 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Establishing α-bromo-γ-butyrolactone as a platform for synthesis of functional aliphatic polyesters – bridging the gap between ROP and SET-LRP†
Peter
Olsén
,
Jenny
Undin
,
Karin
Odelius
and
Ann-Christine
Albertsson
*
Department of Fibre and Polymer Technology, KTH Royal Institute of Technology, SE-100 44, Stockholm, Sweden. E-mail: aila@kth.se
First published on 27th February 2014
Abstract
Utilizing α-bromo-γ-butyrolactone (αBrγBL) as a comonomer with ε-caprolactone (εCL) or L-lactide (LLA) produces copolymers with active and available grafting sites, e.g., for SET-LRP, where the choice of the grafting monomers is limited only by one's imagination. This was deduced by utilizing a wide range of different acrylates of varying polarities and was realized with the aid of a fluorinated alcohol, 2,2,2-trifluoroethanol, which acts as a universal solvent for both the hydrophobic macroinitiators and the grafting monomers. Using αBrγBL successfully provides a simple route to merge the two polymerization methodologies, ROP and SET-LRP. αBrγBL inherently meets all of the prerequisites to act as a platform monomer for the synthesis of functional aliphatic polyesters, i.e., it is inexpensive, available, and able to form isolated grafting sites along the polymer chain. The copolymerization of αBrγBL together with two of the most commonly used cyclic ester monomers, ε-CL and LLA, proceeds with a high degree of control and a linear relationship between the feed ratio of αBrγBL and its composition in the copolymer. The formation of isolated units of αBrγBL in the copolymer is visualized by the reactivity ratios of the copolymerization reactions and confirmed by 13C-NMR spectroscopy. The incorporation of isolated αBrγBL is the feature that makes this class of copolymers unique, and it can be considered to provide a route to the “perfect graft copolymer” with a degradable backbone.
Introduction
In the early era of polymer science, great emphasis was placed on the mere ability to create polymers, whereas today, functionality is the key. Functional polymers open up applications with endless possibilities, where properties can be tailored, altered, and/or maintained over the complete lifetime of the material. In light of this, the focus today is on conferring function to the main chain of the polymer. One class of polymers that is inherently of great value for many applications is aliphatic polyesters; because of their ester functionality, they most often degrade within a reasonable time frame. Unfortunately, many of these monomers lack sites that allow alterations and modifications of the polymer backbone.Therefore, a major scientific focus has been on imparting different functionalities to aliphatic polyesters. The routes pursued range from copolymerization with functional monomers1–4 and post-polymerization modification5,6 to functional initiators,7,8 to name a few. They all have their pros and cons, yet copolymerization with functional monomers offers a high degree of functionality, most often with the retention of a high degree of control. Important aspects to consider when copolymerizing a functional monomer are its reactivity, maintenance of its function, and how the functionality is spread throughout the polymeric chain. The routes for the synthesis of functional monomers appropriate for ring-opening polymerization are, in theory, infinite. In the literature, there exist descriptions of numerous elegant monomers that yield the desired polymeric properties, but they are often hindered by lengthy synthetic routes resulting in low yields at high cost, thereby limiting their applicability to a large scale.
Hence, we felt inspired to find an inexpensive and straightforward monomer that can bestow the desired functionality on commonly used aliphatic polyesters. We therefore turned our attention towards a relatively unexploited family of lactones: the γ-lactones.9 γ-Lactones are a class of monomers with a complicated past; unsubstituted γ-butyrolactone was identified as being unable to polymerize as early as 1932 by Carothers et al.10 Subsequently, there have been numerous attempts and even quantum mechanical calculations reconfirming this statement.11–14 Not considering polymers synthesized under extreme pressure and high temperature or the formation of oligomers,15–19 this statement still holds true.
Many monomers that are unable to homopolymerize, however, can be copolymerized, as shown with γ-butyrolactone, 1,3-dioxane, 1,4-dioxane, etc.20,21 In 1964, Tada et al. performed the first copolymerization of γ-butyrolactone together with the more ring-strained monomer, β-propiolactone.22 γ-Butyrolactone's ability to act as a comonomer was further explored in terms of its thermodynamic and kinetic behavior, and the altered properties of the synthesized copolymers have been documented.16,18,23–29 The main objective for forming these copolymers includes the use of an inexpensive, flexible, and degradable monomer that, together with β-butyrolactone, resembles polymers produced by bacterial fermentation, i.e., polyhydroxybutyrate.23–29 Recently, γ-butyrolactone's ability to form isolated units along the polymer chain at moderate conversion during copolymerization has been highlighted.11,13
Combining ring-opening polymerization with controlled radical graft polymerization offers the ability to specify the graft copolymer for a specific task. Therefore, much effort has been concentrated on developing monomers with these characteristics, starting in 1999, when ε-caprolactone with an attached ATRP initiator at the γ-position was synthesized.1 Others have realized this feature by synthesizing α-substituted ε-caprolactones with chlorine and bromine thus successfully producing radical graft copolymers with degradable backbones.2,30,31 However, factors that are often overlooked include how the monomers with radical initiating sites are dispersed throughout the polymer chain and what the useable grafting monomers are.
To overcome these shortcomings, we turned our attention towards a scarcely used building block, α-bromo-γ-butyrolactone. We believe that this monomer inherently has desirable properties such as being inexpensive and having functionality and low homo-reactivity, together with being two-sided, i.e., both having the ability to be ring-opened while at the same time functioning as a SET-LRP initiator. Its anticipated inability to form a homopolymer should result in isolated sites that are susceptible to SET-LRP, thus providing “the perfect graft copolymer”. The hypothesis is that this building block will provide an easy way to merge SET-LRP with controlled ROP. Our aim is to establish α-bromo-γ-butyrolactone as a platform monomer for the synthesis of functional aliphatic polyesters. This will be achieved by copolymerizing α-bromo-γ-butyrolactone with L-lactide or ε-caprolactone, followed by a sequential grafting of acrylates of varying polarities: n-butyl acrylate, methyl methacrylate, and 2-hydroxyethyl methacrylate, under SET-LRP conditions with the aid of the universal solvent, 2,2,2-trifluoroethanol.
Experimental
Materials
ε-Caprolactone (εCL) (Aldrich) was dried over calcium hydride for at least 24 h and subsequently distilled at reduced pressure under an inert atmosphere prior to use. L-Lactide (LA) (≥99%, Boehringer Ingelheim) was recrystallized twice from toluene (HPLC grade, Fisher Scientific, Germany) and once from dry toluene (99.8%, Sigma-Aldrich, Sweden) and was dried in vacuo for at least 48 h prior to use. α-Bromo-γ-butyrolactone (αBrγBL) (97%, Sigma-Aldrich, Sweden) was dried over molecular sieves (3 Å) and stored under an inert atmosphere prior to use.Stannous octoate (Sn(Oct)2) (Sigma-Aldrich, Sweden) was dried over molecular sieves (3 Å) before use. n-Butyl acrylate (nBuAc) (Alfa Aesar, Germany), methyl methacrylate (MMA) (Merck, Germany), and 2-hydroxyethyl methacrylate (HEMA) (Aldrich, Germany) were purified by passing through aluminum oxide (Merck Chemicals, Germany) prior to use. Tris[2(dimethylamino)ethyl]amine (Me6TREN) (Sigma-Aldrich, Sweden) and Cu(II)Br2 (Sigma-Aldrich, Sweden) were stored under a nitrogen atmosphere prior to use. Benzyl alcohol (≥99%, Sigma-Aldrich, Sweden), chloroform (HPLC grade, Fisher Scientific, Germany), methanol (general purpose grade, Fisher Scientific, Germany), 2,2,2-trifluoroethanol (TFE) (Sigma-Aldrich, Sweden), and chloroform-d (99.8%, with silver foil, Cambridge Isotope Laboratories) were used as received.
Polymerization reactions
After 20 h, the reaction mixture was cooled to room temperature, and the copolymers were dissolved in chloroform and precipitated three consecutive times in methanol. The precipitates were dried under reduced pressure for 4 days.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :catalyst:ligand:monomer ratio of 1:1:2:50. In other 25 mL double-necked round-bottom flasks equipped with three-way valves and septa, the different initiator polymers, poly(εCL-r-αBrγBL) or poly(LLA-r-αBrγBL), that were chosen based on the highest amount of incorporated αBrγBL, were dissolved in TFE. Both systems were cooled in dry ice, and the air was removed by three freeze–vacuum–nitrogen–thaw cycles. The initiator polymers were subsequently transferred into the first round-bottom flask under an inert atmosphere. A 10 cm length of copper wire (Cu(0)) with a diameter of 20 gauge was then added to the flask. The flask was immersed in a thermostatted oil bath at 25 °C. Samples were withdrawn at specific time intervals under a nitrogen atmosphere, and the conversion and molecular weights were determined by 1H-NMR and GPC. The reaction was ended after 15 h. The resulting polymer was dissolved in chloroform and precipitated in cold methanol and then dried at room temperature until all the solvent had evaporated.
:catalyst:ligand:monomer ratio of 1:1:2:50. In other 25 mL double-necked round-bottom flasks equipped with three-way valves and septa, the different initiator polymers, poly(εCL-r-αBrγBL) or poly(LLA-r-αBrγBL), that were chosen based on the highest amount of incorporated αBrγBL, were dissolved in TFE. Both systems were cooled in dry ice, and the air was removed by three freeze–vacuum–nitrogen–thaw cycles. The initiator polymers were subsequently transferred into the first round-bottom flask under an inert atmosphere. A 10 cm length of copper wire (Cu(0)) with a diameter of 20 gauge was then added to the flask. The flask was immersed in a thermostatted oil bath at 25 °C. Samples were withdrawn at specific time intervals under a nitrogen atmosphere, and the conversion and molecular weights were determined by 1H-NMR and GPC. The reaction was ended after 15 h. The resulting polymer was dissolved in chloroform and precipitated in cold methanol and then dried at room temperature until all the solvent had evaporated.
Instruments
000 g mol−1.
Results and discussion
Our aim was to establish α-bromo-γ-butyrolactone (αBrγBL) as a platform monomer for the synthesis of functional aliphatic polyesters. The hypothesis was that αBrγBL, an easily accessible monomer, will inherently act as a bridge between ring-opening polymerization (ROP) and grafting by single electron transfer living radical polymerization (SET-LRP) (Scheme 1). It was also anticipated that αBrγBL, when copolymerized by ring-opening polymerization, would form isolated sites along the copolymer chain. Although αBrγBL was shown to possess all of these features, it has, to our knowledge, previously been neglected in this context, and it has been shown that the unopened αBrγBL by itself can act as a radical initiator for ATRP.32 To fully elucidate the properties of αBrγBL, we concentrated on two different questions: how does αBrγBL copolymerize with commonly used lactones or lactides focusing on the kinetics and the formed macromolecular architecture, and how does it behave during a grafting step via SET-LRP. | ||
| Scheme 1 Synthetic outline for the formation of aliphatic polyester graft copolymers. First, ring-opening polymerization of α-bromo-γ-butyrolactone with ε-caprolactone or L-lactide forms the macroinitiator, followed by the grafting of different acrylates via SET-LRP. | ||
Grafting of acrylates from the aliphatic polyester main chain
There is a great focus in polymer science on joining controlled radical polymerization with controlled ring-opening polymerization. More specifically, we are interested in forming radical graft copolymers using acrylic monomers on a degradable backbone composed of aliphatic polyesters.1,2,30,31,33–35One method that holds vast potential for grafting from aliphatic polyesters is SET-LRP. SET-LRP was originally developed by Percec et al. for the synthesis of polyvinyl chloride and later for polyacrylates and provides a polymerization method with excellent control and high chain end vitality.36–38 Our group has previously grafted various acrylates via SET-LRP from a hydrophilic hemicellulose backbone, where the required polarity of the solvents is more a necessity rather than a drawback.39–42 But the polarity of these solvent restricts the use of aliphatic polyesters such as poly(ε-caprolactone) (PCL) as a polymeric grafting initiator due to its hydrophobicity. However, recently, the same group together with Haddleton et al. resolved this issue by introducing a new class of solvents for SET-LRP, fluorinated alcohols.43–48 These solvents have been coined “universal solvents” for SET-LRP, as they open the possibility of using hydrophobic monomers, and in our case, open the possibility of using a hydrophobic pre-polymer as an initiator.
The copolymerization of α-bromo-γ-butyrolactone (αBrγBL) with εCL or LLA yielded a macroinitiator with active and available grafting sites for SET-LRP. The grafting of the different acrylic monomers, methyl methacrylate (MMA), 2-hydroxyethyl methacrylate (HEMA), and butyl acrylate (nBuAc) from poly(εCL-r-αBrγBL), proceeded in a controlled manner with a linear relationship between the conversion and the molecular weight (Fig. 1). However, when using poly(LLA-r-αBrγBL) as a macroinitiator, the grafting of MMA was accompanied by severe degradation of the main chain (Fig. 1). The exact nature of this degradation behavior is still under investigation. Most likely it is due to the fact that Me6-TREN can act as a transesterification catalyst during grafting, which is further supported by the dispersity evolution of poly(εCL-r-αBrγBL) grafted with both MMA and HEMA. All selected monomers were successfully grafted onto the polymer backbone, thereby highlighting the versatility and ability of αBrγBL to act as a bridge between SET-LRP and ROP, for a wide range of monomers.
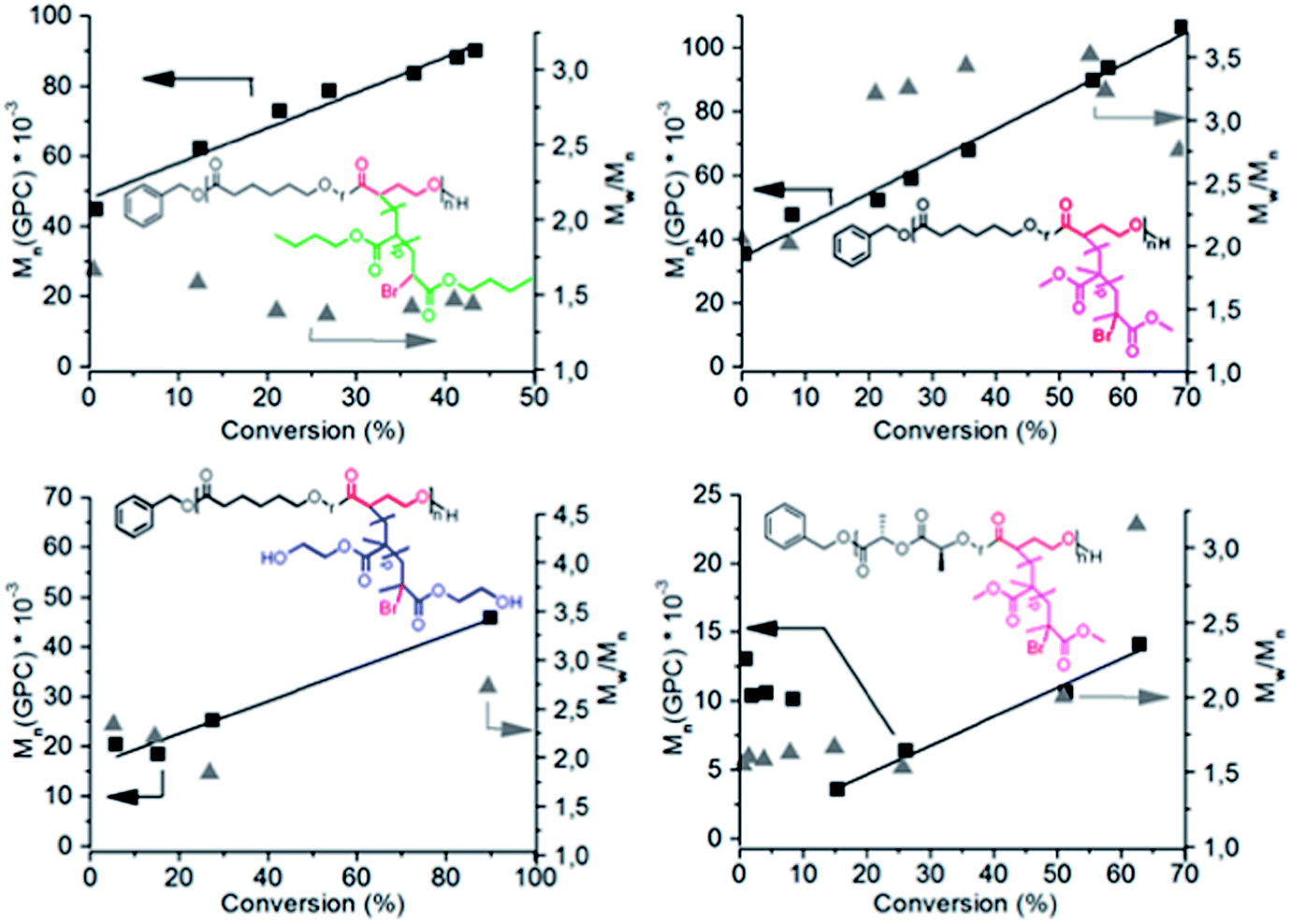 | ||
| Fig. 1 The relationship of the monomer consumption with the molecular weight obtained from GPC of the SET-LRP grafted copolymers of poly(LLA-r-αBrγBL) and poly(εCL-r-αBrγBL). The polymerization reactions were conducted with a Me6-TREN/αBrγBL ratio of 2 and 10 cm of copper wire. | ||
Elucidating the copolymerization behavior of αBrγBL with εCL or LLA
In the 1930s, Carothers et al. stated that “the γ-lactones and other five-membered cyclic esters show no tendency to polymerize, and no corresponding polymers are known”. This was concluded after γBL had been heated both in the presence and absence of catalyst for one year.10 Later, in the end of the 1990s, γBL was polymerized using Al(OiPr)3 as a catalyst, forming short oligomers.16,18 Although oligomers were formed, the original statement is still valid to a large extent, and hence similar behavior is to be expected for αBrγBL. This was also verified in this work when no homopolymerization of αBrγBL was observed after 20 h with Sn(Oct)2 as a catalyst at 110 °C.The chemical structure of αBrγBL suggests that the most suitable ROP catalyst would be a coordination–insertion catalyst. This is based on the notion that any catalyst with a slightly basic character, such as 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), and 4-(dimethylamino)pyridine (DMAP), could lead to an elimination of the bromine moiety at the α-position, hence removing the monomer's ability to further act as an initiator for SET-LRP. Therefore, Sn(Oct)2 was chosen as a catalyst for the copolymerization of αBrγBL with εCL or LLA.
The change in the chemical shift of the α-proton for αBrγBL of the precipitated copolymers of αBrγBL and either εCL or LLA revealed that the monomer had been incorporated into the polymer chain (Fig. 2a and b). The difference in the shift of the α-proton upon copolymerization of αBrγBL depends on which comonomer was used, i.e., εCL or LLA, and the chemical shifts of the α-proton of the αBrγBL unit were δP(αBrγBL-r-εCL) = 4.34 ppm and δP(αBrγBL-r-LLA) = 4.43 ppm when polymerized with εCL and LLA, respectively. This also indicates that there is an absence of homosequences of αBrγBL along the polymer chain, which would lead to the appearance of a peak at the same chemical shift in both copolymers.
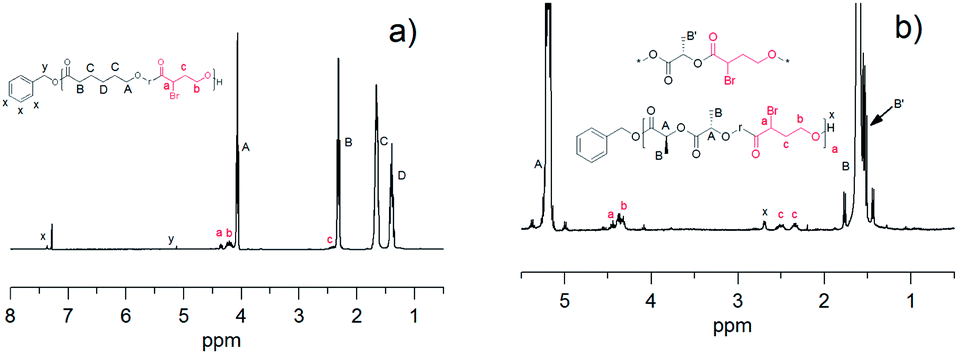 | ||
| Fig. 2 1H-NMR spectra of the copolymers (a) P(εCL-r-αBrγBL) and (b) P(LLA-r-αBrγBL). | ||
Kinetic features of the copolymerization of αBrγBL with εCL or LLA
The kinetic features of the copolymerization of αBrγBL with either εCL or LLA were examined using two main experiments: the amount of monomer incorporation with a varying feed ratio of αBrγBL and the monomer consumption at prolonged reaction times. Although the five-membered lactone ring is considered easy to open, it is also easy to close. Therefore, the intuitive trend would be that the lower the ratio of αBrγBL to the comonomer, the higher the conversion of αBrγBL would be. In other words, the incorporation of the monomer is a matter of statistics. If there exist more reactive monomers in the vicinity of the newly ring-opened αBrγBL that can react with the active chain end, the probability of αBrγBL to be “locked-in” the polymerizing chain is increased.To visualize how the conversion of αBrγBL is affected by the initial feed ratio, several reactions were conducted at a constant monomer-to-initiator ratio of the more reactive monomer, εCL and LLA, [M]/[I] = 400 or 200, where only the ratio of αBrγBL was varied. The notion of a “locking-in” methodology during copolymerization was based on the idea that the most probable addition of αBrγBL occurs at the chain end during the propagation of the chain and not through trans-esterification-based ROP.
The incorporated amount and conversion of αBrγBL during copolymerization with both εCL and LLA follow the expected trends. That is, the higher the feed ratio of αBrγBL to the comonomer, the more units are incorporated into the main chain, and the lower is the monomer's total conversion (Fig. 3a and b). The amount of αBrγBL was determined by 1H-NMR spectroscopy and calculated using the difference in the chemical shifts of the α-proton of the monomer and the formed polymer (Table 1). Composition of the copolymers of αBrγBL and εCL or LLA as a function of varying feed ratios and monomer-to-initiator ratios.
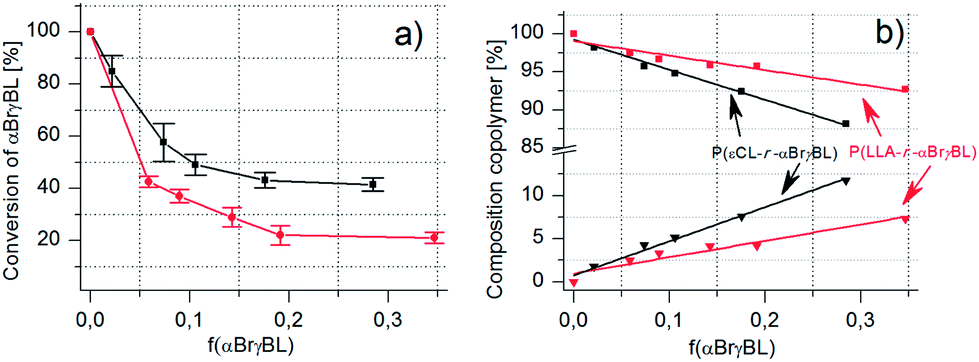 | ||
Fig. 3 (a and b) Monomer consumption and copolymer composition with increased feed ratio of αBrγBL in copolymers with εCL or LLA ( = P(LLA-r-αBrγBL)) and = P(LLA-r-αBrγBL)) and  = (P(εCL-r-αBrγBL)). = (P(εCL-r-αBrγBL)). | ||
| Polymer | M1 | M2 | F(αBrγBL)a | [I]:[M1]:[M2] |
F(αBrγBL)b | M n (GPC) | D |
|---|---|---|---|---|---|---|---|
| a Feed ratio [M2]/[M1] mol%. b Polymer composition ratio [M2]/[M1]. | |||||||
| P(εCL-r-αBrγBL) | εCL | αBrγBL | 0.03 | 1:400:8 |
0.02 | 54800 |
1.61 |
| P(εCL-r-αBrγBL) | εCL | αBrγBL | 0.07 | 1:400:30 |
0.04 | 52700 |
1.57 |
| P(εCL-r-αBrγBL) | εCL | αBrγBL | 0.11 | 1:400:44 |
0.05 | 46000 |
1.71 |
| P(εCL-r-αBrγBL) | εCL | αBrγBL | 0.18 | 1:400:72 |
0.08 | 48100 |
1.69 |
| P(εCL-r-αBrγBL) | εCL | αBrγBL | 0.28 | 1:400:116 |
0.12 | 35600 |
1.68 |
| P(LLA-r-αBrγBL) | LLA | αBrγBL | 0.06 | 1:200:12 |
0.02 | 17300 |
1.28 |
| P(LLA-r-αBrγBL) | LLA | αBrγBL | 0.09 | 1:200:18 |
0.03 | 22600 |
1.20 |
| P(LLA-r-αBrγBL) | LLA | αBrγBL | 0.14 | 1:200:28 |
0.04 | 17000 |
1.15 |
| P(LLA-r-αBrγBL) | LLA | αBrγBL | 0.19 | 1:200:38 |
0.04 | 18900 |
1.18 |
| P(LLA-r-αBrγBL) | LLA | αBrγBL | 0.35 | 1:200:70 |
0.07 | 20100 |
1.17 |
It is possible to incorporate quite a high amount of αBrγBL into the copolymers (up to 12 mol%) (Table 1). This is, however, connected to a low total conversion of αBrγBL during copolymerization (Fig. 3a). The low conversion of αBrγBL would be considered a major drawback if it simply acted as a property-altering monomer, but because its main purpose is to act as an initiator for SET-LRP, the incorporated amount is more than enough. The limited degree of incorporation could even be considered an advantage, i.e., if the conversion of αBrγBL is high, the formation of homosequences is more likely. It has been shown that during the copolymerization of γ-butyrolactone (γBL) and εCL when the conversion exceeds 12%, the block sequences of γBL were formed.13 This result was in contrast to what had previously been shown, that is, the formation of isolated monomers even at conversions as high as 22%.11 Although there is some discrepancy in the numbers, it should be safe to conclude that if the conversion is below 12%, copolymers with isolated γBL units are formed. Hence, the polymerization behavior of γBL is used as a template for the anticipated polymerization behavior of αBrγBL.
During controlled polymerization, control over the dispersity of the formed polymers is of immense importance. For the performed copolymerization reactions, it is evident that when εCL was used as a comonomer, higher dispersities (1.6–1.7) were attained, in contrast to the results for LLA (1.2–1.3). Possible explanations could be either that the aggregation state during bulk polymerization at 110 °C is vital, PCL has a Tm ∼ 60 °C, PLLA has a Tm ∼ 160 °C, or that the difference in reactivity of εCL and LLA results in a different dispersity (Table 1). To elucidate the mechanism of αBrγBL addition, we conducted kinetic experiments where the dependence of Mn, D, monomer consumption, and the composition of αBrγBL and εCL on time were determined (Fig. 4). The reaction conditions chosen were 110 °C, 1 mol% Sn(Oct)2, [MCL]/[I] = 400, and [MCL]/[MαBrγBL] = 2.
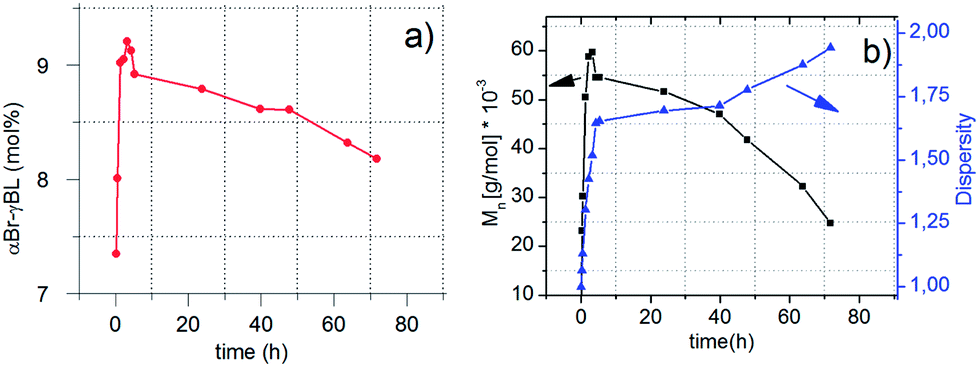 | ||
| Fig. 4 (a and b) Conversion in the copolymerization of αBrγBL and εCL as a function of time (a) and the effects on the number molecular weight (Mn) and dispersity of the copolymer (b). | ||
The number average molecular weight and dispersity of the αBrγBL and εCL copolymers show a clear dependence on the polymerization time (Fig. 4a and b). During the initial stage of the copolymerization, the addition of αBrγBL shows a linear relationship to the conversion of εCL. Hence, the addition of αBrγBL is dependent on having a more reactive monomer, in this case εCL, acting as an end-capping monomer. If there is no more reactive monomer available in the reaction, after the addition of αBrγBL to the propagating chain end, it will ring-close again and resume its monomeric form. When the more reactive monomer is fully consumed, the addition of αBrγBL stops, and the molecular weight decreases rapidly, together with an increase of the dispersity (Fig. 4a and b). The molecular weight behavior of the copolymers is consistent with what has been observed during the copolymerization of γBL and LLA, which was attributed to the occurrence of transesterification reactions.23 In contrast to what was found here, they did not observe any reduction in the amount of γBL in the copolymers with time. The reduction observed here is believed to be a consequence of αBrγBL being more easily transesterified than εCL, resulting in chain ends of αBrγBL that, for thermodynamic reasons, ring-closes to produce the monomeric unit, thus reducing the amount of αBrγBL in the copolymer.
Architectural features of the copolymerization of αBrγBL with εCL and LLA
A good way to describe copolymerization behavior is to use the system-specific reactivity ratios during copolymerization. To calculate these ratios, it is vital to keep the conversion low (often below 35%) in order to obtain accurate values. The restricted ability of αBrγBL to homopolymerize will render a relatively true value of the reactivity ratios even at maximum conversions of the most reactive monomer (i.e., εCL or LLA). Even so, all calculations were performed at a conversion below 20%.The reactivity ratios were calculated using the Fineman and Ross method,49 where the reactivity ratios, r1 = k11/k12 and r2 = k22/k21, are given as the ratios of the rate constants between the four different possible copolymerization reactions. The r1 reactivity ratios of the copolymerization of αBrγBL with εCL and LLA were determined to be 4.4 and 18.5, respectively, whereas the r2 ratios were close to zero for both (Fig. 5a). This can mean two things: the non-existence of the αBrγBL chain-end addition to the monomeric form of αBrγBL, or the reactivity for the addition to εCL is many times higher. If αBrγBL was able to homopolymerize, the latter explanation would result in the formation of “diblock-like” copolymers. However, αBrγBL's inability to homopolymerize yields a solid conclusion of the macromolecular architecture based on the reactivity ratio r2, that is, isolated αBrγBL units are formed throughout the polymer main chain. Although the r2 value for the copolymerization of αBrγBL and LLA does not rule out the formation of homosequences of αBrγBL, the deviation from zero is interpreted as a function of the line regression rather than the system itself.
 | ||
| Fig. 5 (a and b) The Fineman and Ross method was used to calculate the reactivity ratios as shown by the linear fit (a). The 13C-NMR spectrum shows the isolated units of α-bromo-γ-butyrolactone (αBrγBL) along the polymer chain during copolymerization with ε-caprolactone (εCL) (b). | ||
εCL situated at the propagating chain end has a higher reactivity towards αBrγBL during copolymerization than LLA. This is revealed by its r1 value being almost four times smaller than that determined for LLA (Fig. 2 and Table 1). This is in line with what was anticipated for the copolymerization. The difference in reactivity of the propagating chain end originates from a primary hydroxyl being more reactive than a secondary hydroxyl, as shown by the difference in the homopolymerization and copolymerization of εCL and LLA. The rate of homopolymerization of εCL is much higher than that of PLLA in similar systems, although the ring strain is higher for LLA. During the copolymerization of LLA and εCL, this results in a gradient copolymer, where LLA is predominant in the beginning and εCL in the end.50
The existence of isolated units of αBrγBL along the main chain of the copolymers was further verified by 13C-NMR spectroscopy. The carbonyl group is very sensitive to its neighboring units;51 this makes it a valuable tool to verify the architectural features of copolymers. Isolated αBrγBL along the main chain of the εCL copolymer was found as three distinct peaks of equal intensity (Fig. 4b). For copolymers with εCL, there is often assumed triplet behavior, meaning that each carbonyl group is affected by its two neighboring units.18 In summation, the copolymers synthesized are shown to be isolated αBrγBL units along the chain, providing excellent sites for subsequent SET-LRP.
Conclusions
α-Bromo-γ-butyrolactone (αBrγBL) as a comonomer with ε-caprolactone (εCL) or L-lactide (LLA) produces copolymers (poly(εCL-r-αBrγBL) or poly(εCL-r-αBrγBL), respectively) with active and available grafting sites for SET-LRP. The different grafted acrylates range from hydrophobic n-butyl acrylate and methyl methacrylate to hydrophilic 2-hydroxyethyl methacrylate. These copolymerization reactions were accomplished with the aid of a fluorinated alcohol, 2,2,2-trifluoroethanol, which acts as a universal solvent for both the hydrophobic macroinitiator and the grafting monomers. The grafting via SET-LRP from poly(εCL-r-αBrγBL) for all acrylates proceeded in a controlled manner with a linear relationship between the conversion and the molecular weight. This successfully shows that αBrγBL provides a versatile and simple route to merge the two polymerization methodologies, ROP and SET-LRP.The copolymerization of αBrγBL together with two of the most commonly used cyclic ester monomers, ε-CL, and LLA, proceeds with high control, and a linear relationship between the feed ratio of αBrγBL and its composition in the copolymer is observed. During the copolymerization, the consumption of εCL and αBrγBL is linearly related to each other, although the rate is lower for αBrγBL. When the most active comonomer, εCL, is fully consumed, the conversion of αBrγBL stops. We can therefore conclude that the addition of αBrγBL occurs mainly at the active chain end rather than as an effect of transesterification. Its inherent inability to form homo-sequences under ordinary polymerization conditions was observed both in the 13C-NMR spectra, which only displayed peaks originating from isolated αBrγBL units along the polymer chain, and from the calculated reactivity ratios.
We believe that αBrγBL inherently holds all the prerequisites to act as a platform monomer for the synthesis of functional aliphatic polyesters, i.e., it is inexpensive, available, and able to form isolated grafting sites along the polymer chain. The incorporation of isolated αBrγBL is a feature that makes this class of copolymers unique and is considered to provide a route to the “perfect graft copolymer” with a degradable backbone.
Acknowledgements
The authors acknowledge an ERC Advance Grant from PARADIGM (Grant agreement no. 246776), which financially supported this work.Notes and references
- D. Mecerreyes, B. Atthoff, K. A. Boduch, M. Trollsås and J. L. Hedrick, Macromolecules, 1999, 32, 5176–5182 Search PubMed.
- S. Lenoir, R. Riva, X. Lou, C. Detrembleur, R. Jérôme and P. Lecomte, Macromolecules, 2004, 37, 4055–4061 CrossRef CAS.
- F. Coumes, V. Darcos, D. Domurado, S. Li and J. Coudane, Polym. Chem., 2013, 4, 3705–3713 RSC.
- F. Jing and M. Hillmyer, J. Am. Chem. Soc., 2008, 130, 13826–13827 CrossRef CAS PubMed.
- P. Olsén, K. Odelius and A.-C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3039–3045 CrossRef.
- S. Ponsart, J. Coudane and M. Vert, Biomacromolecules, 2000, 1, 275–281 CrossRef CAS PubMed.
- M. Ryner, A. Finne, A. Albertsson and H. R. Kricheldorf, Macromolecules, 2001, 34, 7281–7287 CrossRef CAS.
- P. Dubois, N. Ropson, R. Jérôme and P. Teyssié, Macromolecules, 1996, 29, 1965–1975 CrossRef CAS.
- T. Moore, R. Adhikari and P. Gunatillake, Biomaterials, 2005, 26, 3771–3782 CrossRef CAS PubMed.
- W. H. Carothers, G. L. Dorough and F. J. Van Natta, J. Am. Chem. Soc., 1932, 54, 761–772 CrossRef CAS.
- M. Nishiura, Z. Hou, T. Koizumi, T. Imamoto and Y. Wakatsuki, Macromolecules, 1999, 32, 8245–8251 CrossRef CAS.
- V. V. Burlakov, A. V. Letov, P. Arndt, W. Baumann, A. Spannenberg, C. Fischer, L. I. Strunkina, M. K. Minacheva, Y. S. Vygodskii, U. Rosenthal and V. B. Shur, J. Mol. Catal. A: Chem., 2003, 200, 63–67 CrossRef CAS.
- S. Agarwal and X. Xie, Macromolecules, 2003, 36, 3545–3549 CrossRef CAS.
- K. N. Houk, A. Jabbari, H. K. Hall and C. Aleman, J. Org. Chem., 2008, 73, 2674–2678 CrossRef CAS PubMed.
- A. Nakayama, N. Kawasaki, S. Aiba, Y. Maeda and I. Arvanitoyannis, Polymer, 1998, 39, 1213–1222 CrossRef CAS.
- A. Duda, T. Biela and J. Libiszowski, Polym. Degrad. Stab., 1998, 59, 215–222 CrossRef CAS.
- G. Nobes, R. Kazlauskas and R. Marchessault, Macromolecules, 1996, 9297, 4829–4833 CrossRef.
- A. Duda, S. Penczek, P. Dubois, D. Mecerreyes and R. Jérôme, Macromol. Chem. Phys., 1996, 1273–1283 CrossRef CAS.
- F. Korte and W. Glet, Polym. Lett., 1966, 4, 685–689 CrossRef CAS.
- R. Szymanski, Makromol. Chem., 1991, 2943–2959 CrossRef CAS.
- M. Bednarek, T. Biedron, P. Kubisa and S. Penczek, Makromol. Chem., Macromol. Symp., 1991, 487, 475–487 CrossRef.
- K. Tada, Y. Numata, T. Saegusa and J. Furukawa, Makromol. Chem., 1964, 77, 220–228 CrossRef CAS.
- A. Nakayama, N. Kawasaki, I. Arvanitoyannis, S. Aiba and N. Yamamoto, J. Environ. Polym. Degrad., 1996, 4, 205–211 CrossRef CAS.
- C. Lee, R. Urakawa and Y. Kimura, Macromol. Chem. Phys., 1997, 1120, 1109–1120 CrossRef.
- A. Bhaw-Luximon, D. Jhurry, S. Motala-timol and Y. Lochee, Macromol. Symp., 2006, 60–68 CAS.
- L. Ubaghs, M. Waringo, H. Keul and H. Höcker, Macromolecules, 2004, 37, 6755–6762 CrossRef CAS.
- H. Fukuzaki, Y. Aiba, M. Yoshida, M. Asano and M. Kumakura, Makromol. Chem., 1989, 1153–1559 Search PubMed.
- H. R. Kricheldorf, J. Thomas Mang and M. Jonté, Macromol Chem., 1985, 955–976 CrossRef CAS.
- K. Ito, T. Inoue and Y. Yamashita, Makromol. Chem., 1970, 139, 153–164 CrossRef CAS.
- R. Riva, S. Lenoir, R. Jérôme and P. Lecomte, Polymer, 2005, 46, 8511–8518 CrossRef CAS.
- G. Wang, Y. Shi, Z. Fu, W. Yang, Q. Huang and Y. Zhang, Polymer, 2005, 46, 10601–10606 CrossRef CAS.
- F. Zeng, Y. Shen, S. Zhu and R. Pelton, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3821–3827 CrossRef CAS.
- H. Tian, Z. Tang, X. Zhuang, X. Chen and X. Jing, Prog. Polym. Sci., 2012, 37, 237–280 CrossRef CAS.
- S. Han, H. Wan, D. Lin, S. Guo, H. Dong, J. Zhang, L. Deng, R. Liu, H. Tang and A. Dong, Acta Biomater., 2014, 10, 670–679 CrossRef CAS PubMed.
- S. Guo, Y. Huang, T. Wei, W. Zhang, W. Wang, D. Lin, X. Zhang, A. Kumar, Q. Du, J. Xing, L. Deng, Z. Liang, P. C. Wang, A. Dong and X.-J. Liang, Biomaterials, 2011, 32, 879–889 CrossRef CAS PubMed.
- V. Percec, A. V. Popov, E. Ramirez-Castillo, M. Monteiro, B. Barboiu, O. Weichold, A. D. Asandei and C. M. Mitchell, J. Am. Chem. Soc., 2002, 124, 4940–4941 CrossRef CAS PubMed.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed.
- N. H. Nguyen, M. E. Levere and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 860–873 CrossRef CAS.
- J. Voepel, U. Edlund, A.-C. Albertsson and V. Percec, Biomacromolecules, 2011, 12, 253–259 CrossRef CAS PubMed.
- J. Voepel, U. Edlund and A.-C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2366–2372 CrossRef CAS.
- U. Edlund, C. Rodriguez-Emmenegger, E. Brynda and A.-C. Albersson, Polym. Chem., 2012, 3, 2920–2927 RSC.
- U. Edlund and A.-C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2650–2658 CrossRef CAS.
- S. R. Samanta and V. Percec, Polym. Chem., 2014, 5, 169–174 RSC.
- S. R. Samanta, H.-J. Sun, A. Anastasaki, D. M. Haddleton and V. Percec, Polym. Chem., 2014, 5, 89–95 RSC.
- S. R. Samanta and V. Percec, Polym. Chem., 2014, 5, 169–174 RSC.
- S. R. Samanta, M. E. Levere and V. Percec, Polym. Chem., 2013, 4, 3212–3224 RSC.
- S. R. Samanta, A. Anastasaki, C. Waldron, D. M. Haddleton and V. Percec, Polym. Chem., 2013, 4, 5555–5562 RSC.
- S. R. Samanta, A. Anastasaki, C. Waldron, D. M. Haddleton and V. Percec, Polym. Chem., 2013, 4, 5563–5569 RSC.
- M. Fineman and S. D. Ross, J. Polym. Sci., 1950, 5, 259–265 CrossRef CAS.
- M. Florczak and A. Duda, Angew. Chem., Int. Ed. Engl., 2008, 47, 9088–9091 CrossRef CAS PubMed.
- H. Kricheldorf, J. Jonté and M. Berl, Makromol. Chem., 1985, 38, 25–38 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Equations used for calculation of conversions and compositions, 1H-NMR spectra of nBuAc grafted poly(εCL-r-αBrγBL), and conversion against time from the SET-LRP grafting experiments. See DOI: 10.1039/c4py00148f |
| This journal is © The Royal Society of Chemistry 2014 |