 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimising the enzyme response of a porous silicon photonic crystal via the modular design of enzyme sensitive polymers†
Alexander H.
Soeriyadi‡
ab,
Bakul
Gupta‡
ab,
Peter J.
Reece
c and
J. Justin
Gooding
*ab
aSchool of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: justin.gooding@unsw.edu.au
bAustralian Centre of NanoMedicine, University of New South Wales, Sydney, NSW 2052, Australia
cSchool of Physics, University of New South Wales, Sydney, NSW 2052, Australia
First published on 6th January 2014
Abstract
We describe the immobilization within the pores of a porous silicon photonic crystal of an enzyme degradable polymer network, for optical biosensing. A porous silicon (PSi) rugate filter is a one-dimensional photonic crystal with a high-reflectivity optical resonance that is sensitive to small changes in the refractive index of the pore space permeating through the structure. An enzymatically degradable polymer network was constructed by first “clicking” an antifouling copolymer, poly(oligo ethylene glycol-co-acrylic acid)-N3, to an alkyne functionalized PSi surface via copper(I)-catalysed alkyne–azide cycloaddition (CuAAC) reaction. MMP-2 or MMP-9 specific cleavable peptide sequences, with diamine functional groups, were then added, using a 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide/N-hydroxysuccinimide (EDC/NHS) chemistry to react with the acrylic acid group. The polymer network was completed by further attachment of a sacrificial polymer, poly(hydroxyethyl acrylate-co-N-hydroxysuccinimide ester acrylate). X-ray photoelectron spectroscopy (XPS) and optical reflectivity measurements reveal successful modification of the PSi with the polymer–peptide network. Exposure of the biosensor platform to solutions of matrix metalloproteinases, MMP-2 or MMP-9, caused a change in the average refractive index of the photonic crystal, resulting in a discernible blue shift in the reflectivity spectra. The blue shift indicated the degradation of the polymer network within the porous network. Selective detection of different MMPs was demonstrated, via the use of different peptide sequences, which are selectively digestible by different MMPs, to link the two polymers.
Introduction
There is a need for devices that can detect various biological species for the development of biotechnology and medical diagnostics. For instance, it is important to measure the activity of enzymes like proteinases released from cells for the understanding of fundamental cell biology and biomedical applications.1–3 One class of the proteinases is the matrix metalloproteinases (MMPs) which are known to be released by cells as part of their normal tissue remodelling processes, such as embryonic development and cell migration.4 MMPs have also been implicated in various pathologies, such as inflammation, cardiovascular disease, and are known to be upregulated in almost every type of human cancer.4,5 Currently, the most common proteinase activity measurements are performed by fluorogenic or calorimetric methods with commercially available proteinase assay kits.6 Enzymatic responsive polymers have also been developed as sensing elements in biological devices.7–10 The working principle of most of these systems is based on fluorescent molecules that are connected to a quencher through peptide sequences. Cleavage of the peptide sequences upon exposure to proteinase enzymes gives a concomitant increase in fluorescence.11,12 An example of such a system is demonstrated in the work of Wosnick et al.13 where they conjugated poly(phenylene ethynylene) with fluorescence activation upon treatment with trypsin. In another study by Chien et al.,14 polymer micelles were utilised to observe a colour change upon exposure to MMPs. A further example is shown by Tanaka et al.15 where digestion of a biodegradable polymer by phosphatase enzymes caused the release of fluorescein to measure the enzymatic activity of cell lysate. However, all these approaches require the need for labels. In this regard, porous silicon has emerged in the last decade as a powerful platform for label free and highly sensitive optical biosensing.16–18PSi can be fabricated by the etching of silicon which creates a nanostructured architecture with pores that are perpendicular to the surface.17 These pores can be continuously tuned in size during fabrication to create a one-dimensional photonic crystal that reflects light at well-defined wavelengths. In particular, a rugate filter structure with small full width at half maximum (FWHM) has been used because its narrow reflectance peaks assist in producing sensitive devices.19 The position of the rugate filter resonance is sensitive to the refractive index of the material inside the pores, giving PSi the potential to be a label free biosensing device.19,20 In this paper the nanopores are used as a medium to immobilize a network of enzymatic degradable polymers. As a transducer, PSi translates enzymatic reactions that occur inside the pores into an optical response as a function of the change in refractive index (n) of the material inside the pores.17,21 This is essential for the use of PSi as a biosensor, as any biological or synthetic materials that have a higher refractive index than air or water can be immobilized into the pores of PSi. For instance, organic materials such as peptides and biopolymers have a refractive index in the range of 1.4–1.5. As the materials leave the pores, they are replaced by water (nwater = 1.33) or air (nair = 1), shifting the reflectivity spectrum to lower wavelengths of light, which is referred to as a blue shift.
Previous reports on using rugate filters as biosensors for proteinase detection have been limited to devices that can only detect a range of proteinases.21,22 The lack of specificity of the current ‘state of the art’ in the literature is due to the immobilization of more generic biomolecules such as zein23 (digestible by pepsin which degrades food protein) and gelatin21 (digestible by subtilisin, a non-specific proteinase, and gelatinases). As a result, the current PSi biosensors are limited by the choice of biomolecules that can be immobilized inside the pores. In this present study, the aim was to develop a generic strategy to allow the detection of a specific proteinase (e.g. MMP-2 or MMP-9). Improvement of the specificity is achieved by employing a new modular approach to form enzyme sensitive materials inside the PSi pores. This is carried out by attaching a custom-made polymer–peptide network that is highly tuneable where the peptide linkage provides the selectivity. This innovative strategy will expand the range of capabilities for PSi based optical biosensors to provide a universal platform for enzyme detection systems.
Although there are plenty of examples for polymerization on surfaces,24,25 the modification of copolymer structures, or networks, anchored inside the pores of photonic crystals is still scarce in the literature. One example was shown by Voelcker and co-workers26 where surface initiated polymerization of poly(N-isopropyl acrylamide) inside the pores of porous silicon was achieved through Atom Transfer Radical Polymerization (ATRP) where the initiator was immobilized through silane chemistry on oxidised silicon. However, this method is not suitable for our purpose as the oxide layer within the PSi is not sufficiently stable in aqueous solution, under the timescales of the enzymatic measurements, resulting in signal drift.27 One way to maintain the optical integrity of the PSi is through passivation of the silicon via hydrosilylation.28 Previous studies by us have also shown that hydrosilylation with 1,8-nonadiyne opened up the possibility of further modification of the pores with “click” chemistry.29–31 In this work, building on our surface chemistry,29–31 the PSi was further modified with a synthetic polymer–peptide network. An anti-fouling polymer, P1 poly(oligoethylene glycol acrylate-stat-acrylic acid)N3, and a sacrificial polymer, P2 poly(hydroxyethyl acrylate-stat-N-hydroxysuccinimide ester acrylate), were synthesized via Cu(0)-mediated polymerization.32–34 The enzymatically degradable polymeric network was immobilized within the pores of PSi by first “clicking” P1 to an alkyne modified PSi surface, followed by immobilization of a peptide with diamine functionalities, and then the attachment of P2. These surfaces were then tested for their performance as an optical biosensor.
Materials and methods
Ethyl-2-bromoisobutyrate (EbiB, Aldrich, 98%), copper(II) bromide (Sigma-Aldrich, 99%), tris(2-(dimethylamino)ethyl)amine (Me6TREN) (Sigma-Aldrich, 99%), tetrahydrofuran (THF, Sigma, 99%), monomer acrylic acid N-hydrosuccinimide ester (Sigma-Aldrich, 99%) and dimethyl sulphoxide (DMSO, UNIVAR, AR) were all used as received. Copper wire (diameter = 1.25 mm) was activated by washing in sulfuric acid for 10 min. Tris(2-(dimethylamino)ethyl)amine (Me6TREN) (Sigma-Aldrich, 99%) was used as received. Monomers oligo(ethylene glycol) methyl ether acrylate (OEGA480) and hydroxyethyl acrylate (HEA) (Sigma-Aldrich, 99%) were de-inhibited by percolating over a column of basic alumina (Ajax, AR). Prime grade single-side polished silicon wafers, 100-oriented (〈100〉 ± 0.05°), p-type (boron), 500–550 μm thick, 0.001–0.0015 Ω cm resistivity as provided by suppliers, were obtained from SILTRONIX (Archamps, France).Experimental procedure
Polymerization of P1 and P2
Conversion of polymerization was calculated via NMR. Both OEGA and tBA conversion was calculated by comparing the ratio between the intensity of acrylate bond (CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) and methyl ether bond (O–CH3) and tert-butyl bond (C–C3H9) for OEGA and tBA respectively. The conversion for copolymerization is calculated to be >99%.
CH2) and methyl ether bond (O–CH3) and tert-butyl bond (C–C3H9) for OEGA and tBA respectively. The conversion for copolymerization is calculated to be >99%.
The polymer was purified via dialysis to remove any small molecule impurities. Pure poly(OEGA-stat-tBA) (2 g) was then dissolved in DMF (4.5 mL) and mixed with NaN3 (100 mg) and left to react overnight. The resulting polymer was purified again via dialysis against water and methanol. The presence of the azide group was verified by IR measurements. Finally the polymer was dissolved in dichloromethane (DCM) and mixed with trifluoroacetic acid (TFA) for the deprotection of the tert-butyl group to generate the acrylic acid group. The final polymer was isolated via dialysis and vacuum to remove any solvent residue and analysed via NMR and IR.
Conversion of polymerization was calculated via NMR. Both OEGA and tBA conversion was calculated by comparing the ratio between the intensity of acrylate bond (CHCH2) and alkyl ester bond (O–CH2) and succinimide bond (CH2–CH2) for HEA and NHSA respectively. The conversion for copolymerization was calculated to be 100% for NHSA and 75% for HEA.
Fabrication of a PSi–polymer construct
The first fabrication step to construct the PSi–polymer structure is formation of an alkyne self-assembled monolayer to form surface 1, Scheme 1 by a thermal hydrosilylation method previously described by our group (ESI†).31 It was then followed by the attachment of polymer P1 as explained below.
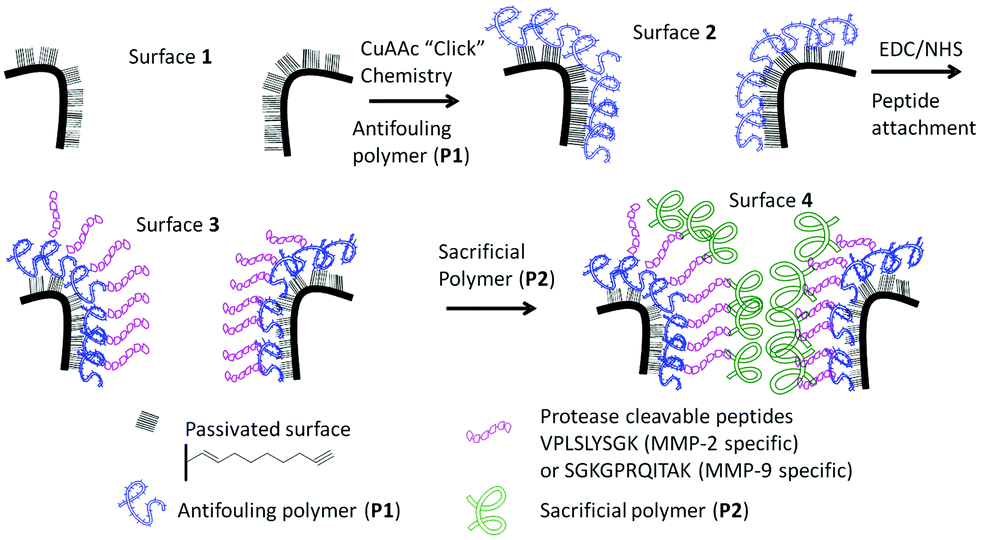 | ||
| Scheme 1 Schematic of the surface modification steps required for the preparation of an enzymatic responsive platform for monitoring proteinase activity. Firstly, an antifouling polymer (P1) was attached to the alkyne functional surface (1) using “click” chemistry to form surface 2. The acid moieties of the polymer were further activated and reacted with the peptide using EDC/NHS chemistry (3). Surfaces incubated with the sacrificial polymer P2 result in the final surface 4. | ||
Proteinase assays
For the proteinase assays, the surface modified samples were placed in a sample with proteinase enzymes in PBS and incubated at 37 °C during the entire duration of measurements. The samples were transferred to the stage and measured under wet conditions at specific designated time intervals. Optical reflectivity spectra were measured using a custom-built optical setup (Fig. S2†).Characterization techniques
Results and discussion
The overall construction of the PSi–polymer platform for proteinase detection is shown in Scheme 1.Polymer synthesis
Two polymers were synthesized for the purpose of this work (Scheme 2). Firstly, the antifouling polymer (P1) developed in this study required several characteristics, (1) an anti-fouling domain, (2) the ability to be functionalized by a peptide, and (3) a functional moiety to allow conjugation to the alkyne terminated PSi surface. In order to achieve these properties, controlled radical polymerization was used for the synthesis of P1. Controlled radical polymerization has proven to be a powerful tool to create complex macromolecular structures with control over molecular weight, topography, functional groups, as well as end groups.35 There are various controlled radical polymerization methods such as Reversible Addition Fragmentation Transfer (RAFT),36 Atom Transfer Radical Polymerization (ATRP),36 Nitroxide Mediated Polymerization (NMP)38 and recently Cu(0)-mediated radical polymerization.33,39 In this work, all polymer synthesis was performed via Cu(0)-mediated polymerization. This polymerization technique was selected because of the ease of the polymerization procedure and its excellent end group fidelity which has been demonstrated previously.34,40 Firstly, P1 was synthesized by statistical copolymerization of OEGA480 and tBA to make poly(OEGA-stat-tBA)Br. As can be seen from Fig. S3,† the polymerization undergoes virtually full conversion (>99%) with excellent molecular weight control (PDI = 1.11, Fig. S4†). The molecular weight obtained from GPC (Mn = 8400 g mol−1) is in good agreement with the molecular weight estimated by NMR (Mn = 7500 g mol−1). The bromine end-group of the polymer was then modified with NaN3 to give poly(OEGA-stat-tBA)N3 followed by the deprotection of the tert-butyl group with trifluoroacetic acid (TFA) to form an acrylic acid group resulting in poly(OEGA-stat-AA)N3. Deprotection of the tert-butyl group to give an acidic group can be observed by NMR spectroscopy by tracking the disappearance of peaks at 1.85 ppm (Fig. 1). Moreover, this modification can be easily followed by FT-IR measurements as shown in Fig. 2 where the azide peak is easily identifiable at a bending vibration of 2120 cm−1 and –OH stretch from hydrogen bonds in the carboxylic group is seen as a broad peak at 3200–3600 cm−1 after deprotection of the tert-butyl group with acid treatment.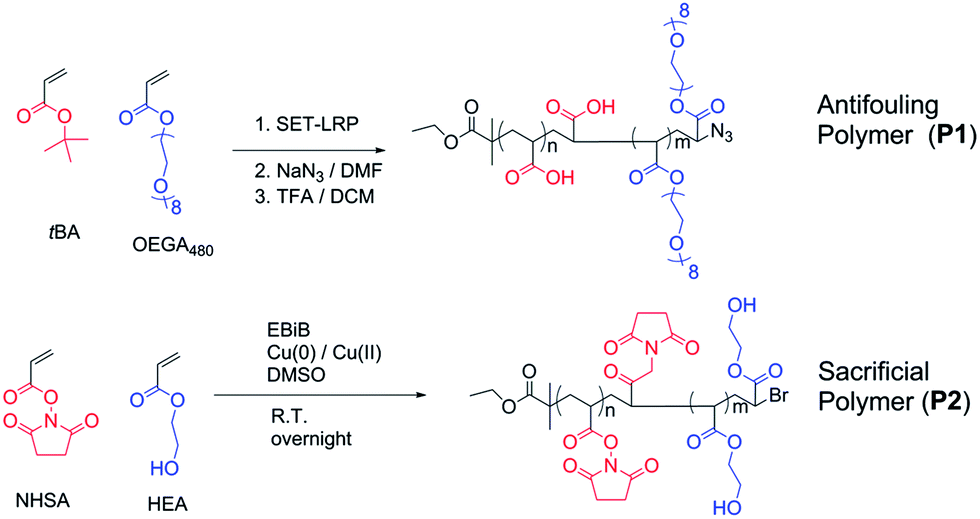 | ||
| Scheme 2 Schematic for the synthesis of the anchored polymer (P1) poly(OEGA-stat-AA)N3 and the sacrificial polymer (P2) poly(HEA-stat-NHSA). Both polymerization reactions were Cu(0)-mediated. The final polymer P1 was obtained through end group modification with sodium azide and deprotection of the tert-butyl group under acidic conditions. | ||
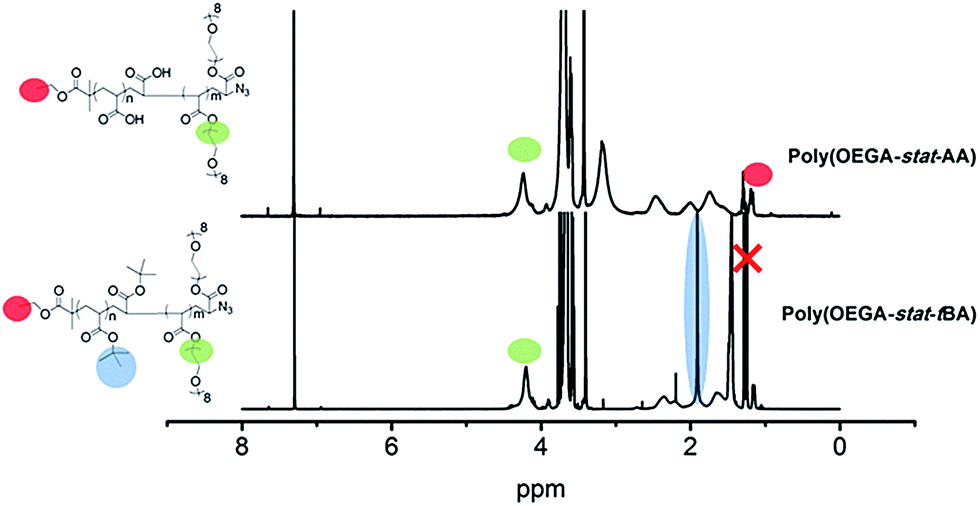 | ||
| Fig. 1 NMR characterization of anchored polymer P1 synthesis before and after deprotection. The NMR spectra show expected peaks at 4.27 ppm and 1.85 ppm corresponding to the functional groups of OEGA and tBA respectively. After deprotection, there is an obvious disappearance of the peak at around 1.85. Note: the red cross denotes solvent peaks. | ||
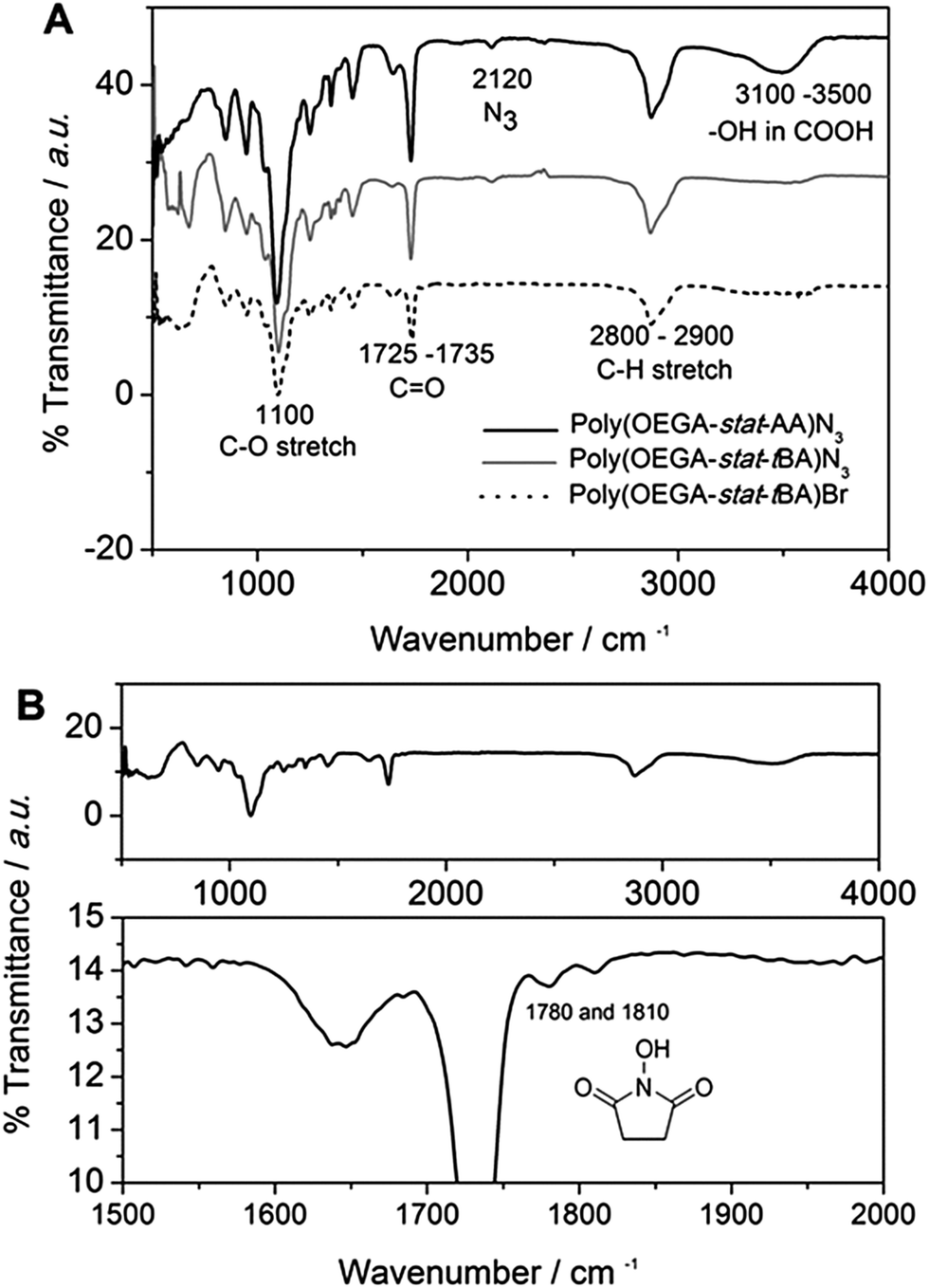 | ||
| Fig. 2 FT-IR spectra of (top, (A)) synthesis and modification of the anchored polymer P1. The spectra show before and after azidation and deprotection of tBA to AA an expected peak at 2120 cm−1 indicating the presence of an azide terminal group and a broad peak at 3100–3500 cm−1 indicating the presence of an acidic group. (bottom, (B)) The sacrificial polymer P2 with two peaks at 1780 and 1810 cm−1 indicating the presence of the N-hydroxysuccinimide ester moiety in the copolymer. | ||
The sacrificial polymer P2 poly(HEA-stat-NHSA) was also synthesized via Cu(0)-mediated polymerization. P2 is a sacrificial unit to leave the pores after cleavage of the proteinase selective peptide by the relevant MMP. Hence it is required to couple to free amine groups of the peptide sequence that are already immobilized onto P1. Therefore, P2 required functional groups that can react with amine groups such as N-hydroxysuccinimide ester (NHS) or pentafluorophenyl group (PFP). The NHS group is chosen over PFP as the pentafluorophenol group which will be liberated through the reaction with amine could be toxic. In order to synthesize P2, N-hydroxysuccinimide ester acrylate (NHSA) was copolymerized with hydroxyethyl acrylate (HEA). The reason for this choice is two-fold, (1) hydroxyethyl acrylate (HEA) is known to be a biocompatible material and (2) the hydroxyethyl acrylate group has a different chemical signature compared with POEGA used in P1 where the greater amount of the O–CO group can be detected by X-ray photoelectron spectroscopy (XPS). The polymerization of HEA and NHSA was followed by NMR, as shown in Fig. S5.† The conversion after 14 h reaction was found to be 100% of NHSA and 75% of HEA. The molecular weight of P2 was calculated from NMR (Mn = 7500 g mol−1) and GPC (Mn = 7200 g mol−1) with a narrow polydispersity index (PDI) = 1.13 (Fig. S6†). The molecular weight obtained from GPC is in good agreement with theoretical calculation and molecular weight obtained from NMR. It is important to note that polymerization of NHSA has been shown to be challenging by Shunmugan et al.41 when using ATRP polymerization as well as RAFT polymerization as demonstrated by D'Agosto et al.42 In this work the copolymerization of hydroxyethyl acrylate (HEA) and N-hydroxysuccinimide ester acrylate was reasonably straightforward and can be useful for other applications. More importantly, the presence of the NHSA group in the polymer is essential for further reaction with the amine group that is the presence in the peptide sequences employed. The presence of this functional group can be clearly seen in proton NMR with peaks at 2.85 ppm which arise from the CH2–CH2 bond of the N-hydroxysuccinimide group (Fig. S5†). The presence of the N-hydroxysuccinimide group is also confirmed by FTIR measurements with two small peaks at 1780 and 1810 cm−1 indicative of the N-hydroxysuccinimide moiety (Fig. 2).
PSi fabrication and modification
Porous silicon was made via anodic etching of p-type Si(100) in ethanolic HF. A rugate filter structure was chosen due to its optical reflectivity character which has a narrow and sharp peak.29Fig. 3A shows the cross-sectional views of the entire 60-layer porous silicon (PSi) rugate filter structure with the pores perpendicular to each other. The rugate filter was formed after anodization using a sinusoidal current density (freshly etched surface). The properties of the porous structure such as porosity and pore sizes can be controlled via the anodization parameters. Fig. 3B shows a top view of the porous structure with the average pore size of 20 nm. Surface modification was then performed to modify the surfaces of the pores to allow passivation of the surface from water penetration and thus protecting the photonic crystal entity from surface oxidation (i.e. formation of Si–Ox from Si).43 This step is essential as the optical reflectivity of the material is highly dependent on the chemistry of the pores.27 Hydrosilylation with 1,8-nonadiyne (Scheme S1†) was chosen as it provides a closely packed self-assembled monolayer that protects the silicon from oxidation as well as possessing remaining alkyne functionalities which provide the opportunity for further modification via copper catalyzed azide–alkyne cycloaddition (CuAAc) “click” chemistry.31
 | ||
| Fig. 3 Scanning electron microscopy: ((A), left) cross-sectional view of a 60-layer PSi rugate filter and ((B), right) top view of the PSi with an average pore size of 27 nm. | ||
The success of the surface modification of the PSi was determined by optical reflectivity measurements and XPS. Firstly, hydrosilylation of the 1,8-nonadiyne on PSi (freshly etched surface → 1, Scheme S1†) resulted in a significant red shift of the reflectivity peaks as seen from Fig. S7.† Previous work by Guan et al. has shown that PSi microparticles passivated via thermal hydrosilylation maintain their optical integrity for up to 7 days in biological environment such as phosphate buffered saline (PBS), human blood, or Dulbecco's modified eagle's medium (DMEM) cell culture media.44 Thus, this surface is suitable for the proteinase assay which is the subject of this study. “Clicking” of the anchored polymer poly(OEGA-stat-AA)N3P1 on an alkyne functional surface was conducted through CuAAc reaction and resulted in further red shift (surface 2, Fig. 4). Peptide immobilization was then conducted via the activation of the carboxylic acid via EDC/NHS chemistry followed by addition of the peptide sequences, VPLSLYSGK peptide for MMP-2 proteinase specific cleavable linker and SGKGPRQITAK for MMP-9 proteinase specific linker. These peptide sequences were chosen based on previous work by Turk et al.45 and Kridel et al.46 which determined the cleavage sites for different MMP proteinases (MMP-2 and MMP-9 respectively) and their kinetics. It was concluded that the linkages between serine (S)–leucine (L) and glutamine (Q)–isoleucine (I) are the most prevalent linkage sites for MMP-2 and MMP-9 proteinase enzymes respectively.45 Both peptide sequences were designed with two free amine functional groups, one was located at the N-terminal of the peptide and the other from alysine (K) with a free amine on the side chain at the C-terminal. This diamine functionality allows statistical cross-linking with the sacrificial polymer P2 poly(HEA-stat-NHSA). Any amine group from the peptide which does not react with –COOH of P1 during the EDC/NHS step may react with the NHSA group of P2. Both the immobilization of the peptide (surface 2 → 3) and the sacrificial polymer P2 (surface 3 → 4) was easily followed by reflectivity measurements as shown in Fig. 4. The total red shift of the peaks was measured to be around 25–30 nm which indicates successful immobilization of the organic compounds inside the pores of the PSi.
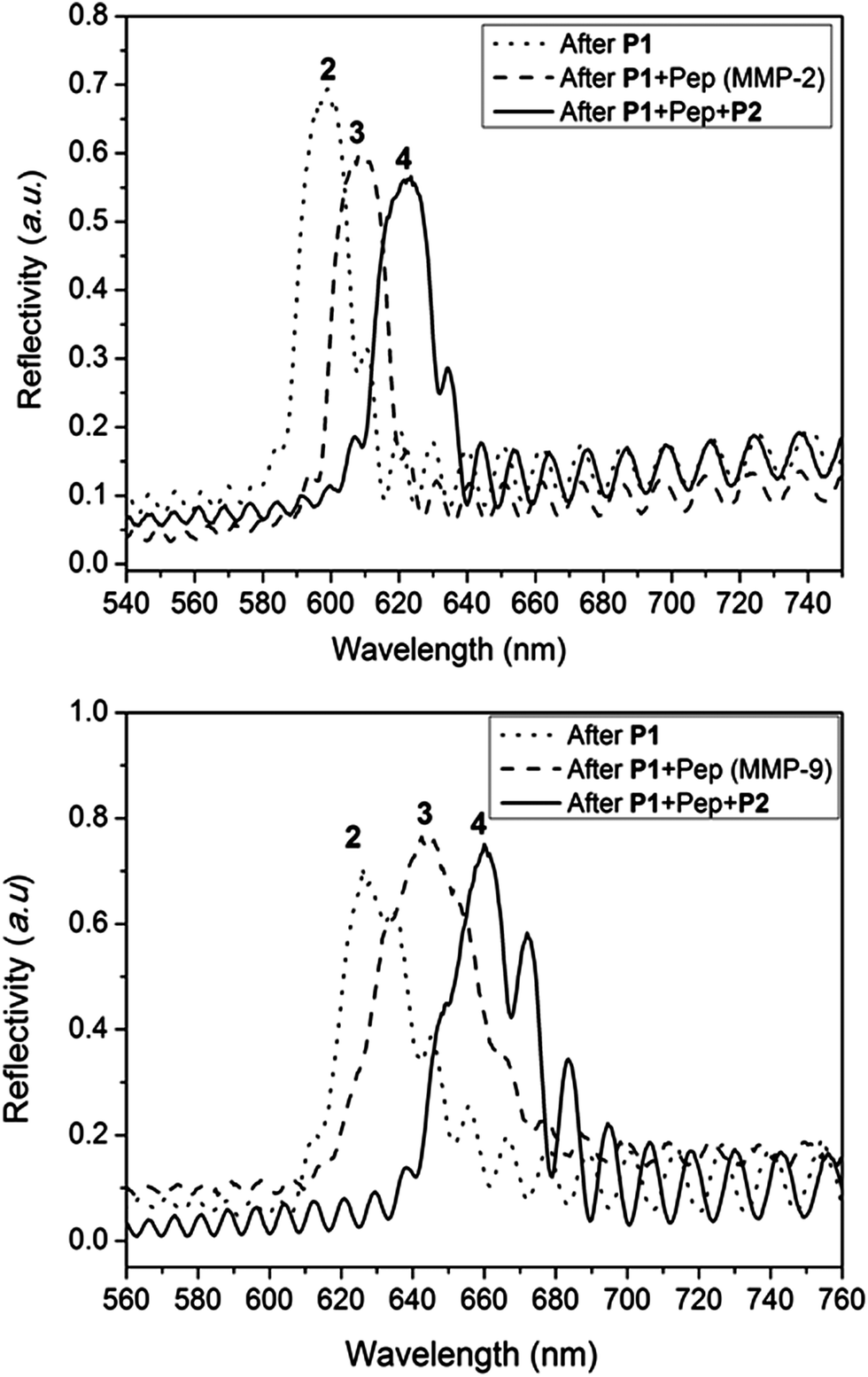 | ||
| Fig. 4 Typical reflectivity measurement of PSi during different stages of surface modification showing reflectivity peaks shifting to a higher wavelength (“red shift”) upon modification with the antifouling polymer P1, the peptide sequence cleavable by MMP-2 or MMP-9 proteinase enzyme, and the sacrificial polymer P2. Surface 2, 3, and 4 refer to the difference surfaces depicted in Scheme 1. All reflectivity measurements were performed under wet conditions (i.e. under aqueous conditions). | ||
For further characterization of the surface modification process, chemical analysis was performed by X-ray photoelectron spectroscopy (XPS). XPS survey spectra (Fig. S8†) show the element at each step of modification. It can be seen from the survey spectra that the expected elements Si, C, O, and F were present in surface 1 but no N was present. N peaks appeared with surfaces 2–4 which indicates successful addition of polymers and peptide onto the pores. Furthermore, Fig. 5 shows narrow scans of C 1s and N 1s of each step of the modification procedures. Surface 1 shows a peak at a binding energy of 285 eV indicative of the carbon bound to carbon (C–C). This signal came from the functionalized alkyne layer. Since there was no elemental nitrogen present on the surface, no nitrogen signal was detected as expected. After attachment of P1 (surface 2), there were new peaks observed at ∼286.8 eV in the C 1s spectrum. This was indicative of oxygen bound to carbon (C–O) which represented the ethylene glycol group of P1. The high-resolution N 1s data (Fig. 5b) showed two peaks centred at 400.58 eV and 403.48 eV with a ratio of around 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, indicating the presence of chemically distinct nitrogen atoms consistent with the formation of a triazole moiety. This strongly suggests the fusion of the azido polymer P1 to the alkyne modified surface.31 After immobilization of the peptide, there was a significant increase in the intensity of the N 1s peak especially at 400.38 eV which is attributed to various nitrogen based peptide bonds in the peptide (mainly amine and amide).19 The best fit to the experimental N 1s emission curve was obtained when this spectral region was deconvoluted into two functions having binding energies of 402.38 eV and 400.38 eV. The ratios of N/C and N/Si were calculated which are increased from 0.004 to 0.014 and 0.015 to 0.057 respectively from surface 2 to 3, indicating more N as the peptide and polymer attached. Finally, the C 1s narrow scan of surface 4 shows a significant increase in peaks at 289.1 eV which can be attributed to ester bonds (O–CO). This is evidence for the attachment of poly(HEA-stat-NHSA) which has a high component of ester group (O–CO) in the HEA bond. It is important to note that from the N 1s narrow scan there is attenuation of the peak at around 403 eV due to the fact that the triazole peaks are small components of the whole construct especially after another layer of polymer is bound. The composition of N compared to C and Si is also increased to 0.019 and 0.066 from 0.014 and 0.057 respectively.
:1, indicating the presence of chemically distinct nitrogen atoms consistent with the formation of a triazole moiety. This strongly suggests the fusion of the azido polymer P1 to the alkyne modified surface.31 After immobilization of the peptide, there was a significant increase in the intensity of the N 1s peak especially at 400.38 eV which is attributed to various nitrogen based peptide bonds in the peptide (mainly amine and amide).19 The best fit to the experimental N 1s emission curve was obtained when this spectral region was deconvoluted into two functions having binding energies of 402.38 eV and 400.38 eV. The ratios of N/C and N/Si were calculated which are increased from 0.004 to 0.014 and 0.015 to 0.057 respectively from surface 2 to 3, indicating more N as the peptide and polymer attached. Finally, the C 1s narrow scan of surface 4 shows a significant increase in peaks at 289.1 eV which can be attributed to ester bonds (O–CO). This is evidence for the attachment of poly(HEA-stat-NHSA) which has a high component of ester group (O–CO) in the HEA bond. It is important to note that from the N 1s narrow scan there is attenuation of the peak at around 403 eV due to the fact that the triazole peaks are small components of the whole construct especially after another layer of polymer is bound. The composition of N compared to C and Si is also increased to 0.019 and 0.066 from 0.014 and 0.057 respectively.
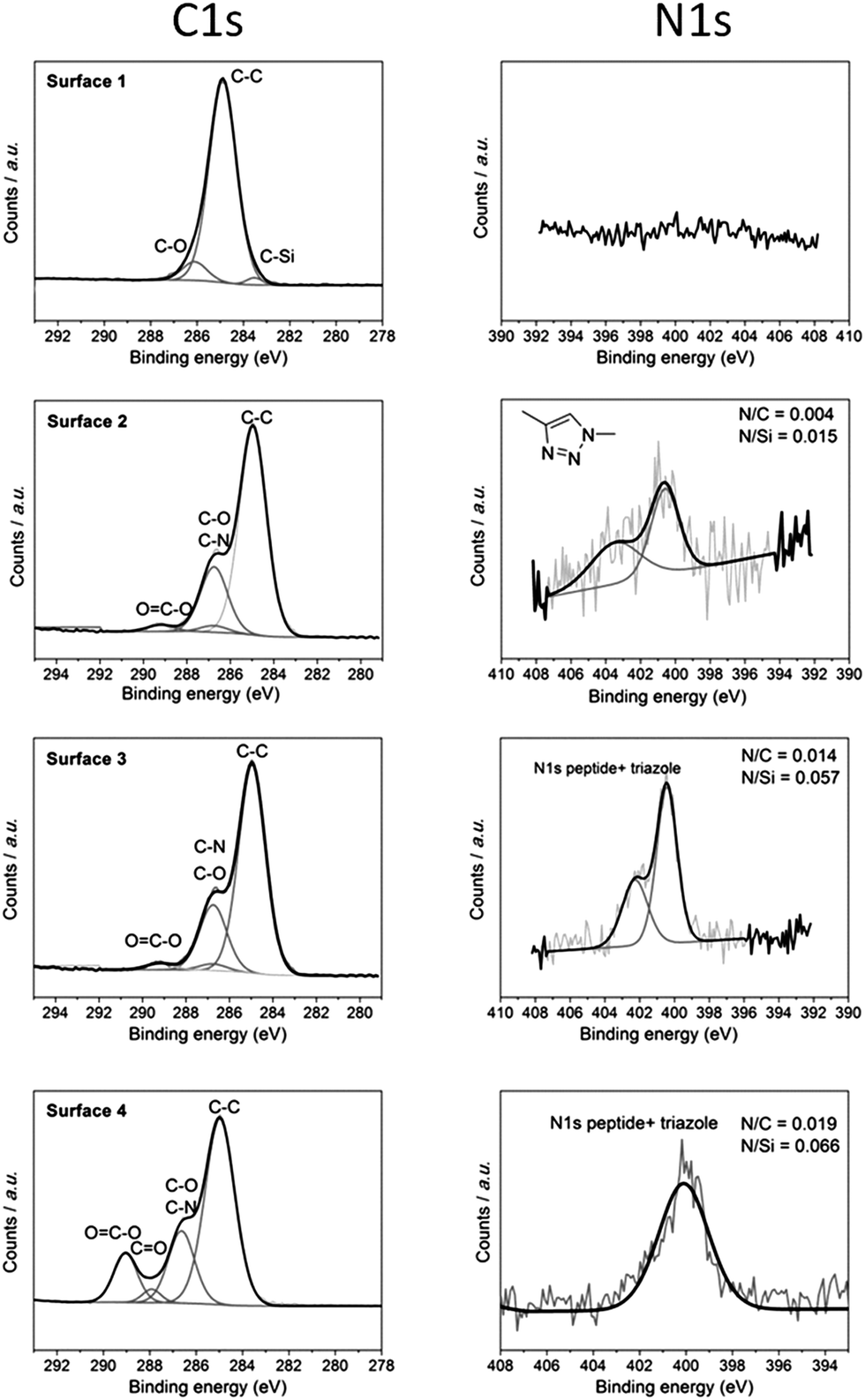 | ||
| Fig. 5 XPS narrow scans for the C 1s and N 1s regions for each of the surfaces 1–4 shown in Scheme 1. Both narrow scans indicate the immobilization of P1, peptide, and P2 with the expected peaks for C–O and O–CO bonds at 286.8 eV and 289.1 eV respectively. The ratio elemental analysis of N/Si and N/C also showed an increase after each modification step as expected. | ||
Proteinase assay
The performance of the modified PSi–polymer construct as an optical biosensor was tested by conducting proteinase assays and measuring the optical shift in the reflectivity spectra of the proteinase incubated surfaces over time. The PSi–polymer chip was incubated with different proteinase enzymes as well as a control (PBS buffer solution). Examples of the reflectivity measurement can be seen in Fig. 6 and S9.† Two different proteinases, MMP-2 and MMP-9, were chosen to test the selectivity of the surface to different proteinases. Upon exposure to proteinase enzymes incubated at 37 °C, cleavage of the peptide bond takes place due to the proteolytic activity of the enzymes as depicted in Fig. 7 (top). As the peptide is cleaved by the proteinases, the amino acid fragments and the sacrificial polymeric material P2 leave the pores bringing the reflectivity shift to the peaks corresponding to surface 2, changing the bulk refractive index of the crystal. This change in refractive index is measured as a “blue shift” in the reflectivity spectra of the PSi sensor.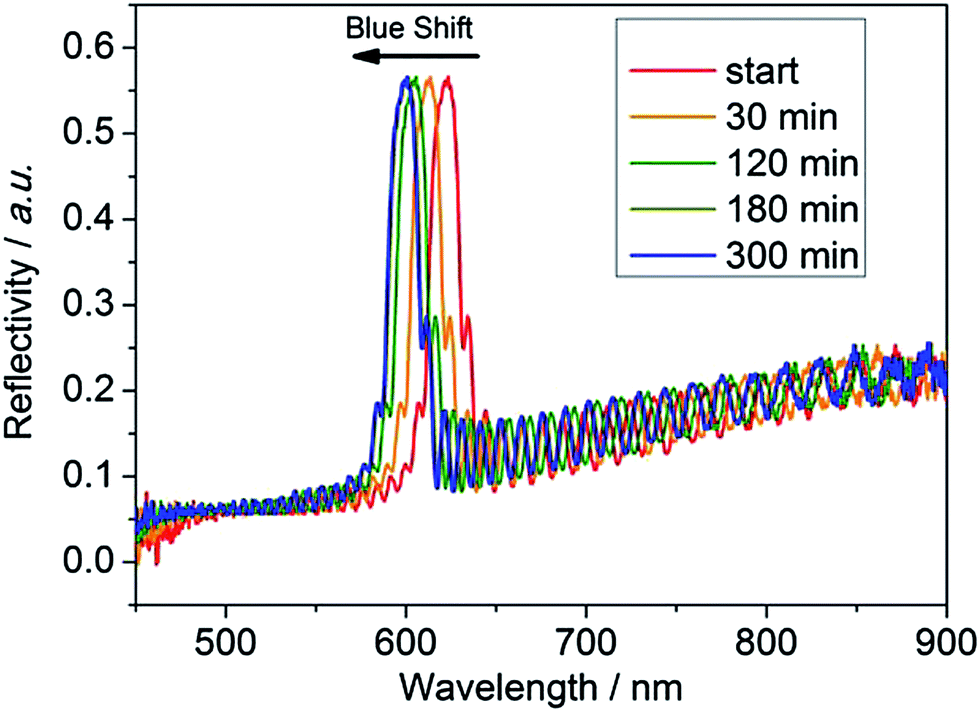 | ||
| Fig. 6 Reflectivity spectrum of the PSi chip with the VPLSLYSGK cleavable polymer network after exposure to the protease enzyme MMP-2 with the reflectivity peaks shifting towards a lower wavelength (“blue shift”) indicating materials leaving the pores. The peaks shift because as the protease digests the organic material over time, the space that is left by the organic material (n ∼ 1.45) is filled by water (n = 1.33), which has a lower refractive index than the organic material. | ||
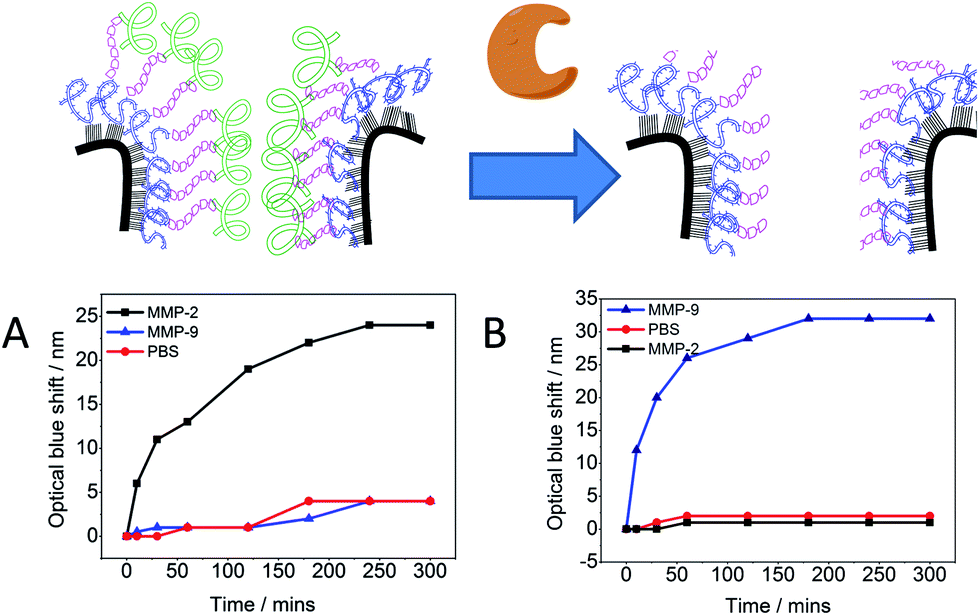 | ||
| Fig. 7 Proteinase assay of the PSi chip modified with P1, P2, and MMP-2 specific cleavable peptide VPLSLYSGK (A) or MMP-9 specific cleavable peptide SGKGPRQITAK (B). The optical blue shift is an indication that peptide cleavage of the material inside the PSi pores led to replacement of the organic material by water. The modified PSi chip only responds upon exposure to a specific enzyme. | ||
The optical detection of proteinase activity was accomplished in 100 nM concentration of proteinase enzyme. Previous work by Kilian et al.21 has shown the potential of using modified PSi as an optical biosensor by immobilizing a peptide and biopolymer gelatin.19,21 In the work presented here, improvements have been made by increasing the total reflectivity shift and the specificity through the formation of the crosslinked polymer network for a given MMP. Fig. 7 demonstrates the specificity of the chip towards specific proteinases as an optical response. It was observed when the chips with specific peptides were incubated with the complementary proteinase (for which the chip was made selective). In control samples with PBS solution negligible optical shift was observed (<5 nm). More importantly, incubation of the modified PSi with a mismatch proteinase also resulted in negligible optical shift (<5 nm). That is the polymer–VPLSLYSGK network was not cleaved under exposure to MMP-9 proteinase and the polymer–SGKGPRQITAK network was not cleaved under exposure to MMP-2 proteinase. The proteinase assay activity was done up to 5 hours as the plot approaches a plateau after around 3–4 hours. This indicates that the enzyme has digested all the “digestible” material inside the pores.
The detection capabilities of this system are an extension of previous work by us as well as Sailor and co-workers.18,19,21 These smart surface chips will have potential as selective and sensitive optical biosensors.18,21 However, the integration of polymer networks have added specificity to the system making it more versatile. For example, one could simply change the peptide sequence tailored to such a sequence cleavable by a given target proteinase.
Conclusion
We have demonstrated a generic approach to optimize the sensing capability of porous silicon through a modular polymer conjugation strategy where the surface of the PSi was first modified with an antifouling polymer, then an enzyme cleavable link was added which bridged the antifouling polymer and a second sacrificial polymer that was lost upon enzyme cleavage of the peptide. Polymers required for this work were synthesized through Cu(0)-mediated polymerization which is suitable to synthesize copolymers poly(OEGA-stat-AA)N3 and poly(HEA-stat-NHSA) with good control. PSi substrates with specific responses to MMP-2 and MMP-9 were prepared through grafting of the copolymers and peptide sequences. Cleavage of the peptide–polymer network by the appropriate proteinase decreases the average refractive index of the photonic crystal resulting in a change in the reflectivity peak to lower wavelengths (blue-shift). The PSi–polymer constructs were shown to have selectivity towards different MMP enzymes. The approach could easily be tailored for different chemical/biochemical moieties, thus increasing the potential of such smart surfaces for biosensor applications. These structures could be easily expanded for other proteinase enzymes by simply changing the specific peptide sequences.References
- J. El-Ali, P. K. Sorger and K. F. Jensen, Nature, 2006, 442, 403–411 CrossRef CAS PubMed.
- J.-H. Park, L. Gu, G. von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Nat. Mater., 2009, 8, 331–336 CrossRef CAS PubMed.
- M. L. Yarmush and K. R. King, Annu. Rev. Biomed. Eng., 2009, 11, 235–257 CrossRef CAS PubMed.
- I. Stamenkovic, Semin. Cancer Biol., 2000, 10, 415–433 CrossRef CAS PubMed.
- M. Egeblad and Z. Werb, Nat. Rev. Cancer, 2002, 2, 161–174 CrossRef CAS PubMed.
- C. Cupp-Enyard, J. Vis. Exp., 2008, 19, 899 Search PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- J. Hu and S. Liu, Macromolecules, 2010, 43, 8315–8330 CrossRef CAS.
- J. Hu, G. Zhang and S. Liu, Chem. Soc. Rev., 2012, 41, 5933–5949 RSC.
- Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289–7325 RSC.
- M. R. Pinto and K. S. Schanze, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7505–7510 CrossRef CAS PubMed.
- S. B. Lowe, J. A. G. Dick, B. E. Cohen and M. M. Stevens, ACS Nano, 2011, 6, 851–857 CrossRef PubMed.
- J. H. Wosnick, C. M. Mello and T. M. Swager, J. Am. Chem. Soc., 2005, 127, 3400–3405 CrossRef CAS PubMed.
- M.-P. Chien, M. P. Thompson, E. C. Lin and N. C. Gianneschi, Chem. Sci., 2012, 3, 2690–2694 RSC.
- K. Tanaka, N. Kitamura and Y. Chujo, Macromolecules, 2010, 43, 6180–6184 CrossRef CAS.
- Y. Y. Li, F. Cunin, J. R. Link, T. Gao, R. E. Betts, S. H. Reiver, V. Chin, S. N. Bhatia and M. J. Sailor, Science, 2003, 299, 2045–2047 CrossRef CAS PubMed.
- M. J. Sailor and J. R. Link, Chem. Commun., 2005, 1375–1383 RSC.
- M. M. Orosco, C. Pacholski and M. J. Sailor, Nat. Nanotechnol., 2009, 4, 255–258 CrossRef CAS PubMed.
- K. A. Kilian, T. Böcking, K. Gaus, M. Gal and J. J. Gooding, ACS Nano, 2007, 1, 355–361 CrossRef CAS PubMed.
- H. Qiao, B. Guan, J. J. Gooding and P. J. Reece, Opt. Express, 2010, 18, 15174–15182 CrossRef CAS PubMed.
- K. A. Kilian, L. M. H. Lai, A. Magenau, S. n. Cartland, T. Böcking, N. Di Girolamo, M. Gal, K. Gaus and J. J. Gooding, Nano Lett., 2009, 9, 2021–2025 CrossRef CAS PubMed.
- A. Jane, R. Dronov, A. Hodges and N. H. Voelcker, Trends Biotechnol., 2009, 27, 230–239 CrossRef CAS PubMed.
- M. M. Orosco, C. Pacholski, G. M. Miskelly and M. J. Sailor, Adv. Mater., 2006, 18, 1393–1396 CrossRef CAS.
- O. Foster, A. H. Soeriyadi, M. R. Whittaker, T. P. Davis and C. Boyer, Polym. Chem., 2012, 3, 2102–2111 RSC.
- K. A. Gunay, N. Schuwer and H.-A. Klok, Polym. Chem., 2012, 3, 2186–2192 RSC.
- R. B. Vasani, S. J. P. McInnes, M. A. Cole, A. M. M. Jani, A. V. Ellis and N. H. Voelcker, Langmuir, 2011, 27, 7843–7853 CrossRef CAS PubMed.
- B. Gupta, Y. Zhu, B. Guan, P. J. Reece and J. J. Gooding, Analyst, 2013, 138, 3593–3615 RSC.
- M. P. Stewart and J. M. Buriak, Angew. Chem., Int. Ed., 1998, 37, 3257–3260 CrossRef CAS.
- S. Ciampi, T. Böcking, K. A. Kilian, J. B. Harper and J. J. Gooding, Langmuir, 2008, 24, 5888–5892 CrossRef CAS PubMed.
- B. Guan, S. Ciampi, G. Le Saux, K. Gaus, P. J. Reece and J. J. Gooding, Langmuir, 2010, 27, 328–334 CrossRef PubMed.
- S. Ciampi, T. Böcking, K. A. Kilian, M. James, J. B. Harper and J. J. Gooding, Langmuir, 2007, 23, 9320–9329 CrossRef CAS PubMed.
- Q. Zhang, P. Wilson, Z. Li, R. McHale, J. Godfrey, A. Anastasaki, C. Waldron and D. M. Haddleton, J. Am. Chem. Soc., 2013, 135, 7355–7363 CrossRef CAS PubMed.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS PubMed.
- K. Matyjaszewski and J. Spanswick, Mater. Today, 2005, 8, 26–33 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- D. J. Siegwart, J. K. Oh and K. Matyjaszewski, Prog. Polym. Sci., 2012, 37, 18–37 CrossRef CAS PubMed.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- F. Nyström, A. H. Soeriyadi, C. Boyer, P. B. Zetterlund and M. R. Whittaker, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5313–5321 CrossRef.
- C. Boyer, A. H. Soeriyadi, P. B. Zetterlund and M. R. Whittaker, Macromolecules, 2011, 44, 8028–8033 CrossRef CAS.
- R. Shunmugam and G. N. Tew, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5831–5843 CrossRef CAS.
- F. D'Agosto, M.-T. Charreyre and C. Pichot, Macromol. Biosci., 2001, 1, 322–328 CrossRef CAS.
- K. A. Kilian, T. Böcking, S. Ilyas, K. Gaus, W. Jessup, M. Gal and J. J. Gooding, Adv. Funct. Mater., 2007, 17, 2884–2890 CrossRef CAS.
- B. Guan, A. Magenau, K. A. Kilian, S. Ciampi, K. Gaus, P. J. Reece and J. J. Gooding, Faraday Discuss., 2011, 149, 301–317 RSC.
- B. E. Turk, L. L. Huang, E. T. Piro and L. C. Cantley, Nat. Biotechnol., 2001, 19, 661–667 CrossRef CAS PubMed.
- S. J. Kridel, E. Chen, L. P. Kotra, E. W. Howard, S. Mobashery and J. W. Smith, J. Biol. Chem., 2001, 276, 20572–20578 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3py01638b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |