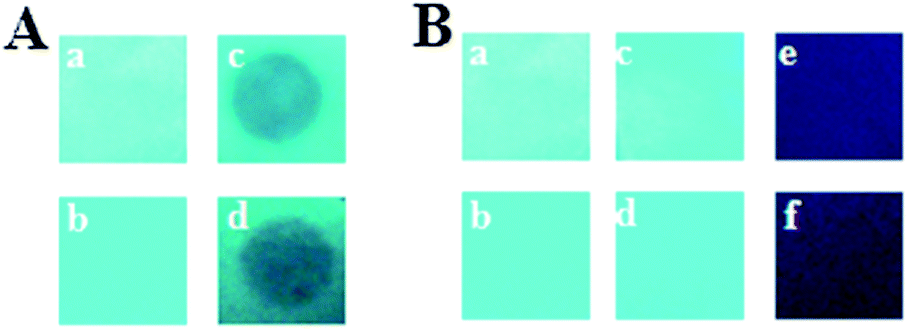 | ||
| Fig. 5 Paper strip tests. (A) Vapor-mode detection of TNB: test strips before (a and b) and after (c and d) placing the strips on top of a glass vial containing solid TNB for 5 min; for PCzTPE (a and c) or PCzTPE0.5 (b and d), respectively. (B) Solution-mode detection of TNB: test strips before (a and b) and after dipping the strips into pure THF (c and d) and into a 10−4 M TNB solution in THF (e and f); for PCzTPE (a, c and e) or PCzTPE0.5 (b, d and f), respectively. | ||
Conclusions
In summary, two poly(3,6-carbazole)s with AIE-active tetraphenylethylene (TPE) side chains have been successfully synthesized. The polymers combine the electron-deficient character of the polycarbazole backbone and the AIE behavior of the TPE containing side chains. So, both polymers PCzTPE and PCzTPE0.5 show distinct AIE properties. For sensing of nitroaromatic explosives PL quenching experiments were carried out. Aggregated PCzTPE0.5 shows amplified PL quenching during trinitrobenzene (TNB) addition in THF–water mixtures (1:9, v/v) with a maximum Stern–Volmer quenching constant of 1.26 × 106 M−1. Solid-state paper strips with deposits of both polymers show TNB-induced PL quenching, both towards TNB vapor or TNB solution thus demonstrating promising practical application potential in solid state sensors for nitroaromatic explosives. Other possible applications (e.g. in OLEDs) are currently tested.
Acknowledgements
W.D. would like to thank for financial support from the fellowship program of the China Scholarship Council. A.P.C. would like to thank the DAAD for a PhD fellowship. Thanks to Anke Helfer for performing the GPC, TGA and DSC analyses and to João Pina for the measurement of solid state photoluminescence quantum yields.Notes and references
- M. Belletete, J. Bouchard, M. Leclerc and G. Durocher, Macromolecules, 2005, 38, 880 CrossRef CAS.
- R. Jakubiak, C. J. Collison, W. C. Wan and L. Rothberg, J. Phys. Chem. A, 1999, 103, 2394 CrossRef CAS.
- M. Grell, D. D. C. Bradley, X. Long, T. Chamberlain, M. Inbasekaran, E. P. Woo and M. Soliman, Acta Polym., 1998, 49, 439 CrossRef CAS.
- J. Wang, Y. Zhao, C. Dou, H. Sun, P. Xu, K. Ye, J. Zhang, S. Jiang, F. Li and Y. Wang, J. Phys. Chem. B, 2007, 111, 5082 CrossRef CAS.
- S.-F. Lim, R. H. Friend, I. D. Rees, J. Li, Y. Ma, K. Robinson, A. B. Holmes, E. Hennebicq, D. Beljonne and F. Cacialli, Adv. Funct. Mater., 2005, 15, 981 CrossRef CAS.
- C.-W. Wu, C.-M. Tsai and H.-C. Lin, Macromolecules, 2006, 39, 4298 CrossRef CAS.
- A. Kraft, A. C. Grimsdale and A. B. Holmes, Angew. Chem., Int. Ed., 1998, 37, 402 CrossRef.
- S. Setayesh, A. C. Grimsdale, T. Weil, V. Enkelmann, K. Müllen, F. Meghdadi, E. J. W. List and G. Leising, J. Am. Chem. Soc., 2001, 123, 946 CrossRef CAS.
- S. Hecht and J. M. J. Frechet, Angew. Chem., Int. Ed., 2001, 40, 74 CrossRef CAS.
- L. Chen, S. Xu, D. McBranch and D. Whitten, J. Am. Chem. Soc., 2000, 122, 9302 CrossRef CAS.
- C. Fan, S. Wang, J. W. Hong, G. C. Bazan, K. W. Plaxco and A. J. Heeger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6297 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- B. Z. Tang, X. Zhan, G. Yu, P. P. S. Lee, Y. Liu and D. Zhu, J. Mater. Chem., 2001, 11, 2974 RSC.
- Z. Xie, B. Yang, W. Xie, L. Liu, F. Shen, H. Wang, X. Yang, Z. Wang, Y. Li, M. Hanif, G. Yang, L. Ye and Y. Ma, J. Phys. Chem. B, 2006, 110, 20993 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC.
- R. Hu, J. W. Y. Lam, Y. Liu, X. Zhang and B. Z. Tang, Chem.–Eur. J., 2013, 19, 5617 CrossRef CAS PubMed.
- S. Yin, Q. Peng, Z. Shuai, W. Fang, Y. H. Wang and Y. Luo, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 205409 CrossRef.
- Q. Peng, Y. Yi, Z. Shuai and J. Shao, J. Am. Chem. Soc., 2007, 129, 9333 CrossRef CAS.
- X. Fan, J. Sun, F. Wang, Z. Chu, P. Wang, Y. Dong, R. Hu, B. Z. Tang and D. Zou, Chem. Commun., 2008, 2989 RSC.
- J. W. Grate, Chem. Rev., 2008, 108, 726 CrossRef CAS PubMed.
- J.-S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 5321 CrossRef CAS.
- J.-S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 11864 CrossRef CAS.
- S. W. Zhang and T. M. Swager, J. Am. Chem. Soc., 2003, 125, 3420 CrossRef CAS PubMed.
- M. E. Walsh, Talanta, 2001, 54, 427 CrossRef CAS.
- Y. Jehuda, Mass Spectrom. Rev., 1982, 1, 257 CrossRef.
- J. C. Mathurin, T. Faye, A. Brunot and J. C. Tabet, Anal. Chem., 2000, 72, 5055 CrossRef CAS.
- J. M. Sylvia, J. A. Janni, J. D. Klein and K. M. Spencer, Anal. Chem., 2000, 72, 5834 CrossRef CAS.
- E. Wallis, T. M. Griffin, N. Popkie Jr, M. A. Eagan, R. F. McAtee, D. Vrazel and J. McKinly, Proc. SPIE, 2005, 5795, 54 CrossRef CAS.
- G. A. Eicaman and J. A. Stone, Anal. Chem., 2004, 1, 390 Search PubMed.
- M. Krausa and K. Schorb, J. Electroanal. Chem., 1999, 461, 10 CrossRef CAS.
- H. Nie, Y. Zhao, M. Zhang, Y. Ma, M. Baumgarten and K. Müllen, Chem. Commun., 2011, 47, 1234 RSC.
- D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537 CrossRef CAS PubMed.
- C. McDonagh, C. S. Burke and B. D. MacCraith, Chem. Rev., 2008, 108, 400 CrossRef CAS PubMed.
- X. Liu, Y. Xu and D. Jiang, J. Am. Chem. Soc., 2012, 134, 8738 CrossRef CAS.
- S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef CAS PubMed.
- Y. Salinas, R. Martinez-Manez, M. D. Marcos, F. Sancenon, A. M. Costero, M. Parra and S. Gil, Chem. Soc. Rev., 2012, 41, 1261 RSC.
- S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef CAS PubMed.
- H. Li, H. Wu, E. Zhao, J. Li, J. Z. Sun, A. Qin and B. Z. Tang, Macromolecules, 2013, 46, 3907 CrossRef CAS.
- W. Wu, S. Ye, R. Tang, L. Huang, Q. Li, G. Yu, Y. Liu, J. Qin and Z. Li, Polymer, 2012, 53, 3163 CrossRef CAS PubMed.
- P. Lu, J. W. Y. Lam, J. Liu, C. K. W. Jim, W. Yuan, N. Xie, Y. Zhong, Q. Hu, K. S. Wong, K. K. L. Cheuk and B. Z. Tang, Macromol. Rapid Commun., 2010, 31, 834 CrossRef CAS PubMed.
- W. Wu, S. Ye, L. Huang, L. Xiao, Y. Fu, Q. Huang, G. Yu, Y. Liu, J. Qin, Q. Li and Z. Li, J. Mater. Chem., 2012, 22, 6374 RSC.
- W. Wu, S. Ye, G. Yu, Y. Liu, J. Qin and Z. Li, Macromol. Rapid Commun., 2012, 33, 164 CrossRef CAS PubMed.
- S. Grigalevicius, L. Ma, G. Qian, Z. Xie, M. Forster and U. Scherf, Macromol. Chem. Phys., 2007, 208, 349 CrossRef CAS.
- S. A. Patil, U. Scherf and A. Kadashchuk, Adv. Funct. Mater., 2003, 13, 609 CrossRef CAS.
- B. Souharce, C. J. Kudla, M. Forster, J. Steiger, R. Anselmann, H. Thiem and U. Scherf, Macromol. Rapid Commun., 2009, 30, 1258 CrossRef CAS PubMed.
- M. Banerjee, S. J. Emond, S. V. Lindeman and R. Rathore, J. Org. Chem., 2007, 72, 8054 CrossRef CAS PubMed.
- F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2012, 51, 8012 CrossRef CAS PubMed.
- D. Hu, G. Cheng, P. Lu, H. Liu, F. Shen, F. Li, Y. Lv, W. Dong and Y. Ma, Macromol. Rapid Commun., 2011, 32, 1467 CrossRef CAS PubMed.
- F. Galbrecht, T. W. Bünnagel, U. Scherf and T. Farrell, Macromol. Rapid Commun., 2007, 28, 387 CrossRef CAS.
- C. Y. K. Chan, Z. Zhao, J. W. Y. Lam, J. Liu, S. Chen, P. Lu, F. Mahtab, X. Chen, H. H. Y. Sung, H. S. Kwok, Y. Ma, I. D. Williams, K. S. Wong and B. Z. Tang, Adv. Funct. Mater., 2012, 22, 378 CrossRef CAS.
- J. Li, J. Liu, J. W. Y. Lam and B. Z. Tang, RSC Adv., 2013, 3, 8193 RSC.
- J. Wang, J. Mei, W. Yuan, P. Lu, A. Qin, J. Sun, Y. Ma and B. Z. Tang, J. Mater. Chem., 2011, 21, 4056 RSC.
| This journal is © The Royal Society of Chemistry 2014 |