 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater-dispersible cationic polyurethanes containing pendant trialkylphosphoniums†
Musan
Zhang
,
Sean T.
Hemp
,
Mingqiang
Zhang
,
Michael H.
Allen
Jr.
,
Richard N.
Carmean
,
Robert B.
Moore
and
Timothy E.
Long
*
Department of Chemistry, Macromolecules and Interfaces Institute, Virginia Tech, Blacksburg, VA 24061-0212, USA. E-mail: telong@vt.edu; Fax: +1-540-231-8517; Tel: +1-540-231-2480
First published on 6th March 2014
Abstract
Novel trialkylphosphonium ionic liquids chain extenders enabled the successful synthesis of poly(ethylene glycol)-based, cationic polyurethanes with pendant phosphoniums in the hard segments (HS). Aqueous size exclusion chromatography (SEC) confirmed the charged polyurethanes, which varied the phosphonium alkyl substituent length (ethyl and butyl) and cationic HS content (25, 50, 75 mol%), achieved high absolute molecular weights. Dynamic mechanical analysis (DMA) demonstrated the triethylphosphonium (TEP) and tributylphosphonium (TBP) polyurethanes displayed similar thermomechanical properties, including increased rubbery plateau moduli and flow temperatures. Fourier transform infrared spectroscopy (FTIR) emphasized the significance of ion–dipole interaction on hydrogen bonding. Atomic force microscopy (AFM), small-angle X-ray scattering (SAXS), and wide-angle X-ray diffraction (WAXD) supported microphase separated morphologies in the trialkylphosphonium polyurethanes, despite the presence of ionic interactions. Sorption isotherm experiments revealed the TEP polyurethane exhibited the highest water vapor sorption profile compared to the TBP, which displayed similar water sorption profiles to the noncharged analogue. The phosphonium polyurethanes displayed significantly improved tensile strain; however, lower tensile stress of the TEP polyurethane was presumably due to absorbed water. In addition to physical characterizations, we also explored the trialkylphosphonium polyurethanes as nucleic acid delivery vectors. The phosphonium polyurethanes bound DNA at low charge ratios, and the polyplexes exhibited enhanced colloidal stability under physiological salt conditions.
Introduction
The critical fields of biomedical technology and high performance materials demand innovative macromolecular design for multifunctional applications. The literature extensively describes the success of ion-containing polymers, including polyelectrolytes and ionomers, to meet the challenges of emerging technologies from water purification membranes, conductive thin films, and electro-active actuators to antimicrobial bioadhesives, tissue scaffolds, and drug delivery vehicles.1,2 Phosphonium-containing polymers are relatively unexplored compared to their ammonium analogues.3 However, phosphonium cations afford many advantages resulting from the phosphorous electronic distribution, which dictates electronegativity, nucleophilicity, and polarizability.4,5 A subtle atomic modification from nitrogen to phosphorus in quaternary salts results in significant differences in properties including electrostatic strength, ionic conductivity, thermal stability, and chemical resistivity. Phosphonium-containing salts and polymers displayed enhanced resistance to the deleterious Hoffman elimination, which is often observed for quaternary ammoniums. Recently, our research initiative to explore ammonium-alternative, cationic structures led to the first report of phosphonium polyelectrolytes and phosphonium diblock copolymers as macromolecular nucleic acid delivery vehicles.6,7Many researchers have elucidated the significance of noncovalent interactions, including electrostatics and hydrogen bonding, on nanoscale morphologies of polymers with various architectures. Random copolymers of n-butyl acrylate and trialkylphosphonium styrenics demonstrated tunable thermomechanical properties depending on the alkyl length and counteranion.8 In many instances, hydrogen bonded self-assembly produced synergistic effects with ionic interactions, which resulted in unique microphase separated, nanostructured morphologies.9,10 The versatility of the phosphonium cation and significance of morphology extend beyond bulk polymer properties. Diblock copolymers containing a phosphonium complexation block and poly(ethylene glycol)-based (PEG) stabilizing block produced colloidally stable polymer–DNA polyplexes most likely due to the formation of complex solution structure.6,7
The impact of electrostatic interactions on the morphology of segmented copolymers, have been described in segmented copolyesters,11 polyurethanes,12,13 poly(arylene ethers),14 and ionenes.15 Polyurethanes are ideal segmented copolymers to probe the interactions between electrostatics and hydrogen bonding. Several studies reported the electrostatic interactions in charged polyurethanes disrupted the hydrogen bonding, which eliminated the microphase separated morphology necessary to maintain suitable mechanical properties.16 Although the electrostatic interactions partially restored the mechanical properties lost from disrupted hydrogen bonding, a sacrifice in the tensile properties was often observed.
Quaternary nitrogens represent the most common example of cationic polyurethanes, and only one example currently exists for phosphonium polyurethanes.12 This manuscript describes water-dispersible, phosphonium-containing polyurethanes synthesized using novel phosphonium ionic liquid chain extenders. Aqueous size exclusion chromatography (SEC) confirmed high absolute molecular weight polymers. Dynamic mechanical analysis (DMA) supported the presence of a microphase separated morphology, which was further confirmed using complementary characterization techniques including atomic force microscopy (AFM), small-angle X-ray scattering (SAXS), and wide-angle X-ray diffraction (WAXD). Fourier transform infrared spectroscopy (FTIR) confirmed the phosphonium polyurethanes maintained a hydrogen bonded physical network. Thermogravimetric sorption analysis probed the water absorption properties, which provided valuable information for interpretation of the tensile behavior. Our results indicated that ion–dipole interactions promoted more cohesive, microphase separated networks and suppressed hydrogen bonding to the polar PEG SS leading to undesired phase-mixing. Compelling preliminary nucleic acid delivery investigations further demonstrated versatility of the phosphonium polyurethanes for future biological applications.
Experimental section
Materials
Triethylphosphine (99%), tri-n-butylphosphine (anhydrous, 99%), and 1,4-butanediol (BD, anhydrous, 99.5%) were purchased from Sigma-Aldrich and used as received. Poly(ethylene glycol) (PEG, Sigma-Aldrich, Mn = 2000 g mol−1) was dried at 60 °C under reduced pressure (10 mmHg) for 18 h. Anhydrous N,N-dimethylformamide (DMF, 99.8%) was obtained from Fischer Scientific. Dibutyltin dilaurate (DBTDL, Sigma-Aldrich, 95%) catalyst solution was prepared at 1 wt% in anhydrous tetrahydrofuran (THF). Dicyclohexylmethane-4,4′-diisocyanate (HMDI, 99.5%), which was graciously donated from Bayer Material Science, was used without further purification. 2-(Bromomethyl)-2-methyl-1,3-propanediol was synthesized according to procedures described in the literature and dried at 40 °C under reduced pressure (10 mmHg) for 12 h.17Trialkylphosphonium diol synthesis
The synthesis of tri-n-butyl(1,3-dihydroxypropyl)phosphonium bromide serves as a representative example; the triethylphosphonium diol was prepared similarly. 2-(Bromomethyl)-2-methyl-1,3-propanediol (1.05 molar excess) was charged into a flame-dried, 100 mL, round-bottomed flask with a magnetic stir bar and purged with dry N2. Tri-n-butylphosphine was added using dry syringe techniques, and the reaction proceeded at 100 °C for 2 days under vigorous stirring. The initial, biphasic reaction was homogenous after completion. Unreacted reagents were vacuum distilled from the reaction at 100 °C (<1 mmHg) until volatile reactants no longer evolved. The product was washed multiple times with diethyl ether and dried at 90 °C under reduced pressure (0.5 mmHg) for 24 h. Caution should be used when washing the product with diethyl ether.Polyurethane synthesis
The following procedure for 75 mol% HS tri-n-butylphosphonium (TBP(75)) polyurethane serves as a representative polymerization. Dry PEG (2.5 mmol) was charged into a flame-dried, three-necked, round-bottomed flask equipped with an addition funnel, nitrogen inlet, and overhead mechanical stirrer and purged thoroughly with N2. The DBTDL catalyst solution (50 ppm) and HMDI (10 mmol) were syringed into the addition funnel. The reaction flask was first heated to 70 °C, and the DBTDL and HMDI mixture was added after the PEG thoroughly melted. The reaction continued under constant stirring for 5 h at 70 °C. The dry chain extender, tributylphosphonium diol (7.5 mmol), was charged into a separate flame-dried, round-bottomed flask and purged with N2. Dry DMF (∼10 mL) was cannulated into the flask. The dissolved solution was transferred into the addition funnel and slowly added drop-wise to the reaction. The addition funnel was washed with additional anhydrous DMF and added to the reaction to achieve a final 10 wt% polymer concentration. The reaction continued for 1 day, and DMF was gently removed at 35 °C under reduced pressure (0.1 mmHg) to obtain a concentrated polyurethane solution. The solution was precipitated into diethyl ether and washed several times. The polyurethanes were dried under reduced pressure (10 mmHg) for 1 day at room temperature and 2 days at 50 °C.Polyurethane films were solvent coated using a micrometer film applicator onto silicon-coated Mylar™ substrates. The coating thickness of the polyurethane solution (10 wt% in 90/10 (w/w%) chloroform/methanol) was 0.1 mm for all samples. The films were dried at 25 °C for 1 day, followed by 1 day at 25 °C (10 mmHg), and 1 day at 80 °C (10 mmHg).
Characterization
All monomers and polymers were dried under reduced pressure (0.5 mmHg) at 80 °C overnight prior to characterization. 1H NMR spectroscopy (Varian Inova, 400 MHz, D2O) and mass spectrometry (Agilent 6220 LC-TOF-MS) confirmed monomer composition. Dynamic light scattering (DLS, Malvern Zetasizer NanoZS) confirmed the absence of polymer aggregation in the aqueous mobile phase (54/23/23 (v/v/v%) water/methanol/acetic acid with 0.1 M sodium acetate). Aqueous size exclusion chromatography (SEC) provided absolute molecular weights using a Water 1515 Isocratic HPLC Pump and Waters 717plus Autosampler with Waters 2414 refractive index and Wyatt MiniDAWN MALLS detectors (flow rate 0.8 mL min−1). A Wyatt Optilab T-rEX differential refractometer measured offline dn/dc values at 658 nm and 35 °C. A TA Instruments thermogravimetric analyzer Q50 determined the temperature (Td) at 5% weight loss of monomers and polymers (N2, 25 to 600 °C, ramp 10 °C min−1). Differential scanning calorimetry (DSC, TA Instruments, Q1000) measured monomer and polymer thermal transitions from heat/cool/heat experiments (N2, −80 to 200 °C, ramp 10 °C min−1). A TA Instruments Q5000 thermogravimetric sorption analyzer (TGA-SA) performed sorption isotherm experiments at 25 °C on polyurethane samples (75 mol% HS) using a 5% relative humidity (RH) step procedure from 0–95% RH. Each RH step proceeded for 2 h or until the weight stabilized (<0.01% change over 10 min). An instrumental pre-drying method was applied to each sample before the experiment at 50 °C and 0% RH. Dynamic mechanical analysis (DMA, TA Instruments, Q800) revealed PU thermomechanical properties from temperature ramp experiments under an oscillatory tension mode (−100 to 180 °C, ramp 3 °C min−1, 1 Hz). A 5500R Instron universal testing instrument performed tensile testing at a cross-head speed of 50 mm min−1; the polyurethane tensile stress, % elongation, and Young's moduli are reported based on an average of five specimens. Fourier transform infrared (FTIR) spectroscopy (Varian ATR-FTIR) evaluated the extent of hydrogen bonding at ambient conditions (128 scans, background subtraction, and ATR corrected). Atomic force microscopy (Veeco Multimode AFM) probed the polyurethane surface morphology under tapping mode (tip spring constant 42 N m−1, free air amplitude 3.00 V, tapping to free air amplitude ratio = 0.6).SAXS and WAXD were performed using a Rigaku S-Max 3000 3 pinhole SAXS system, equipped with a rotating anode emitting X-ray with a wavelength of 0.154 nm (Cu Kα). Scattering from a silver behenate standard was used to calibrate the sample-to-detector distance. For SAXS, the sample-to-detector distance was 1603 mm and for WAXD, the sample-to-detector distance was 82.5 mm. Two-dimensional SAXS patterns were obtained using a fully integrated 2D multiwire, proportional counting, gas-filled detector, with an exposure time of 1 h. SAXS data had been corrected for sample thickness, sample transmission and background scattering. WAXD two-dimensional diffraction patterns were obtained using an image plate, with an exposure time of 1 h. All SAXD/WAXD data were analyzed using the SAXSGUI software package to obtain radically integrated SAXS/WAXD intensity versus scattering vector q (SAXS), where q = (4π/λ)sin(θ), θ is one half of the scattering angle and λ is the wavelength of X-ray or 2θ (WAXD) profiles.
Results and discussion
Water-dispersible, phosphonium polyurethanes were successfully generated using novel trialkylphosphonium diol chain extenders. An efficient, one-step, nucleophilic substitution reaction between 2-(bromomethyl)-2-methyl-1,3-propanediol and trialkylphosphine produced trialkyl(1,3-dihydroxypropyl)phosphonium bromide in high yields (Scheme 1). Both triethylphosphonium (TEP) and tributylphosphonium (TBP) diols exhibited high thermal stabilities above 370 °C. The TEP diol displayed a glass transition temperature (Tg) and melting point at −40 and 124 °C, respectively. In contrast, the amorphous TBP diol revealed a single Tg at −33 °C, which classified this monomer as a room temperature ionic liquid. Trialkylphosphonium diols that contain primary hydroxyls served as efficient chain extenders to afford cationic polyurethanes with pendant phosphonium groups in the HS. The rationale for the pendant phosphonium position was to presumably promote facile ionic interactions. | ||
| Scheme 1 Synthesis of trialkyl(1,3-dihydroxypropyl)phosphonium bromide. | ||
Scheme 2 illustrates the two-step synthesis of triethylphosphonium (TEP(x)) and tri-n-butylphosphonium (TBP(x)) polyurethanes, where x represents the mol% HS. Chain extension of the HMDI-endcapped PEG prepolymer with trialkylphosphonium diols afforded cationic phosphonium polyurethanes. Dynamic light scattering of the phosphonium polyurethanes confirmed the absence of polymer aggregation in the aqueous SEC solvent (Fig. S3†). Fig. 1 displays representative aqueous SEC light scattering traces for various phosphonium polyurethanes. Table 1 summarizes the absolute molecular weights using experimentally determined dn/dc values. Possible inefficient SEC separation presumably accounted for the low polydispersity; however, all polyurethanes exhibited Mn > 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 with monomodal distributions.
000 g mol−1 with monomodal distributions.
 | ||
| Scheme 2 Synthesis of trialkylphosphonium polyurethanes. | ||
 | ||
| Fig. 1 Representative aqueous SEC light scattering chromatograms of phosphonium PU (54/23/23 water/methanol/acetic acid (v/v/v%), 0.1 M sodium acetate). | ||
| PU | M n (g mol−1) | M w/Mna | HS mol% | HS wt% |
|---|---|---|---|---|
| a Aqueous SEC-MALLS detector unless otherwise indicated. b DMF 0.05 M LiBr SEC-MALLS detector. | ||||
| BD(50)b | 20300 |
1.54 | 50 | 26 |
| BD(75)b | 22100 |
2.05 | 75 | 43 |
| TEP(25) | 46500 |
1.19 | 25 | 21 |
| TEP(50) | 55800 |
1.30 | 50 | 31 |
| TEP(75) | 25400 |
1.37 | 75 | 53 |
| TBP(25) | 50200 |
1.17 | 25 | 22 |
| TBP(50) | 38500 |
1.28 | 50 | 33 |
| TBP(75) | 46500 |
1.10 | 75 | 56 |
Table 2 provides a summary the polyurethane thermal properties. The trialkylphosphonium polyurethanes displayed similar 5% weight thermal weight loss (Td5%) to the noncharged analogues at equivalent HS mol%. The phosphonium-containing polyurethanes offered higher thermal stabilities relative to ammonium-based polyurethanes, which showed evidence of Hoffman elimination at 180 °C.18,19 The phosphonium polyurethanes exhibited a Tg at approximately −55 °C corresponding to the PEG SS. The BD(75) PEG melting temperature (Tm) at 35 °C was higher than the Tm (∼9 °C) observed for TEP(75) and TBP(75). However, at lower HS content, the phosphonium polyurethanes exhibited similar melting temperatures to noncharged controls. Thermal analysis suggested both the noncharged analogue and phosphonium polyurethanes possessed a microphase separated morphology.
| PU | T d5% (°C) | T g(SS)b (°C) | T c(SS)b (°C) | T m(SS)b (°C) |
|---|---|---|---|---|
| a TGA: N2, 10 °C min−1. b DSC: Tg and Tm determined from 2nd heating cycle, Tc from cooling cycle, ND: crystallization temperatures not detected using DSC. | ||||
| BD(50) | 310 | −50 | −4 | 36 |
| BD(75) | 311 | −52 | 13 | 31 |
| TEP(25) | 328 | −52 | −2 | 36 |
| TEP(50) | 321 | −55 | −8 | 34 |
| TEP(75) | 303 | −50 | ND | 9 |
| TBP(25) | 337 | −52 | −4 | 36 |
| TBP(50) | 330 | −54 | −9 | 33 |
| TBP(75) | 324 | −56 | −9 | 11 |
DMA probed the thermomechanical properties of noncharged and phosphonium polyurethanes containing 75 mol% HS. Fig. 2 reflects at least three major thermal transitions in the storage modulus and tanδ curves as a function of temperature. The lowest transition at −51 °C corresponded to the PEG Tg. The second transition at approximately 1 °C indicated the melting of the PEG SS. The noncharged BD(75) displayed a broad melting transition at higher temperatures compared to the phosphonium polyurethanes. Both TEP(75) and TBP(75) consistently displayed higher storage moduli between the Tg and Tm transitions compared to BD(75). The third transition was attributed to the HS Tg and the disruption of hydrogen bonding interactions. In the noncharged BD(75), the rubbery plateau terminal region occurred at 60 °C. In sharp contrast, TEP(75) and TBP(75) demonstrated higher storage moduli and an extended rubbery plateau compared to BD(75). Interestingly, the phosphonium polyurethane alkyl length did not affect the rubbery plateau modulus, which suggested the physical crosslink density related to the hard segment content on electrostatic interactions. TBP(75) displayed two distinct transitions associated with hydrogen bonding dissociation and viscous flow at 110 °C and 160 °C, respectively. The sharp contrast in the thermomechanical properties of the noncharged analogue and phosphonium polyurethanes suggested that the phosphonium charge promoted a more well-defined, microphase separated morphology, which resulted in higher storage moduli and extended rubbery plateaus. Painter and coworkers reported hydrogen bonded urethane N–H and ether oxygen occurred more frequently in polyurethanes containing a more polar SS such as PEG, which accounted for more phase-mixing.20 The incorporation of phosphonium cations into the polyurethane HS functioned synergistically with hydrogen bonding interactions, which led to improved microphase separation and enhanced thermomechanical properties compared to the noncharged control. Goddard and Copper observed similar trends in ammonium polyurethanes and concluded the cationic ammonium group promoted hydrogen bonding and microphase separation.21
 | ||
| Fig. 2 Dynamic mechanical analysis of noncharged and phosphonium PU (75 mol% HS). Left and right arrows indicate curves for the storage modulus and tanδ, respectively. | ||
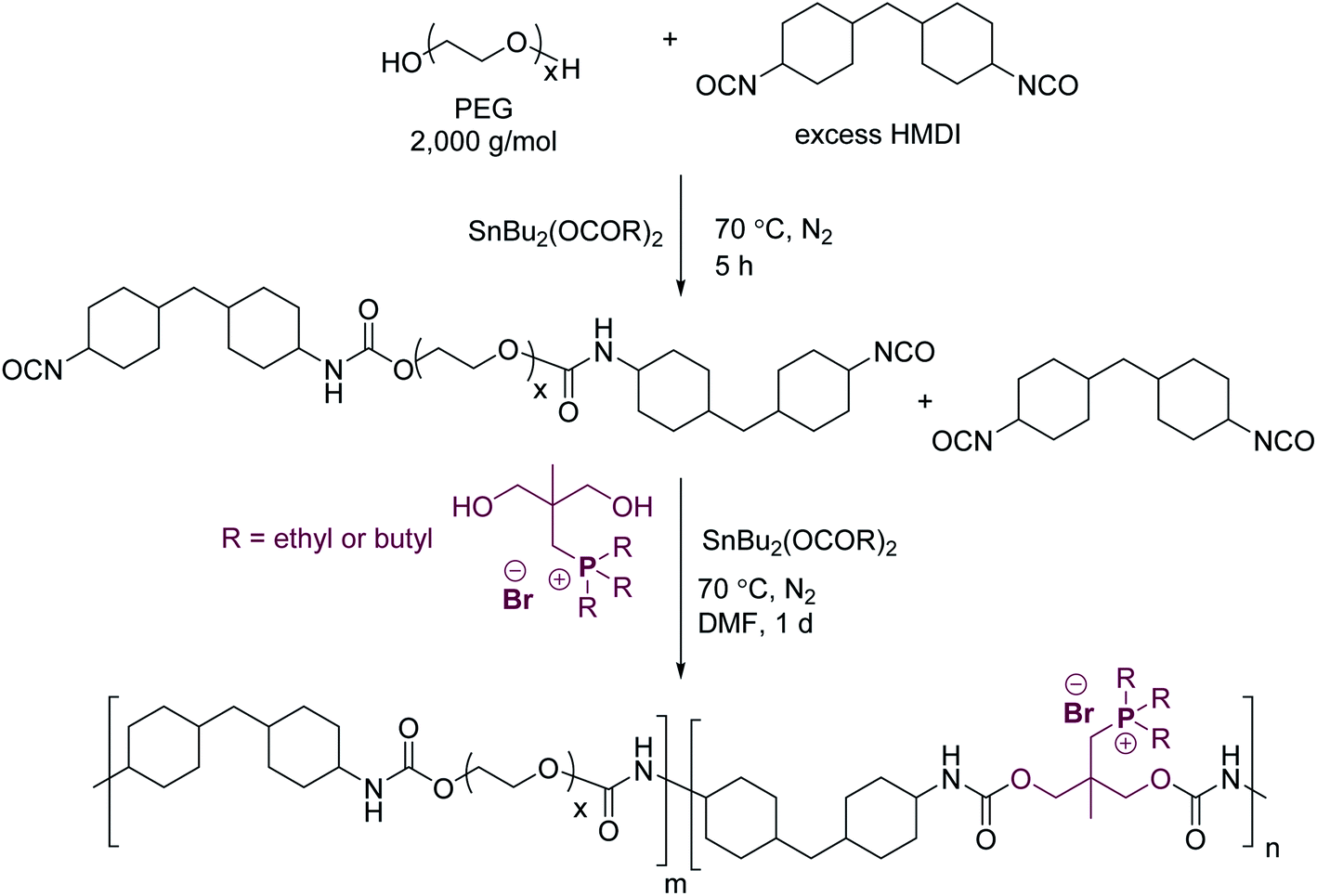
Fourier transform infrared spectroscopy (FTIR) probed various hydrogen bonding interactions in noncharged and phosphonium polyurethanes using the urethane N–H (∼3100–3550 cm−1) and carbonyl (∼1650–1800 cm−1) stretching regions (Fig. 3). Noncharged polyurethanes typically display two main N–H absorption bands associated with the “free” N–H stretch at 3500 cm−1 and hydrogen bonded N–H at lower wavenumbers.22 Several investigations reported on the complex hydrogen bonding interactions arising from multiple proton donor–acceptor pairs.23–25 In the noncharged BD(75), the two primary hydrogen bond acceptors are urethane carbonyls and PEG ether oxygens (Fig. 3a). The BD(75) FTIR spectra displays a small shoulder at 3326 cm−1 and a dominant stretch at 3300 cm−1 corresponding to the N–H–ether oxygen and N–H–urethane carbonyl interactions, respectively. BD(75) demonstrated a greater extent of N–H–urethane carbonyl hydrogen bonding due to stronger intermolecular interactions. The phosphonium polyurethanes demonstrated an additional broad shoulder at 3260 cm−1, which corresponded with hydrogen bonded urethane N–H and bromide anion. Several investigations on cationic polyurethanes observed similar N–H bands.16,18,19,26,27 More importantly, unlike urethane N–H and ether oxygens hydrogen bonding, which promoted phase-mixing, the ion–dipole interactions in phosphonium polyurethanes occurred preferentially within the HS matrix and provided an explanation for the improved thermomechanical behavior.
 | ||
| Fig. 3 FTIR spectra of noncharged and phosphonium polyurethanes: (a) N–H stretch between 3000–3500 cm−1 and (b) carbonyl stretch between 1600–1750 cm−1. | ||
The FTIR carbonyl spectral region provides additional characterization of the molecular hydrogen bonding environment and supports similar trends observed in the N–H region (Fig. 3b). Polyurethanes with high SS content display a low-frequency absorbance at 1730–1740 cm−1 commonly assigned to free carbonyls.22,24,28,29 As the hydrogen bond alignment increases though ordering within the HS matrix, the strength of the hydrogen bonding interactions improves, which result in lower frequency carbonyl absorbance. In conventional polyurethanes, disordered hydrogen bonding are weaker interactions that typically occurred within the phase-separated interface, SS matrix, or phase-mixed regions.20 BD(75) revealed two distinct carbonyl environments of similar intensities at 1712 and 1695 cm−1 corresponding to the disordered and weakly associating, ordered hydrogen bonded urethane carbonyls, respectively. In contrast, phosphonium polyurethanes displayed a single peak at 1700 cm−1 suggesting a distinct carbonyl environment related to weakly associating, ordered carbonyls within the HS matrix. The slightly higher peak value in the phosphonium polyurethanes likely resulted from the alkyl steric hindrance which may reduce the hydrogen bonding order compared to the linear butanediol.
Atomic force microscopy elucidated the influence of phosphonium cations on the polymer surface morphology. Both the noncharged and phosphonium polyurethanes contained irregularly structured, hard domains (light colored) dispersed within the SS matrix (dark colored) (Fig. 4).30,31 BD(75) exhibited the least defined microphase separated morphology, suggesting evidence of phase-mixing particularly at the HS–SS interface. In contrast, microphase separation improved for the phosphonium polyurethanes from the ethyl to butyl substituents. The HS–SS interface becomes increasingly distinct relative to the noncharged control. Furthermore, the TBP(75) hard phase appeared smaller and more percolated throughout the low-Tg matrix, suggesting a higher degree of HS interconnectivity.
 | ||
| Fig. 4 Atomic force microscopy phase images: (a) BD(75), (b) TEP(75), and (c) TBP(75) polyurethanes. The scale bars represent 200 nm. | ||
SAXS provided complementary analysis of the bulk morphology to confirm the presence of microphase separation. Table 3 summarizes the interdomain spacing from SAXS analysis for BD(75), TEP(75), and TBP(75). Both the noncharged and phosphonium polyurethanes exhibited a single, broad intensity peak characteristic of non-uniform microphase-separated morphology often observed in segmented block copolymers (Fig. S6†).30,32–36 The peak position at maximum scattering intensity, qmax, relates to the interdomain spacing, d, according to Bragg's law:

| PU | q (nm−1) | d (nm) |
|---|---|---|
| a SAXS: peak position determined at maximum scattering intensity: d = 2π/q. | ||
| BD(75) | 0.52 | 12 |
| TEP(75) | 0.55 | 12 |
| TBP(75) | 0.58 | 11 |
The SAXS results indicated the trialkylphosphonium functionality did not impact the interdomain spacing, which were similar to the BD(75) noncharged analogue. The difference in the scattering intensity for TEP(75) was most likely due to scattering contrast. The WAXD analysis confirmed the presence of PEG crystallinity in BD(75), which was not observed in the phosphonium-containing polyurethanes (Fig. S7†).
Tensile testing indicated that the phosphonium polyurethanes offered significantly improved mechanical properties compared to neutral controls (Table 4). Both TEP(75) and TBP(75) had 100% increase in tensile strains compared to BD(75) (Fig. 5). The semicrystalline PEG phase in BD(75) possibly reduced SS flexibility, which resulted in lower elongations. In the phosphonium polyurethanes, the combination of an amorphous, low-Tg phase and improved microphase separation likely contributed to increased elongations and Young's moduli. Higher Young's moduli in the phosphonium polyurethanes reflected greater hard domain cohesive strength.19,37 Furthermore, evidence of modest strain hardening, associated with ordered alignment of the HS domains during deformation, further supported a well-defined, microphase separated morphology.
| PU | Young's modulus (MPa) | Stress at break (MPa) | Strain at break (%) |
|---|---|---|---|
| a Tensile: crosshead speed 50 mm min−1; values reported as an average of five samples. | |||
| BD(75) | 10.0 ± 0.5 | 7.1 ± 0.3 | 354 ± 35 |
| TEP(75) | 21.0 ± 5.6 | 4.6 ± 0.7 | 862 ± 13 |
| TBP(75) | 134 ± 16 | 10.8 ± 1.0 | 857 ± 20 |
 | ||
| Fig. 5 Representative tensile stress–strain curves of noncharged and phosphonium polyurethanes. Arrow designates the stress and strain at break for BD(75). | ||
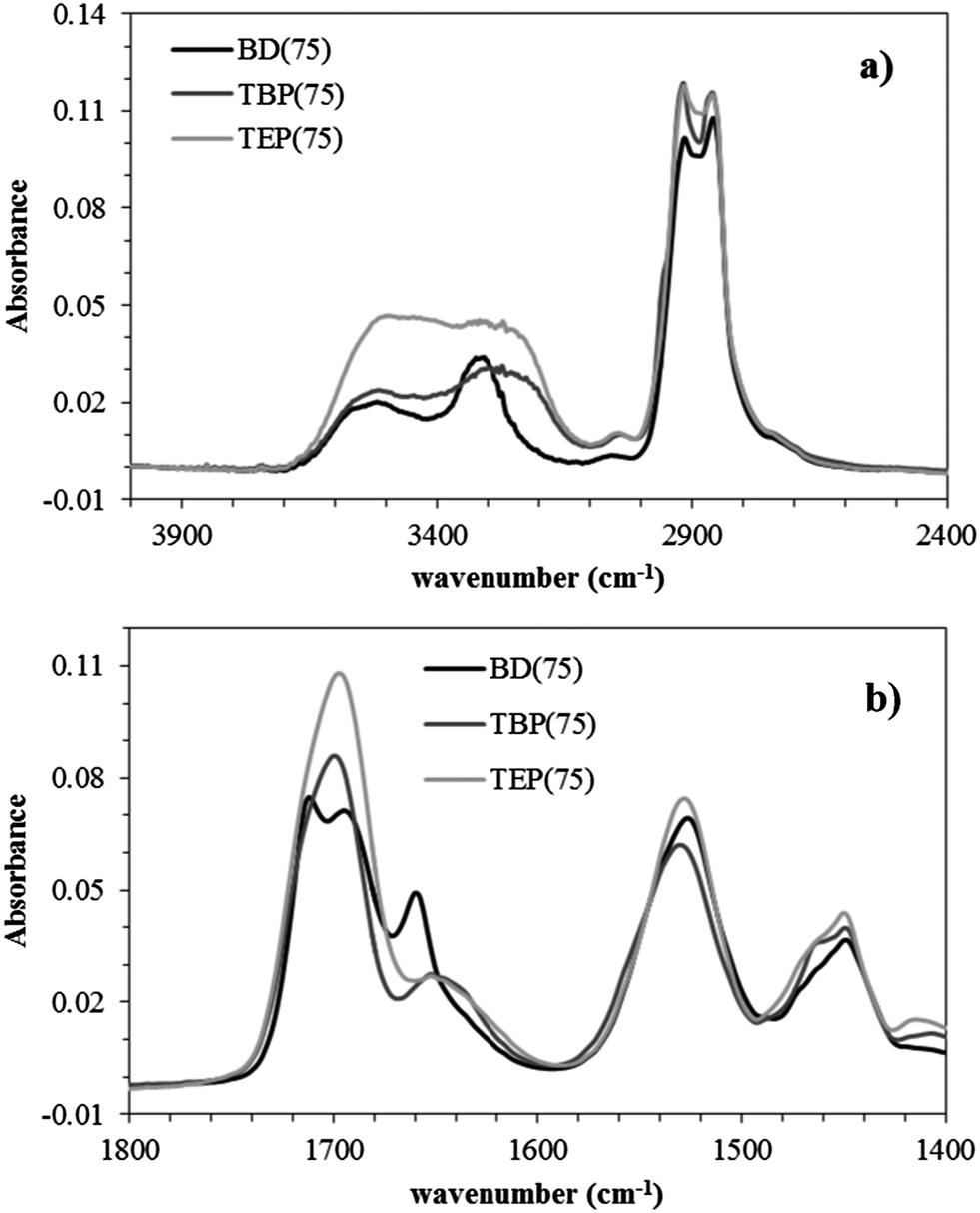
Tensile analyses also supported the impact of alkyl length on mechanical properties. The TBP(75) displayed a distinct yield point corresponding to plastic deformation of the hydrogen bonded, highly interconnected hard phase. In contrast, the TEP(75) displayed a noticeably lower tensile stress compared to both TBP(75) and BD(75). This observation led to an investigation of the polyurethane water sorption behavior. Fig. 6 illustrates the sample weight% as a function of %RH during a thermogravimetric water sorption experiment. The TEP(75) clearly exhibited the highest water absorption at every %RH, and the TBP(75) displayed a similar water absorption profile to the BD(75). These results suggested the TBP(75) was less hygroscopic than the TEP(75) due to the longer butyl chains. The higher absorbed water content in TEP(75) presumably functioned as a HS plasticizer, which lowered the tensile stress. Further investigations of the water sorption kinetics will provide complementary information on the effect of the phosphonium alkyl length. Previous investigations reported increased tensile strengths and reduced strains in ionic polyurethanes when the extent of electrostatic interactions exceeded hydrogen bonding associations.38,39 Our results demonstrated that the phosphonium polyurethanes possessed desirable synergistic interactions, which significantly contributed to the overall improved mechanical performance.
 | ||
| Fig. 6 Sorption isotherm of noncharged and phosphonium polyurethanes (75 mol% HS) at 25 °C. | ||
A desirable feature of the phosphonium polyurethanes was their spontaneous water-dispersibility and formation of non-aggregated, nanoparticles in solution. DLS investigations showed the phosphonium polyurethanes with longer butyl substituents produced larger nanoparticles in solution compared to ethyl analogues (Fig. S4†). Furthermore, the size of the particle decreased with increasing HS content. Typically, ionic polymers in salt-free aqueous conditions exhibit large-scale aggregation due to hydrophobic interactions.40 The absence of aggregation as shown by DLS strongly suggests that these phosphonium polyurethanes may exhibit interesting solution morphologies similar to other block copolymer surfactants and previously described polyurethane ionomers.41 Further investigations on the long-term colloidal stability and characterization of the solution structures are necessary to confirm our hypothesis.
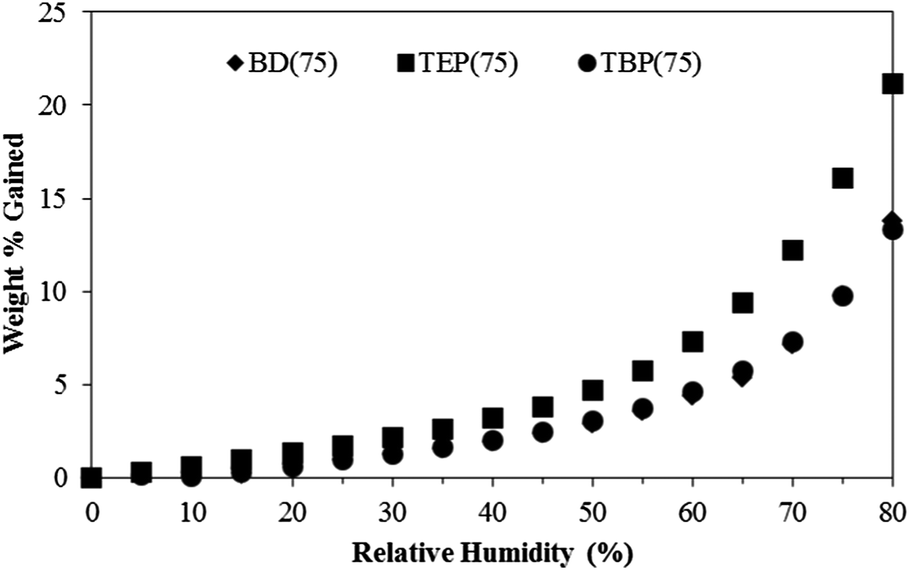
The water-dispersible phosphonium polyurethanes demonstrated potential as nonviral, macromolecular gene delivery vehicles. DNA binding gel shift assays confirmed DNA binding at low charge (+/−) ratios, where (+) represents the number of positive polymer cationic sites and (−) represents the number of negative DNA phosphate groups (Fig. 7). As expected, the +/− ratio at complete DNA binding decreased from 4 to 2 with increasing HS content due to a higher cationic content. In general, DLS investigations found that the size of the polyplexes systematically decreased with increasing HS content, which presumably related to stronger DNA–polymer interactions with increasing cationic charge density. Furthermore, DLS studies verified the polyplexes formed from TBP(50) and TBP(75) maintained particle stability over 24 h in physiological salt solutions (Fig. 8). However, TBP(25) showed an increase in the particle size attributed to the release of DNA after 24 h from weaker binding. These preliminary investigations indicated that phosphonium polyurethanes serve as promising vectors for nucleic acid delivery.
 | ||
| Fig. 7 DNA binding gel shift assay of cationic polyurethanes with various TBP mol% content. Arrows designate +/− ratio at complete DNA binding. | ||
 | ||
| Fig. 8 Polyplex stability (+/− ratio = 4) over 24 h in salt solution, Dulbecco's modified Eagle's media (DMEM). | ||
Conclusions
Cationic polyurethanes possess several desirable advantages including improved microphase separation and mechanical performance for a variety of applications from high performance engineering thermoplastics to innovative biomaterials. This manuscript reports the synthesis of cationic polyurethanes bearing pendant trialkylphosphoniums using novel ionic liquid, phosphonium chain extenders. Polyurethane compositions varied the HS content and alkyl substituent to elucidate their influence on thermomechanical properties and morphology. The phosphonium polyurethanes demonstrated significantly higher rubbery plateau moduli, increased flow temperature from 60 to 160 °C, and evidence of microphase separation. FTIR spectroscopy revealed the phosphonium polyurethanes maintained hydrogen bonding, and hydrogen bonding to anions within the HS most likely served as the predominant driving force for microphase separation. Morphological investigations employed both AFM and SAXS, and confirmed microphase separated morphologies with more defined HS features in tributylphosphonium polyurethane compared to the triethyl analogue. The enhanced microphase separation led to improved tensile stress and strain properties compared to the noncharged control. Preliminary nucleic acid delivery studies verified the TBP polyurethanes bound DNA at low charge ratios, and DLS confirmed increasing polyplex stability with higher cationic charge. Our investigations demonstrated the phosphonium polyurethanes offered desirable properties for a broad range of emerging applications.Acknowledgements
This work is supported in part by the U.S. Army Research Laboratory and the U.S. Army Research Office under the Army Materials Center of Excellence Program, contract W911NF-06-2-0014. This material is also based upon work supported in part by the US Army Research Office under Grant no. W911NF-07-1-0452 Ionic Liquids in Electro-active Devices (ILEAD) MURI. This material is partially based upon work supported by the National Science Foundation under Grant no. DMR-0923107. The authors thank Bayer Material Science for providing the dicyclohexylmethane-4,4′-diisocyanate monomer. We extend our gratitude to Amanda Hudson and Jennifer Ludwig for their collaborations on this project and Professor Garth Wilkes for insightful discussions. In addition, the authors thank the Virginia Tech Institute of Critical Technology and Applied Sciences (ICTAS) Nanoscale Characterization and Fabrication Laboratory (NCFL) for atomic force microscopy characterization.References
- J. Yuan and M. Antonietti, Polymer, 2011, 52(7), 1469–1482 CrossRef CAS.
- G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377(1–2), 1–35 CrossRef CAS.
- S. T. Hemp, M. Zhang, M. Tamami and T. E. Long, Polym. Chem., 2013, 4(12), 3582–3590 RSC.
- W. Jaeger, J. Bohrisch and A. Laschewsky, Prog. Polym. Sci., 2010, 35(5), 511–577 CrossRef CAS.
- S.-W. Wang, W. Liu and R. H. Colby, Chem. Mater., 2011, 23(7), 1862–1873 CrossRef CAS.
- S. T. Hemp, M. H. Allen, M. D. Green and T. E. Long, Biomacromolecules, 2011, 13(1), 231–238 CrossRef PubMed.
- S. T. Hemp, A. E. Smith, J. M. Bryson, M. H. Allen and T. E. Long, Biomacromolecules, 2012, 13(8), 2439–2445 CrossRef CAS PubMed.
- S. Cheng, M. Zhang, T. Wu, S. T. Hemp, B. D. Mather, R. B. Moore and T. E. Long, J. Polym. Sci., Part A: Polym. Chem., 2012, 50(1), 166–173 CrossRef CAS.
- B. D. Mather, M. B. Baker, F. L. Beyer, M. D. Green, M. A. G. Berg and T. E. Long, Macromolecules, 2007, 40(13), 4396–4398 CrossRef CAS.
- S. T. Hemp and T. E. Long, Macromol. Biosci., 2012, 12(1), 29–39 CrossRef CAS PubMed.
- M. Zhang, M. Zhang, R. B. Moore and T. E. Long, Polymer, 2013, 54(14), 3521–3528 CrossRef CAS.
- S. R. Williams, W. Wang, K. I. Winey and T. E. Long, Macromolecules, 2008, 41(23), 9072–9079 CrossRef CAS.
- R. Gao, M. Zhang, N. Dixit, R. B. Moore and T. E. Long, Polymer, 2012, 53(6), 1203–1211 CrossRef CAS.
- H. Ghassemi, D. J. Riley, M. Curtis, E. Bonaplata and J. E. McGrath, Appl. Organomet. Chem., 1998, 12(10–11), 781–785 CrossRef CAS.
- M. Tamami, S. R. Williams, J. K. Park, R. B. Moore and T. E. Long, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4159–4167 CrossRef CAS.
- D. Dieterich, W. Keberle and H. Witt, Angew. Chem., Int. Ed. Engl., 1970, 9(1), 40–50 CrossRef CAS.
- J. Mindemark and T. Bowden, Polymer, 2011, 52(25), 5716–5722 CrossRef CAS.
- R. J. Goddard and S. L. Cooper, Macromolecules, 1995, 28(5), 1390–1400 CrossRef CAS.
- S.-A. Chen and W.-C. Chan, J. Polym. Sci., Part B: Polym. Phys., 1990, 28(9), 1499–1514 CrossRef CAS.
- M. M. Coleman, K. H. Lee, D. J. Skrovanek and P. C. Painter, Macromolecules, 1986, 19(8), 2149–2157 CrossRef CAS.
- R. J. Goddard and S. L. Cooper, J. Polym. Sci., Part B: Polym. Phys., 1994, 32(8), 1557–1571 CrossRef CAS.
- C. M. Brunette, S. L. Hsu and W. J. MacKnight, Macromolecules, 1982, 15(1), 71–77 CrossRef CAS.
- C. P. Christenson, M. A. Harthcock, M. D. Meadows, H. L. Spell, W. L. Howard, M. W. Creswick, R. E. Guerra and R. B. Turner, J. Polym. Sci., Part B: Polym. Phys., 1986, 24(7), 1401–1439 CrossRef CAS.
- V. W. Srichatrapimuk and S. L. Cooper, J. Macromol. Sci., Part B: Phys., 1978, 15(2), 267–311 CrossRef.
- S. C. Yoon, Y. K. Sung and B. D. Ratner, Macromolecules, 1990, 23(20), 4351–4356 CrossRef CAS.
- H. A. Al-Salah, H. X. Xiao, J. A. McLean Jr and K. C. Frisch, J. Polym. Sci., Part A: Polym. Chem., 1988, 26(6), 1609–1620 CrossRef CAS.
- D. Dieterich, Prog. Org. Coat., 1981, 9(3), 281–340 CrossRef CAS.
- G. A. Senich and W. J. Mac Knight, A Dynamic Mechanical Study of Phase Segregation in Toluene Diisocyanate, Block Polyurethanes, in Multiphase Polymers, American Chemical Society, 1979, vol. 176, pp. 97–127 Search PubMed.
- V. V. Zharkov, A. G. Strikovsky and T. E. Verteletskaya, Polymer, 1993, 34(5), 938–941 CrossRef CAS.
- M. Zhang, R. B. Moore and T. E. Long, J. Polym. Sci., Part A: Polym. Chem., 2012, 50(18), 3710–3718 CrossRef CAS.
- R. S. McLean and B. B. Sauer, Macromolecules, 1997, 30(26), 8314–8317 CrossRef CAS.
- S. Velankar and S. L. Cooper, Macromolecules, 1998, 31(26), 9181–9192 CrossRef CAS.
- S. A. Visser and S. L. Cooper, Macromolecules, 1991, 24(9), 2576–2583 CrossRef CAS.
- D. J. Yarusso and S. L. Cooper, Macromolecules, 1983, 16(12), 1871–1880 CrossRef CAS.
- G. L. Wilkes and J. A. Emerson, J. Appl. Phys., 1976, 47(10), 4261–4264 CrossRef CAS.
- S. Das, I. Yilgor, E. Yilgor and G. L. Wilkes, Polymer, 2008, 49(1), 174–179 CrossRef CAS.
- B. K. Kim, S. Y. Lee and M. Xu, Polymer, 1996, 37(26), 5781–5793 CrossRef CAS.
- J. A. Miller, K. K. S. Hwang and S. L. Cooper, J. Macromol. Sci., Part B: Phys., 1983, 22(2), 321–341 CrossRef.
- C.-Z. Yang, T. G. Grasel, J. L. Bell, R. A. Register and S. L. Cooper, J. Polym. Sci., Part B: Polym. Phys., 1991, 29(5), 581–588 CrossRef CAS.
- M. Hara and J. L. Wu, Macromolecules, 1988, 21(2), 402–407 CrossRef CAS.
- B. K. Kim, Colloid Polym. Sci., 1996, 274(7), 599–611 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, 1H NMR spectroscopy, FT-IR, DLS, TGA-SA, SAXS, WAXD, DSC analysis. See DOI: 10.1039/c3py01779f |
| This journal is © The Royal Society of Chemistry 2014 |