 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe power of light in polymer science: photochemical processes to manipulate polymer formation, structure, and properties
Shunsuke
Chatani
a,
Christopher J.
Kloxin
b and
Christopher N.
Bowman
*ac
aDepartment of Chemistry and Biochemistry, University of Colorado, UCB 215, Boulder, Colorado 80309, USA. E-mail: Christopher.Bowman@colorado.edu
bDepartment of Material Science and Engineering and Department of Chemical and Biomolecular Engineering, University of Delaware, Newark, Delaware 19716, USA
cDepartment of Chemical and Biological Engineering, Materials Science and Engineering Program, University of Colorado, UCB 596, Boulder, Colorado 80309, USA
First published on 24th December 2013
Abstract
As the demand for polymeric materials transitions towards the need for customizable, high value, specialty polymeric materials, the ability to use light to initiate various physicochemical changes in polymers represents one of the most powerful and rapidly evolving approaches. Whether for polymer formation, polymer modification, shape change, or inducing smart material responses, light has the unique capacity for enabling 4D manipulation of each of those processes. Given the simple, 3D ability to focus light on a targeted voxel and the even simpler ability to turn a light on and off to facilitate temporal control, light has been used widely in various polymer modifications. Further, in addition to the ability to enhance the control of various reactive processes, due to the much greater energy available in a photon as compared to the thermal energy available, light enables chemical processes to occur at ambient conditions that are otherwise inaccessible without heating. In particular, within the polymer chemistry field, light has been used to cause bond formation, bond degradation, and isomerization, with subsequent reactions including polymerization, polymer degradation, polymer functionalization, and responsive changes in properties of smart materials. Here, this article attempts to provide a fundamental basis for the various photochemical processes implemented in polymer systems, followed by selected examples of that implementation in various polymerization, functionalization, degradation, and other reactions.
Introduction
The use of light to control the formation, structure, and biochemomechanical properties of polymeric materials represents one of the most powerful paradigms in polymer science. As discussed in the remainder of this article, with the advent of customizable, high value, specialty polymeric materials, the ability to fine-tune the polymer structure and properties, particularly with spatiotemporal control, has seen increasing utilization. Light is used widely in both fundamental evaluation of formation–structure–property relationships; is used to form, degrade, functionalize, and crosslink polymers; and it is used in a vast array of applications stretching from traditional photolithographic applications to in vivo polymerized biomedical applications.Advantages of photochemical processes
Light has at least three distinct advantages over other analogous methods used in polymer chemistry. First and most critically, processes that are unable to occur energetically on reasonable timescales at ambient temperature occur rapidly when photoinitiated. The energy in a mole of photons at 365 nm is 130 times more than kT (i.e., the thermal energy available to activate a reaction) at 25 °C while at 254 nm, the even higher energy photons have 190 times more energy than kT at 25 °C. Thus, chemical reaction processes such as isomerization, cyclization, and bond breaking are readily initiated upon irradiation and subsequent absorption. In appropriate compounds, these processes do not readily occur at ambient temperature because of an insufficient amount of energy available to overcome the activation barrier whereas the absorption of a photon is sufficient to populate energetic states that lead to these various reactions due to the higher energy available from typical UV and shorter wavelength visible irradiation. Ultimately, this aspect of photochemical reactions means that mixtures of materials with all of the necessary components for causing the desired reaction may be present for extended times at ambient, or even mildly elevated, temperatures without reacting and subsequently react rapidly at those same conditions upon exposure to the appropriate light source.This behavior leads to the second attribute that renders photochemical processes desirable in polymer science – the temporal control afforded by the simplicity of being able to turn a light on and off. Photochemical processes are activated simply by opening a shutter or switching on a light and require little in the way of harsh environmental conditions, solvents or toxic components to initiate the reaction. In fact, one of the primary driving forces for implementing photochemical reactions is the ability to eliminate solvents from the reaction mixture, as enabled by the shelf-life stability and ability to shutter the process. In a related aspect, typical light sources for photoinitiating processes are also widely available, depending on the need for specific wavelengths and intensities. The recent development of LED light sources in the UV and blue visible range has enabled an even broader range of polymer scientists and engineers to use photochemically initiated processes. Further, being able to react at ambient conditions and formulate a complete mixture of reaction components far in advance of it being reacted implies that for coatings, biomedical implants, and other applications, “greener” processes that eliminate the need for any solvents are readily developed. It should be noted that while nearly all photoinitiated processes are started by turning a light on, extinguishing the light source may or may not stop a process that was photochemically initiated. Most radical polymerizations, photoisomerization, and photoinduced cyclization reactions will cease immediately or at least rapidly in the absence of light. In contrast, many photocatalytic reactions such as the generation of Cu(I) in the copper-catalyzed azide–alkyne reaction1–6 or the photogeneration of acids and bases that catalyze various reactions7–9 will proceed long after the light is extinguished.
Finally, in addition to ambient initiation and temporal control, one of the most powerful advantages of photochemical processes is the ability to control the desired reaction spatially. With simple masking or patterned illumination that is possible with light sources coupled to mirror-arrays such as digital light projectors, two-dimensional control of photochemical initiation is achieved; however, it is also possible to focus light within a volume to achieve three-dimensional reaction control. This focused light leads to an intensity profile within the focal volume that may be many orders higher than anywhere else in the material. Thus, with one or both of these approaches it is possible not only to control when a photochemical process occurs but it is also possible to control where that photochemical process occurs. The spatiotemporal control of photochemical polymer processes historically has been exploited in numerous lithography applications, from chemically amplified photoresists to printing plates to stereolithography (or additive manufacturing), each requiring the formation of or physicochemical changes to the polymer in the exposed regions as compared to the unexposed regions.
Photochemical reaction types
While the general advantages of photochemical processes are great and drive the types of applications that are considered, there is also a broad range of generic reaction types that may be photochemically initiated. In principle, the first law of photochemistry dictates that only absorbed photons can be effective on causing chemical changes. As such, the primary photochemical event involves direct reaction catalysis by the photon and generally a maximum of one primary photochemical event will occur per absorbed photon. However, as illustrated in Fig. 1, a number of primary photochemical reactions are possible, and an even broader range of events may be catalyzed by the species so-generated. Direct reaction within the absorbing molecule and photosensitization, in which a portion of the absorbed energy is transferred by one of a variety of means to another molecule, are both possible.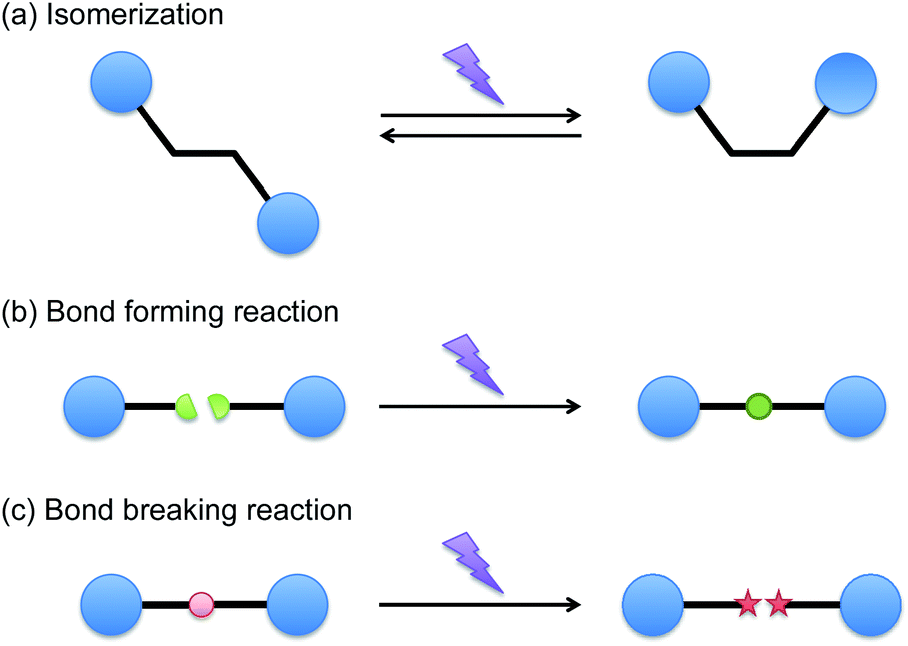 | ||
| Fig. 1 Representative primary photochemical reactions; isomerization, bond forming and bond breaking reactions. | ||
Broadly, the primary photochemical reactions can be classified as isomerization reactions, bond forming reactions, or bond breaking reactions. In considering the photoisomerization reactions, cis–trans isomerization reactions are commonly used to alter light absorption, to probe the molecular structure of polymers, to manipulate phase behavior in self-assembled systems, to cause or actuate macroscopic shape changes of polymers and to promote or inhibit chemical activation of a species. Shorter wavelengths of light are typically used to induce the trans-to-cis isomerization while longer wavelengths of light are used to revert back to the typically more stable trans isomer; the thermal back reaction also will generally lead to the trans isomer. In the bond forming reactions, the most common bond formation reactions involve cycloaddition reactions. For example, [2 + 2] cycloaddition reactions in compounds such as cinnamates have been used to form crosslinks in prepolymers for many decades.10 In both of these approaches, a significant weakness that arises in these reactions is the broad limitation that in each of these systems only a single bond is formed or molecular structure isomerized as a result of each photon that is absorbed. Since typical photon fluxes from a 365 nm light source will be on the order of 3 mM photons per s (assuming a monochromatic light source with an output of 10 mW cm−2 and a sample thickness of 100 microns), the maximum reaction rate would only be on that same order. This low reaction rate thus makes these approaches infeasible unless a high intensity light source, a long reaction time, or a system that is very sensitive to small changes in reaction extent is used.
In contrast, bond breaking reactions have traditionally been used to achieve very high overall reaction rates through the generation of a reactive intermediate species via the primary photochemical event. Photochemically initiated bond cleavage reactions have taken the form of deprotection, homolytic cleavage, and reverse cyclization reactions. While each of these reactions remains fundamentally limited by the feature that only a single primary photochemical event can occur for each photon, here, frequently, the species generated as a result of that reaction are initiating species such as radicals or catalysts that have taken the form of anions, cations, or various metallic species. These catalytic species are then able to catalyze the desired overall reaction at a rate that is many orders of magnitude faster than the underlying photon flux, enabling reactions to occur on practical timescales with reasonable light exposure conditions.
Applications and history
Ultimately, this combination of advantages – ambient, energy efficient, solventless processing that is controllable in a spatiotemporal manner – and the aforementioned array of types of photochemical reactions have enabled a broad swath of applications that implement photochemical polymer processes. While ancient and historical applications of photochemical processes in polymers included ship sealants, mummification, and photography, the modern history of photoinitiated processes in polymers began in the 1940s, when the first patents issued for a UV-cured ink based on unsaturated polyesters.11 Broadly, from the application perspective, one can consider that there are applications of photochemical processes in microelectronics, stereolithography, biotechnology and biomedicine, energy, optics, photonics, coatings and thin film formation, UV-curable inks, responsive materials, adhesives, and in green chemicals development and processing. In each of these areas, one or more of the distinct advantages of photochemical initiation is used as an enabling feature for that application.The remainder of this article first provides a brief introduction to photochemistry followed by representative examples of each of the types of photochemical reactions that have been used to form or transform polymer systems. Finally, the article discusses representative examples of the implementation of these reactions in various applications.
Photochemistry
As noted above, a photon needs to be absorbed for any subsequent photochemical reaction to occur in a molecule. Several aspects are necessary for a photochemical reaction to occur: (i) overlap must exist between the spectral emission of the light source and the absorption spectra of the targeted chromophore, (ii) the absorbed photon must have sufficient energy to enable the desired reaction, and (iii) the energy must ultimately lead to appropriate orbital transitions in the target molecule to cause the desired reaction. Formulations used in photoinitiated processes thus must contain a photosensitive molecule that has an absorption in the wavelength range in which the process is to be conducted. For an effective reaction to occur, a photon is absorbed either directly by a molecule that also contains the targeted reactive functional group (e.g., photoisomerization and photocoupling reactions), by a molecule that generates reactive intermediates (e.g., generation of radical/ionic intermediates for polymerization/crosslinking reactions), or by a photosensitizing molecule that interacts with other molecules in the formulation while in its excited state (i.e., photosensitization) to cause either one of the former reactions. As noted, generation of reactive intermediates which are capable of promoting numerous transformations has the additional benefit of a high degree of secondary amplification of the primary photochemical event, which makes these types of reactions proceed much more rapidly. For example, in the case of radical chain-growth photopolymerizations, it is known that one photon may trigger the growth of a polymer chain of up to 105 monomeric units,12 and some of these types of polymerizations require less than one second of light exposure to proceed to complete polymerization.Light absorption of a single absorbing species is readily described by the Beer–Lambert law:
| A = εcd = −log(I/I0) | (1) |
| IA = (I0 − I) | (2) |
For each possible photochemical process, a wavelength dependent quantum yield, φ, exists where the quantum yield represents the fraction of the photons absorbed that lead to the desired molecular transformation. For a single absorbing species, the overall (initiation) rate of the desired primary photochemical reaction, Ri, is then determined for a single absorbing species from:
| Ri = φIA = φ(I0 − I) | (3) |
As the irradiation source (i.e., incident light intensity), molar absorptivity, and quantum yield may be strong functions of the wavelength, the total photon absorption and reaction rate are calculated by integrating over the absorption spectra of the chromophore in combination with the emission spectra of the irradiation source. It should be noted that a process efficiency ranging from 0–1 is also often included when secondary reactions occur. This approach is particularly common for reactions such as radical initiation where the initiation rate for polymerization is the product of initiating efficiency of the primary radicals, quantum yield, and absorbed photon flux. In such a case, the efficiency classically represents the fraction of photoinitiator radicals that successfully react with a monomer unit to start the polymerization rather than undergoing unsuccessful side reactions such as recombination or primary radical termination.
It is worth noting that while it is apparent that strong absorption is necessary for rapid reaction rates in photochemical systems, light absorption also represents one of the greatest limitations of photochemical processes in polymers. Since photoinduced processes only occur where the light reaches (or where intermediates can subsequently diffuse), opaque, highly absorptive and thick polymer films generally give rise to strong gradients in the light intensity and thus the reaction rate. For reactions such as polymerization, these light intensity gradients lead to significantly different material properties on the incident and dark sides of the film, depending on the specific conditions. In some reactions, such as the photocleavage of a nitrobenzyl ether moiety,13 photodarkening occurs, where the photochemical product has a higher attenuation than the reactant. In contrast, photobleaching can lead to a greater depth of cure than predicted based on the initial absorption of the material.14,15 The lack of a reaction in strongly attenuated regions as well as an inability to penetrate thick, colored, or opaque substrates represents one of the most critical shortcomings for photoinduced processes in polymers.
Photon absorption promotes an electron from its ground state (S0) to its excited singlet state (S1), which is described in a typical Jablonski diagram (Fig. 2).16 In the absence of any photochemical processes, S1, representing the excited singlet state of the chromophore, either deactivates to S0 by fluorescence emission or through internal conversion by heat dissipation. Alternatively, it undergoes intersystem crossing (ISC) to a triplet state (T1). T1 either deactivates to S0 by phosphorescence emission or by ISC. These photophysical processes are in competition with photochemical processes, which include the various targeted reactions described in Fig. 1 and mostly occur from the excited triplet state T1, encompassing isomerization, coupling, and bond cleavage reactions.
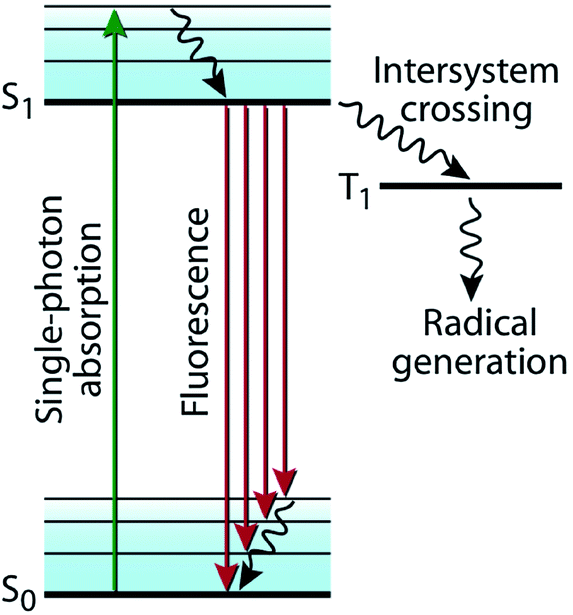 | ||
| Fig. 2 Jablonski diagram. The excited singlet state (S1) undergoes intersystem crossing (ISC) to a triplet state (T1), which leads to various photochemical reactions (e.g., radical generation as presented here). (Image adapted from ref. 16.) | ||
Isomerization
One of the simplest photoinduced transformations to be implemented in polymers is the photoinduced isomerization reaction, for example in chromophores such as azobenzene.Isomerizable chromophores, such as the azobenzene shown in Fig. 3, can be incorporated covalently as side chains or within the polymer backbone or they can be included physically by dissolving the chromophore into the polymer. For these isomerizations, a wavelength and intensity-dependent photostationary state representing a fraction of the molecules in each state is established upon exposure to light where different wavelengths can preferentially be used to bias the photostationary state to either the cis or trans configuration. A thermal back reaction of varying rate is also present and leads to the reconversion of the molecule back to the thermodynamically stable state in the absence of irradiation. Reactions of this type have been used to induce phase transitions in self-assembled systems,17 generate responsive, actuating polymer films,18–35 create surface patterns,26,27,36,37 modulate optical properties of polymers,38 generate photoswitchable biomacromolecules,39–44 control activation/deactivation of catalysts,45,46 among others.
 | ||
| Fig. 3 Photoinduced isomerization of azobenzene. | ||
Specifically, azobenzene-containing polymer networks have been investigated as a stimuli-responsive material that could convert light energy to macroscopic work. Finkelmann et al. developed azobenzene-containing liquid-crystalline elastomers (azo-LCEs) that exhibited photomechanical volume change upon UV irradiation.17 This behavior was attributed to the isothermal, order–disorder transition of the LC phase that was induced by photoisomerization of azobenzene side chains. As an extension of this work, the groups of Ikeda,18–22 Broer23–27 and White28–35 reported azobenzene-containing liquid-crystalline polymer networks (azo-LCNs) that demonstrate photomechanical behavior and in some systems, even at a temperature below their glass transition temperatures, i.e., in their glassy state.31–35 Unlike azo-LCEs, the photomechanical response of azo-LCNs is due to reorientation of the azobenzene mesogens that results from linearly polarized light irradiation, through which the azobenzenes realign perpendicular to the polarization direction.
Similarly, photoinduced surface relief (PSR) formation induced by photoisomerization has been shown as a method to form rewritable, micro- to nanoscale surface patterns based on photoisomerization in polymers.36 Liquid crystal alignment films are proposed as one of the unique applications of PSR structure,37 where the rewritable feature of PSR facilitates liquid crystal in-plane switching and realignment. Modulation of optical properties is another aspect of photoisomerization,38 and holographic gratings have been fabricated using azobenzene-containing polymers as a means for enabling controllable refractive index changes.
Photoswitchable biomacromolecules have also been extensively investigated39–43 to promote both fundamental understanding of biomacromolecule structures/functions and applications such as DNA-based nanodevices. Recent studies by Asanuma et al. developed and evaluated a photon-fueled DNA nanodevice that operates by selective DNA hybridization.44 Two different azobenzene-based chromophores that isomerize under different wavelengths of light were implemented within the DNA structure, and the effect of isomerization was effectively amplified via selective dissociation of complementary double stranded DNA.
Photoisomerizable control of activation/deactivation of catalysts that are used in various polymer and polymerization reactions has also been demonstrated. Photoswitching of the basicity of an azobenzene-containing piperidine base was achieved by modulating the steric hindrance around the nitrogen atom via photoisomerization of azobenzene moieties (Fig. 4).45 Peters et al. have shown that the reaction rates of a Henry reaction changed significantly before and after irradiation. Another advantage of this catalyst is that the catalytic activity could be turned either on or off depending on the wavelength of the light exposure that could be used either to protect or deprotect the amine. Thus, this system exhibits complete temporal control of the reaction where light can be used not only to start the reaction but can also be used to remove the reactive base and turn the reaction off as well. Photoswitching of Lewis acidity has also been shown by using azobenzene-containing catecholboranes.46 One of the advantages of these strategies is chemical amplification, since the absorption of a single photon will release one catalyst molecule that is capable of catalyzing multiple chemical events, thus dramatically enhancing the overall efficiency of the photoinduced process.
 | ||
| Fig. 4 Photoisomerization of the azobenzene moiety changes the steric hindrance around the basic nitrogen. Thus, the basicity is modulated by switching the azobenzene configuration. (Scheme adapted with permission from ref. 45. Copyright 2008 Wiley VCH.) | ||
Bond forming reactions
A second common photochemical transformation that has been used in polymer systems is the photoinduced cyclization, or bond forming reaction. While not widely used, these reactions have a long history in polymer chemistry and have found several unique niches into which they have been successfully applied. The most common of these reactions is the [2 + 2] cycloaddition reaction, particularly involving cinnamate-functionalized monomers and polymers, as illustrated schematically in Fig. 5.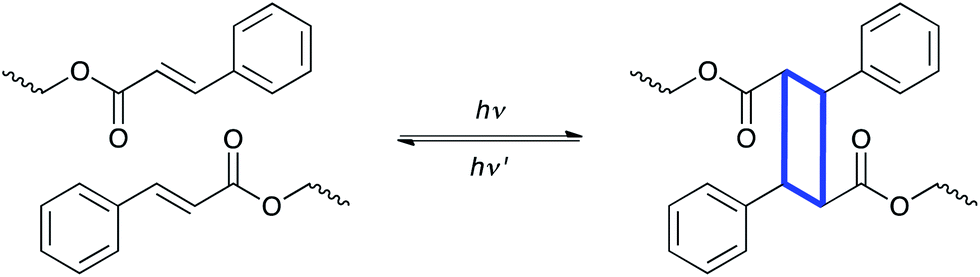 | ||
| Fig. 5 [2 + 2] cycloaddition reaction of cinnamate containing polymers, which forms new carbon–carbon bonds in the material. | ||
In the 1950s, poly(vinyl cinnamate) coupling reactions were used to crosslink between cinnamoyl moieties.10 This approach represented the earliest photocrosslinkable polymer, focused on for its potential for use in negative photoresists. Since that time, cinnamate derivatives as well as chalcone, bis-maleimides, anthracene derivatives, and cyclized polyisoprene have been shown to proceed via photoinduced cycloaddition reactions.11 These types of reactions have been applied to several fields including liquid crystal alignment,47,48 self-healing,49,50 shape memory materials51,52 and collagen crosslinking.53 In particular, Langer and coworkers developed a light-induced shape memory polymer using cinnamic acid to enable photoreversible crosslinking.51 After deforming a cinnamoyl functionalized material from a permanent shape to a temporary shape, >260 nm light was used to induce the cycloaddition reaction between two cinnamates to form new crosslinks that preserved the desired temporary shape. The photoresponsive crosslinks comprising the cyclic structures were reversibly cleaved by exposure to light of <260 nm to recover the original permanent shape. The ability to fix the temporary shape for these materials at temperatures below that of traditional thermoresponsive shape memory polymers was a significant advantage, as no increase or change in temperature was necessary to either store the temporary shape or to return the object to its permanent shape. The relatively low reaction efficiency (one photon per one reaction event) as well as the short wavelengths needed for cleavage are potential drawbacks due to limited penetration depths and limited response times. Nevertheless, this approach is versatile and powerful as an alternative fixation mechanism for shape memory polymers.
Bond breaking reactions
In addition to the retrocyclization reaction highlighted in the cinnamate-based polymer example above, numerous other bond-breaking reactions are induced by exposure to light. In particular, retrocyclization reactions (Fig. 5, reverse reaction); deprotection; the formation of radical, acid/cationic, base/anionic intermediates (Fig. 6); and many other bond breaking reactions such as the formation of active metal catalysts have all been achieved by light exposure of appropriate compounds. The formation of radical intermediates has been also attained by photoredox reactions using light-active transition metal catalysts. Given the nature of these reactions, they are often used to generate reactive intermediates or catalysts for further reaction. As such, they have been used to initiate radical polymerizations, thiol–ene and thiol–yne functionalization reactions, cationic polymerization, cation-mediated chemically amplified photoresists, and in photoinitiating the copper-catalyzed azide–alkyne cycloaddition reaction. The implementation of photoinduced bond breaking reactions is far more prevalent than either of the other two reactions in polymer chemistry and has been broadly used to make polymers, control polymer structure, biologically or chemically functionalize polymers, and to degrade polymers – all with the ability for spatiotemporal control of the overall process. | ||
| Fig. 6 Selected examples of photoinduced bond breaking reactions. (a) Deprotection chemistry of o-nitrobenzyl groups. (b) Formation of radicals via Norrish type I (direct cleavage) process, which is a unimolecular homocleavage from the excited state of light sensitive molecules. (c) Formation of radicals via a Norrish type II (sensitization) process, in which a sensitizer abstracts hydrogen from a co-initiator (e.g., amines) to generate radicals. (d) Formation of acidic protons by photolysis of photoacid generators. (e) o-Nitrobenzyl groups are also used to generate bases. A primary amine, o-nitrobenzylaldehyde and carbon dioxide are all formed from o-nitrobenzylcarbamates by light irradiation. (Scheme adapted with permission from ref. 9. Copyright 1991 American Chemical Society.) | ||
Photoinduced deprotection
As an example of photoinduced deprotection chemistry, photolabile functional groups such as o-nitrobenzyl groups have been extensively investigated as a tool to spatiotemporally control degradation of hydrogels.54 Anseth et al. have pioneered in this area and reported a photodegradable hydrogel that contains o-nitrobenzyl groups in the polymer network, which could be degraded by UV, visible or two-photon IR light to modify its properties both spatially and temporally.55 This approach enables photoinduced control of both cell growth and differentiation. Kasko and co-workers have recently shown that selective degradation could be obtained from a single hydrogel by using various modified o-nitorobenzyl groups.56–58 They demonstrated that the rate of degradation and the activating wavelengths could be controlled based on the structures of the o-nitrobenzyl moieties, which were subsequently applied to specific stem cell release,56 sequential release of three different therapeutics (Fig. 7)58 and controlled release of bioactive molecules57 from hydrogels. | ||
| Fig. 7 Sequential release of different molecules using various o-nitrobenzyl linkers. (a) Three different macromers with dyes attached to each via various o-nitrobenzyl linkers were incorporated into a hydrogel. (b) Sequential irradiation of 436, 405 and 365 nm light to the hydrogel leads to sequential, controlled release of each dye, due to the difference in degradation kinetics of the o-nitrobenzyl moieties. (Image adapted with permission from ref. 58. Copyright 2012 American Chemical Society.) | ||
Photoinduced generation of radicals
One of the most common implementations of light induced processes in polymer chemistry is that of radical initiated photopolymerization reactions. These reactions are used to form lithographic materials, UV-curable inks, coatings, dental materials, tissue engineering matrices, adhesives and many others. Photogenerated radicals have also been used recently to initiate the copper-catalyzed azide alkyne cycloaddition reaction in which the photogenerated radicals subsequently reduce Cu(II) to Cu(I) which catalyzes the reaction.2,3,59Broadly, there are two types of radical photoinitiating systems: Norrish type I (direct cleavage), which undergoes unimolecular bond cleavage to generate radicals, and Norrish type II (sometimes referred to as sensitization though the interaction here is often not simply energy transfer as in classical photosensitization), which undergoes a bimolecular reaction in which the excited state of the absorbing molecule interacts with a second molecule to generate radicals. Type I and II reactions are more commonly used for UV and visible light initiation, respectively, since the energy provided by visible light is not sufficient for breaking most chemical bonds. Examples of the two radical generation routes are illustrated in Fig. 6b and c.
The initiation rate for a typical radical photoinitiator system that generates two identical primary radicals can be written as (cf.eqn (3)):
| Ri = 2fφI0(1 − 10ε[I]d) | (4) |
Ri = 2fφI0[I]εd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(10) ln(10) | (5) |
Multiphoton processes have been shown to be particularly useful for generating radicals by light with three dimensional control (Fig. 8a).66 Because the photochemical initiation rate depends on the square of the light intensity for two photon processes, high intensity light sources with relatively low photon energy (i.e., longer wavelength) are needed and the chemical events occur only within a volume that absorbs multiple photons simultaneously. Utilization of lasers allows photoinitiated chemical events like polymerization and crosslinking reaction to be controlled in three dimensions for such two photon processes.
 | ||
| Fig. 8 Processes to go beyond the diffraction limit. (a) Multiphoton processes occur upon absorption of two photons of relatively low energy, long wavelength light (e.g., IR) to cause excitation of a UV sensitive molecule, which enables controlled initiation of the reaction in three dimensions. (b) STED (stimulated emission depletion) type processes consist of two different events; one is initiation and the other is either inhibition or STED, to force the reaction to occur in a confined volume, below the diffraction limit corresponding to the initiating wavelength. | ||
In contrast, as inspired by stimulated emission depletion (STED) fluorescence microscopy, a combination of two single photon processes, using one wavelength and molecule for photoinitiation and a separate wavelength–molecule combination for photoinhibition/deactivation, was used to initiate polymerization reactions with exceptionally high resolution, enabling three-dimensional photopatterning below the diffraction limit (Fig. 8b). Several systems, for example, pulsed light initiation and continuous light deactivation by one color,67 or initiation and inhibition separately controlled by two colors68 have been reported, both of which generated patterns much smaller than the wavelengths of light used.
Photoinitiated radical polymerization is undoubtedly the most widespread use of a photoinitiated process in polymer science. Its ability to transform immediately from liquid monomer to a solid, typically crosslinked material with spatial and temporal control and nearly no release of volatile organic compounds (VOC) has pushed this technology to the forefront of UV-curable inks, coatings, adhesives and lithographic applications where the high speed achievable in photopolymerizations is of great advantage while the ability for ambient temperature polymerization enables applications in stereospecific polymers (e.g. syndiotactic polymers69), liquid crystalline polymers and biomaterials.
Herein, recent examples of exciting work done with unique free radical photopolymerization are discussed. Ma et al.70 have reported photodirected wrinkle formation on a surface of an elastomeric film (Fig. 9). A strained elastomer was radically crosslinked only at the surface by light attenuation using a photoabsorber to control the depth of penetration, which generated a large difference in modulus and shrinkage of the elastomeric film and its skin layer to form wrinkles. Wrinkle wavelength was controlled by light dose, and the wrinkle pattern was controlled by masking the irradiation source, which clearly indicates the exceptional advantages of photoinduced reactions for controlling material properties.
 | ||
| Fig. 9 Wrinkle formation on the surface of an elastomeric film. (a) The matrix was based on an off-stoichiometric thiol-Michael addition reaction between thiols and acrylates with excess acrylates. (b) The film surface was further crosslinked by photoinitiated radical polymerization under strain. Due to a photoabsorber embedded in the matrix, crosslinking happened only at the surface to form a skin layer with a high modulus that leads to wrinkle formation upon release of the strain. (Image adapted with permission from ref. 70. Copyright 2013 American Chemical Society.) | ||
Traditional radical photopolymerization systems consist of photoinitiators and monomers that have two or more carbon–carbon double bonds. Radicals propagate through carbon–carbon double bonds in a short time period to form a crosslinked polymer network that is inalterable in shape and exhibits a glass transition temperature that may be far above ambient or curing conditions. Although each reaction within the overall photopolymerization is quite simple, consisting of photoinitiation, propagation, termination and chain transfer reactions, the rapidly evolving polymer characteristics, particularly the species mobility and crosslink density, make the overall reaction quite complex.
Photoinduced generation of ion-based reactive intermediates
While radical-induced photopolymerizations represent the most widely used photochemical reactions, the generation of other reactive intermediates via photoinduced reactions has also been critical in enabling significant advances in polymer chemistry. In particular, the nature of radicals and radical polymerizations is such that radicals are short-lived, the polymerizations are strongly inhibited by oxygen, and the conversion of monomer into polymer leads to significant shrinkage and shrinkage-induced stress. As such, a great deal of emphasis has been placed on photo-generating ionic species.71 Ionic species would not be sensitive to the same termination or inhibition reactions and would be capable, with appropriate design of the overall system, of achieving significant continued polymerization after the irradiation has ceased (i.e., continued dark reactions). As such, the synthesis and implementation of photoinitiating systems that lead to the formation of acids and bases has received extensive attention.Acid/cation generation
Since Crivello and co-workers first described the use of iodonium and sulfonium salts as photoinitiators for cationic polymerizations, also known as photoacid generators (PAGs) in late 1970's,7,8 a variety of new onium salts with high quantum yields for the formation of cations have been developed.72 These photoacid generators have seen broad use in the development of chemically amplified photoresists73 as well as in the development of cationic photopolymerization reactions.7,74 Cationic photopolymerizations have several distinct advantages, such as a lack of inhibition by oxygen, low shrinkage and shrinkage induced stress due to the use of ring opening monomers, good adhesion to inorganic substrates and a substantial post cure effect due to the longer life of the propagating cation center. Epoxy and vinyl ether-based monomers are the most commonly used, and they have been shown to be less toxic than many (meth)acrylate monomers used for radical photopolymerization.A typical photoacid generating system, i.e., a diaryliodonium salt, is presented in Fig. 6d. Photoinduced decomposition and subsequent reaction of the radical cation with solvent or monomer leads to the generation of a Bronsted acid, H+, which initiates the acid catalyzed reaction of interest, whether polymerization or one of many other desired acid-catalyzed reactions. Since many of the initial onium salt photoinitiators worked most efficiently upon UV irradiation in the lower wavelength region from 200–300 nm, photoinitiators that could generate an acid efficiently upon exposure to longer wavelengths have been a focus. In particular, free radical promoted cationic polymerization has been one approach to developing a visible light sensitive photoacid generator. In one example, the photoinitiator system consists of camphorquinone in combination with a benzyl alcohol to generate free radicals upon visible light exposure, after which the radicals subsequently participate in the free radical induced decomposition of the diaryliodonium salt to form the acid and catalyze the polymerization of a variety of epoxy monomers.75
Among the many applications for photoacids, photoresists and nanoimprint lithography materials76,77 are the most significant. Today's semiconductor industry relies on photoresists, which are a thin protecting layers on silicon substrates that is photopatterned and then selectively removed. Cationic photoresists are implemented in both negative (e.g. polymerization of epoxides) and positive resists (e.g. deprotection or depolymerization reactions). Among these photoresists, the advent of chemically amplified photoresists represents one of the most significant advancements made in this field as it provided high sensitivity and excellent feature replication. This broad approach is possible since the absorption of a single photon leads to a single proton being generated, where each proton is then capable of catalyzing several hundred deprotection reactions, demonstrating the enabling character of chemical amplification. Advantages for cationic photopolymerization, such as a tolerance to oxygen, low viscosity of the monomers and low shrinkage via ring-opening epoxide reactions make it highly suitable for forming micro- to nano- scale patterns with high reproducibility as needed in nanoimprint lithography.76,77
Base/anion generation
Anionic polymerization or any other base-catalyzed reaction has not been as widely developed or explored as either the photoinduced radical or cationic systems.78 However, there are a wide range of reactions that could be catalyzed by a base, including urethane formation from alcohols and isocyanates, ring opening reaction of epoxies by nucleophilic species and Michael addition reactions, and several of these reactions are important in industrial applications. Most base-catalyzed processes are inert to oxygen and moisture, which is a great advantage for applications in coatings, in contrast to radical and cationic photopolymerizations due to their inhibition by oxygen and moisture, respectively. The living character of base-catalyzed reactions, like their acid counterparts, also assures that dark cure is useful to reach high conversions.The primary limitation on the implementation of base catalyzed reactions is a lack of suitable photoinitiating systems. Most photoinitiators, or photolatent bases, that are reported to date suffer either from low catalytic activity, long-term stability or solubility in the organic media, or limited availability. However, in the last decade there have been a large number of new concepts for photolatent bases. Dietliker et al. introduced photolatent tertiary amines by using photoinduced steric release or photoinduced oxidative introduction of an amidine double bond.79 α-Amino ketone photoinitiators, as an example of the former, have a significant difference in polythiol/polyisocyanate mixture pot life between sterically hindered (0.33% catalyst doubles the viscosity in 18 h) and unhindered (0.15% catalyst doubles the viscosity in less than 2 min) amines. For the latter photoinitiator, they have shown that the Michael addition reaction and the thiol/epoxy reaction occurred rapidly upon UV irradiation with no reaction occurring in the absence of any irradiation.
Selected implementations of light induced processes
While the field of photoinitiated reactions in polymer science is too vast to review comprehensively in a single review, here, we attempt to highlight specific examples and implementations of photochemical processes used to control the formation, structure, and properties of polymers. These examples are chosen to represent emerging opportunities, span the types of photochemical reactions, and cover a range of potential application fields and types of polymers.Light enabled control of living radical polymerizations
The use of light in living or controlled radical polymerization has received a great deal of interest as recently highlighted in a review by Yamago et al.80 One of the earliest approaches to photochemically controlling radical polymerization was developed by Otsu using the photoiniferter (initiator-chain transfer-terminator) concept.81 Otsu et al. photopolymerized vinyl monomers in the presence of a dithiocarbamate, which functioned as the photoiniferter. The dithiocarbamate homolytically cleaves the C–S bond during irradiation to generate the radicals that start polymerization. Subsequently, a propagating radical reacts with dithiocarbamyl radicals to re-generate a dormant dithiocarbamate species at the end of the polymer chain. Block copolymers, star polymers and amphiphilic polymers have all been formed using this method though significant issues arose when trying to control the polymerization of many common monomer types through this reaction.More recent efforts have attempted to control radical polymerization by photochemistry. Taking atom transfer radical polymerization (ATRP) as an example, several approaches including photochemical generation of the active Cu(I) catalyst from Cu(II),82,83 utilization of a photoredox catalysts to activate dormant species84 and utilization of photoiniferters to generate dormant species that could be activated by light irradiation85,86 were each shown to facilitate temporal control of controlled radical polymerizations. The resulting polymers possess reinitiatable ends that may be extended to form block copolymers or other controlled architecture polymers. In one example of using a photoredox catalyst, fac-Ir(ppy)3 was successfully employed in ATRP with ethyl 2-bromo-2-phenylacetate as an initiator under irradiation of visible light. 50 ppm of the catalyst gave well controlled poly(methyl methacrylate) with Mn ranging from 2900 to 22900 with low polydispersity Mw/Mn ranging from 1.19–1.25, depending on the conditions. Interestingly, photopolymerization only proceeded concurrent with light irradiation and could be turned on/off repeatedly, indicative of the living character of this reaction.84 Photo-controlled polymerization using trithiocarbonate moieties has also been shown as an effective approach. Zhou et al.87 reported the synthesis of telechelic polymers starting from a trithiocarbonate-centered initiator that possesses norbornene groups on each end (Fig. 10). The norbornene end-groups reacted with trifunctional tetrazines via a Diels–Alder reaction to form a gel, which enables the polymer chains to be grown in-between crosslinks with spatiotemporal control. This approach opens up a methodology to photocontrol network densities and/or monomer compositions in a crosslinked material.
 | ||
| Fig. 10 Photocontrolled modification of network crosslink densities. (A) A crosslinked gel was formed via a Diels–Alder reaction between norbornene and tetrazine functionalized monomers. (B) A polymerization reaction involving trithiocarbonate moieties was initiated by light irradiation, which enables the network densities and/or monomer compositions to be altered spatioselectively. (Image adapted with permission from ref. 87. Copyright 2013 Wiley VCH.) | ||
Photoinitiated click reactions
A rapidly evolving focus of research in polymer science has been on the implementation of click chemistry methods for forming and manipulating polymers. The click concept, as originally proposed by Sharpless,88 focused on developing and using a highly capable, small set of chemical reactions that were characterized by high efficiency and yield, orthogonality with other reactions, readily obtained starting materials, stereospecificity, and a robustness that enables them to proceed rapidly at ambient conditions. These reactions have been widely explored in various aspects of polymer science89–91 though recently a drive has been initiated to develop and use click reactions that can be photoinitiated. The marriage of photochemical initiation and click reactions is ideal: photochemical initiation provides for spatiotemporal control and the click reaction proceeds nearly ideally once initiated. Two primary reactions, thiol–ene/yne and the CuAAC as discussed in depth below, have been the focus of these efforts but other photoinitiated click reactions have also been explored.92Photoinitiated thiol–ene and thiol–yne coupling reactions
Radical mediated coupling of thiols and reactive carbon–carbon double bonds, i.e., the thiol–ene reaction, has a long history since its discovery in the early 20th century though its prominence has risen significantly recently due to its consideration as one of the click-type reactions.93–96 The thiol–ene reaction has attributes of a click reaction that include achieving quantitative yields, requiring only small concentrations of relatively benign catalysts, having rapid reaction rates with reactions occurring either in bulk or in environmentally benign solvents over a large concentration range, requiring essentially no clean up, being insensitive to ambient oxygen and water, yielding a single regioselective product, and the ready availability of a range of both thiol and ene-functional reactants. Due to the step-growth character of the thiol–ene polymerization reaction, it is also advantageous in that it does not suffer from many of the problems that classical chain-growth (meth)acrylate photopolymerizations have, including relatively high shrinkage and stress and the formation of heterogeneous polymer networks. The ability to form the thiol–ene reaction by light exposure makes this reaction even more robust, allowing it to achieve quantitative conversions under even the mildest of conditions with spatiotemporal control that makes it amenable to a vast range of applications, which include microfluidic devices,97,98 functional beads and fibers,99–101 surface modification,98,102 photolithography,103,104 high performance crosslinked polymers,102,105 hydrogels106–108 and biomaterials109,110 applications. There have been several comprehensive reviews of thiol–ene radical chemistry and polymerizations, which have thoroughly covered the reaction mechanism and traditional applications.94–96,111 Herein, we focus on recent advances in photoinitiated thiol–ene reactions.An ability to overcome oxygen inhibition, rapid curing, step growth network formation and reaching high conversion with minimal shrinkage all make thiol–ene systems suitable for forming microfluidics devices. Contact liquid photolithographic polymerization (CLiPP) using thiol–ene formulations has been reported to possess much higher feature fidelity with shorter curing times in features with high aspect ratios (up to 10) compared to multifunctional acrylates.97 Off-stoichiometric thiol–ene reactions were applied to microfluidic devices to fabricate patterned wettability via photoinitiated thiol–ene reaction with excess thiols and ene-functionalized small molecules (Fig. 11).98 This approach is one example that takes advantage of the step growth character and highly efficient click reaction features. In addition to being used to make microfluidic devices, thiol–ene/yne reactions have been performed in several microfluidic device geometries as well. Monodisperse functional beads were synthesized by droplet formation in microfluidic device followed by radical photopolymerization of these droplets. Various types of monomers could be incorporated to give a wide range of Tg's (from 0 to 44 °C), as well as various amount of functional groups such as amines and hydroxyls.99 Unique fibers with complex intersection geometries were also reported using microfluidics system.100,101
 | ||
| Fig. 11 An off-stoichiometric thiol–ene reaction was used to (a) fabricate a microfluidic device and for (b) surface modification. This example clearly shows the advantages of thiol–ene reactions that arise from their step-growth addition character and the availability of vast arrays of monomers that are used on combination with the control that light initiation affords. (Image adapted from ref. 98.) | ||
Recently, Adzima et al. have demonstrated thiol–ene based photocrosslinking of a Diels–Alder network by thiol–ene reaction between thiols and Diels–Alder adducts that renders the typically thermoreversible Diels–Alder network to be irreversible.103 It was combined with two-photon techniques to write a 3D pattern in a solid substrate that could be realized and isolated from the solid matrix by retro-Diels–Alder depolymerization at elevated temperatures. Griesser et al.104 have similarly performed a two-photon induced thiol–ene reaction to photo-crosslink ROMP derived polymers, which clearly indicates the versatility of the thiol–ene reaction as a means to photo-crosslink double bonds in linear/crosslinked polymers.
Photoinitiated CuAAC reaction
Since its discovery in 2002,112,113 the copper-catalyzed alkyne–azide cycloaddition reaction, also known as the CuAAC reaction, has emerged in a wide range of synthetic approaches and applications such as bioconjugation, polymer synthesis, dendrimer synthesis, surface modification and particle fabrication. The reaction, often referred to as the click reaction, has a high yield, relatively high reaction rate, orthogonality, and utilizes mild reaction conditions.114,115 However, the Cu(I) catalysts needed for this reaction have typically been generated in situ by the chemical reduction of Cu(II) to Cu(I) or been directly added as a Cu(I) salt. These approaches to catalyzing the reaction did not afford spatial or temporal control of the reaction, which is crucial for the generation of functional materials and many biological applications. To overcome this limitation and improve the implementation, robustness and control of the CuAAC reaction, extensive work has been done by several groups to realize a photoinitiatable CuAAC reaction (photo-CuAAC, Fig. 12). Popik et al.116 were the first group to develop a means for photoinitiating the azide–alkyne cycloaddition reaction as they synthesized structures that decomposed into cyclooctynes upon exposure to light through decarbonylation of cyclopropenones. Once formed, the cyclooctynes rapidly undergo Cu-free azide–alkyne coupling. This process involves a complex synthetic methodology as well as the limitation that only a single reaction product is formed for each photon absorbed. | ||
| Fig. 12 Photo-CuAAC reaction mediated by (a) photoinduced metal ligand charge transfer and (b) copper reduction by photoinitiated radicals. Image adapted with permission from ref. 92. Copyright 2013 Wiley VCH. | ||
To preserve the photoinitiation capability developed by Popik and enhance the reaction capabilities, several groups have sought to create approaches in which the Cu(I) catalytic species are generated photochemically in situ from Cu(II) species. As early as 2006, Ritter and König explored photoinitiating the CuAAC reaction by photogeneration of the Cu(I) catalyst using an electron donor and redox-active chromophore, reducing Cu(II) to Cu(I) in situ.4 Further, Tasdelen et al. performed a photoinitiated CuAAC reaction by utilizing a photoinduced electron transfer reaction in which Cu(I) species were generated by UV irradiation of Cu(II)–ligand complexes.5 Adzima et al. were the first to apply a photo-CuAAC scheme to polymerization simply by adding a radical photoinitiator where the radicals formed during light exposure reduced Cu(II) to Cu(I).59 They enabled spatiotemporal control of both polymer functionalization and hydrogel formation via the photo-click reaction. Subsequently, Gong et al.117 developed a novel Cu(II) containing photoinitiator with great solubility in both organic and aqueous media, and this complex readily triggers not only the photo-CuAAC reaction but also ATRP upon visible light irradiation. Controlled cell attachment to a patterned PEG-based hydrogel has been achieved by a combination of the photo-CuAAC reaction with an azide-functionalized polymer and a PEG-tetraalkyne. This approach has also been applied in polymer synthesis such as conjugating polymer fragments to form block copolymers in solution.2 Patterned hydrogels were reported using a combination of the CuAAC reaction and an addition of PEG-based radical to alkynes, which were both initiated by photogenerated radicals.6 Most recently, Gong et al. developed a bulk photopolymerization method based on multifunctional alkynes and azides using the photo-CuAAC reaction. The polymer network thus formed was highly crosslinked with a high glass transition temperature attributed to the extensive triazole linkages formed by the CuAAC reaction.3
Responsive polymer films
While light is able to induce polymerization, polymer functionalization, crosslinking and many other attributes during the creation of a polymer, it is also capable of causing a variety of property changes after a material has already been formed. The field of smart, responsive polymers118–120 is rapidly expanding with the opportunity for photosensitive polymers to play a major role. Here, examples of polymer films are presented that respond to light exposure by changing shape, topography, actuating, and/or degrading.Chemically crosslinked networks are generally described as thermosets, in recognition that these networks are permanent, intractable and inert thus can be neither melted nor molded. However, recent approaches combine the desirable attributes of conventional thermosets with the dynamics of controllably reversible bond structures have yielded covalent adaptable networks (CANs).121,122 Addition–fragmentation chain transfer (AFT) capable moieties such as allyl sulfides123 or trithiocarbonates124 (Fig. 13a) were recently incorporated in CANs. Photoinitiated radical generation, as achieved simply through the inclusion of a radical photoinitiator during polymer formation, causes addition–fragmentation processes to occur throughout the film. These bond exchange reactions, illustrated in Fig. 13a, occur only when and where exposure to light occurs, causing an elastic to plastic transition and ultimately the polymer flows in those regions. This approach has been used to achieve photoinduced plasticity,123 photolithographically defined features,125 photoinduced film bending and folding (photo-origami)126 and even photoinitiated healing.127 Bond reshuffling between thiuram disulfides has been utilized in polymer networks to enable self-healing upon visible light irradiation (Fig 13b). There is no need to add additional radical generators to this system since the disulfide bond of the thiuram disulfide moiety readily cleaves under light.127
 | ||
| Fig. 13 Selected examples of photocontrolled covalent adaptable networks (CANs). (a) Photogenerated radical (i.e., carbon radical) reacts with trithiocarbonate moiety via reversible addition fragmentation chain transfer process. Scheme adapted with permission from ref. 124. Copyright 2012 John Wiley and Sons (b) Disulfide moiety of thiuram disulfides are cleaved by visible light irradiation to generate thiyl radicals, which have capabilities of reshuffling the thiuram disulfide bond. (Scheme adapted with permission from ref. 127. Copyright 2011 Wiley VCH.) | ||
Photoinduced isomerization reactions, especially for example, of azobenzene groups, have been extensively investigated for introducing changes of shape, topography and physical properties in polymer films as described in the previous section.
Light-to-heat energy conversion has also been employed to cause folding of shape memory polymer films.128 Pre-strained polystyrene, which is also known as Shrinky-Dinks, has been utilized with black ink patterning and local absorption of light to cause shape recovery only in selected (i.e., black ink patterned) area surfaces, which generates patterned relaxation and shrinkage that caused the film to fold. Herein, light was used as a selective, patternable heat source for the material and no chemical reactions were required in the folding process.
In another approach to creating responsive films, several groups have explored the development of photochemical techniques for inducing degradation of the polymer structure. Classically, polymer photodegradation, i.e., chain scission and/or crosslinking of polymers as triggered by photon absorption, has been well known to alter polymer properties, and this degradation has long limited the ability of many polymer films to be used in environments with outdoor sun exposure.129 Photodegradation of this type involves several pathways. Radicals generated by photochemical processes trigger degradation synergistically in combination with air, water, organics, temperature variations and mechanical stresses. Traditionally, then, polymer photodegradation has been viewed as a highly undesirable feature of polymer films; however, for many applications, spatiotemporal control of photodegradation would be a significant advantage. Thus, several approaches have been developed to design materials with the controlled ability to degrade in response to light exposure. Photocleavable protecting groups like o-nitrobenzyl groups are commonly used for inducing degradation upon light irradiation, as described in the previous section.
In another more indirect approach to photodegradation, Fairbanks et al. synthesized disulfide-linked hydrogels with tunable degradation.130 The hydrogels were composed of 4-armed PEG chains terminated with thiol groups, which formed disulfide linked hydrogels upon oxidation. These gels were loaded with a radical photoinitiator, which facilitated rapid gel photodegradation when the photogenerated radicals cause cleavage of the disulfide crosslinks in the material. The photodegradation mechanism was also exploited as a novel means to fabricate self-healing hydrogels based on the disulfide exchange reaction. Interestingly, the extent of photodegradation was easily tuned simply by adjusting the irradiation dose.
Conclusions and future perspectives
The utilization of light to induce chemical transformations to create, rearrange, or cleave chemical bonds provides a powerful toolset for the synthesis, modification, and manipulation of polymeric materials. The fundamental concepts of absorption and attenuation are universal for all photochemical processes and define the limits on sample geometry and irradiation conditions. A critical distinction is whether the photochemical process is a single photon-single reaction event or one that catalyzes or induces a cascade of reactions. The latter strategy significantly reduces the number of photons required to induce a chemical reaction, as highlighted above in the generation of active species such as radicals, acids, and bases.The spatiotemporal and photoorthogonal control of photochemical transformations enable sequential reactions to be triggered when and, where desired, and as well as in a specific order. While these attributes are critical in several traditional photochemical technologies, such as photolithography of microchips, they are primed to be implemented in new materials syntheses and modification in next generation applications spanning nano- to biotechnology. New photocleavable moieties and photo-enabled click chemical strategies will provide an unprecedented control over macromolecular construction of polymer architectures.
Acknowledgements
The authors would like to acknowledge National Science Foundation CHE 1214109 and CBET 1264298 for providing funding for this research.Notes and references
- B. J. Adzima, Y. Tao, C. J. Kloxin, C. a. DeForest, K. S. Anseth and C. N. Bowman, Nat. Chem., 2011, 3, 258 CrossRef PubMed.
- M. A. Tasdelen, G. Yilmaz, B. Iskin and Y. Yagci, Macromolecules, 2012, 45, 56 CrossRef CAS.
- T. Gong, B. J. Adzima, N. H. Baker and C. N. Bowman, Adv. Mater., 2013, 25, 2024 CrossRef CAS PubMed.
- S. C. Ritter and B. König, Chem. Commun., 2006, 4694 RSC.
- M. A. Tasdelen and Y. Yagci, Tetrahedron Lett., 2010, 51, 6945 CrossRef CAS PubMed.
- R. T. Chen, S. Marchesan, R. A. Evans, K. E. Styan, G. K. Such, A. Postma, K. M. McLean, B. W. Muir and F. Caruso, Biomacromolecules, 2012, 13, 889 CrossRef CAS PubMed.
- J. V. Crivello and J. H. W. Lam, Macromolecules, 1977, 10, 1307 CrossRef CAS.
- J. V. Crivello and J. H. W. Lam, J. Polym. Sci., Part A: Polym. Chem., 1979, 17, 977 CAS.
- J. F. Cameron and J. M. J. Frechet, J. Am. Chem. Soc., 1991, 113, 4303 CrossRef CAS.
- L. M. Minsk, US Pat., 2,725,377, 1955.
- J.-P. Fouassier, Radiation Curing in Polymer Science and Technology: Fundamentals and Methods, Springer, 1993, Vol. 1 Search PubMed.
- J. Kloosterboer, Adv. Polym. Sci., 1988, 84, 1 CrossRef CAS.
- C. P. Holmes, J. Org. Chem., 1997, 62, 2370 CrossRef CAS PubMed.
- J. T. Cabral and J. F. Douglas, Polymer, 2005, 46, 4230 CrossRef CAS PubMed.
- J. A. Warren, J. T. Cabral and J. F. Douglas, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 72, 021801 CrossRef.
- T. F. Scott, C. J. Kloxin, D. L. Forman, R. R. McLeod and C. N. Bowman, J. Mater. Chem., 2011, 21, 14150 RSC.
- H. Finkelmann, E. Nishikawa, G. G. Pereira and M. Warner, Phys. Rev. Lett., 2001, 87, 015501 CrossRef CAS.
- T. Ikeda, M. Nakano, Y. Yu, O. Tsutsumi and A. Kanazawa, Adv. Mater., 2003, 15, 201 CrossRef CAS.
- Y. Yu, M. Nakano, A. Shishido, T. Shiono and T. Ikeda, Chem. Mater., 2004, 16, 1637 CrossRef CAS.
- M. Yamada, M. Kondo, J. Mamiya, Y. Yu, M. Kinoshita, C. J. Barrett and T. Ikeda, Angew. Chem., Int. Ed., 2008, 47, 4986 CrossRef CAS PubMed.
- M. Yamada, M. Kondo, R. Miyasato, Y. Naka, J. Mamiya, M. Kinoshita, A. Shishido, Y. Yu, C. J. Barrett and T. Ikeda, J. Mater. Chem., 2009, 19, 60 RSC.
- M. Kondo, M. Sugimoto, M. Yamada, Y. Naka, J. Mamiya, M. Kinoshita, A. Shishido, Y. Yu and T. Ikeda, J. Mater. Chem., 2010, 20, 117 RSC.
- K. D. Harris, R. Cuypers, P. Scheibe, C. L. van Oosten, C. W. M. Bastiaansen, J. Lub and D. J. Broer, J. Mater. Chem., 2005, 15, 5043 RSC.
- C. L. van Oosten, D. Corbett, D. Davies, M. Warner, C. W. M. Bastiaansen and D. J. Broer, Macromolecules, 2008, 41, 8592 CrossRef CAS.
- C. L. van Oosten, C. W. M. Bastiaansen and D. J. Broer, Nat. Mater., 2009, 8, 677 CrossRef CAS PubMed.
- D. Liu, C. W. M. Bastiaansen, J. M. J. den Toonder and D. J. Broer, Angew. Chem., Int. Ed., 2012, 51, 892 CrossRef CAS PubMed.
- D. Liu, C. W. M. Bastiaansen, J. M. J. den Toonder and D. J. Broer, Macromolecules, 2012, 45, 8005 CrossRef CAS.
- T. J. White, N. V. Tabiryan, S. V. Serak, U. A. Hrozhyk, V. P. Tondiglia, H. Koerner, R. A. Vaia and T. J. Bunning, Soft Matter, 2008, 4, 1796 RSC.
- T. J. White, S. V. Serak, N. V. Tabiryan, R. A. Vaia and T. J. Bunning, J. Mater. Chem., 2009, 19, 1080 RSC.
- S. Serak, N. Tabiryan, R. Vergara, T. J. White, R. A. Vaia and T. J. Bunning, Soft Matter, 2010, 6, 779 RSC.
- K. M. Lee, M. L. Smith, H. Koerner, N. Tabiryan, R. A. Vaia, T. J. Bunning and T. J. White, Adv. Funct. Mater., 2011, 21, 2913 CrossRef CAS.
- K. M. Lee, N. V. Tabiryan, T. J. Bunning and T. J. White, J. Mater. Chem., 2012, 22, 691 RSC.
- K. M. Lee, D. H. Wang, H. Koerner, R. A. Vaia, L.-S. Tan and T. J. White, Angew. Chem., Int. Ed., 2012, 51, 4117 CrossRef CAS PubMed.
- M. R. Shankar, M. L. Smith, V. P. Tondiglia, K. M. Lee, M. E. McConney, D. H. Wang, L.-S. Tan and T. J. White, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18792 CrossRef CAS PubMed.
- J. J. Wie, K. M. Lee, M. L. Smith, R. A. Vaia and T. J. White, Soft Matter, 2013, 9, 9303 RSC.
- N. K. Viswanathan, D. Y. Kim, S. Bian, J. Williams, W. Liu, L. Li, L. Samuelson, J. Kumar and S. K. Tripathy, J. Mater. Chem., 1999, 9, 1941 RSC.
- X. T. Li, A. Natansohn and P. Rochon, Appl. Phys. Lett., 1999, 74, 3791 CrossRef CAS PubMed.
- A. S. Matharu, S. Jeeva and P. S. Ramanujam, Chem. Soc. Rev., 2007, 36, 1868 RSC.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422 RSC.
- G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2006, 45, 4900 CrossRef CAS PubMed.
- C. Dohno, S. Uno and K. Nakatani, J. Am. Chem. Soc., 2007, 129, 11898 CrossRef CAS PubMed.
- J. Andersson, S. Li, P. Lincoln and J. Andréasson, J. Am. Chem. Soc., 2008, 130, 11836 CrossRef CAS PubMed.
- H. Asanuma, T. Ito, T. Yoshida, X. Liang and M. Komiyama, Angew. Chem., Int. Ed., 1999, 38, 2393 CrossRef CAS.
- H. Nishioka, X. Liang, T. Kato and H. Asanuma, Angew. Chem., Int. Ed., 2012, 51, 1165 CrossRef CAS PubMed.
- M. V. Peters, R. S. Stoll, A. Kühn and S. Hecht, Angew. Chem., Int. Ed., 2008, 47, 5968 CrossRef CAS PubMed.
- N. Kano, J. Yoshino and T. Kawashima, Org. Lett., 2005, 7, 3909 CrossRef CAS PubMed.
- M. Obi, S. Morino and K. Ichimura, Jpn. J. Appl. Phys., 1999, 38, 145 Search PubMed.
- O. Yaroshchuk and Y. Reznikov, J. Mater. Chem., 2012, 22, 286 RSC.
- C. Chung, Y. Roh, S. Cho and J. Kim, Chem. Mater., 2004, 16, 3982 CrossRef CAS.
- J. Ling, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2011, 21, 18373 RSC.
- A. Lendlein, H. Jiang, O. Jünger and R. Langer, Nature, 2005, 434, 695 CrossRef PubMed.
- A. Garle, S. Kong, U. Ojha and B. M. Budhlall, ACS Appl. Mater. Interfaces, 2012, 4, 645 CAS.
- C.-M. Dong, X. Wu, J. Caves, S. S. Rele, B. S. Thomas and E. L. Chaikof, Biomaterials, 2005, 26, 4041 CrossRef CAS PubMed.
- P. M. Kharkar, K. L. Kiick and A. M. Kloxin, Chem. Soc. Rev., 2013, 42, 7335 RSC.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59 CrossRef CAS PubMed.
- D. R. Griffin and A. M. Kasko, J. Am. Chem. Soc., 2012, 134, 13103 CrossRef CAS PubMed.
- D. R. Griffin, J. L. Schlosser, S. F. Lam, T. H. Nguyen, H. D. Maynard and A. M. Kasko, Biomacromolecules, 2013, 14, 1199 CrossRef CAS PubMed.
- D. R. Griffin and A. M. Kasko, ACS Macro Lett., 2012, 1, 1330 CrossRef CAS.
- B. J. Adzima, Y. Tao, C. J. Kloxin, C. A. DeForest, K. S. Anseth and C. N. Bowman, Nat. Chem., 2011, 3, 256 CrossRef CAS PubMed.
- N. B. Cramer, S. K. Reddy, M. Cole, C. Hoyle and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5817 CrossRef CAS.
- N. B. Cramer, J. P. Scott and C. N. Bowman, Macromolecules, 2002, 35, 5361 CrossRef CAS.
- C. Decker, C. Bianchi, F. Morel, S. Jönsson and C. E. Hoyle, Macromol. Chem. Phys., 2000, 201, 1493 CrossRef CAS.
- C. Decker, F. Morel, S. Jönsson, S. Clark and C. Hoyle, Macromol. Chem. Phys., 1999, 200, 1005 CrossRef CAS.
- C. W. Miller, E. S. Jönsson, C. E. Hoyle, K. Viswanathan and E. J. Valente, J. Phys. Chem. B, 2001, 105, 2707 CrossRef CAS.
- C. Hoyle, K. Viswanathan, S. Clark, C. W. Miller, C. Nguyen, S. Jönsson and L. Shao, Macromolecules, 1999, 32, 2793 CrossRef CAS.
- C. N. LaFratta, J. T. Fourkas, T. Baldacchini and R. A. Farrer, Angew. Chem., Int. Ed., 2007, 46, 6238 CrossRef CAS PubMed.
- L. Li, R. R. Gattass, E. Gershgoren, H. Hwang and J. T. Fourkas, Science, 2009, 324, 910 CrossRef CAS PubMed.
- T. F. Scott, B. A. Kowalski, A. C. Sullivan, C. N. Bowman and R. R. McLeod, Science, 2009, 324, 913 CrossRef CAS PubMed.
- S.-H. Shim, M.-K. Ham, J. Huh, Y.-K. Kwon and Y.-J. Kwark, Polym. Chem., 2013, 4, 5449 RSC.
- S. J. Ma, S. J. Mannino, N. J. Wagner and C. J. Kloxin, ACS Macro Lett., 2013, 2, 474 CrossRef CAS.
- M. Shirai and M. Tsunooka, Prog. Polym. Sci., 1996, 21, 1 CrossRef CAS.
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245 CrossRef CAS.
- H. Ito, Adv. Polym. Sci., 2005, 172, 37 CrossRef CAS.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4241 CrossRef CAS.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 866 CrossRef CAS.
- X. Cheng, L. J. Guo and P.-F. Fu, Adv. Mater., 2005, 17, 1419 CrossRef CAS.
- L. J. Guo, Adv. Mater., 2007, 19, 495 CrossRef CAS.
- K. Suyama and M. Shirai, Prog. Polym. Sci., 2009, 34, 194 CrossRef CAS PubMed.
- K. Dietliker, R. Hüsler, J.-L. Birbaum, S. Ilg, S. Villeneuve, K. Studer, T. Jung, J. Benkhoff, H. Kura, A. Matsumoto and H. Oka, Prog. Org. Coat., 2007, 58, 146 CrossRef CAS PubMed.
- S. Yamago and Y. Nakamura, Polymer, 2013, 54, 981 CrossRef CAS PubMed.
- T. Otsu, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2121 CrossRef CAS.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Rapid Commun., 2011, 32, 58 CrossRef CAS PubMed.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Chem. Phys., 2011, 212, 2036 CrossRef CAS.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850 CrossRef CAS PubMed.
- S.-H. Qin, D.-Q. Qin and K.-Y. Qiu, New J. Chem., 2001, 25, 893 RSC.
- Y. Kwak and K. Matyjaszewski, Macromolecules, 2010, 43, 5180 CrossRef CAS.
- H. Zhou and J. A. Johnson, Angew. Chem., Int. Ed., 2013, 52, 2235 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nystro, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- M. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52, 5930 CrossRef CAS PubMed.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540 CrossRef CAS PubMed.
- A. B. Lowe, Polym. Chem., 2010, 1, 17 RSC.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745 RSC.
- J. F. Ashley, N. B. Cramer, R. H. Davis and C. N. Bowman, Lab Chip, 2011, 11, 2772 RSC.
- C. F. Carlborg, T. Haraldsson, K. Öberg, M. Malkoch and W. van der Wijngaart, Lab Chip, 2011, 11, 3136 RSC.
- R. A. Prasath, M. T. Gokmen, P. Espeel and F. E. Du Prez, Polym. Chem., 2010, 1, 685 RSC.
- D. A. Boyd, A. R. Shields, P. B. Howell and F. S. Ligler, Lab Chip, 2013, 13, 3105 RSC.
- D. A. Boyd, A. R. Shields, J. Naciri and F. S. Ligler, ACS Appl. Mater. Interfaces, 2013, 5, 114 CAS.
- S. Mongkhontreerat, K. Öberg, L. Erixon, P. Löwenhielm, A. Hult and M. Malkoch, J. Mater. Chem. A, 2013, 1, 13732 CAS.
- B. J. Adzima, C. J. Kloxin, C. A. Deforest, K. S. Anseth and C. N. Bowman, Macromol. Rapid Commun., 2012, 33, 2092 CrossRef CAS PubMed.
- T. Griesser, A. Wolfberger, U. Daschiel, V. Schmidt, A. Fian, A. Jerrar, C. Teichert and W. Kern, Polym. Chem., 2013, 4, 1708 RSC.
- O. D. McNair, B. J. Sparks, A. P. Janisse, D. P. Brent, D. L. Patton and D. A. Savin, Macromolecules, 2013, 46, 5614 CrossRef CAS.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005 CrossRef CAS.
- H. Shih, A. K. Fraser and C.-C. Lin, ACS Appl. Mater. Interfaces, 2013, 5, 1673 CAS.
- H. Shih and C.-C. Lin, Macromol. Rapid Commun., 2013, 34, 269 CrossRef CAS PubMed.
- J. D. McCall and K. S. Anseth, Biomacromolecules, 2012, 13, 2410 CrossRef CAS PubMed.
- W. M. Gramlich, I. L. Kim and J. A. Burdick, Biomaterials, 2013, 34, 9803 CrossRef CAS PubMed.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355 RSC.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed.
- M. Meldal, Macromol. Rapid Commun., 2008, 29, 1016 CrossRef CAS.
- A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769 CrossRef CAS PubMed.
- T. Gong, B. J. Adzima and C. N. Bowman, Chem. Commun., 2013, 49, 7950 RSC.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101 CrossRef PubMed.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278 CrossRef CAS PubMed.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14 CrossRef CAS PubMed.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643 CrossRef CAS PubMed.
- C. N. Bowman and C. J. Kloxin, Angew. Chem., Int. Ed., 2012, 51, 4272 CrossRef CAS PubMed.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615 CrossRef CAS PubMed.
- D. Leung and C. N. Bowman, Macromol. Chem. Phys., 2012, 213, 198 CrossRef CAS.
- C. J. Kloxin, T. F. Scott, H. Y. Park and C. N. Bowman, Adv. Mater., 2011, 23, 1977 CrossRef CAS PubMed.
- J. Ryu, M. D'Amato, X. Cui, K. N. Long, H. Jerry Qi and M. L. Dunn, Appl. Phys. Lett., 2012, 100, 161908 CrossRef PubMed.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew. Chem., 2011, 123, 1698 CrossRef.
- Y. Liu, J. K. Boyles, J. Genzer and M. D. Dickey, Soft Matter, 2012, 8, 1764 RSC.
- J. F. Rabek, Polymer Photodegradation: Mechanisms and Experimental Methods, Springer, 1995 Search PubMed.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |