 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pluronic® block-copolymers in medicine: from chemical and biological versatility to rationalisation and clinical advances
Anaïs
Pitto-Barry
* and
Nicolas P. E.
Barry
*
Department of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: A.Pitto-Barry@warwick.ac.uk; N.Barry@warwick.ac.uk
First published on 24th March 2014
Abstract
This mini-review highlights the latest advances in the chemistry and biology of Pluronic® triblock copolymers. We focus on their applications in medicine, as drug delivery carriers, biological response modifiers, and pharmaceutical ingredients. Examples of drug delivery systems and formulations currently in clinical use, clinical trials or preclinical development are highlighted. We also discuss the role that Pluronic® copolymers may play in the innovative design of new nanomedicines in the near future.
 Anaïs Pitto-Barry | Anaïs Pitto-Barry was born in Lyon in 1984. She received her MSc degree in Chemistry from the Ecole Nationale Supérieure de Chimie de Rennes, France in 2008. She graduated with her PhD in 2012 from the Université de Neuchâtel, Switzerland in Supramolecular Chemistry. She then joined the group of Professor O'Reilly at the University of Warwick with a postdoctoral fellowship funded by the Swiss National Science Foundation, before being funded by the EPSRC. |
 Nicolas P. E. Barry | Nicolas Barry received his MSc degree in Chemistry from the Université de Rennes, France in 2008. He obtained his PhD in Organometallic Chemistry under the supervision of Professors Süss-Fink and Therrien at the Université de Neuchâtel, Switzerland, during 2008 to 2011. He then moved to the University of Warwick, as a Swiss National Science Foundation postdoctoral fellow in the laboratory of Professor Sadler, from 2011 to 2013. In 2014, he took up a Leverhulme Early Career fellowship at the University of Warwick to begin independent research. |
Introduction
The design and fabrication of nanosized objects by using nanotechnology tools have recently resulted in a number of conceptual advances, and now directly impact other fields, such as neuroscience, medicine, and energy.1–4 The last decades have seen the emergence of new nanomaterials and the development of safer and more effective medicines (nanomedicines) opening up new perspectives for addressing social and environmental critical issues.5 Among those, synthetic polymer therapeutics are of particular interest in medicine, owing to their synthetic versatility as well as their tuneable and unique properties.6 A number of biologically active polymer–drug conjugates and polymeric formulations, such as micelles, hydrogels and polymer-coated nanoparticles, are currently in clinical development.7 The most commonly used polymers for applications in medicine include polyethylene glycol (PEG), poly(D,L-lactide-co-glycolide) (PLGA), poly(lactic acid) (PLA), poly(glutamic acid) (PGA), poly(caprolactone) (PCL), N-(2-hydroxypropyl)-methacrylate (HPMA) copolymers, and poly(amino acids).8,9 The wide utilisation of amphiphilic block copolymers in nanomedicine not only stems from their attractive fundamental properties useful in solving drug delivery problems, but also from possible intrinsic biological activity.The class of ABA triblock copolymers commercially available as Pluronic® (non-proprietary name “poloxamers”) offers a pool of more than 50 amphiphilic,10 water-soluble and polymorphic materials (A = hydrophilic block poly(ethylene oxide) (PEO) and B = hydrophobic block poly(propylene oxide) (PPO); Fig. 1). The original manufacturer BASF introduced a specific nomenclature for those copolymers consisting of a letter indicating the morphism of each copolymer (liquid (L), paste (P), and flake (F)) followed by one or two figures referring to the Pluronic® grid shown in Fig. 1,11,12 and providing 1/300 of the molar mass of the PPO block per unimer. The last digit shows one-tenth of the molar mass percentage of PEO content per unimer. As an example, P123 and P103 are both pastes, having the same molar mass percentage of PEO per unimer (30%) with longer PO units for P123.
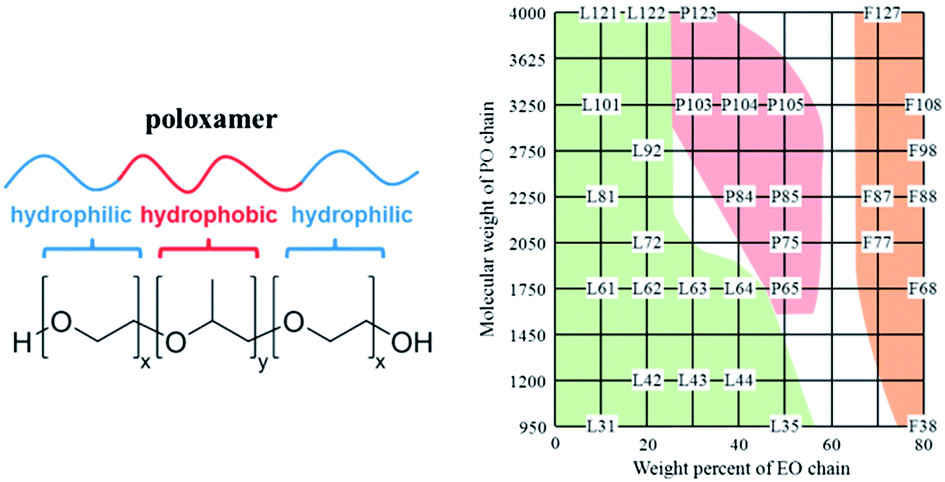 | ||
| Fig. 1 Molecular structure of a Pluronic® triblock copolymer (left) and the Pluronic® grid adapted from ref. 11 and 12 (right; colour code: physical state of copolymers under ambient conditions: green = liquid; red = paste; orange = flake). | ||
The physical and chemical properties of Pluronic® copolymers can be finely tuned by modifying the molar mass ratio between the PEO and PPO blocks (from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 to 8:2), which directly modifies the in vivo properties and interactions with cells and cell membranes, and provides high potential for the design of innovative nanomedicines and new biomaterials.
:9 to 8:2), which directly modifies the in vivo properties and interactions with cells and cell membranes, and provides high potential for the design of innovative nanomedicines and new biomaterials.
In this article we review the properties that make Pluronic® block copolymers unique nanotechnology tools and highlight some areas of the current interest in their biochemistry. We focus especially on the relationship between the physical and chemical properties of Pluronics® and their utilisation in medicine as drug delivery systems, pharmaceutical ingredients and biological response modifiers. Formulations currently in clinical use, clinical trials or preclinical development are highlighted and discussed based on the latest significant advances.
Pluronics® as nanocarriers for drug delivery
In the early 1960s, the emergence of lipid vesicles13 had been a conceptual advance that paved the way for the development of nanomedicines. Such development led to the approval for clinical use of a dozen of nanotechnology therapeutic products. Among those, nanoparticles made of polymers (NPs) are of particular interest owing to their synthetic versatility as well as their tuneable properties. Their utilisation as drug delivery systems originates from their ability to navigate the drug cargo in vivo environment, combined with potential targeting and control of the drug concentration and distribution at the tissue, cell, and subcellular levels.8 They also provide control of drug solubility, by either increasing the aqueous solubility of highly lipophilic drugs or decreasing the solubility of molecules which might otherwise be rapidly excreted, increase the circulation time in blood compared to the corresponding free drug, and may also allow multidrug delivery and theranostics.4As early as 1994, it was demonstrated that nanospheres, synthesised from amphiphilic copolymers composed of two biocompatible blocks (including a polyethylene glycol(PEG)-block), exhibit dramatically increased blood circulation times and low liver accumulation in mice.14 This discovery was followed by numerous reports of PEGylated NPs with various architectures,15 and eventually led to PEG being listed as “Generally Recognised as Safe” (GRAS) by the FDA, and to the clinical translation of a number of PEGylated NPs.8 A number of PEG-containing block copolymers were then developed and among them, the amphiphilic and polymeric characters of Pluronic® block copolymers have raised growing interest for the design of drug delivery systems. In water, and above the critical micelle concentration, these triblock copolymers self-assemble in core–shell micelles comprising hydrophobic PPO blocks as core and hydrated PEO blocks as corona.16 Such polymeric micelles (typically smaller than 100 nm) possess a core–shell structure and poorly water-soluble drugs can be incorporated into the PPO hydrophobic core and protected from inactivation in biological media.
The interest in using poloxamer polymers as drug delivery systems is not recent, and the first anticancer micellar formulation to reach clinical evaluation was actually a mixture of the anticancer drug doxorubicin (Dox; Fig. 2) with Pluronics® L61 and F127 micelles (SP1049C, currently developed by Supratek Pharma Inc.).17 The recent result of a phase II study of SP1049C in patients with advanced adenocarcinoma of the oesophagus and gastroesophageal junction shows a notable antitumor activity and favourable safety profile for SP1049C in such cancers.18 Currently, Supratek Pharma Inc. indicates that SP1049C is in clinical phase 3-stage study for the treatment of cancers that are resistant to doxorubicin. SP1049C has been designated as an orphan drug for carcinoma of the oesophagus and in gastric cancers in the U.S. and is ready to enter a pivotal international clinical phase 3 study. In 2013, Kabanov, a co-founder of Supratek Pharma, Inc., and co-workers reported the ability of SP1049C to deplete cancer stem cells (highly tumorigenic cells with high proliferation potential that are believed to drive tumor growth and progression) and decrease tumorigenicity and aggressiveness of cancer cells in vivo, opening up a potential broader spectrum of action for SP1049C (e.g. in leukemia and breast cancer).19
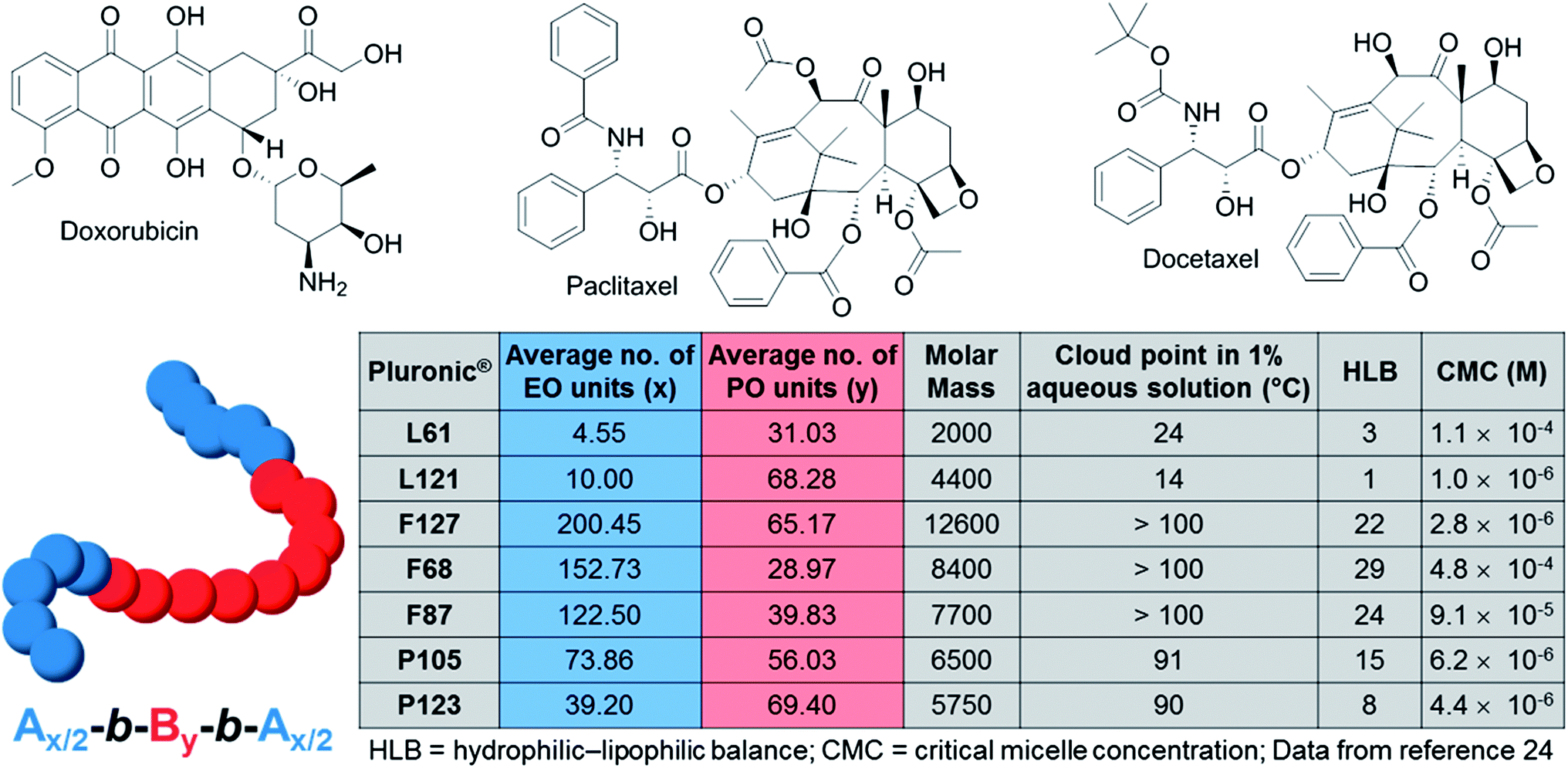 | ||
| Fig. 2 Molecular structures of doxorubicin, paclitaxel, docetaxel and physicochemical characteristics of some of the discussed Pluronic® block copolymers. | ||
The encapsulation of Dox in the Pluronic® micelles shifts its biodistribution, which leads to a better accumulation of the micellar drug in the tumors compared to the free drug, a superior antitumor activity over Dox in a wide range of doxorubicin-sensitive and -resistant human solid and hematopoietic malignancies, and also delays the emergence of Dox-resistance mechanisms. Importantly, the Pluronic® copolymers not only induce the modulation of Dox pharmacokinetic parameters, but are also directly involved in the biological effect of the formulation. Indeed, the P-glycoprotein function in the cell membrane of Dox-resistant cancer cells is disrupted by Pluronic® L61 which also selectively depletes cellular ATP.20
Other clinically used anticancer drugs have been recently encapsulated in Pluronic® copolymer micelles. Docetaxel (DTX; Fig. 2), a semi-synthetic analog of paclitaxel exhibits potency against a variety of solid tumors but is impaired by poor aqueous solubility, rapid phagocytic activity, renal clearance, and non-selective distribution. DTX is clinically used in a formulation with the non-ionic surfactant Tween 80 (polysorbate 80).21 This formulation, named Taxotere®, was approved by the FDA in 2004. However, Taxotere® leads to serious side effects, such as hypersensitivity, nephrotoxicity, neurotoxicity, and incompatibility with common polyvinyl chloride intravenous administration sets.22 In 2013,23 Fang and co-workers demonstrated that DTX encapsulated in Pluronic® P105 and F127 mixed micelles displays a higher cytotoxicity towards A549-Taxol resistant cancer cells than Taxotere® injections. Moreover, 1.85× longer blood time circulation, 3.82× larger area under the plasma concentration–time curve and in vivo therapeutic improvement were observed with DTX-loaded P105/F127 mixed micelles compared to Taxotere®. Nonetheless, further studies will be required for evaluating the toxicity and potential side-effects induced by this Pluronic® formulation.
In 2002,24 Kabanov and co-workers comprehensively reviewed the utilisation of Pluronic® block copolymers for gene delivery. Their ability to increase expression of genes delivered into cells using non-viral vectors, to increase the transfection with adenovirus and lentivirus vectors, to conjugate with polycations, to condense DNA and to form polyplexes, and the regional increase of naked DNA expression in tumors raised considerable attention for gene delivery applications. Since then, a number of promising results have been reported, including the novel poloxamer Synperonic® F108 which enhances lentiviral transduction over the best-in-class polybrene-assisted transduction,25 and a series of small-sized polyethylenimine-conjugated Pluronic® copolymers (PCMs) that enhances the delivery of an antisense phosphorodiamidate morpholino oligomer (PMO) in vitro and in dystrophic mdx mice.26
Pluronics® as biological response modifiers
As briefly exemplified in the previous section, Pluronic® block copolymer drug delivery systems not only are capable of encapsulating drugs and modifying their pharmacokinetic properties, but also are potent biological response modifiers able to sensitise multidrug resistant (MDR) cancer cells and to enhance drug transport across cellular barriers.27 Pluronic® copolymer effects in MDR cells are believed to be due to several important biological mechanisms, including microviscosity modification of the cellular membrane,28 inhibition of the mitochondrial chain and ATP level depletion in cancer and barrier cells (one of the biological modes of action of doxorubicin-containing Pluronic® micelles SP1049C),29 inhibition of drug efflux transporters, such as P-glycoprotein and multidrug resistance proteins,30 pro-apoptotic signalling enhancement and anti-apoptotic defence decrease in the MDR cell,31 inhibition of the glutathione/glutathione S-transferase detoxification system,32 and abolishment of drug retention in cytoplasmic vesicles.33The release of cytochrome C induction and the increase of reactive oxygen species (ROS) levels in the cytoplasm due to mitochondrial disruption in MDR cells by Pluronic® P85 copolymers have also been recently demonstrated by Kabanov and co-workers.34 Differential metabolic responses to Pluronic® in MDR and non-MDR cells and selective respiration inhibition in MDR cell mitochondria were particularly shown.34 The mitochondrial disruption in MDR cells results in ROS level increase and release of cytochrome C (Fig. 3), which leads to the enhancement of the drug-induced apoptosis and contributes to potent chemosensitisation of MDR tumours by the Pluronic® P85 block copolymer.
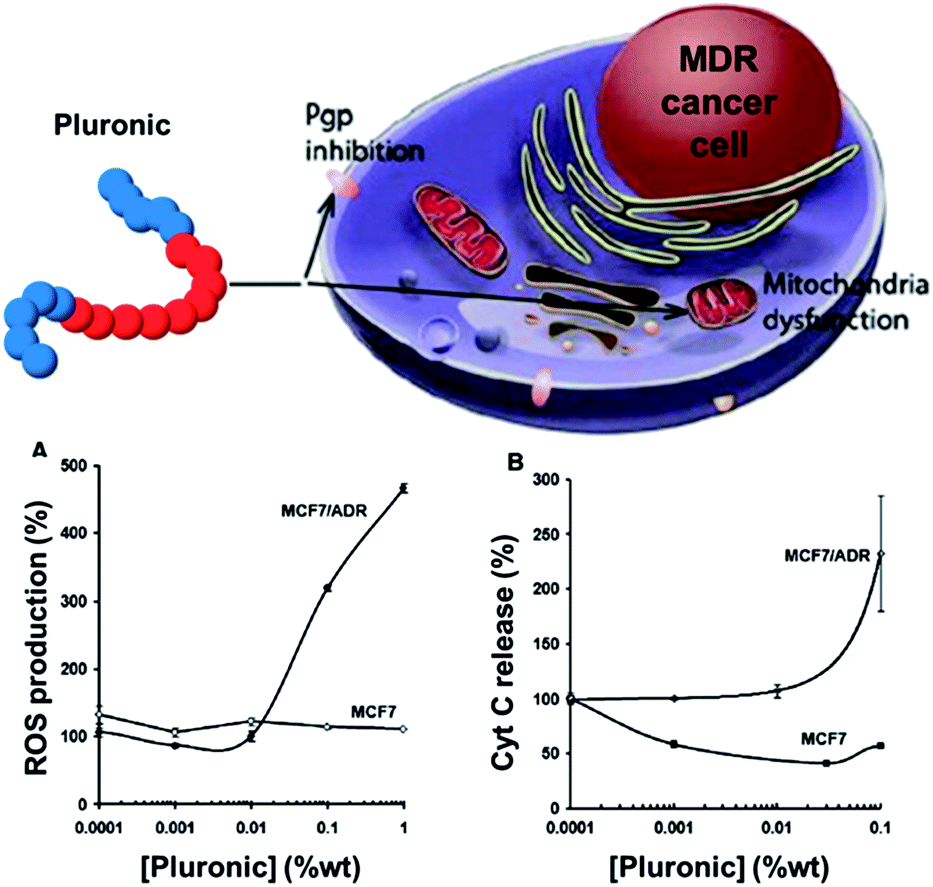 | ||
| Fig. 3 Effect of Pluronic® P85 on (A) ROS production and (B) cytochrome C release in MCF7 and multidrug resistant MCF7/ADR cells. Adapted from ref. 34. | ||
Oxidative stress caused by the generation of ROS is a particularly effective method of killing cancer cells.35 ROS are produced in a wide range of physiological processes, in particular by mitochondria, and act as a ‘double-edged sword’ in cell health. On one hand, they play beneficial roles in cellular signalling; on the other hand, uncontrolled excessive production of ROS, or a diminished ability of cells to scavenge ROS, gives rise to oxidative stress and subsequent damage to various cellular components, including proteins, lipids and DNA. Cancer cells being often under increased oxidative stress compared to normal cells, an element of selectivity is achieved by further increasing the level of oxidative stress.
Remarkably, the effect of Pluronic® block copolymers on cancer cells dramatically depends on their chemical compositions and on the molar mass ratio between the PEO and PPO blocks (a quantified description of the key physicochemical properties of some Pluronics® is shown in Fig. 2). For instance, Pluronic® L61 (with an intermediate length of the PPO block and a relatively hydrophobic structure) was found to be the most efficient poloxamer inhibitor of the drug efflux systems in MDR cells but does not impact the P-glycoprotein function or cellular ATP levels,36 whilst Pluronics® with long PPO blocks (e.g. F127, P123, L121) and high hydrophobicity strongly modify the microviscosity of plasma membranes and potently inhibit the P-glycoprotein function, but suffer from poor cellular uptake. Such dramatic differences in the Pluronic® biological mechanisms offer a rich pool of possible unimers and a combination of unimers that may be used for designing NPs with finely tuned and optimised properties, as both drug delivery systems and biological response modifiers.
Pluronics® as surfactants, and gelation and coating agents for pharmaceutical formulation
Pluronic® block copolymers are polymorph materials that cover a range of gelation states from liquid to paste and solid, depending on the molar mass ratios between the PEO and PPO blocks (see the Pluronic® grid in Fig. 1). This structural versatility makes them particularly attractive emulsifying, solubilising, and dispersing ingredients for pharmaceutical formulation.37 Pluronic® copolymers are listed as excipients in the U.S. and British pharmacopoeias and are widely used in a variety of clinical applications.38,39 Some poloxamer-based devices are also clinically utilised in the U.S. and in Europe, for instance the Pluronic® 407 gel (LeGoo® from Pluromed Inc.) used as off-pump coronary bypass (rapid gel injection after arteriotomy of the abdominal aorta in order to occlude the vessel – Poloxamer 407 – becomes solid within a few seconds).40 A number of clinical trials now involve Pluronic® polymer containing formulations (Table 1).| Industry/sponsor | Pluronic® containing formulation | Use | Stage |
|---|---|---|---|
| Supratek Pharma Inc. | SP1049C: Doxorubicin + Pluronics® L61 and F127 | Advanced esophageal adenocarcinoma | Phase III |
| Mast Therapeutics, Inc. | Purified Poloxamer 188 (based on Pluronic® F68) | Vaso-occlusive crisis | Phase III |
| British Columbia Cancer Agency | Topical amitriptyline 2%, ketamine 1%, and lidocaine 5% in Pluronic® lecitine organogel | Neuropathic pain secondary to radiation therapy | Phase III completed |
| CoDa Therapeutics, Inc. | Nexagon®: Pluronic® gel | Persistent corneal epithelial defects | Phase II |
| Sancilio and Company, Inc. | SC401B (Pluronic® F87 as a surfactant) | Severe hypertriglyceridemia | Phase III |
| Valentis | VLTS-934 (Pluronic®Poloxamer 188) | Peripheral vascular disease | Phase II completed |
The amphiphilicity and surfactant properties of Pluronic® triblock copolymers make them particularly suitable for polymer decoration of up-conversion nanoparticles (UCNPs). UCNPs polymer coating is a technology that has shown promise for increasing the water-solubility, and stability of UCNPs and for functionalising their surface. A range of methods have been investigated for achieving stable and efficient decorations, including ligand exchange,41 ligand oxidation,42 electrostatic adsorption,43 and surface grafting.44
In 2012,45 the Pluronic® F127 was used as an oil-to-water phase transfer for coating UCNPs. Such oil-to-water phase transfer coating was exemplified with NaYF4:Yb,Er(Tm) nanoparticles (see Fig. 4 for the principle of this method). The Pluronic® decoration allowed the production of highly stable up-conversion nanoparticles with strong luminescence properties, low general toxicity and high biocompatibility, which opens new perspectives for the application of UCNPs as bio-imaging agents.
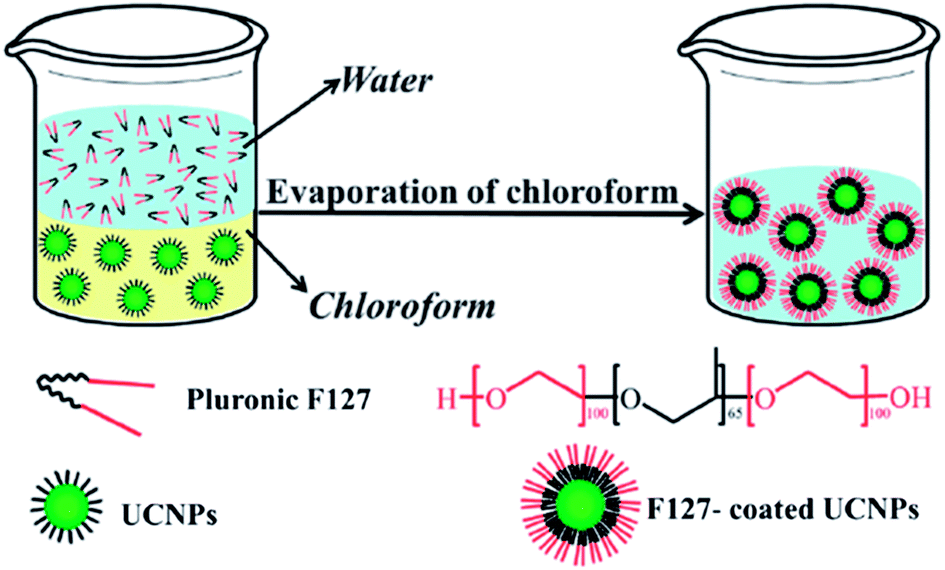 | ||
| Fig. 4 Schematic illustration of UNCP coating through oil-to-water phase transfer. Reproduced from ref. 45 with permission from The Royal Society of Chemistry. | ||
Functionalised Pluronics® and innovative nanocarriers
The studies summarised in this review all point out the importance of the chemical composition of the Pluronic® block copolymer for the physicochemical and biological properties of the resulting Pluronic® formulation. Hydrophobicity and polymorphism are for instance crucial parameters for their biological mode of action. The majority of poloxamer formulations undergoing clinical trials relies either on their physical state properties (e.g. gels for surgery applications, incorporation as surfactants) or on the molar mass and size of the self-assembled micelles (typically between 10 and 100 nm). Indeed, to date, both clinically validated therapeutic and imaging NPs usually target cancer cells in a passive way,4 and poloxamer NPs do not make exception. Such a passive drug targeting is achieved by taking advantage of the enhanced permeability and retention (EPR) effect in tumour tissues.46 Tumour vasculature is highly disorganised as compared to the vasculature in normal tissues, and the vascular endothelium in tumours proliferates rapidly and discontinuously. Nevertheless, the delivery of the active pharmaceutical ingredient (API) into specific intracellular sites in cancer cells requires active targeting. Actively targeted NPs may be internalised through clathrin-dependent endocytosis pathways, caveolin-assistance, cell-adhesion-molecule directed or lipid-raft-associated mechanisms, leading to endosome formation, which ultimately leads to lysosomes.47The structural versatility of poloxamers and the relatively simple chemical structure of these ABA copolymers make them particularly suitable for chemical transformation and functionalisation. For instance, a series of cationic functionalised Pluronic® copolymers was obtained by grafting polyethylenimine with nonionic amphiphilic surfactant polyether-Pluronic®.48 They have been shown to be promising DNA and small interfering RNA/microRNA delivery systems. Paclitaxel (Taxol®; Fig. 2), one of the most used chemotherapy drugs in the treatment of a range of cancers including ovarian, breast and non-small cell lung cancers, has also recently been encapsulated into Pluronic® F68 micelles.49 These micelles, further immobilised with a glycol chitosan/heparin composite, benefit from both passive targeting via the EPR effect and active targeting achieved by the specific interactions between the fibrinogen-derived products in the tumor tissue and the heparin on the glycol chitosan/heparin composite.50 In addition to being an elegant example of NP double targeting of cancer cells and to potentially providing an alternative to the FDA approved breast anticancer paclitaxel-loaded PEG-PLA micelles (Genexol-PM; PLA = poly-lactide), it also illustrates the versatility of the Pluronic® block copolymers and the rich pool of NPs synthetically accessible from such copolymers.
The ability of Pluronic® block copolymers to provide steric stabilisation to lyotropic liquid crystalline particles might also be of particular interest for the design of innovative drug delivery systems.51–53 Nanostructured cubic lyotropic liquid crystalline colloidal particles (or Cubosomes®)54 possess an internal ordered structure made of lipid bilayers and water channels.55 They have been proven both efficient drug vectors (via controlled release through diffusion) and suitable for intravenous administration (being dispersed and stabilised as sub-micron sized nanostructured particles).56–58 Despite this promise, the stabilisation of such Cubosomes® remains challenging, and only a few stabilisers have been reported.59,60 Among these, Pluronic® F127 has been demonstrated to be the best known liquid crystal stabiliser.61 Nonetheless, the need to use a high concentration (up to 1 wt% of the total sample mass) to properly disperse the Cubosomes® and the possible disruption and destabilisation of the internal liquid crystalline structure of the Cubosomes® (Fig. 5) are two limitations for the utilisation of Pluronic® F127 as a stabiliser.61
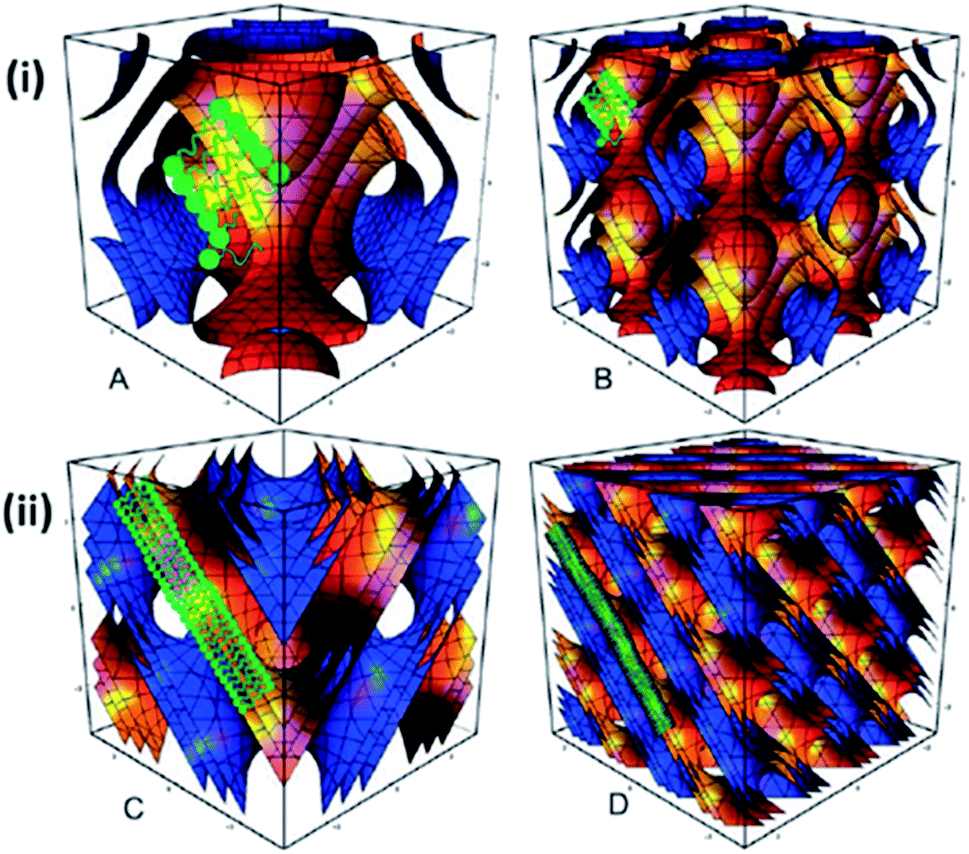 | ||
| Fig. 5 Disruption and destabilisation of the internal liquid crystalline structure from Pn3m (double diamond) to Im3m (primitive) due to the utilisation of Pluronic® F127 for the dispersion of bulk monoolein. The unit cell structure of (i) Im3m (A and B) and (ii) Pn3m (C and D) cubic phases. Reproduced from ref. 62 with permission from The Royal Society of Chemistry. | ||
Recently,62 by using high throughput methodologies to prepare and characterise the dispersions of monoolein and phytantriol, the capability of a range of Pluronics® for stabilising Cubosomes® was investigated. In this work, Boyd, Drummond, and co-workers showed that the hydrophilic-lipophilic balance (HLB), molar mass and cloud point of Pluronic® are crucial physicochemical properties for the stabilisation of liquid crystalline cubic phase particles. It was demonstrated that Pluronic® F108 both maintains colloidal stability, and preserves the integrity of the internal liquid crystalline structure of the Cubosomes®, making it an excellent candidate as a lyotropic liquid crystalline particle stabiliser and opening up new perspectives for the design of innovative drug and biomedical imaging agent encapsulation systems. Furthermore, this study particularly highlights the importance of the relationship between Pluronic® physical and chemical properties and utilisation in medicine.
Conclusions
It is clear that the utilisation of nanotechnology tools for the design of polymer nanoparticles, although being still an emergent concept, has already shown tangible promise in medicine. Pluronic® block copolymers are particularly suitable for the exploration of bio-inspired, bioengineered and biomimetic NPs. In parallel with the design of new Pluronic®-based drug delivery systems and Pluronic®-containing formulations, great effort has also been devoted to elucidating their role in the activity enhancement of the encapsulated drug, their biological mode of action and metabolism.We have tried to focus here particularly on recent advances in the utilisation of Pluronic® block copolymers as drug delivery systems, biological response modifiers, and pharmaceutical ingredients and illustrated them with formulations currently undergoing clinical trials. These examples demonstrate that there is a wide scope for designing innovative Pluronic® formulations, either by functionalising the poloxamer with vectors on its surface which can target specific cell receptors, or by combining different types of poloxamers to finely tune the biological properties of the resulting mixed micelles.
Such a chemical and biological versatility suggests future fundamental advances in the design of Pluronic®-based nanomedicines, for instance by encapsulating hydrophobic molecules with unusual modes of actions. We have recently shown the importance of metals in medicine,63 and how nanotechnology may help to shape the future of medicinal inorganic chemistry,4 and we believe that Pluronic® block copolymers are called to play a significant role in this future.
Acknowledgements
We thank the Leverhulme Trust (Early Career Fellowship no. ECF-2013-414 to NPEB), the University of Warwick (Grant no. RDF 2013-14 to NPEB) and EPSRC (EP/G004897/1 to APB) for support. We also thank Peter Sadler for having kindly commented on the manuscript.Notes and references
- A. P. Alivisatos, A. M. Andrews, E. S. Boyden, M. Chun, G. M. Church, K. Deisseroth, J. P. Donoghue, S. E. Fraser, J. Lippincott-Schwartz, L. L. Looger, S. Masmanidis, P. L. McEuen, A. V. Nurmikko, H. Park, D. S. Peterka, C. Reid, M. L. Roukes, A. Scherer, M. Schnitzer, T. J. Sejnowski, K. L. Shepard, D. Tsao, G. Turrigiano, P. S. Weiss, C. Xu, R. Yuste and X. Zhuang, ACS Nano, 2013, 7, 1850–1866 CrossRef CAS PubMed
.
- A. M. Andrews, A. Schepartz, J. V. Sweedler and P. S. Weiss, J. Am. Chem. Soc., 2013, 136, 1–2 CrossRef PubMed
- O. Neumann, A. S. Urban, J. Day, S. Lal, P. Nordlander and N. J. Halas, ACS Nano, 2012, 7, 42–49 CrossRef PubMed
- N. P. E. Barry and P. J. Sadler, ACS Nano, 2013, 7, 5654–5659 CrossRef CAS PubMed
- P. S. Weiss, ACS Nano, 2013, 7, 2873–2874 CrossRef CAS PubMed
- A. R. Kirtane and J. Panyam, Nat. Nanotechnol., 2013, 8, 805–806 CrossRef CAS PubMed
- O. C. Steinbach, Ther. Delivery, 2014, 5, 113–118 CrossRef CAS
- N. Kamaly, Z. Xiao, P. M. Valencia, A. F. Radovic-Moreno and O. C. Farokhzad, Chem. Soc. Rev., 2012, 41, 2971–3010 RSC
- S. Acharya and S. K. Sahoo, Adv. Drug Delivery Rev., 2011, 63, 170–183 CrossRef CAS PubMed
- http://worldaccount.basf.com/wa/NAFTA/Catalog/ChemicalsNAFTA/pi/BASF/Brand/pluronic .
- M. Kurahashi, K. Kanamori, K. Takeda, H. Kaji and K. Nakanishi, RSC Adv., 2012, 2, 7166–7173 RSC
- P. Alexandridis and T. Alan Hatton, Colloids Surf., A, 1995, 96, 1–46 CrossRef CAS
- A. D. Bangham, M. M. Standish and J. C. Watkins, J. Mol. Biol., 1965, 13, 238–252 CrossRef CAS PubMed
- R. Gref, Y. Minamitake, M. T. Peracchia, V. Trubetskoy, V. Torchilin and R. Langer, Science, 1994, 263, 1600–1603 CAS
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef CAS PubMed
- W. Brown, K. Schillen, M. Almgren, S. Hvidt and P. Bahadur, J. Phys. Chem., 1991, 95, 1850–1858 CrossRef CAS
-
J. H. Collet, in Handbook of Pharmaceutical Excipients. American Pharmaceutical, ed. R. C. Rowe, P. Sheskey and P. J. Weller, American Pharmaceutical Association, Washington, DC, 2003, pp. 447–450 Search PubMed
- J. Valle, A. Armstrong, C. Newman, V. Alakhov, G. Pietrzynski, J. Brewer, S. Campbell, P. Corrie, E. Rowinsky and M. Ranson, Invest. New Drugs, 2011, 29, 1029–1037 CrossRef CAS PubMed
- D. Y. Alakhova, Y. Zhao, S. Li and A. V. Kabanov, PLoS One, 2013, 8, e72238 CAS
- E. V. Batrakova, S. Li, W. F. Elmquist, D. W. Miller, V. Y. Alakhov and A. V. Kabanov, Br. J. Cancer, 2001, 85, 1987–1997 CrossRef CAS PubMed
- M. C. Bissery, G. Nohynek, G. J. Sanderink and F. Lavelle, Anticancer Drugs, 1995, 6, 339–355 CrossRef CAS PubMed
- H. Gelderblom, J. Verweij, K. Nooter, A. Sparreboom and E. L. Cremophor, Eur. J. Cancer, 2001, 37, 1590–1598 CrossRef CAS PubMed
- L. Chen, X. Sha, X. Jiang, Y. Chen, Q. Ren and X. Fang, Int. J. Nanomed., 2013, 8, 73–84 Search PubMed
- A. V. Kabanov, E. V. Batrakova and V. Y. Alakhov, J. Controlled Release, 2002, 82, 189–212 CrossRef CAS PubMed
- I. Höfig, M. J. Atkinson, S. Mall, A. M. Krackhardt, C. Thirion and N. Anastasov, J. Gene Med., 2012, 14, 549–560 CrossRef PubMed
- M. Wang, B. Wu, P. Lu, C. Cloer, J. D. Tucker and Q. Lu, Mol. Ther., 2013, 21, 210–216 CrossRef CAS PubMed
- E. V. Batrakova and A. V. Kabanov, J. Controlled Release, 2008, 130, 98–106 CrossRef CAS PubMed
- E. Batrakova, S. Li, S. Vinogradov, V. Alakhov, D. Miller and A. Kabanov, J. Pharmacol. Exp. Ther., 2001, 299, 483–493 CAS
- E. Batrakova, S. Li, V. Alakhov and A. Kabanov, Polym. Prepr., 2000, 41, 1639–1640 CAS
- A. Kabanov, E. Batrakova and V. Alakhov, Adv. Drug Delivery Rev., 2002, 54, 759–779 CrossRef CAS PubMed
- T. Minko, E. V. Batrakova, S. Li, Y. Li, R. I. Pakunlu, V. Y. Alakhov and A. V. Kabanov, J. Controlled Release, 2005, 105, 269–278 CrossRef CAS PubMed
- E. V. Batrakova, S. Li, V. Y. Alakhov, W. F. Elmquist, D. W. Miller and A. V. Kabanov, Pharm. Res., 2003, 20, 1581–1590 CrossRef CAS
- A. Venne, S. Li, R. Mandeville, A. Kabanov and V. Alakhov, Cancer Res., 1996, 56, 3626–3629 CAS
- D. Y. Alakhova, N. Y. Rapoport, E. V. Batrakova, A. A. Timoshin, S. Li, D. Nicholls, V. Y. Alakhov and A. V. Kabanov, J. Controlled Release, 2010, 142, 89–100 CrossRef CAS PubMed
- J. Watson, Open Biol., 2013, 3, 120144 CrossRef PubMed
- E. Batrakova, S. Li, V. Alakhov, D. Miller and A. Kabanov, J. Pharmacol. Exp. Ther., 2003, 304, 845–854 CrossRef CAS PubMed
- E. R. Gariepy and J. C. Leroux, Eur. J. Pharm. Biopharm., 2004, 58, 409–426 CrossRef PubMed
-
I. R. Schmolka, Poloxamers in the Pharmaceutical Industry, CRC Press, Boca Ratin FL, 1991 Search PubMed
- E. V. Batrakova, S. Li, A. M. Brynskikh, A. K. Sharma, Y. Li, M. Boska, N. Gong, R. L. Mosley, V. Y. Alakhov, H. E. Gendelman and A. V. Kabanov, J. Controlled Release, 2010, 143, 290–301 CrossRef CAS PubMed
- A. Gucu, I. Cavusoglu, C. Eris, F. Toktas, T. Goncu and A. Ozyazicioglu, J. Cardiothorac. Surg., 2013, 8, 16 CrossRef PubMed
- L. Xing, T. Yang, Y. Yang, C. Xu and F. Li, Biomaterials, 2010, 31, 7078–7085 CrossRef PubMed
- Z. Chen, H. Chen, H. Hu, M. Yu, F. Li, Q. Zhang, Z. Zhou, T. Yi and C. Huang, J. Am. Chem. Soc., 2008, 130, 3023–3029 CrossRef CAS PubMed
- J. Zhou, M. Yu, Y. Sun, X. Zhang, X. Zhu, Z. Wu, D. Wu and F. Li, Biomaterials, 2011, 32, 1148–1156 CrossRef CAS PubMed
- G. Jiang, J. Pichaandi, N. J. J. Johnson, R. D. Burke and F. C. J. M. van Veggel, Langmuir, 2012, 28, 3239–3247 CrossRef CAS PubMed
- Z. Wu, C. Guo, S. Liang, H. Zhang, L. Wang, H. Sun and B. Yang, J. Mater. Chem., 2012, 22, 18596–18602 RSC
- T. Konno, H. Maeda, K. Iwai, S. Tashiro, S. Maki, T. Morinaga, M. Mochinaga, T. Hiraoka and I. Yokoyama, Eur. J. Cancer Clin. Oncol., 1983, 19, 1053–1065 CrossRef CAS PubMed
- L. M. Bareford and P. W. Swaan, Adv. Drug Delivery Rev., 2007, 59, 748–758 CrossRef CAS PubMed
- W. Fan, X. Wu, B. Ding, J. Gao, Z. Cai, W. Zhang, D. Yin, X. Wang, Q. Zhu, J. Liu, X. Ding and S. Gao, Int. J. Nanomed., 2012, 7, 1127–1138 CAS
- S. H. Yuk, K. S. Oh, S. H. Cho, S. Y. Kim, S. Oh, J. H. Lee, K. Kim and I. C. Kwon, Mol. Pharmaceutics, 2011, 9, 230–236 CrossRef PubMed
- K. S. Oh, J. Y. Song, S. H. Cho, B. S. Lee, S. Y. Kim, K. Kim, H. Jeon, I. C. Kwon and S. H. Yuk, J. Controlled Release, 2010, 148, 334–350 CrossRef PubMed
- C. Cervin, P. Vandoolaeghe, C. Nistor, F. Tiberg and M. Johnsson, Eur. J. Pharm. Sci., 2009, 36, 377–385 CrossRef CAS PubMed
- B. J. Boyd, D. V. Whittaker, S. M. Khoo and G. Davey, Int. J. Pharm., 2006, 309, 218–226 CrossRef CAS PubMed
- M. Malmsten, J. Dispersion Sci. Technol., 2007, 28, 63–72 CrossRef CAS
- S. Engstrom, Lipid Technol., 1990, 2, 42–45 Search PubMed
- C. J. Drummond and C. Fong, Curr. Opin. Colloid Interface Sci., 1999, 4, 449–456 CrossRef CAS
- K. W. Lee, T. H. Nguyen, T. Hanley and B. J. Boyd, Int. J. Pharm., 2009, 365, 190–199 CrossRef CAS PubMed
- G. Liu, C. E. Conn, L. J. Waddington, S. T. Mudie and C. J. Drummond, Langmuir, 2010, 26, 2383–2391 CrossRef CAS PubMed
- M. J. Moghaddam, L. d. Campo, L. J. Waddington and C. J. Drummond, Soft Matter, 2010, 6, 5915–5929 RSC
- B. J. Boyd, Y. D. Dong and T. Rades, J. Liposome Res., 2009, 19, 12–28 CrossRef CAS PubMed
- M. Uyama, M. Nakano, J. Yamashita and T. Handa, Langmuir, 2009, 25, 4336–4338 CrossRef CAS PubMed
- A. Yaghmur and O. Glatter, Adv. Colloid Interface Sci., 2009, 147–148, 333–342 CrossRef CAS PubMed
- J. Y. T. Chong, X. Mulet, L. J. Waddington, B. J. Boyd and C. J. Drummond, Soft Matter, 2011, 7, 4768–4777 RSC
- N. P. E. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC
| This journal is © The Royal Society of Chemistry 2014 |