 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
RAFT polymerization of hydroxy-functional methacrylic monomers under heterogeneous conditions: effect of varying the core-forming block
L. P. D.
Ratcliffe
a,
A.
Blanazs†
a,
C. N.
Williams
b,
S. L.
Brown
b and
S. P.
Armes
*a
aDepartment of Chemistry, The University of Sheffield, Dainton Building, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK. E-mail: s.p.armes@shef.ac.uk
bScott Bader Company Limited, Wollaston, Wellingborough, Northamptonshire, NN29 7RL, UK
First published on 6th March 2014
Abstract
Statistical copolymerization of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of a water-miscible monomer (2-hydroxyethyl methacrylate, HEMA) with a water-immiscible monomer (4-hydroxybutyl methacrylate, HBMA) has been conducted in water via reversible addition–fragmentation chain transfer (RAFT) polymerization using a water-soluble poly(glycerol monomethacrylate) macromolecular chain transfer agent (PGMA macro-CTA). In principle, such a hybrid formulation might be expected to be intermediate between RAFT dispersion polymerization and RAFT emulsion polymerization. Under such circumstances, it is of particular interest to examine whether both monomers are actually consumed and, if so, whether their rates of reaction are comparable. Given the water-solubility of both the PGMA macro-CTA and the free radical azo initiator, it is perhaps counter-intuitive that the water-immiscible HBMA is initially consumed significantly faster than the water-miscible HEMA, as judged by 1H NMR studies of this copolymerization. However, both comonomers are eventually almost fully consumed at 70 °C. A detailed phase diagram has been constructed for this RAFT formulation that enables reproducible syntheses of various pure copolymer morphologies, including spheres, worms and vesicles. It is emphasized that utilizing a 1:1 HEMA/HBMA molar ratio produces a core-forming statistical copolymer block that is isomeric with the poly(2-hydroxypropyl methacrylate) (PHPMA) core-forming block previously synthesized via RAFT aqueous dispersion polymerization (see A. Blanazs et al., Macromolecules, 2012, 45, 5099–5107). Hence it is rather remarkable that the thermo-responsive behavior of PGMA–P(HBMA-stat-HEMA) statistical block copolymer worm gels differs qualitatively from that exhibited by PGMA–PHPMA diblock copolymer worm gels.
:1 molar ratio of a water-miscible monomer (2-hydroxyethyl methacrylate, HEMA) with a water-immiscible monomer (4-hydroxybutyl methacrylate, HBMA) has been conducted in water via reversible addition–fragmentation chain transfer (RAFT) polymerization using a water-soluble poly(glycerol monomethacrylate) macromolecular chain transfer agent (PGMA macro-CTA). In principle, such a hybrid formulation might be expected to be intermediate between RAFT dispersion polymerization and RAFT emulsion polymerization. Under such circumstances, it is of particular interest to examine whether both monomers are actually consumed and, if so, whether their rates of reaction are comparable. Given the water-solubility of both the PGMA macro-CTA and the free radical azo initiator, it is perhaps counter-intuitive that the water-immiscible HBMA is initially consumed significantly faster than the water-miscible HEMA, as judged by 1H NMR studies of this copolymerization. However, both comonomers are eventually almost fully consumed at 70 °C. A detailed phase diagram has been constructed for this RAFT formulation that enables reproducible syntheses of various pure copolymer morphologies, including spheres, worms and vesicles. It is emphasized that utilizing a 1:1 HEMA/HBMA molar ratio produces a core-forming statistical copolymer block that is isomeric with the poly(2-hydroxypropyl methacrylate) (PHPMA) core-forming block previously synthesized via RAFT aqueous dispersion polymerization (see A. Blanazs et al., Macromolecules, 2012, 45, 5099–5107). Hence it is rather remarkable that the thermo-responsive behavior of PGMA–P(HBMA-stat-HEMA) statistical block copolymer worm gels differs qualitatively from that exhibited by PGMA–PHPMA diblock copolymer worm gels.
Introduction
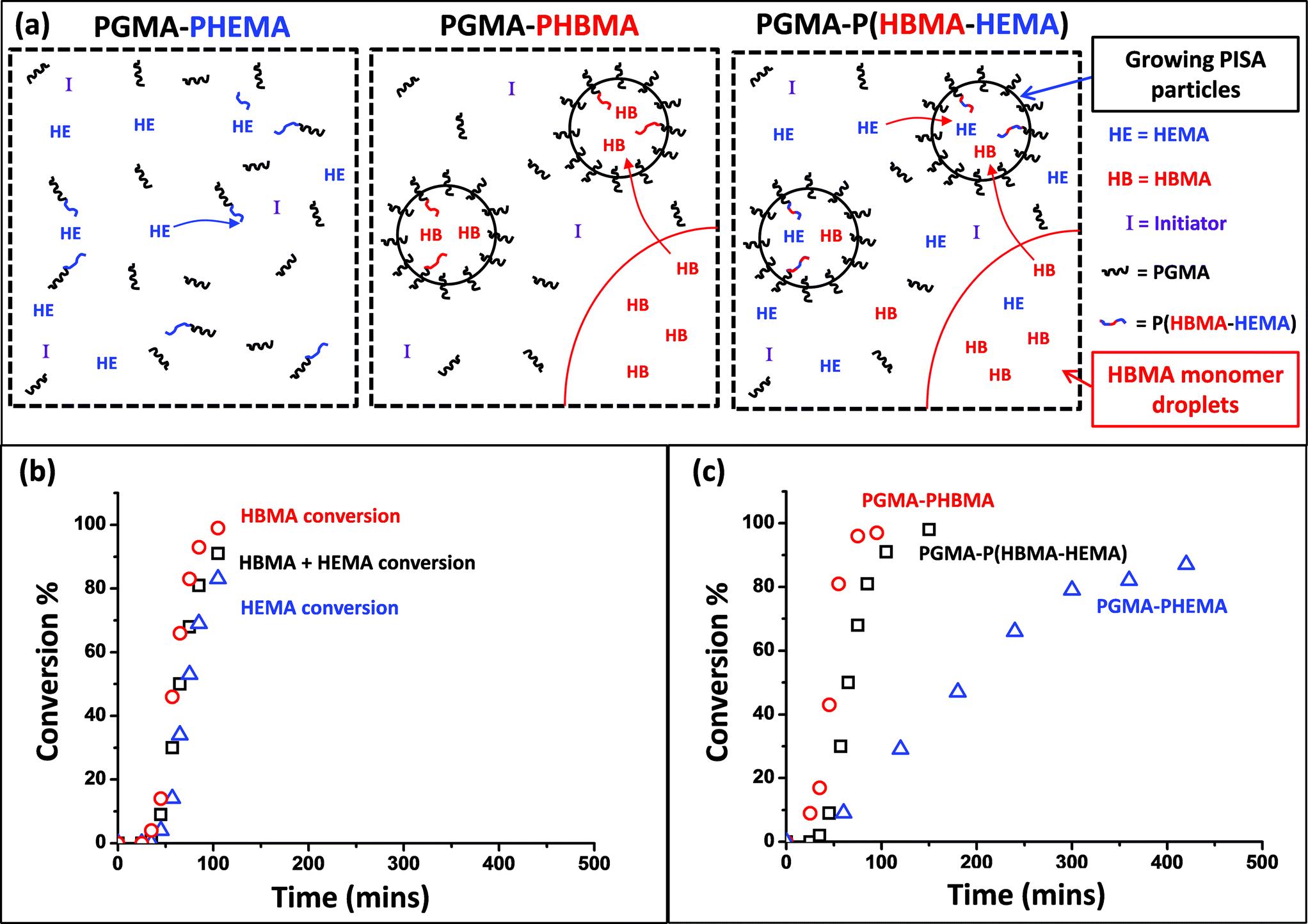
AB diblock copolymers can self-assemble in a solvent that is selective for one of the blocks to form a wide range of morphologies, including spheres,1–3 worms,4–10 cylinders11 and vesicles.6,12,13 In principle, these nano-sized structures could be utilized as bio-delivery vehicles,14,15 nano-reactors,16 inorganic/organic templates,17 or as polymeric surfactants.18 However, their generation usually involves a post-polymerization processing step such as a solvent switch or pH switch,2 or thin film rehydration19–21 that is typically conducted in dilute solution (<1%). This is a severe restriction for many potential commercial applications.Over the last two decades, the development of living radical polymerization techniques such as nitroxide-mediated polymerization,22 atom transfer radical polymerization23,24 and reversible addition–fragmentation chain transfer (RAFT) polymerization25 has revolutionized synthetic polymer chemistry. These radical-based chemistries have enabled the design of a remarkably wide range of functional polymers, including many examples of block copolymer architectures.26–28 In particular, RAFT polymerization has been recently utilized in the context of polymerization-induced self-assembly (PISA) to prepare a range of colloidally stable block copolymer nano-objects at relatively high copolymer concentrations (up to 25%).29–31 PISA syntheses can be conducted under dispersion polymerization conditions and this approach has been demonstrated to be rather generic: highly efficient syntheses have been conducted in water,32–42 alcoholic solvents43–54 or n-alkanes.55–58 Given its low cost, non-toxicity, non-flammability and high heat capacity, water offers particular advantages as a solvent for such RAFT PISA syntheses. However, there are relatively few vinyl monomers that meet the criterion for an aqueous dispersion polymerization: the monomer should be water-miscible but the corresponding polymer must be water-insoluble. For example, An and co-workers have reported the use of either N-isopropyl acrylamide59 or 2-methoxyethyl acrylate36 as a core-forming block for the preparation of thermo-sensitive spherical nanoparticles/nanogels.38,60 However, the prototypical monomer for RAFT aqueous dispersion polymerization is 2-hydroxypropyl methacrylate (HPMA).33,39–42,61–63 An alternative approach is RAFT aqueous emulsion polymerization, as developed by Charleux and co-workers64–72 and Hawkett and co-workers.73,74 In principle, such formulations are applicable to a wide range of water-immiscible vinyl monomers, such as styrene, methyl methacrylate or n-butyl acrylate. PISA formulations based on either RAFT aqueous dispersion polymerization or RAFT aqueous emulsion polymerization have been utilized to prepare low polydispersity block copolymers in the form of near-monodisperse spheres,32,68,69,73,75 polydisperse worms,63,64 polydisperse vesicles61,72,76 or framboidal vesicles.77 In each case, a RAFT macromolecular chain transfer agent (macro-CTA) acts as a water-soluble steric stabilizer, while the growing hydrophobic core-forming block drives in situ self-assembly.
In the present work, we have explored a new type of RAFT PISA formulation that involves growing a water-insoluble statistical copolymer as the core-forming block from a water-soluble poly(glycerol monomethacrylate) (PGMA) macro-CTA. The two comonomers were deliberately selected to be water-miscible (2-hydroxyethyl methacrylate, HEMA) and water-immiscible (4-hydroxybutyl methacrylate, HBMA). For such a hybrid formulation, it was not immediately obvious whether (co)polymerization would initially proceed via aqueous dispersion polymerization or aqueous emulsion polymerization. Indeed, given their differing solubilities, it was also not clear whether both comonomers would be efficiently polymerized in such syntheses (see Scheme 1). The minimum HEMA/HBMA molar ratio for ensuring water miscibility (at 20% w/w solids at 70 °C) was determined to be 6.0. Aqueous dispersion polymerization occurs at or above this critical value. On the other hand, a 1:1 HEMA/HBMA molar ratio corresponds to aqueous emulsion polymerization conditions, since a substantial fraction of HBMA monomer remains water-immiscible. If both comonomers are fully consumed using this latter protocol, the resulting statistical copolymer is actually isomeric with the poly(2-hydroxypropyl methacrylate) (PHPMA) core-forming block used in the majority of the literature examples of RAFT aqueous dispersion polymerization reported to date.33,39–42,61–63 Given that the PHPMA block also confers thermo-sensitivity that leads to a reversible worm-to-sphere morphological transition,62,63 it is clearly of interest to examine whether an isomeric core-forming block comprising a 1:1 HEMA/HBMA statistical copolymer also exhibits such thermo-sensitive behaviour.
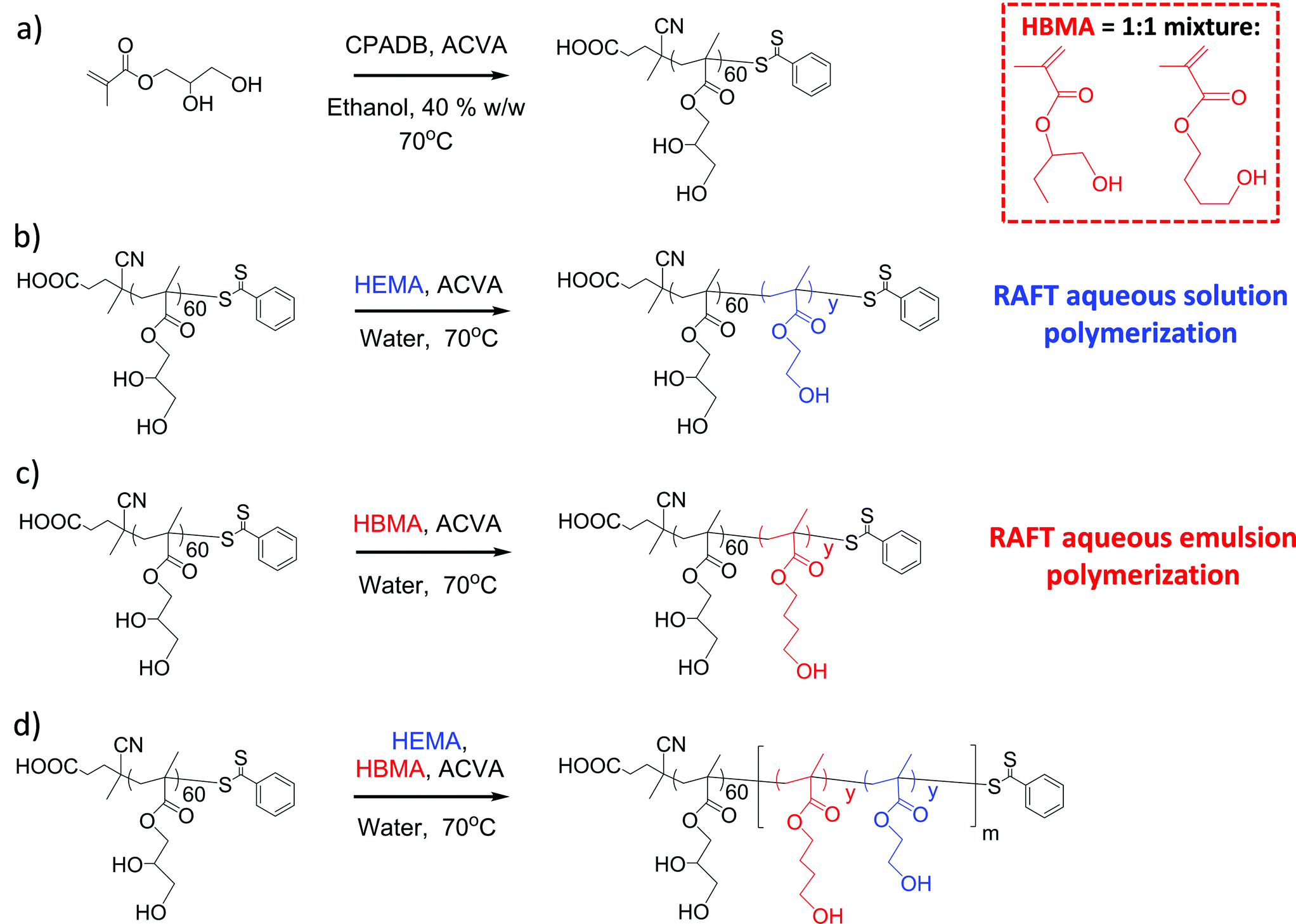 | ||
| Scheme 1 (a) Synthesis conditions used to prepare a PGMA60 macro-CTA, (b) PGMA60–PHEMAy diblock copolymer, (c) PGMA60–PHBMAy diblock copolymer and (d) PGMA60–P(HBMAy–HEMAy)m statistical diblock copolymer. A CTA/ACVA molar ratio of 4.0 was used in all cases. | ||
Experimental
Materials
2-Hydroxypropyl methacrylate (HPMA, 97%), 4,4′-azobis(4-cyanopentanoic acid) (ACVA; V-501; 99%), 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPADB) and 4-hydroxybutyl methacrylate (HBMA, 94% consisting of a 1:1 mixture of 4-hydroxybutyl methacrylate and 1-hydroxybutan-2-yl methacrylate) were purchased from Sigma Aldrich UK and used as received. HPLC analysis of the HPMA monomer indicated a dimethacrylate impurity of around 0.10 mol%. Glycerol monomethacrylate (GMA; 99.8%, ∼0.06 mol% dimethacrylate) and 2-hydroxyethyl methacrylate (HEMA, >99% Ultra grade, ∼0.10 mol% dimethacrylate) were donated by GEO Specialty Chemicals (Hythe, UK) and used without further purification. Deuterated methanol (CD3OD) was purchased from Goss Scientific (Nantwich, UK). All solvents used in this work were HPLC grade and purchased from Fisher Scientific (Loughborough, UK).
Copolymer characterization
000 g mol−1, K = 2.094 × 10−3, α = 0.642). Chromatograms were analyzed using Varian Cirrus GPC software.
Synthesis and purification of PGMA60 macro-CTA
A typical protocol for the synthesis of PGMA60 macro-CTA is given below: to a round-bottomed flask containing CPADB RAFT agent (3.43 mmol, 0.96 g), GMA monomer (0.24 mol, 38.44 g) and anhydrous ethanol (1.28 mol, 59.4 g) was added to afford a target degree of polymerization (DP) of 70. To this, ACVA initiator (0.69 mmol, 0.19 g, CTA/ACVA molar ratio = 5.0) was added and the resulting pink solution was sparged with N2 for 20 minutes, before the sealed flask was immersed into an oil bath set at 70 °C. After 2.5 h (conversion 75% as judged by 1H NMR) the polymerization was quenched by immersion of the flask in an ice bath and opening it to air. The polymerization solution was then precipitated into a ten-fold excess of chloroform and washed three times in the precipitation solvent before being placed under high vacuum for three days at 40 °C. 1H NMR analysis indicated a DP of 60 for this PGMA macro-CTA. Taking into account the target DP of 70 and the conversion of 75%, this suggests a CTA efficiency of 88%. DMF GPC analysis indicated Mn and Mw/Mn values of 17000 g mol−1 and 1.08, respectively.
RAFT synthesis of PGMA60–PHBMA75 diblock copolymer
A typical protocol for the synthesis of PGMA60–PHBMA75 diblock copolymer is as follows: PGMA60 macro-CTA (0.200 g, 0.021 mmol) was added to a 25 mL round-bottomed flask, followed by HBMA monomer (0.247 g, 1.56 mmol), and water (4.03 g, to make a 10% w/w solution). ACVA was then added (1.06 mg, 5.10 μmol, CTA/ACVA molar ratio = 4.0) and the solution was sparged with N2 for 30 minutes. The flask was sealed and immersed in an oil bath set at 70 °C. The reaction solution was magnetically stirred for 16 h to ensure complete monomer conversion and the polymerization was subsequently quenched by exposure to air and cooling the flask to 20 °C.RAFT synthesis of PGMA60–PHEMA75 diblock copolymer
A typical protocol for the synthesis of PGMA60–PHEMA75 diblock copolymer is as follows: PGMA60 macro-CTA (0.220 g, 0.023 mmol) was added to a 25 mL round-bottomed flask, followed by HEMA monomer (0.223 g, 1.7 mmol) and water (4.00 g, to make a 10% w/w solution). ACVA was then added (1.55 mg, 0.0056 mmol, CTA/ACVA molar ratio = 4.0) and the solution was sparged with N2 for 30 minutes. The flask was sealed and immersed in an oil bath set at 70 °C. The reaction solution was then magnetically stirred for 16 h to ensure complete monomer conversion and the polymerization was subsequently quenched by exposure to air and cooling the flask to 20 °C.RAFT synthesis of PGMA60–P(HBMAy-stat-PHEMA(6)y) diblock copolymer
In addition to the reactant concentrations, the targeted composition of the core-forming block (i.e. the initial HEMA/HBMA molar ratio) determines whether the copolymerization proceeds via emulsion or dispersion polymerization. Using a 1:1 HEMA/HBMA molar ratio results in an aqueous emulsion polymerization, as judged by visual inspection (turbid solution owing to the presence of monomer emulsion droplets). A typical protocol for the synthesis of PGMA60–P(HBMA75-stat-HEMA75) statistical diblock copolymer is as follows: PGMA60 macro-CTA (0.150 g, 0.015 mmol) was added to a 25 mL round-bottomed flask, followed by HEMA monomer (0.152 g, 1.17 mmol), HBMA monomer (0.185 g, 1.17 mmol), and water (4.39 g, to produce 10% w/w total solids). ACVA was then added (1.06 mg, 0.0038 mmol, macro-CTA/ACVA molar ratio = 4.0) and the solution was sparged with N2 for 30 minutes. The flask was sealed and immersed in an oil bath set at 70 °C and stirred for 16 h to ensure complete monomer conversion. The polymerization was quenched by exposure to air and cooling the flask to 20 °C.
Results and discussion
The synthesis of sterically-stabilized diblock copolymer nanoparticles in aqueous media using a water-soluble PGMA macro-CTA requires chain extension with a suitably hydrophobic core-forming block. The HPMA monomer previously utilized for this purpose is water-soluble up to 10% w/v at 70 °C and forms a water-insoluble polymer when polymerized at this temperature. Thus HPMA is a rare example of a commodity monomer that is suitable for an aqueous dispersion polymerization formulation.32,39,41,76 In the present work, two methacrylic monomers were considered as alternatives to HPMA. 2-Hydroxyethyl methacrylate (HEMA is water-miscible in all proportions and this monomer produces a water-swellable/weakly hydrophobic homopolymer.78 In contrast, 4-hydroxybutyl methacrylate (HBMA; a 1:1 mixture of 1-hydroxybutan-2-yl methacrylate and 4-hydroxybutyl methacrylate, see Scheme 1) is only water-miscible up to 2.0% w/v at 70 °C and produces a water-insoluble homopolymer. Thus, HEMA is potentially suitable for RAFT aqueous dispersion polymerization, whereas HBMA is more likely to lead to a RAFT aqueous emulsion polymerization formulation.64–72 These hypotheses were confirmed by visual inspection: the initial PGMA + HEMA formulation was an optically transparent homogeneous solution, whereas the initial PGMA + HBMA formulation was turbid owing to the presence of micrometer-sized emulsion droplets.
A PGMA macro-CTA was synthesized using 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid, as this commercially available RAFT agent is known to give well-controlled polymerizations for methacrylic monomers.79 The reaction conditions for this PGMA macro-CTA synthesis (target DP = 70) are summarized in Scheme 1a. A CTA/initiator molar ratio of 5.0 was selected to ensure a sufficiently fast but controlled homopolymerization. The reaction was quenched after 2.5 h and 1H NMR studies indicated 75% conversion. The crude macro-CTA was subsequently purified by precipitation into chloroform and dried to afford a final DP of 60 with 88% CTA efficiency. This PGMA macro-CTA had an Mn of 17000 and an Mw/Mn of 1.08, as judged by DMF GPC using PMMA standards (see Fig. 1a).
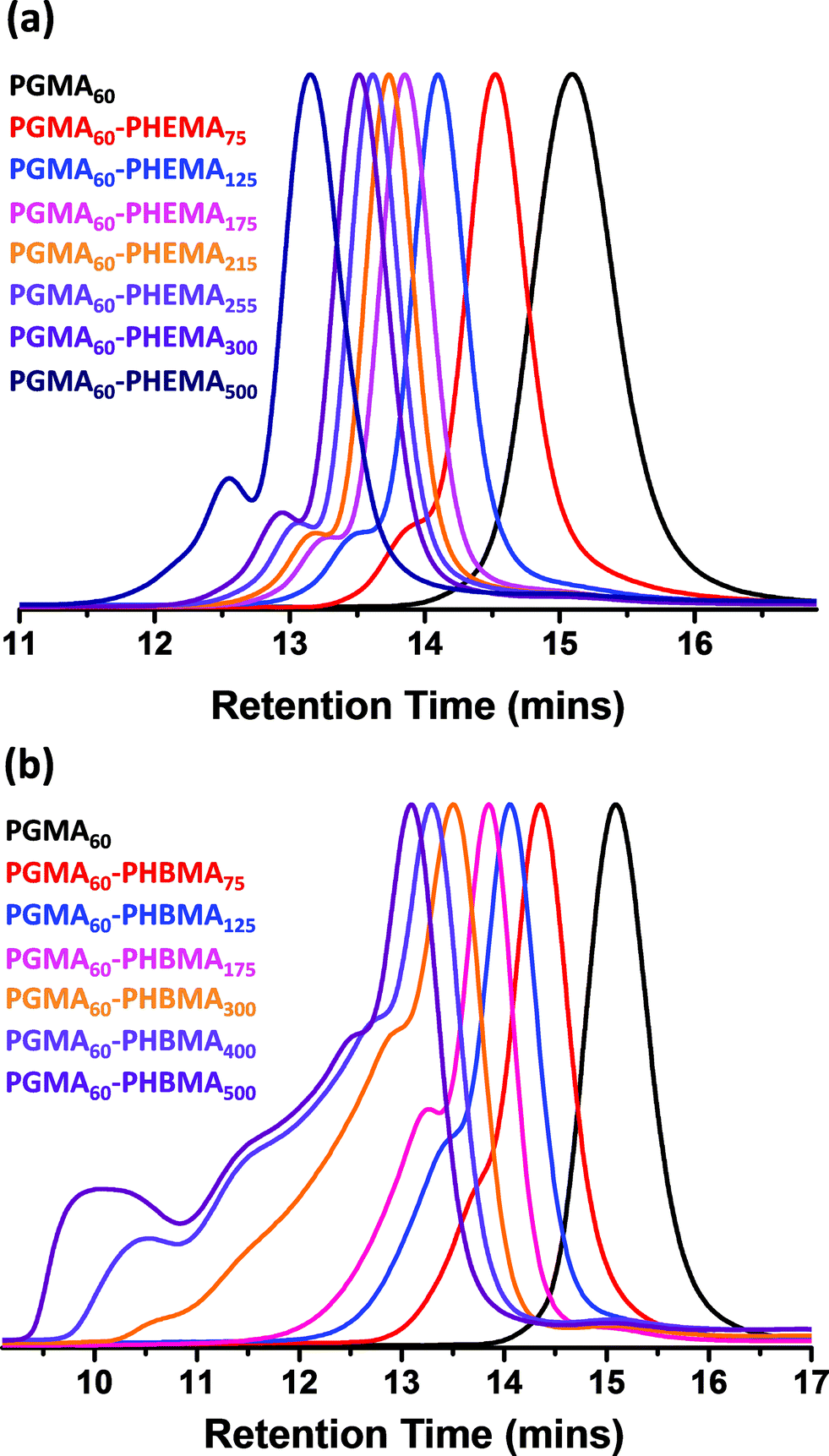 | ||
| Fig. 1 DMF GPC traces obtained for a series of (a) PGMA60–PHEMAy diblock copolymers and (b) PGMA60–PHBMAy diblock copolymers synthesized at 10% w/w solids at 70 °C in aqueous media. A macro-CTA/ACVA molar ratio of 4.0 was used in all cases. | ||
The PGMA macro-CTA was subsequently chain-extended with HEMA (see Scheme 1b). All polymerizations were conducted at 10% w/w solids, unless otherwise stated. A range of DPs were targeted for the PHEMA block, with DMF GPC molecular weights shown in Fig. 1a. All polymerizations attained more than 99% conversion as judged by 1H NMR and minimal macro-CTA contamination was observed in all cases, which indicated high blocking efficiencies. A high molecular weight shoulder was observed, which becomes more prominent when targeting higher PHEMA DPs. This feature is most likely due to the small amount of dimethacrylate impurity present in HEMA monomer (∼0.10 mol% according to HPLC data provided by the manufacturer).80,81 However, some degree of termination by combination under monomer-starved conditions cannot be excluded. The PGMA–PHEMA diblock copolymer molecular weight increased linearly with target DP and all copolymers had relatively low polydispersities (Mw/Mn ≤ 1.28 for PHEMA target DPs up to 500; see Table 1). However, DLS and TEM studies suggested that self-assembly did not occur in any of these syntheses. Phase separation was observed at a target PHEMA DP of greater than or equal to 215, indicating colloidally unstable dispersions at room temperature. It was concluded that PHEMA is an unsuitable core-forming block for successful RAFT aqueous dispersion polymerization, since it is not sufficiently hydrophobic to induce effective in situ self-assembly.
| Entry no. | Targeted block composition | M n (g mol−1) | M w/Mn |
|---|---|---|---|
| 1 | PGMA60 | 17000 |
1.08 |
| 2 | PGMA60–PHEMA75 | 26800 |
1.15 |
| 3 | PGMA60–PHEMA125 | 39600 |
1.11 |
| 4 | PGMA60–PHEMA175 | 47700 |
1.12 |
| 5 | PGMA60–PHEMA215 | 53600 |
1.13 |
| 6 | PGMA60–PHEMA255 | 59000 |
1.16 |
| 7 | PGMA60–PHEMA300 | 65900 |
1.17 |
| 8 | PGMA60–PHEMA500 | 91300 |
1.28 |
| 9 | PGMA60–PHBMA75 | 33000 |
1.16 |
| 10 | PGMA60–PHBMA125 | 44600 |
1.25 |
| 11 | PGMA60–PHBMA175 | 57000 |
1.32 |
| 12 | PGMA60–PHBMA300 | 87000 |
2.27 |
| 13 | PGMA60–PHBMA400 | 115300 |
4.65 |
| 14 | PGMA60–PHBMA500 | 146000 |
9.50 |
The rather more hydrophobic HBMA monomer was also homopolymerized using the PGMA macro-CTA. As previously mentioned, the relatively low water solubility of this monomer ensured that such syntheses were conducted under RAFT aqueous emulsion polymerization conditions (see chain extension Scheme 1c). Again, high conversions were attained (>99% as judged by 1H NMR) and only a rather small fraction of PGMA macro-CTA remained unreacted according to DMF GPC studies (see Fig. 1). Much higher polydispersities were observed for this formulation compared to the PHEMA homopolymerizations, particularly when targeting higher PHBMA DPs (Table 1). However, this does not actually mean that RAFT control has been lost in such syntheses: the HBMA monomer contains a relatively high level of dimethacrylate impurity, which inevitably leads to some degree of branching/cross-linking.61,80,81 This hypothesis was confirmed by purifying a batch of HBMA monomer via column chromatography (using silica as a stationary phase). As expected, RAFT aqueous emulsion polymerizations conducted using this purified monomer produced substantially lower polydispersities (see Fig. 2).
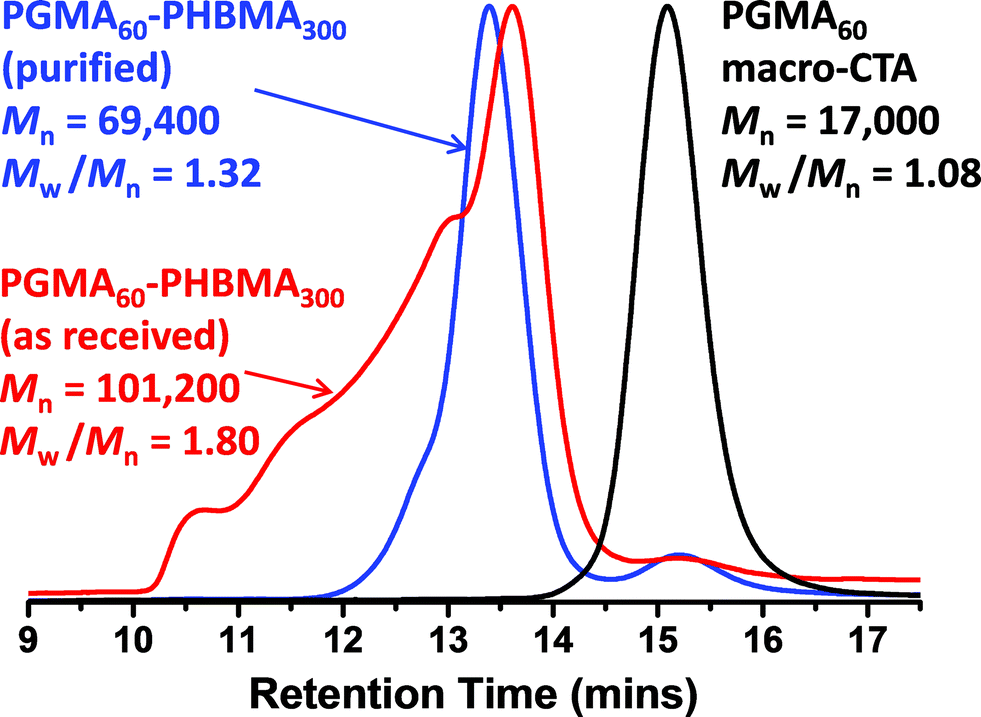 | ||
| Fig. 2 DMF GPC curves obtained for PGMA60–PHBMA300 diblock copolymers synthesized at 10% w/w solids at 70 °C via RAFT aqueous emulsion polymerization using HBMA monomer (as received) and the same batch of monomer after purification via column chromatography to remove dimethacrylate impurities. A macro-CTA/ACVA molar ratio of 4.0 was used in both cases. | ||
In contrast to the PGMA–PHEMA formulation, DLS studies of the diluted PGMA–PHBMA dispersions confirmed successful particle formation (see Fig. 3). The onset of self-assembly appears to correspond to a DP of approximately 75 for the core-forming PHBMA block, which produces spheres with a mean hydrodynamic diameter of 26 nm (polydispersity, PDI = 0.269). Larger particles were obtained when higher PHBMA DPs were targeted, up to a diameter of 95 nm (PDI = 0.044) for PGMA60–PHBMA500. When the solids concentration was increased to 25% w/w, almost no change in size was observed (99 nm diameter, PDI = 0.084) compared to the equivalent block copolymer synthesized at 10% w/w solids. Sphere diameters estimated from TEM images were consistent with DLS measurements (see Fig. 3). The polymerizing reaction mixtures became increasingly turbid up to a PHBMA DP of 300, at which point they appeared milky-white (almost opaque). The final copolymer dispersions were colloidally stable, although small amounts of sediment could be observed when targeting higher PHBMA DPs. It also became more difficult to dissolve the final diblock copolymers in either methanol or DMF above a PHBMA DP of 300. This is most likely because of the higher degree of cross-linking indicated by GPC analysis (see Fig. 2). There was also no change in the spherical particle morphology observed for these dispersions, which is in striking contrast to the corresponding PGMA–PHPMA formulation.33,61,63
 | ||
| Fig. 3 TEM images obtained for PGMA60–PHBMAy diblock copolymers prepared at 10% w/w solids at 70 °C, unless otherwise stated. Target diblock compositions, DLS intensity-average diameters and polydispersities are indicated for each dispersion. A macro-CTA/ACVA molar ratio of 4.0 was used in all cases. Final HBMA conversions exceeded 99% for all polymerizations. | ||
It was postulated that other diblock copolymer morphologies might be accessed (e.g. worms and vesicles) by using a statistical mixture of PHBMA and PHEMA as the core-forming block in order to balance their respective hydrophobic/hydrophilic character. As HEMA contains one CH2 unit less than HPMA and HBMA contains an extra CH2 unit, a 1:1 molar ratio should produce a core-forming block that was essentially isomeric to PHPMA, providing that both comonomers were fully consumed. However, this also meant that the copolymerizations would be conducted under essentially emulsion conditions (i.e. in the presence of monomer droplets).
Firstly, the relative rates at which the individual comonomers became incorporated into the statistical copolymer core-forming block were studied. A PGMA60–P(HBMA268–HEMA268) diblock copolymer was targeted and the statistical copolymerization of HBMA with HEMA was conducted at 70 °C and periodically sampled over several hours for 1H NMR spectroscopy studies. Dilution of the aqueous reaction solution using CD3OD led to dissolution of all components. The total comonomer conversion (98% within 2.5 h) was readily determined by monitoring the disappearance of one of the vinyl signals at 6.17 ppm. This was assigned to the single proton that is cis to the methacrylic ester for both HEMA and HBMA monomers and is labelled (a) in Fig. 4. Consumption of HEMA monomer alone was monitored by following the disappearance of signal (b), which is assigned to the two oxyethylene protons next to the ester group. The total comonomer conversion and this HEMA conversion were used to calculate the HBMA conversion by difference. The initial spatial location of the HEMA and HBMA comonomers in this copolymerization is shown schematically in Fig. 5a.
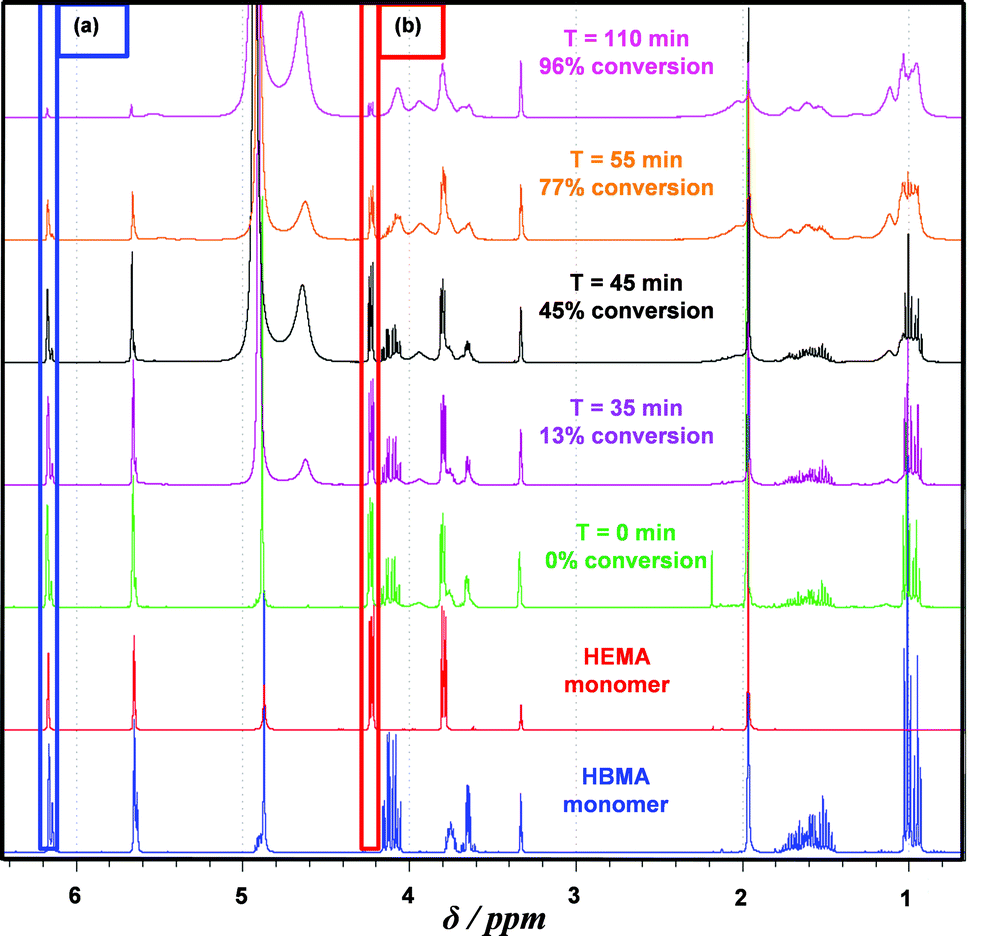 | ||
| Fig. 4 1H NMR spectra recorded at various reaction times for a PGMA60–P(HBMA268–HEMA268) diblock copolymer synthesized via RAFT statistical copolymerization of HBMA and HEMA at 10% w/w solids at 70 °C using a macro-CTA/ACVA molar ratio of 4.0. Conversions were calculated by monitoring the relative reduction of the vinyl proton signal (a) at 6.18 ppm compared to the (co)polymer signals at 3.55–4.25 ppm, which represents disappearance of both vinyl monomers. Consumption of HEMA monomer alone can be determined by monitoring the relative reduction of signal (b) at 4.22 ppm compared to the same (co)polymer signals. | ||
 | ||
| Fig. 5 (a) Schematic representation of the initial spatial location of the HEMA and HBMA comonomers in the RAFT synthesis of PGMA60–P(HBMAy–HEMAy) diblock copolymer nanoparticles. A macro-CTA/ACVA molar ratio of 4.0 was used in all cases. Monomer conversion vs. time curves (as judged by 1H NMR) for HBMA (red circles), (HBMA + HEMA (black squares) and HEMA (blue triangles). (b) Overall comonomer conversion data obtained for PGMA60–P(HBMA268–HEMA268) at 10% w/w solids at 70 °C and the individual comonomer conversions for the statistical copolymerization of HEMA and HBMA. (c) Comparison of the conversion vs. time curves obtained for the same PGMA60–P(HBMA268–HEMA268) synthesis, with PGMA60–PHBMA536 prepared by RAFT emulsion homopolymerization of HBMA at 10.8% w/w solids and PGMA60–PHEMA536 prepared by RAFT solution polymerization of HEMA at 9.2% w/w solids at 70 °C. In these latter two formulations, the % w/w monomer concentration has been adjusted to maintain the same molar concentration. | ||
Inspecting the conversion vs. time plot shown in Fig. 5b (and the corresponding first-order rate plot in Fig. 6a), HBMA initially reacts significantly faster than HEMA (e.g. 14% vs. 4% conversion after 45 min). However, these two comonomers are essentially fully consumed over the course of the copolymerization. This indicates that the copolymer core-forming block ultimately comprises approximately the same comonomer composition as that originally targeted. Given that HEMA is fully water-miscible, and hence located in the same aqueous phase as the macro-CTA and the water-soluble initiator, it is perhaps counter-intuitive that this monomer is actually consumed more slowly than the largely water-immiscible HBMA in the initial stages. On the other hand, Charleux and co-workers have reported relatively fast rates of polymerization for various water-immiscible vinyl monomers when using RAFT aqueous emulsion polymerization formulations.67–69 For example, the homopolymerization of methyl methacrylate using a water-soluble macro-CTA was essentially complete within 2 h at 80 °C.71 Thus it seems that the rate of RAFT aqueous solution/dispersion polymerization of HEMA is significantly less than the rate of RAFT aqueous emulsion polymerization of HBMA.
 | ||
| Fig. 6 Semi-logarithmic plot of monomer conversion vs. time determined by 1H NMR analysis of periodically-sampled aqueous reaction mixtures at 70 °C using a macro-CTA/ACVA molar ratio of 4.0 for: (a) overall rate for the RAFT statistical copolymerization of a 1:1 HBMA/HEMA molar ratio when targeting PGMA60–P(HBMA268–HEMA268) at 10% w/w solids (black squares), and the corresponding individual rates of polymerization of HBMA (red circles) and HEMA (blue triangles) determined for the same copolymerization. (b) Overall rate of RAFT statistical copolymerization for the same PGMA60–P(HBMA268–HEMA268) formulation compared to the overall rates for the RAFT emulsion homopolymerization of HBMA (targeting PGMA60–PHBMA536 at 10.8% w/w solids) and RAFT solution homopolymerization of HEMA (targeting PGMA60–PHEMA536 at 9.2% w/w total solids). (c) Evolution of intensity-average particle diameter (black squares) and count rate (kcps, red triangles) over time for a periodically-sampled PGMA60–P(HBMA268–HEMA268) statistical copolymerization prepared at 10% w/w solids, as determined by DLS studies. The onset of micellar nucleation was observed at approximately 45 min, and was accompanied by a pronounced increase in the scattered light intensity. | ||
To further investigate the relative rate of comonomer incorporation, PGMA60–PHBMA536 and PGMA60–PHEMA536 diblock copolymers were also synthesized where the target DP of the core-forming block was fixed, but the overall solids concentration of each formulation was adjusted accordingly. This was to compensate for the differing monomer masses and hence keep the total number of moles of vinyl monomer(s) constant. All other reaction conditions were identical to those used for the statistical copolymerization of HBMA with HEMA in order to ensure a meaningful comparison.
Comparing Fig. 5c with 5b, the rate of homopolymerization of HBMA is similar to that of its statistical copolymerization with HEMA. However, the rate of homopolymerization of HEMA (87% in 7 h) is clearly substantially slower than its statistical copolymerization with HBMA (89% in 2.5 h). This suggests that the HEMA comonomer eventually partitions into the growing PHBMA-rich micellar nuclei, which are generated during the early stages of the reaction (see schematic representation in Fig. 5a). This higher local HEMA monomer concentration might be expected to produce an increased rate of copolymerization. Indeed, when the same (co)monomer conversion data is replotted to fit a first-order rate equation (see Fig. 6a) a pronounced rate enhancement (for both comonomers) is observed after approximately 45 min. This corresponds to the onset of micellar nucleation for this formulation, as indicated by the DLS studies shown in Fig. 6c. At shorter reaction times, only very low count rates were observed, indicating that particle nucleation had yet to occur. A similar rate enhancement was reported by Blanazs et al.61 for the dispersion polymerization of HPMA, where a PGMA macro-CTA was also used as the reactive steric stabilizer block. In contrast, no such rate enhancement was observed for the equivalent PGMA60–PHEMA536 formulation shown in Fig. 6b, since the PHEMA chains never become sufficiently hydrophobic to ensure effective polymerization-induced self-assembly.
A range of PGMA60–P(HBMAy–HEMAy) statistical diblock copolymers (where y = 50 to 175) were subsequently prepared at 10% w/w solids to assess the morphology of the final nano-objects obtained at full conversion. The GPC curves obtained for this series of copolymers are shown in Fig. 7. The high molecular weight shoulder is much more prominent than that observed for the equivalent PGMA60–PHEMA2y diblock copolymers (see Fig. 1 and Table 1). This was expected, because the HBMA monomer contains a significantly higher level of dimethacrylate cross-linker compared to HEMA monomer. This is reflected in the copolymer polydispersities, which increase from 1.15 for PGMA60–P(HBMA50–HEMA50) up to 1.49 for PGMA60–P(HBMA150–HEMA150). Moreover, these polydispersities are much lower than those obtained for the equivalent PGMA60–PHBMA2y diblock copolymers (see Fig. 1 and Table 1). In all cases high conversions (>99%) were achieved within 16 h, as judged by 1H NMR spectroscopy. This copolymer series produced a range of nano-objects, as judged by TEM and DLS studies (see Fig. 8).
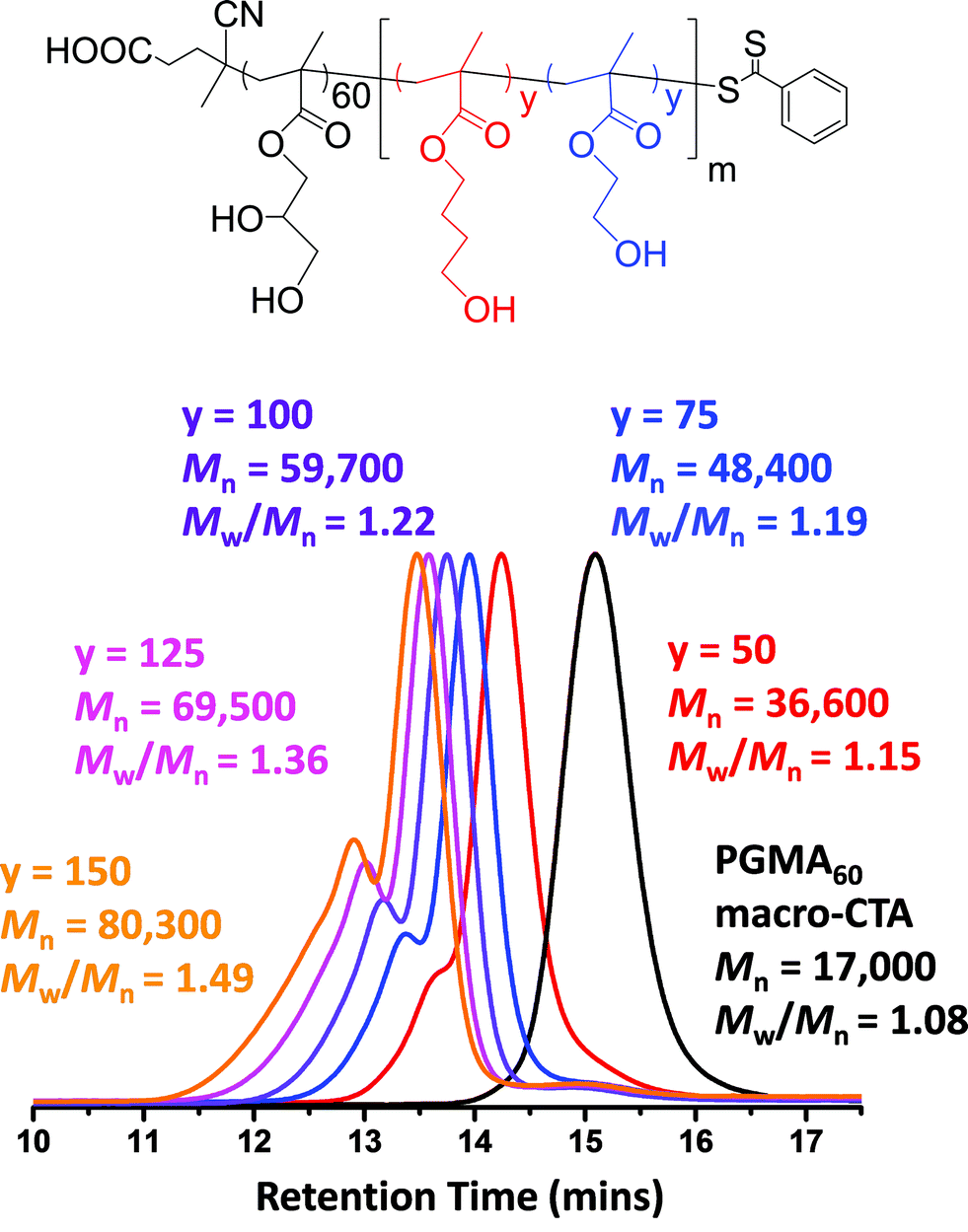 | ||
| Fig. 7 DMF GPC data obtained for a PGMA60 macro-CTA and a series of PGMA60–P(HBMAy–HEMAy) statistical diblock copolymers synthesized at 10% w/w solids at 70 °C. A macro-CTA/ACVA molar ratio of 4.0 was used in all cases. All copolymerizations reached high conversions (> 99%), as judged by 1H NMR spectroscopy. | ||
 | ||
| Fig. 8 TEM images obtained for PGMA60–P(HBMAy–HEMAy) statistical diblock copolymers synthesized at 10% w/w solids at 70 °C. A macro-CTA/ACVA molar ratio of 4.0 was used in all cases. Target diblock compositions, DLS intensity-average diameters and polydispersities are indicated for each dispersion. Final comonomer conversions exceeded 99% for each formulation. | ||
The PGMA60–P(HBMA50–HEMA50) statistical diblock copolymer dispersion was only weakly turbid and exhibited a purely spherical morphology. A mixed phase of spheres and worms was observed for PGMA60–P(HBMA75–HEMA75), which was also slightly more viscous than the other samples. PGMA60–P(HBMA85–HEMA85) and PGMA60–P(HBMA115–HEMA115) also contained worms as well as spheres, but PGMA60–P(HBMA115–HEMA115) contained larger spheres/vesicles. Mostly larger vesicular nano-objects were obtained for PGMA60–P(HBMA125–HEMA125) diblock copolymer, with very few worms.
The 10% w/w copolymer dispersions became noticeably more turbid when targeting longer core-forming blocks up to PGMA60–P(HBMA125–HEMA125), which corresponds to a vesicular morphology (see TEM images in Fig. 8). These vesicles are smaller and less polydisperse in size than those reported by Blanazs et al.33,61 for PGMA–PHPMA diblock copolymers. For example, targeting a core-forming DP of 125 resulted in a DLS intensity-average diameter of 89 nm with a PDI of 0.078. When the target DP was increased to 150, the particle diameter was 115 nm (PDI = 0.067). Finally, for a DP of 175 the particle diameter was 101 nm (PDI = 0.05). These DLS values are in reasonable agreement with the particle diameters observed by TEM. Further characterization using small-angle X-ray scattering (SAXS) to confirm the vesicular morphology is planned for future studies. When these copolymerizations were conducted at higher copolymer concentrations, comparable molecular weight data was obtained for copolymers of the same target core DP (compare Fig. 7 with Table 2). However, the range of nano-objects observed by TEM were more akin to those previously reported for the prototypical PGMA–PHPMA formulation.33,61 In particular, much larger vesicles were obtained compared to those formed at 10% w/w solids, which leads to the characteristic ‘buckling’ of the vesicle membrane under the ultrahigh vacuum conditions required for TEM studies. A detailed phase diagram was subsequently constructed (see Fig. 9) using a large batch of the PGMA60 macro-CTA, which has similar generic features to those previously reported by our group.33 Well-defined spheres and polydisperse vesicles can be obtained at all copolymer concentrations investigated by targeting either a relatively short or a relatively long core-forming P(HBMAy–HEMAy) block, respectively. It is emphasized that this phase diagram contains some kinetically-trapped morphologies (e.g. spheres formed at 10% w/w solids) as well as equilibrium morphologies (e.g. vesicles at 20% w/w solids).33 Nevertheless, it serves as an important ‘road map’ for the reproducible synthesis of pure copolymer morphologies.
:1 molar ratio. A macro-CTA/ACVA molar ratio of 4.0 was used in all cases. All polymerizations proceeded to high (>99%) conversion, as judged by 1H NMR spectroscopy
| Entry no. | Solids content % w/w | Targeted block composition | M n (g mol−1) | M w/Mn |
|---|---|---|---|---|
| 1 | 12.5 | PGMA60–P(HBMA95–HEMA95) | 62000 |
1.33 |
| 2 | 13.5 | PGMA60–P(HBMA95–HEMA95) | 64900 |
1.29 |
| 3 | 17.5 | PGMA60–P(HBMA85–HEMA85) | 57600 |
1.31 |
| 4 | 15 | PGMA60–P(HBMA40–HEMA40) | 33000 |
1.13 |
| 5 | 15 | PGMA60–P(HBMA50–HEMA50) | 37000 |
1.14 |
| 6 | 15 | PGMA60–P(HBMA65–HEMA65) | 41300 |
1.18 |
| 7 | 15 | PGMA60–P(HBMA75–HEMA75) | 47000 |
1.18 |
| 8 | 15 | PGMA60–P(HBMA90–HEMA90) | 50600 |
1.28 |
| 9 | 15 | PGMA60–P(HBMA100–HEMA100) | 57500 |
1.27 |
| 10 | 15 | PGMA60–P(HBMA110–HEMA110) | 59100 |
1.39 |
| 11 | 15 | PGMA60–P(HBMA125–HEMA125) | 66500 |
1.38 |
| 12 | 15 | PGMA60–P(HBMA150–HEMA150) | 77000 |
1.46 |
| 13 | 20 | PGMA60–P(HBMA40–HEMA40) | 32200 |
1.13 |
| 14 | 20 | PGMA60–P(HBMA50–HEMA50) | 36600 |
1.15 |
| 15 | 20 | PGMA60–P(HBMA60–HEMA60) | 40300 |
1.16 |
| 16 | 20 | PGMA60–P(HBMA70–HEMA70) | 44900 |
1.19 |
| 17 | 20 | PGMA60–P(HBMA80–HEMA80) | 47200 |
1.26 |
| 18 | 20 | PGMA60–P(HBMA90–HEMA90) | 50900 |
1.26 |
| 19 | 20 | PGMA60–P(HBMA100–HEMA100) | 57300 |
1.26 |
| 20 | 20 | PGMA60–P(HBMA110–HEMA110) | 55400 |
1.38 |
| 21 | 20 | PGMA60–P(HBMA125–HEMA125) | 66400 |
1.37 |
| 22 | 20 | PGMA60–P(HBMA150–HEMA150) | 75600 |
1.42 |
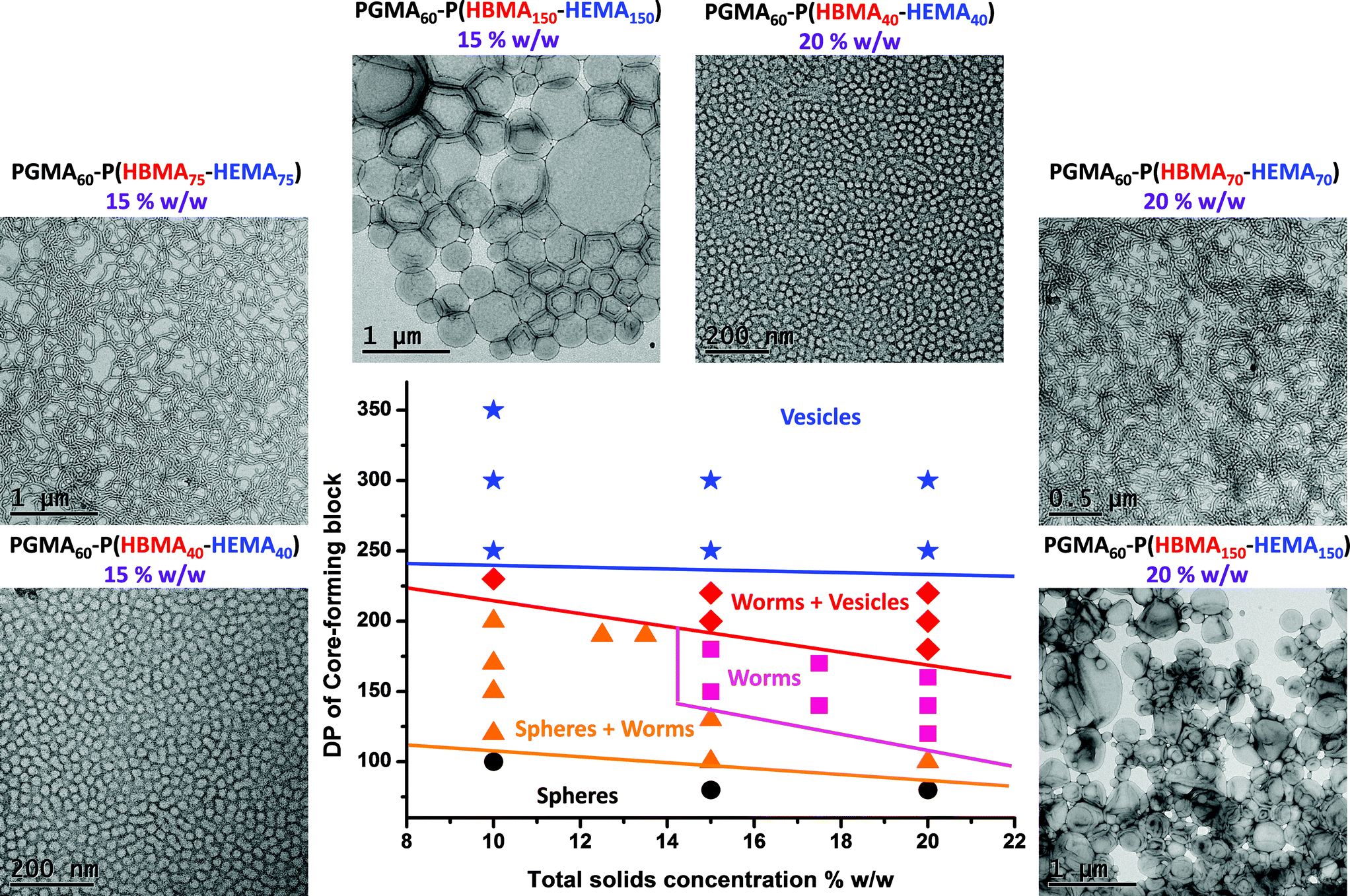 | ||
| Fig. 9 Phase diagram constructed for PGMA60–P(HBMAy–HEMAy) statistical diblock copolymers prepared at 70 °C by RAFT copolymerization of HBMA and HEMA at a 1:1 molar ratio. A macro-CTA/ACVA molar ratio of 4.0 was used in all cases. Selected TEM images are shown for specific diblock compositions and copolymer concentrations, confirming that pure sphere, worm and vesicle morphologies can be obtained under appropriate conditions. | ||
In contrast, a pure worm phase is only accessible at higher copolymer concentrations, although it is significantly broader (encompassing around 50–60 monomer repeat units at 20% w/w) than the worm phase identified for the PGMA–PHPMA formulation.62,63 Copolymers that lie within the pure worm phase (and also some of those located in the surrounding mixed phases) form soft, free-standing gels (as judged by a tube inversion test) of varying turbidity at 20 °C. PGMA–PHPMA diblock copolymer gels exhibit thermo-responsive behavior, with reversible degelation occurring on cooling to around 5–10 °C because of a worm-to-sphere transition associated with a subtle increase in the degree of hydration of the core-forming PHPMA block, see Fig. 10a.63 Remarkably, these new PGMA60–P(HBMAy–HEMAy) copolymer worm gels exhibit qualitatively different thermo-sensitivity, see Fig. 10b and 10c.
 | ||
| Fig. 10 Thermo-responsive behavior observed for (a) PGMA60–PHPMA160, (b) PGMA60–P(HBMA80–HEMA80) and (C) PGMA60–P(HBMA50–HEMA50) copolymer worm gels prepared at 70 °C and 15% w/w solids by RAFT (co)polymerization (the latter copolymer worms were prepared as described by Verber et al.,62 except that 2-cyano-2-propyl dithiobenzoate (CPDB) was used as a RAFT CTA in this example). A macro-CTA/ACVA molar ratio of 4.0 was used in all cases. The digital photographs indicate the thermo-reversible behavior that is observed on cooling to 5 °C for 1 h. | ||
For example, a PGMA60–P(HEMA80–HBMA80) copolymer at 15% w/w solids forms a free-standing gel at 20 °C and remains in this state on cooling to 4 °C for 1 h, see Fig. 10b. Given that PHPMA is isomeric with a 1:1 HBMA/HEMA statistical block of the same mean DP, this non-responsive behavior is wholly unexpected. Moreover, a PGMA60–P(HEMA50–HBMA50) copolymer at the same concentration is a viscous fluid at 20 °C, but becomes a free-standing gel on cooling to 4 °C for 1 h, see Fig. 10c. This behavior is entirely complementary to that shown in Fig. 10a. Thus it appears that a range of gelation behavior (both non-responsive and thermo-responsive) can be obtained for these new statistical block copolymers depending on their precise block compositions. Accordingly, detailed rheological studies of these new copolymer worm gels have been undertaken, which will be reported elsewhere in due course.
Conclusions
A prototypical PGMA–PHPMA RAFT aqueous dispersion polymerization formulation has been extended by varying the nature of the core-forming block. Replacing PHPMA block with PHEMA did not result in particle formation, since the latter block has insufficient hydrophobic character to drive in situ self-assembly. In contrast, targeting a PHBMA core-forming block allowed the synthesis of well-defined spherical micelles of tunable size via RAFT aqueous emulsion polymerization. However, this formulation does not allow the production of worm-like or vesicular morphologies and the diblock copolymer chains exhibit relatively high polydispersities because the HBMA monomer contains a dimethacrylate impurity. A 1:1 HEMA/HBMA molar ratio was subsequently used to produce a range of diblock copolymer nano-objects via a RAFT aqueous emulsion PISA formulation. Such a statistical copolymer core-forming block is actually isomeric with the previously reported PHPMA core-forming block.32 The statistical copolymerization proceeded rapidly (98% comonomer conversion within 2.5 h at 70 °C). Closer analysis via1H NMR spectroscopy indicated that HBMA is consumed faster than HEMA in the early stages of the copolymerization. Thus, to a first approximation, once nucleation occurs the initial reaction mainly involves RAFT aqueous emulsion polymerization of the water-immiscible HBMA. The HEMA then becomes partitioned into the growing water-insoluble cores and is efficiently copolymerized along with the remaining HBMA. These observations are somewhat counter-intuitive given that the relatively unreactive HEMA monomer is co-located in the aqueous phase with the water-soluble PGMA macro-CTA and azo initiator. However, it is certainly consistent with the observation that the RAFT aqueous solution homopolymerization of HEMA proceeds much more slowly than the RAFT aqueous emulsion homopolymerization of HBMA. The resulting PGMA60–P(HBMAy-stat-HEMAy) diblock copolymers had relatively low polydispersities compared to the corresponding PHBMA core-forming block and this new RAFT PISA formulation allowed access to the full range of copolymer morphologies (spheres, worms and vesicles), as judged by TEM and DLS studies. A detailed phase diagram was constructed by systematic variation of the copolymer concentration and the mean degree of polymerization of the core-forming block. Pure sphere, worm and vesicle phases were obtained for syntheses conducted at higher copolymer concentrations. In particular, the thermo-sensitive gelation behavior of PGMA60–P(HBMAy-stat-HEMAy) worms differs qualitatively from that previously reported for the isomeric PGMA–PHPMA diblock copolymer worm formulation.
Acknowledgements
We thank EPSRC and the Scott Bader company for funding a PhD studentship for LPDR. Scott Bader is also thanked for permission to publish this work. SPA acknowledges receipt of a five-year ERC Advanced Investigator grant (PISA 320372).Notes and references
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- L. Zhang and A. Eisenberg, Polym. Adv. Technol., 1998, 9, 677–699 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. II, 1976, 72, 1525–1568 RSC.
- N. Petzetakis, D. Walker, A. P. Dove and R. K. O'Reilly, Soft Matter, 2012, 8, 7408–7414 RSC.
- C. Price, Pure Appl. Chem., 1983, 55, 1563–1572 CrossRef CAS.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CAS.
- Y.-Y. Won, H. T. Davis and F. S. Bates, Science, 1999, 283, 960–963 CrossRef CAS.
- Z. Li, E. Kesselman, Y. Talmon, M. A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98–101 CrossRef CAS PubMed.
- X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS PubMed.
- H. Cui, Z. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647–650 CrossRef CAS PubMed.
- J. Raez, I. Manners and M. A. Winnik, J. Am. Chem. Soc., 2002, 124, 10381–10395 CrossRef CAS PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- M. Massignani, C. LoPresti, A. Blanazs, J. Madsen, S. P. Armes, A. L. Lewis and G. Battaglia, Small, 2009, 5, 2424–2432 CrossRef CAS PubMed.
- M. H. Stenzel, Chem. Commun., 2008, 3486–3503 RSC.
- C. LoPresti, H. Lomas, M. Massignani, T. Smart and G. Battaglia, J. Mater. Chem., 2009, 19, 3576–3590 RSC.
- J. Du and R. K. O'Reilly, Soft Matter, 2009, 5, 3544–3561 RSC.
- S. J. Holder and N. A. J. M. Sommerdijk, Polym. Chem., 2011, 2, 1018–1028 RSC.
- K. Matyjaszewski, Y. Gnanou and L. Leibler, Macromolecular Engineering: Precise Synthesis, Materials Properties, Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2007 Search PubMed.
- D. R. Arifin and A. F. Palmer, Biomacromolecules, 2005, 6, 2172–2181 CrossRef CAS PubMed.
- A. K. Brannan and F. S. Bates, Macromolecules, 2004, 37, 8816–8819 CrossRef CAS.
- P. P. Ghoroghchian, G. Li, D. H. Levine, K. P. Davis, F. S. Bates, D. A. Hammer and M. J. Therien, Macromolecules, 2006, 39, 1673–1675 CrossRef CAS PubMed.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- P. B. Zetterlund, Y. Kagawa and M. Okubo, Chem. Rev., 2008, 108, 3747–3794 CrossRef CAS PubMed.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS PubMed.
- J. Jagur-Grodzinski, React. Funct. Polym., 2001, 49, 1–54 CrossRef CAS.
- M. J. Monteiro and M. F. Cunningham, Macromolecules, 2012, 45, 4939–4957 CrossRef CAS.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Soft Matter, 2012, 8, 3753–3767 Search PubMed.
- Y. Li and S. P. Armes, Angew. Chem., Int. Ed., 2010, 49, 4042–4046 CrossRef CAS PubMed.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- J. Rieger, C. Grazon, B. Charleux, D. Alaimo and C. Jérôme, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2373–2390 CrossRef CAS PubMed.
- C. Grazon, J. Rieger, N. Sanson and B. Charleux, Soft Matter, 2011, 7, 3482–3490 RSC.
- G. Liu, Q. Qiu, W. Shen and Z. An, Macromolecules, 2011, 44, 5237–5245 CrossRef CAS.
- W. Shen, Y. Chang, G. Liu, H. Wang, A. Cao and Z. An, Macromolecules, 2011, 44, 2524–2530 CrossRef CAS.
- G. Liu, Q. Qiu and Z. An, Polym. Chem., 2012, 3, 504–513 RSC.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2011, 28, 914–922 CrossRef PubMed.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2013, 29, 7416–7424 CrossRef CAS PubMed.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707–15713 CrossRef CAS PubMed.
- S. Sugihara, S. P. Armes, A. Blanazs and A. L. Lewis, Soft Matter, 2011, 7, 10787–10793 RSC.
- C.-Q. Huang and C.-Y. Pan, Polymer, 2010, 51, 5115–5121 CrossRef CAS PubMed.
- W. Cai, W. Wan, C. Hong, C. Huang and C.-Y. Pan, Soft Matter, 2010, 6, 5554–5561 RSC.
- W.-D. He, X.-L. Sun, W.-M. Wan and C.-Y. Pan, Macromolecules, 2011, 44, 3358–3365 CrossRef CAS.
- C.-Q. Huang, Y. Wang, C.-Y. Hong and C.-Y. Pan, Macromol. Rapid Commun., 2011, 32, 1174–1179 CrossRef CAS PubMed.
- E. R. Jones, M. Semsarilar, A. Blanazs and S. P. Armes, Macromolecules, 2012, 45, 5091–5098 CrossRef CAS.
- M. Semsarilar, E. R. Jones, A. Blanazs and S. P. Armes, Adv. Mater., 2012, 24, 3378–3382 CrossRef CAS PubMed.
- J. M. Lee, B. H. Lee and S. Choe, Polymer, 2006, 47, 3838–3844 CrossRef CAS PubMed.
- J. R. McKee, V. Ladmiral, J. Niskanen, H. Tenhu and S. P. Armes, Macromolecules, 2011, 44, 7692–7703 CrossRef CAS.
- P. J. Saikia, J. M. Lee, K. Lee and S. Choe, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 872–885 CrossRef CAS PubMed.
- W.-M. Wan and C.-Y. Pan, Polym. Chem., 2010, 1, 1475–1484 RSC.
- D. Zehm, L. P. D. Ratcliffe and S. P. Armes, Macromolecules, 2012, 46, 128–139 CrossRef.
- M. Bathfield, F. D'Agosto, R. Spitz, M.-T. Charreyre, C. Pichot and T. Delair, Macromol. Rapid Commun., 2007, 28, 1540–1545 CrossRef CAS PubMed.
- L. Houillot, C. Bui, C. l. Farcet, C. Moire, J.-A. Raust, H. Pasch, M. Save and B. Charleux, ACS Appl. Mater. Interfaces, 2010, 2, 434–442 CAS.
- L. Houillot, C. Bui, M. Save, B. Charleux, C. Farcet, C. Moire, J.-A. Raust and I. Rodriguez, Macromolecules, 2007, 40, 6500–6509 CrossRef CAS.
- G. Zheng and C. Pan, Macromolecules, 2005, 39, 95–102 CrossRef.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- Z. An, Q. Shi, W. Tang, C.-K. Tsung, C. J. Hawker and G. D. Stucky, J. Am. Chem. Soc., 2007, 129, 14493–14499 CrossRef CAS PubMed.
- W. Shen, Y. Chang, G. Liu, H. Wang, A. Cao and Z. An, Macromolecules, 2011, 44, 2524–2530 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- R. Verber, A. Blanazs and S. P. Armes, Soft Matter, 2012, 8, 9915–9922 RSC.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
- S. Boissé, J. Rieger, K. Belal, A. Di-Cicco, P. Beaunier, M.-H. Li and B. Charleux, Chem. Commun., 2010, 46, 1950–1952 RSC.
- I. Chaduc, W. Zhang, J. Rieger, M. Lansalot, F. D'Agosto and B. Charleux, Macromol. Rapid Commun., 2011, 32, 1270–1276 CrossRef CAS PubMed.
- M. Manguian, M. Save and B. Charleux, Macromol. Rapid Commun., 2006, 27, 399–404 CrossRef CAS PubMed.
- J. Rieger, F. Stoffelbach, C. Bui, D. Alaimo, C. Jérôme and B. Charleux, Macromolecules, 2008, 41, 4065–4068 CrossRef CAS.
- F. Stoffelbach, L. Tibiletti, J. Rieger and B. Charleux, Macromolecules, 2008, 41, 7850–7856 CrossRef CAS.
- W. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 7584–7593 CrossRef CAS.
- X. Zhang, J. Rieger and B. Charleux, Polym. Chem., 2012, 3, 1502–1509 RSC.
- W. Zhang, F. D'Agosto, P.-Y. Dugas, J. Rieger and B. Charleux, Polymer, 2013, 54, 2011–2019 CrossRef CAS PubMed.
- W. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2012, 45, 4075–4084 CrossRef CAS.
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191–2204 CrossRef CAS.
- D. E. Ganeva, E. Sprong, H. de Bruyn, G. G. Warr, C. H. Such and B. S. Hawkett, Macromolecules, 2007, 40, 6181–6189 CrossRef CAS.
- I. Chaduc, A. Crepet, O. Boyron, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2013, 46, 6013–6023 CrossRef CAS.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Langmuir, 2011, 28, 1196–1205 CrossRef PubMed.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Macromolecules, 2012, 45, 5081–5090 CrossRef CAS.
- J. V. M. Weaver, I. Bannister, K. L. Robinson, X. Bories-Azeau, S. P. Armes, M. Smallridge and P. McKenna, Macromolecules, 2004, 37, 2395–2403 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- J. Rosselgong and S. P. Armes, Macromolecules, 2012, 45, 2731–2737 CrossRef CAS.
- J. Rosselgong, S. P. Armes, W. R. S. Barton and D. Price, Macromolecules, 2010, 43, 2145–2156 CrossRef CAS.
Footnote |
| † Present address: BASF SE, GMV/P – B001, 67056 Ludwigshafen, Germany. |
| This journal is © The Royal Society of Chemistry 2014 |