 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis via RAFT polymerization of thermo- and pH-responsive random copolymers containing cholic acid moieties and their self-assembly in water†
Sunirmal
Pal
,
Saswati
Ghosh Roy
and
Priyadarsi
De
*
Polymer Research Centre, Department of Chemical Sciences, Indian Institute of Science Education and Research-Kolkata, PO: BCKV Main Office, Mohanpur-741252, Nadia, West Bengal, India. E-mail: p_de@iiserkol.ac.in
First published on 25th October 2013
Abstract
Here we report the design and synthesis of polymers containing cholic acid pendant groups, a major bile acid synthesized in the liver from cholesterol. Random copolymers derived from 2-(methacryloyloxy)ethyl cholate (MAECA) with polyethylene glycol methyl ether methacrylate (PEGMA) and N,N-dimethylaminoethyl methacrylate (DMAEMA) have been synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization. Compositions of co-monomers in the copolymers were determined by 1H NMR spectroscopy, which were further used to determine reactivity ratios of the monomers using the extended Kelen–Tüdös method at high conversions. Copolymers from MAECA with PEGMA (P(MAECA-co-PEGMA)) series displayed lower critical solution temperatures (LCSTs) up to 23% MAECA content in the copolymer, which was studied by UV-visible spectroscopy. Both thermo- and pH-responsiveness were observed for the P(MAECA-co-DMAEMA) copolymers with an MAECA unit in the copolymer up to 11.8%. The critical aggregation concentration (CAC) of water soluble copolymers was determined by fluorescence spectroscopy. Self-assembly of random copolymers was investigated by 1H NMR spectroscopy, dynamic light scattering (DLS), atomic force microscopy (AFM) and scanning electron microscopy (SEM), which indicated micellar structures in water from cholic acid based random copolymers. The polymeric aggregates were further explored for their drug encapsulation capabilities in aqueous medium using nile red as a hydrophobic model compound.
Introduction
Bile acids are cholesterol-derived naturally occurring amphiphilic molecules, which are essential for digestion and absorption of fats and fat-soluble vitamins in the small intestine to adjust cholesterol levels in the body.1 Bile acid derivatives have been used for cholesterol gallstone dissolution,2 treating primary biliary cirrhosis,3 antiviral and antifungal properties, gene therapy, radiopharmaceuticals, etc.4 Supramolecular gels from salts of bile acid derivatives have immense application potential in biomedical and materials science.5 Driven by the aim to develop bile acid containing polymeric materials, a great deal of research has been dedicated to the incorporation of bile acid residues as cores, pendant groups and end groups, and also in the backbone via polycondensation reactions.6 For preparation of side-chain bile acid containing polymers, most of the reports used the conventional free radical polymerization (FRP) technique, where uncontrolled molecular weights and broad molecular weight distributions (polydispersity index, PDI) were obtained. Recently, much attention has been devoted to the controlled radical polymerization (CRP)7 of bile acid based vinyl monomers, where it is possible to achieve targeted molecular weights, narrow molecular weight distributions and well-defined architectures.8 A comb-shaped cholic acid containing polymer, poly[methacryloyl tri(ethylene glycol) cholanoate], was synthesized by atom transfer radical polymerization (ATRP).9 Novel biomimetic surfactants based on cholesterol as the hydrophobic head group and poly[2-(methacryloyloxy)ethyl phosphorylcholine] as the hydrophilic segment were prepared by ATRP and their micellar behaviour in an aqueous medium was investigated.10 A series of multishape memory copolymers with a polynorbornene main chain, and cholic acid and triethylene glycol side groups were synthesized through ring opening metathesis polymerization (ROMP).11 The glass transition temperature (Tg) of these copolymers was tunable over a temperature range from −58 to 176 °C.Among various CRP methods (ATRP, nitroxide-mediated radical polymerization and reversible addition–fragmentation chain transfer (RAFT) polymerization), RAFT is the most versatile technique.12,13 Amphiphilic diblock copolymers, in which the hydrophobic block was a cholesteryl-based smectic liquid-crystalline polymer and the hydrophilic poly(N,N-diethylacrylamide) block, or a copolymer containing acrylic acid moieties and poly(ethylene oxide) side chains were synthesized by RAFT polymerization.14 To date thermo- and pH-sensitive copolymers containing bile acid derivatives have been synthesized by the FRP process and their phase separation behaviour in aqueous solutions and thermosensitivity were studied.15 However, to the best of our knowledge there is no report on bile acid containing random copolymer synthesis via a CRP route. CRP provides preparation of well-defined polymers with controlled molecular weight, polydispersity, chain architecture, etc., which are difficult to achieve by FRP. Herein we have synthesized a 2-(methacryloyloxy)ethyl cholate (MAECA) monomer and copolymerized using a RAFT technique with polyethylene glycol methyl ether methacrylate (PEGMA) and N,N-dimethylaminoethyl methacrylate (DMAEMA) to produce thermosensitive poly(2-(methacryloyloxy)ethyl cholate-co-polyethylene glycol methyl ether methacrylate) (P(MAECA-co-PEGMA)), and dual thermo- and pH-responsive poly(2-(methacryloyloxy)ethyl cholate-co-N,N-dimethylaminoethyl methacrylate) (P(MAECA-co-DMAEMA)) copolymers. We have chosen cholic acid because the most abundant bile salts in humans are cholate, chenodeoxycholate and deoxycholate.16 Depending on the hydrophobic MAECA content in the copolymer chain, these bile acid containing copolymers self-assemble to spherical micellar structures in aqueous solution, which could be used for guest molecule encapsulation. Random copolymer based self-assembled nano-structures are advantageous over amphiphilic block copolymers because they can be achieved in a single polymerization step unlike block copolymers, which are generally used to generate nano-structured assemblies.17
Experimental
Materials
2-Hydroxyethyl methacrylate (HEMA, 97%), 4-dimethylaminopyridine (DMAP, 99%), anhydrous N,N-dimethylformamide (DMF, 99.9%), nile red (NR), dicyclohexylcarbodiimide (DCC, 99%), pyrene (Py, 98%) and cholic acid (CA, ≥98%) were purchased from Sigma and used without further purification. PEGMA (average Mn = 300 g mol−1) and DMAEMA (98%) were purchased from Sigma and purified prior to polymerization by passing through the basic alumina column to remove inhibitors. 4-Cyano-4-(thiobenzylthio)pentanoic acid (CTP),18 4-cyano-4-(dodecylsulfanylthiocarbonyl)sulfanylpentanoic acid (CDP),19 2-dodecylsulfanylthiocarbonylsulfanyl-2-methylpropionic acid (DMP)20 and benzyl dithiobenzoate (BDB)21 were synthesised as reported elsewhere. CDCl3 (99.9% D), D2O (99.9% D) and DMSO-d6 (99.9% D) were purchased from Cambridge Isotope Laboratories, Inc. 2,2′-Azobisisobutyronitrile (AIBN, Sigma, 98%) and solvents such as hexanes, petroleum ether, tetrahydrofuran (THF), dichloromethane (DCM), etc. were purified by standard procedures.Instruments and analysis
The molecular weights and PDIs of polymers were measured by gel permeation chromatography (GPC) using poly(methyl methacrylate) (PMMA) calibration in DMF at 35 °C. The GPC system consists of a Waters 515 HPLC pump, Waters 2414 RI detector, and two HSPgel columns. The 1H NMR study was conducted on a Bruker AvanceIII 500 spectrometer. Positive mode electrospray ionization mass spectrometry (ESI-MS) was performed on a Q-Tof Micro YA263 high resolution mass spectrometer (Waters Corporation). Hydrodynamic diameter values were determined using a dynamic light scattering (DLS) (Zetasizer Nano ZS, Malvern Instrument Ltd., UK) instrument equipped with a He–Ne laser beam at 658 nm. Polymer solutions (1.0 mg mL−1) were filtered through a 0.45 μm syringe filter prior to measurement and each experiment was repeated four times to obtain the average value. UV-vis measurements were done on a Perkin-Elmer Lambda 35 UV-vis spectrophotometer. Emission spectra were recorded using a Horiba Jobin Yvon Fluoromax-3 instrument. The FT-IR spectrum was recorded on KBr pellets using a Perkin-Elmer Spectrum 100 FT-IR spectrometer. The Tg values were measured using a Mettler Toledo DSC1 STARe differential scanning calorimeter (DSC) at a 10 °C min−1 heating rate in a nitrogen atmosphere. Polymers were cooled from room temperature to −70 °C, then heated to +120 °C and again cooled to −70 °C at 10 °C min−1. The Tg was taken from the third segment of the experiment. Thermogravimetric analysis (TGA) was carried out using a Mettler Toledo TGA/SDTA 851e instrument at a heating rate of 10 °C min−1 in a nitrogen atmosphere. Atomic force microscopy (AFM) images were recorded on an NT-MDT NTEGRA Prima Scanning Probe Microscope operated in semi-contact mode. An aliquot of sample solution (0.1 mg mL−1) was drop cast on a glass slide at room temperature, dried and analyzed by AFM. Scanning electron microscopy (SEM) measurements were performed on a Carl Ziess Sigma SEM instrument.Monomer synthesis
MAECA was synthesised by esterification condensation reaction of CA and HEMA in the presence of DCC and DMAP (Scheme S1†). The coupling reaction was carried out in dry THF under a dry N2 atmosphere. DCC (7.57 g, 36.7 mmol) in 20 mL THF was placed in a 250 mL double necked round bottom flask containing a magnetic stir bar. To this solution, first CA (15.0 g, 36.7 mmol) in 80 mL THF and then DMAP (0.45 g, 3.67 mmol) in 20 mL THF were added under a N2 atmosphere. Then, the system was placed in an ice-water bath and HEMA (4.77 g, 36.7 mmol) was added dropwise to the reaction mixture under stirring. The ice-water bath was removed after 30 min and the reaction mixture was stirred for 28 h at room temperature. The reaction mixture was filtered to separate insoluble N,N′-dicyclohexylurea (DCU) and the filtrate was concentrated by rotary evaporation to remove THF. Then, it was dissolved in ethyl acetate and washed with 0.1 N HCl (100 mL × 3), saturated NaHCO3 (100 mL × 3) and brine solution (100 mL × 2). The organic layer was dried over anhydrous Na2SO4 and the solvent was removed by rotary evaporation. Finally, it was purified by column chromatography using hexanes–ethyl acetate as the mobile phase (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, v/v) with a yield of 50%. 1H NMR (Fig. S1,† CDCl3, δ, ppm): 6.13 and 5.60 (2H, s, 1-H), 4.31–4.35 (4H, m, 5 & 6-H), 3.98 (1H, s, 7′-H),11 3.85 (1H, s, 12′-H),11 3.47 (1H, s, 3′-H),11 1.95 (3H, s, 3-H), 0.98 (3H, d, 21′-H), 0.89 (3H, s, 19′-H) and 0.68 (3H, s, 18′-H). ESI-MS (Fig. S2†): observed m/z for [M + Na+] 543.02.
:3, v/v) with a yield of 50%. 1H NMR (Fig. S1,† CDCl3, δ, ppm): 6.13 and 5.60 (2H, s, 1-H), 4.31–4.35 (4H, m, 5 & 6-H), 3.98 (1H, s, 7′-H),11 3.85 (1H, s, 12′-H),11 3.47 (1H, s, 3′-H),11 1.95 (3H, s, 3-H), 0.98 (3H, d, 21′-H), 0.89 (3H, s, 19′-H) and 0.68 (3H, s, 18′-H). ESI-MS (Fig. S2†): observed m/z for [M + Na+] 543.02.
RAFT polymerization of MAECA
Typically, MAECA (0.20 g, 0.38 mmol), CTP (4.28 mg, 15.4 μmol), AIBN (0.504 mg, 3.07 μmol; from the stock solution of AIBN in DMF) and 0.8 g DMF were taken in a 20 mL septa sealed glass vial equipped with a magnetic stir bar. The reaction vial was purged with dry N2 for 20 min and placed in a preheated reaction block at 70 °C. After a predetermined time, the polymerization was quenched by cooling in an ice-water bath and exposed to air. The polymer (PMAECA) was precipitated using hexanes. The polymer was redissolved in THF and precipitated from hexanes and this process was repeated 5 times. The precipitate was dried under high vacuum at 40 °C for 10 h. Monomer conversions were calculated gravimetrically based on the monomer feed.Synthesis of statistical copolymers
A typical copolymerization procedure was as follows: MAECA (0.190 g, 0.365 mmol), PEGMA (0.110 g, 0.365 mmol), CTP (3.40 mg, 12.2 μmol), AIBN (0.40 mg, 2.43 μmol; 0.08 g solution of 10.4 mg AIBN in 2.09 g DMF) and DMF (1.2 g) were placed in a 20 mL septa sealed vial equipped with a magnetic stir bar, purged with dry N2 for 20 min and placed in a preheated reaction block at 70 °C for 5 h. The feed ratios of monomers were varied to obtain copolymers of various compositions. Copolymerization reactions were quenched and copolymers were purified as described for the homopolymers.Determination of LCST
The turbidity of copolymer aqueous solutions (2 mg mL−1) was measured using a UV-vis spectrophotometer by monitoring the % transmittance (%T) changes at λ = 500 nm.22 The sample was filtered through a 0.45 μm membrane filter, transferred to a quartz cuvette, placed in a UV-vis spectrometer and warmed from 20 to 80 °C. At the measurement temperature the sample was equilibrated for 6 min and the %T value at 500 nm was determined. The LCST was defined as the temperature where a reduction of 50% T of the polymer solution was observed.Determination of critical aggregation concentration (CAC) by fluorescence spectroscopy23
Pyrene was used as a hydrophobic fluorescent probe to measure the CAC of copolymers in aqueous medium. A stock pyrene solution in acetone was added to different 50 mL volumetric flasks. Then, 4.0 mL of aqueous solution containing different concentrations of copolymers was added to the flasks to make the final pyrene concentration in each flask 10−6 mol L−1. These pyrene containing flasks were kept open for 24 h at room temperature for the complete evaporation of acetone. Fluorescence spectra of these solutions were recorded (at excitation wavelength 339 nm) from 360 to 450 nm. The fluorescence intensity ratio of I383/I373 was plotted against the polymer concentration. The CAC was obtained from the intersection of two tangent plots of the intensity ratio I383/I373versus the polymer concentration.Results and discussion
Synthesis of MAECA
MAECA was synthesised by a single step coupling reaction of commercially available CA and HEMA in the presence of DCC and DMAP at room temperature (Scheme S1†). The molecular structure of MAECA was identified by 1H NMR, FT-IR and ESI-MS spectroscopy. The 1H NMR spectrum of MAECA matched well with the proposed chemical structure (Fig. S1†). The experimental molecular mass m/z 543.02 obtained from ESI-MS analysis matched well with the calculated molecular mass of [MAECA + Na+] = 543.69 (Fig. S2†). The FT-IR spectrum of MAECA showed the characteristic carbonyl group C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, double bond CH2CH– and the –OH signals at 1724, 1638 and 3404 cm−1, respectively (Fig. S3†). All these characterizations proved the successful formation of MAECA.
O, double bond CH2CH– and the –OH signals at 1724, 1638 and 3404 cm−1, respectively (Fig. S3†). All these characterizations proved the successful formation of MAECA.
RAFT polymerization of MAECA
CTAs have an obvious influence during the controlled RAFT polymerization of vinyl monomers. It was reported that both trithiocarbonates and dithioesters can be used for the RAFT polymerization of methacrylates with good control over both molecular weight and PDI.24 Herein, in order to find a suitable CTA, we polymerized MAECA in the presence of four different CTAs, such as DMP, CDP, BDB and CTP at 70 °C in DMF solvent using AIBN as a radical initiator at [MAECA]:[CTA]:[AIBN] = 25:1:0.2. Table 1 shows that BDB and DMP resulted polymers, where number average molecular weights determined from GPC (Mn,GPC) are much higher compared to the theoretical molecular weights (Mn,theo) based on monomer conversion. Also, for these two CTAs we noticed broad molecular weight distributions. The polymers from the reactions with CDP and CTP exhibited narrow PDIs (<1.35) and the Mn,GPC values match well with the corresponding Mn,theo values. We selected CTP for further investigations because with this CTA, it was easy to determine number average molecular weights by NMR (Mn,NMR) from the comparison of phenyl chain end protons to the main chain protons (vide infra).
:[CTA] ratios at 70 °C in DMF
| CTA | [M]:[CTA]:[I] |
Time (min) | Conv.a (%) | M n,theo (g mol−1) | M n,NMR (g mol−1) | M n,GPC (g mol−1) | PDId |
|---|---|---|---|---|---|---|---|
| a Determined by gravimetric analysis on the basis of the amount of monomer feed. b M n,theo = ([MAECA]/[CTA] × molecular weight (MW) of MAECA × conversion) + (MW of CTA). c Calculated by 1H NMR chain-end analysis. d Obtained by GPC. | |||||||
| BDB | 25:1:0.2 |
330 | 81 | 10700 |
— | 58000 |
1.94 |
| DMP | 25:1:0.2 |
330 | 64 | 8600 | — | 45500 |
1.67 |
| CDP | 25:1:0.2 |
330 | 85 | 11350 |
— | 14300 |
1.34 |
| CTP | 25:1:0.2 |
330 | 85 | 11350 |
12700 |
9700 | 1.20 |
| CTP | 25:1:0.1 |
450 | 69 | 9250 | 9100 | 10700 |
1.14 |
| CTP | 25:1:0.3 |
210 | 67 | 9000 | 9400 | 9700 | 1.15 |
| CTP | 15:1:0.2 |
330 | 81 | 6600 | — | 6500 | 1.14 |
| CTP | 40:1:0.2 |
330 | 57 | 12150 |
12800 |
10500 |
1.18 |
| CTP | 60:1:0.2 |
330 | 51 | 16200 |
14700 |
17000 |
1.32 |
To explore the controllability of the RAFT technique for the polymerization of MAECA, we performed polymerization reactions at different [MAECA]/[CTP] ratios such as 15:1, 25:1, 40:1 and 60:1, while the [CTP]/[AIBN] ratio was kept constant at 5:1. The GPC traces of four homopolymers in Fig. 1A show major unimodal peaks and chromatograms shifted towards lower elution volume with increasing [MAECA]/[CTP] ratios. We observed narrow PDIs (<1.32) and a liner relationship between Mn,GPC and Mn,theo values for these polymers (Fig. 1B and Table 1). Note that in the high molecular weight region of the GPC traces, a small shoulder was observed for all the PMAECA homopolymers, which could be due to the partial recombination of propagating polymer chains. Also, formation of aggregates during the GPC experiments in polar DMF solvent may give rise to a shoulder in higher molecular weight regions. The influence of the initiator concentration was also studied in which the molar feed ratio of CTP to initiator was varied from 1:0.1 to 1:0.3, while the [MAECA]/[CTP] ratio was maintained at 25:1. Generally, the rate of polymerization depends on the availability of radicals for initiation and Table 1 shows that the polymerization rate increases with decreasing [CTP]/[AIBN] ratio, as expected.
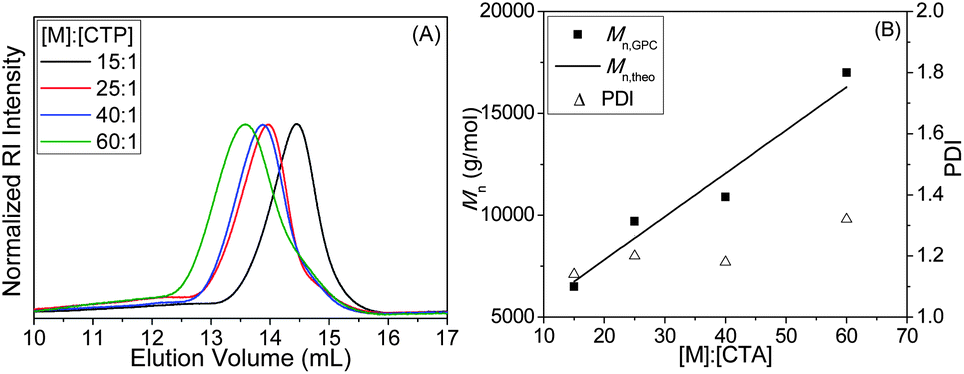 | ||
| Fig. 1 (A) GPC traces of PMAECA as a function of [MAECA]/[CTP] ratios and (B) variation of Mn and PDI with the [MAECA]/[CTP] ratios for the RAFT polymerization of MAECA at 70 °C in DMF. | ||
The structure of PMAECA was confirmed by 1H NMR spectroscopy. In the 1H NMR spectrum of PMAECA in Fig. 2A, the vinyl peaks of the MAECA monomer completely disappeared, and signals at 3.93–4.34 (4H, 5 & 6-H), 3.78 (1H, 7′-H), 3.60 (1H, 12′-H), 3.17 (1H, 3′-H), 0.76–0.98 (9H, 3, 21′, 19′-H), 0.58 (3H, 18′-H), 1.60–1.82 (main chain methylene protons) and 0.75–0.99 ppm (main chain methyl protons) were observed. The phenyl moiety of the CTP unit was incorporated into the polymer chain ends, which showed characteristic weak resonance signals at 7.44–7.84 ppm. The Mn,NMR value of PMAECA was determined by considering the integration value of phenyl protons (7.44–7.84 ppm) at the polymer chain ends and –C(O)–O–CH2–CH2–O–C(O)– protons at 3.93–4.34 ppm from the MAECA units. The Mn,NMR values are summarized in Table 1, which match reasonably well with the Mn,theo values indicating PMAECA chain ends with the fragments of CTP as expected according to the typical RAFT mechanism.
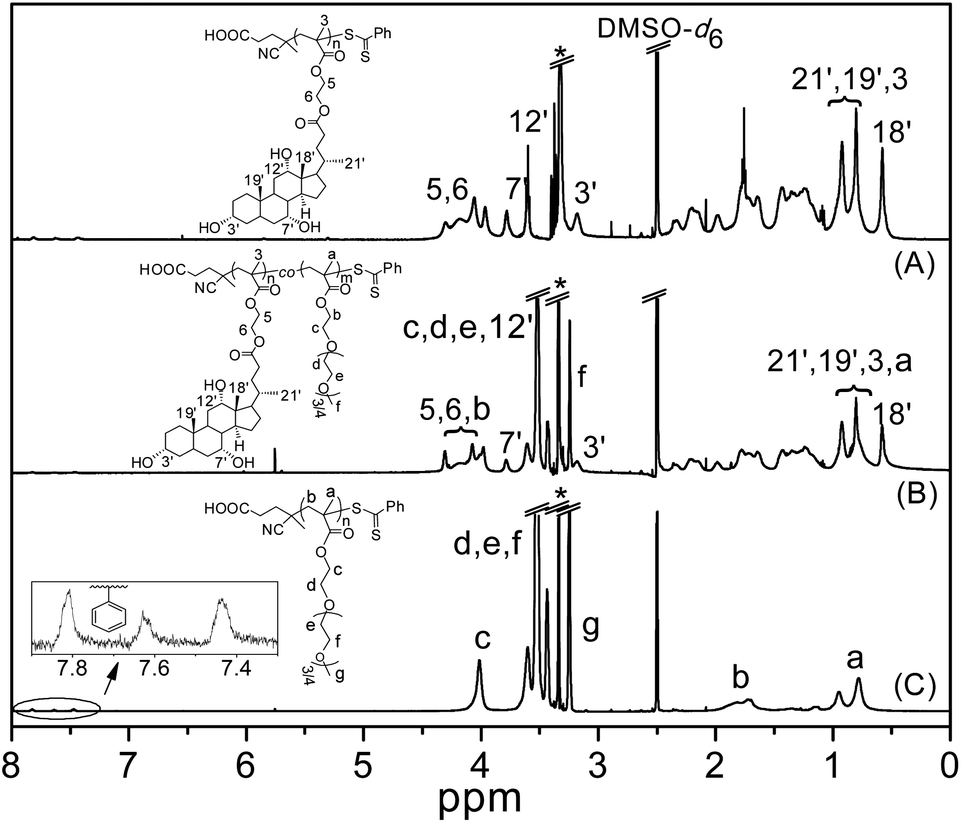 | ||
| Fig. 2 1H NMR spectra of (A) PMAECA, (B) CP5, and (C) PPEGMA in DMSO-d6. | ||
To further demonstrate the retention of dithioester CTA moieties at the polymer chain ends, the chain extension experiment was carried out using PMAECA–macroCTA. PMAECA (Mn,GPC = 6500 g mol−1, PDI = 1.14) was used for the polymerization of MAECA at 70 °C in DMF at [MAECA]:[PMAECA–macroCTA]:[AIBN] = 50:1:0.2. We obtained 77% monomer conversion after 260 min and GPC traces of PMAECA-b-PMAECA shifted toward higher molecular weight side after the chain extension (Fig. S4†). The Mn,GPC = 41000 g mol−1 for PMAECA-b-PMAECA is somewhat higher than Mn,theo = 26550 g mol−1 (PDI = 1.48) and GPC trace is bimodal indicating bimolecular terminations. However, the Mn,NMR = 30100 g mol−1 value is close to the theoretical value with ∼88% CTA chain ends. This result suggests that most of the chain ends of PMAECA are capped with the fragments of CTP and could be used further for block copolymer synthesis.
To investigate the effect of cholate moieties on the solution behaviour of copolymers containing PEGMA (a thermoresponsive monomer) or DMAEMA (a dual thermo- and pH-responsive monomer) units, several RAFT copolymerizations of MAECA with PEGMA and DMAEMA were carried out using [co-monomers]:[CTP]:[AIBN] = 60:1:0.2 in DMF at 70 °C (Scheme 1). We synthesised six copolymers from each of the MAECA–PEGMA and MAECA–DMAEMA series with the MAECA to PEGMA or DMAEMA feed ratios of 5:95; 10:90; 20:80; 30:70; 40:60 and 50:50. Note that CTP showed controlled RAFT polymerization of PEGMA25 and DMAEMA.26 All copolymers were soluble in DMF, THF and DMSO and few of them were soluble in aqueous medium depending on the percentage of PEGMA or DMAEMA hydrophilic units in the copolymer chain. Solubilities of all the polymers in various organic solvents and water are listed in Table S1.† To name copolymers, P(MAECA-co-PEGMA/DMAEMA), we followed the following rules (Table 2): two letters CP and CD indicate MAECA–PEGMA and MAECA–DMAEMA series, respectively; 1, 2, 3, 4, 5 and 6 stand for the different feed compositions of the MAECA monomer.
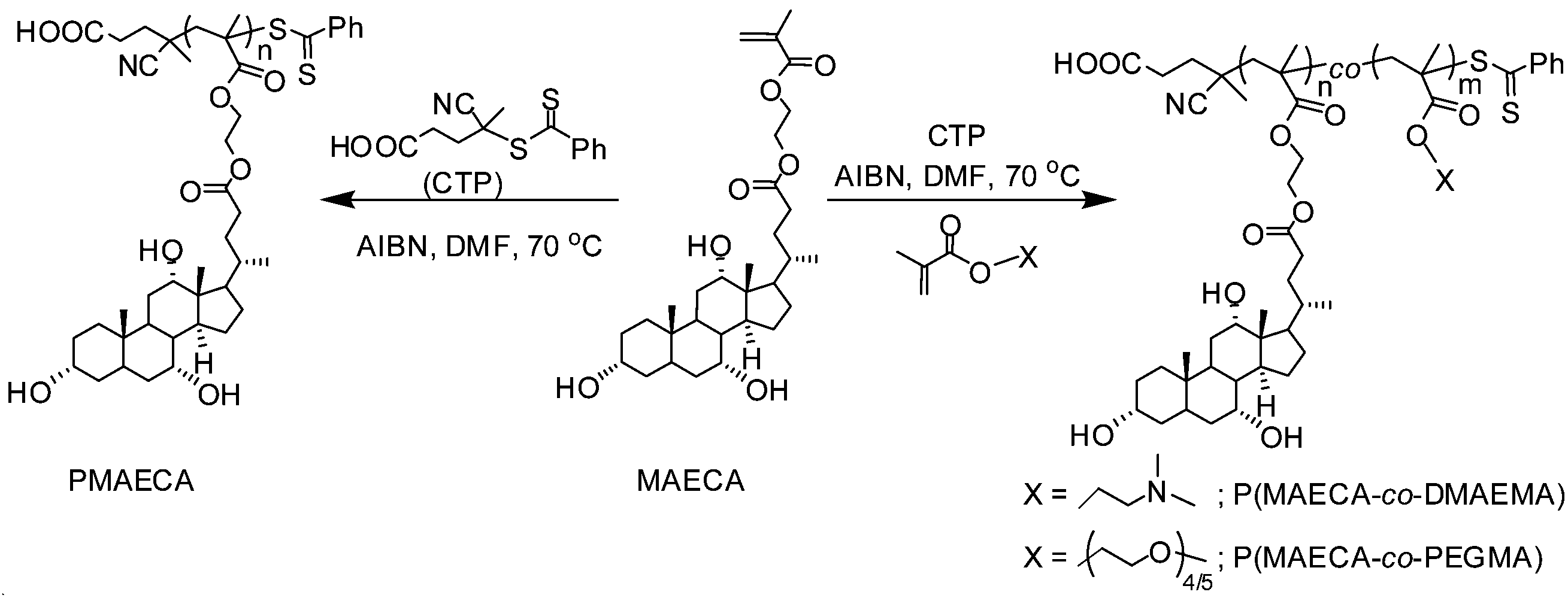 | ||
| Scheme 1 Synthesis of cholic acid based homopolymers and statistical copolymers. | ||
| Polymer | Time (min) | Conv.b (%) | Feed | Copolymer 1H NMR | M n,theo (g mol−1) | M n,NMR (g mol−1) | M n,GPC (g mol−1) | PDId | T g (°C) |
|---|---|---|---|---|---|---|---|---|---|
|
a [Monomer]/[CTP][AIBN] = 60:1:0.2.
b Determined by gravimetric analysis on the basis of the amount of monomer feed.
c Calculated by the 1H NMR study.
d Obtained by GPC.
e Measured using DSC.
|
|||||||||
| Molar fraction of MAECA | |||||||||
| MAECA–PEGMA series | |||||||||
| PPEGMA | 210 | 67 | 0.000 | 0.000 | 12350 |
12500 |
12800 |
1.15 | −56 |
| CP1 | 225 | 83 | 0.05 | 0.055 | 15800 |
13700 |
17000 |
1.10 | −49 |
| CP2 | 210 | 65 | 0.100 | 0.120 | 13000 |
14800 |
20300 |
1.20 | −39 |
| CP3 | 240 | 70 | 0.200 | 0.230 | 15000 |
15100 |
21800 |
1.25 | −20 |
| CP4 | 270 | 76 | 0.300 | 0.340 | 17400 |
16200 |
25400 |
1.26 | 6 |
| CP5 | 300 | 75 | 0.400 | 0.447 | 18200 |
18600 |
27900 |
1.28 | 28 |
| CP6 | 300 | 78 | 0.500 | 0.542 | 19800 |
21000 |
32200 |
1.26 | 35 |
| PMAECA | 330 | 51 | 1.000 | 1.000 | 16200 |
16400 |
17000 |
1.32 | 148 |
| MAECA–DMAEMA series | |||||||||
| PDMAEMA | 390 | 63 | 0.000 | 0.000 | 6200 | 7400 | 7100 | 1.14 | 18 |
| CD1 | 365 | 69 | 0.05 | 0.059 | 7700 | 8900 | 11000 |
1.20 | 30 |
| CD2 | 360 | 76 | 0.100 | 0.118 | 9400 | 8400 | 10750 |
1.48 | 48 |
| CD3 | 360 | 58 | 0.200 | 0.238 | 8800 | 10700 |
14900 |
1.29 | 73 |
| CD4 | 360 | 69 | 0.300 | 0.343 | 12100 |
14200 |
20500 |
1.33 | 90 |
| CD5 | 360 | 78 | 0.400 | 0.450 | 15300 |
18600 |
27700 |
1.30 | 103 |
| CD6 | 360 | 74 | 0.500 | 0.540 | 16000 |
19900 |
31400 |
1.30 | 113 |
The 1H NMR spectra of copolymers were recorded in DMSO-d6 to determine % of co-monomer units in the copolymer chain by integration of the resonance signals related to each monomeric unit (Fig. 2). The 1H NMR spectrum of PPEGMA (Fig. 2C) showed resonance signals at 1.61–1.94 ppm due to the main chain methylene protons, and α-methyl protons appeared at 0.69–1.02 ppm. The side chain methylene protons –C(O)–O–CH2– and –C(O)–O–CH2–CH2–O–(CH2–CH2–O)3/4–CH3 appeared at 4.01 and 3.41–3.65 ppm, respectively. The peak centred at 3.25 ppm was assigned to the –O–CH3 protons, and weak resonance signals appeared at 7.40–7.83 ppm were due to presence of the benzodithioate group (from CTP) at the chain ends. The presence of MAECA units in the copolymers was confirmed from the 1H NMR spectra for this series (Fig. 2B). The compositions of copolymers were determined from the integration ratio of intensities of –OCH3 protons at 3.25 ppm from PEGMA units to the –C(O)–O–CH2–CH2–O–C(O)– methylene protons at 3.93–4.34 ppm from the MAECA fragment (after subtracting the peak area contributed by the PEGMA units in this region). The 1H NMR spectrum of PDMAEMA (Fig. S5†) showed signals at 3.98, 3.42, 2.18, 1.67–1.89 and 0.73–1.01 ppm for –C(O)–O–CH2–, –C(O)–O–CH2–CH2–, –N(CH3)2, main chain methylene and α-methyl protons, respectively. Compositions of P(MAECA-co-DMAEMA) copolymers were calculated from the ratio of peak areas from the –N(CH3)2 protons at 2.18 ppm from DMAEMA units to the –C(O)–O–CH2–CH2–O–C(O)– methylene protons at 3.93–4.34 ppm from MAECA moieties (after subtracting the contribution from –C(O)–O–CH2– methylene protons at 3.98 ppm from DMAEMA units). The compositions of the copolymers determined by 1H NMR are given in Table 2. In all copolymers, we observed characteristic resonance signals in the aromatic region (7.44–7.84 ppm) due to the terminal phenyl moiety at the end of the polymer chains. The Mn,NMR values for PPEGMA and PDMAEMA were determined by comparing the integration areas from the terminal phenyl group at 7.44–7.84 ppm to the side chain –OCH3 protons at 3.25 ppm from PEGMA units and –N(CH3)2 protons at 2.18 ppm from DMAEMA units, respectively. The Mn,NMR values of P(MAECA-co-PEGMA) copolymers were calculated by comparing the integration values of chain end phenyl protons at 7.44–7.84 ppm to the characteristic protons at 3.25 ppm (–OCH3) from PEGMA units and –C(O)–O–CH2–CH2–O–C(O)– protons at 3.93–4.34 ppm from MAECA units. For these copolymers, Mn,NMR = [(DPMAECA × MMAECA) + (DPPEGMA × MPEGMA) + molecular weight of CTP], where DPn and M are the number average degrees of polymerization and molecular weight of the monomer, respectively. Similarly, Mn,NMR values of the P(MAECA-co-DMAEMA) copolymers were determined and all Mn,NMR values are summarized in Table 2. The Mn,GPC and PDI values were determined for all copolymers from the GPC analysis (Table 2). The P(MAECA-co-PEGMA) copolymers showed unimodal RI traces with a small peak at higher molecular weight side due to the formation of dead chains via bimolecular termination reactions (Fig. S6A†). Substantial bimolecular terminations were noticed in the case of the P(MAECA-co-DMAEMA) copolymer series (Fig. S6B†). Although Mn,NMR values match well with the Mn,theo values predicted from stoichiometry and conversion (Table 2), Mn,GPC values are somewhat higher compared to the Mn,theo values and the discrepancy may be mostly due to the different hydrodynamic volume between copolymers and PMMA. Differences between Mn,NMR and Mn,theo values are higher in the case of P(MAECA-co-DMAEMA) copolymers since we observed higher bimolecular terminations (Fig. S6B†) in this series compared to the P(MAECA-co-PEGMA) copolymers (Fig. S6A†).
Copolymerization of two monomers with different properties allows the synthesis of different products by variations in the relative amounts of the two monomer units in the copolymer chain. Since knowledge of reactivity ratios allows prediction of copolymer compositions at a particular monomer feed ratio, the reactivity ratios of MAECA (rMAECA), PEGMA (rPEGMA) and DMAEMA (rDMAEMA) were determined for the copolymerization of MAECA with PEGMA and DMAEMA. To determine reactivity ratios, we have used the extended Kelen–Tüdös (extended K–T) method,27 which is applicable for living controlled copolymerization at higher conversions.28 Using monomer feed compositions, co-monomer contents in the copolymer (determined by NMR in Table 2) and conversion data, the reactivity ratios were determined by the extended K–T method. The rMAECA = 1.62 and rPEGMA = 0.71 for the MAECA–PEGMA series, and rMAECA = 1.35 and rDMAEMA = 0.65 for the MAECA–DMAEMA system were obtained, respectively. These results indicated random distribution of monomer units in both the copolymerization series and the copolymer will be moderately enriched with MAECA units.29 Although all three monomers are methacrylate derivatives, the reactivity of the bulky MAECA monomer is almost double compared to PEGMA or DMAEMA. A similar trend of the reactivity ratio was observed for the copolymerization of tert-butyl methacrylate with the bulky first-generation dendronized monomer carrying an ethylene glycol-based dendron.30
Thermal analysis
The thermal stability was studied by TGA (Fig. 3A and S7A†), which suggested the high stability of PMAECA (up to 320 °C) towards thermal decomposition. The thermal stability of P(MAECA-co-PEGMA) and P(MAECA-co-DMAEMA) copolymers decreased with the decrease of MAECA content in the copolymer. The homopolymers PMAECA, PPEGMA and PDMAEMA displayed a Tg of 148, −59 and 18 °C, respectively. The Tg value for PDMAEMA matches well with the previous report.31 Very low Tg for PPEGMA was observed due to the presence of flexible ethylene glycol units in the side chains and this Tg value is close to the previously reported Tg = −60 °C.32 Interestingly, PMAECA showed very high Tg because of the side chain bulky cholate moieties with –OH groups. Although, high Tg values (e.g., 192 °C) were reported for polymers containing CA moieties,33 we observed Tg = 148 °C for PMAECA due to the presence of an additional ethylene oxide group before the CA moiety, which added some flexibility to the polymer chains.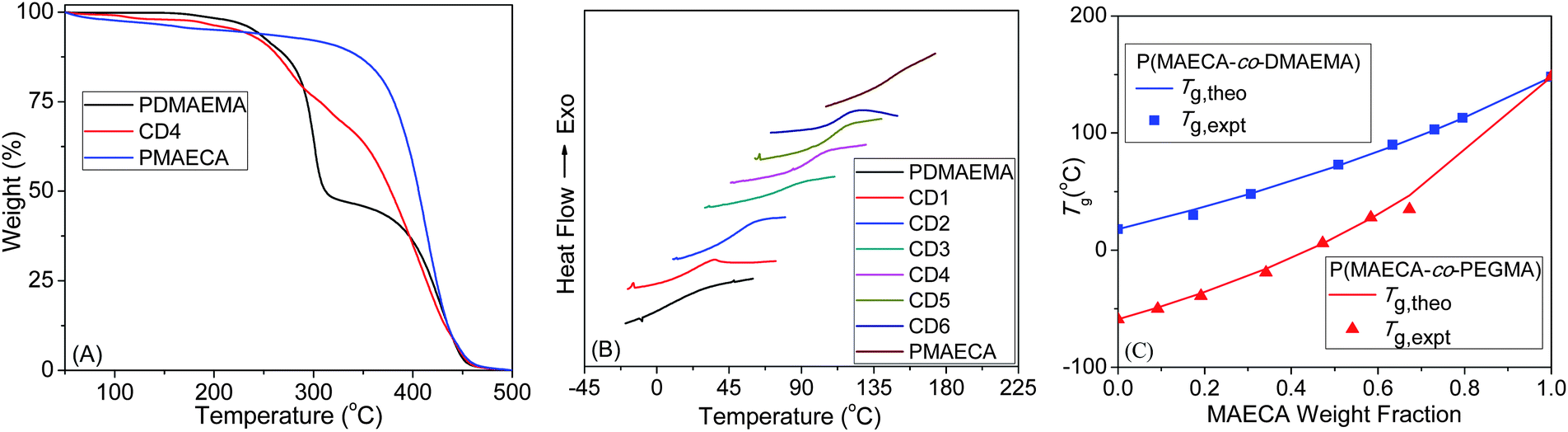 | ||
| Fig. 3 (A) TGA thermograms, (B) DSC traces and (C) comparison of the experimental Tg values to the theoretical Tg values calculated from the Fox relationship. | ||
For the copolymers, Fig. 3B and S7B† show a single Tg value in between the corresponding Tg values for the homopolymers (Table 2), which confirmed homogeneous nature of the copolymers and MAECA forms a copolymer in every composition with PEGMA or DMAEMA. The Fox equation34 was used to calculate the theoretical Tg values (Tg,theo) of copolymers in the bulk state and Tg,theo values are plotted in Fig. 3C against the MAECA composition in the copolymer. Experimental Tg values (Tg,expt) from the DSC study are also plotted in Fig. 3C as a function of the weight fractions of MAECA in two copolymer series, and we observed an excellent correlation between Tg,expt and Tg,theo.
Solution behaviour of side-chain cholate copolymers
Since PPEGMA is a thermoresponsive polymer and PDMAEMA is both temperature and pH-sensitive, we decided to study solution properties of all copolymers at different temperatures and solution pHs. Both thermo- and pH-responsive properties were determined by recording %T using UV-vis spectroscopy at 500 nm. Fig. 4A shows typical %T against temperature curves for CP1, CP2, CP3 and PPEGMA. The homopolymer PPEGMA showed LCST at 72 °C at 2 mg mL−1 concentration, while the LCST of 68 °C was reported for this polymer.35 The LCST of a polymer depends on molecular weight, solution concentration, chain end structure, etc.,36 and the above difference could originate from these effects. For the CP1, CP2 and CP3 copolymers, as expected, LCST values decrease with the increase of the molar fraction of the hydrophobic MAECA content in the copolymer. The LCST values were found to be 61, 56 and 42 °C for CP1, CP2 and CP3, while MAECA contributions in the copolymer chains were 5.5, 12 and 23%, respectively. Significant cloudiness of CP4 was observed for the aqueous solution of CP4 even at close to freezing temperature of water and this copolymer did not illustrate a distinct LCST. The phase transition of CP4 may be troubled by the restricted movement of the side chain ethylene oxide groups due to the surrounding aggregation of hydrophobic components.37 Also, high aggregation tendency of cholate moieties leads to the breakage of hydrogen-bonding from solvated PEGMA units. Note that the LCST values of PPEGMA and copolymers were independent of pH in the studied range 5.5 to 8.5, confirming that PEGMA and MAECA are not pH-dependent units.38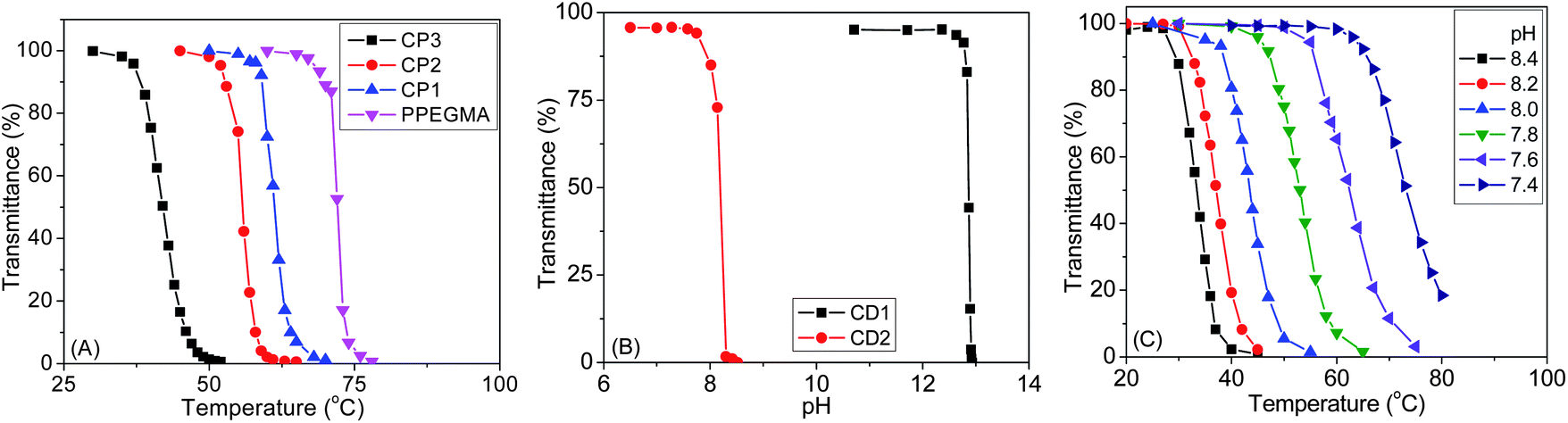 | ||
| Fig. 4 Phase transitions of (A) P(PMAECA-co-PEGMA) series at pH 6, (B) CD1 and CD2 at 25 °C, and (C) CD1 at different pHs. | ||
In the P(MAECA-co-DMAEMA) series, only CD1 and CD2 were soluble in water, where MAECA units in the copolymer chains were 5.9 and 11.8%, respectively. In order to determine the pH responsiveness of PDMAEMA, CD1 and CD2, their aqueous solutions were analyzed as a function of pH at 25 °C. PDMAEMA showed an LCST of 52 °C at pH 7.4 and pH-responsiveness at pH 12.9 at 25 °C as a result of complete deprotonation, and these values match well with the literature.39
The phase transition pH is reduced with the increasing MAECA content in the copolymer and at 25 °C we determined pH-responsiveness at 12.8 and 8.2 for CD1 and CD2, respectively (Fig. 4B). These results indicate that the hydrophobic MAECA units facilitate the aggregation of DMAEMA units in these copolymers at lower pH. The LCST of CD1 and CD2 was studied at different pHs and we determined hydrophilic to hydrophobic transitions for CD1 at 73, 63, 53, 44, 37 and 33 °C at pH values 7.4, 7.6, 7.8, 8.0, 8.2 and 8.4, respectively (Fig. 4C). Similarly, the LCSTs of 54, 43 and 31 °C were determined for CD2 at pH 7.4, 7.6 and 7.8, respectively (Fig. S8†). The transmittance decreased slowly for CD2 (Fig. S8†) at all pHs, indicating disturbed deprotonation of tertiary amine groups with increasing cholate moiety in the copolymer. The turbidity of polymers in aqueous solution depends on pH, involving hydrophobic interactions between cholate moieties and electrostatic repulsion between protonated amino groups. Amino groups are protonated below the responsive pH at a particular temperature, resulting in electrostatic repulsion among them and increased solubility. However, the above results indicate that the LCST can be tuned at a particular solution pH by employing different molar factions of monomer units in the copolymer. The above phase transitions were reversible indicating uninterrupted protonation and deprotonation of the tertiary amine functional groups in the DMAEMA moiety.
Self-assembly behaviour of random copolymers
The self-assembly of random copolymers to different morphologies such as micelles, vesicles, and other shapes in a selective medium is not so common compared to the block copolymers. Since amphiphilic random copolymers contain hydrophobic cholate side chains and hydrophilic PEGMA or DMAEMA units, herein we studied self-assembly characteristics of CP1, CP2, CP3 and CP4 from the P(MAECA-co-PEGMA) series, and CD1 and CD2 from the P(MAECA-co-DMAEMA) category in aqueous medium by fluorescence spectroscopy using pyrene as a fluorescent probe. Fig. S9A† shows the emission spectra of pyrene as a function of CP2 concentration, where the intensity of fluorescence gradually increased with increasing CP2 concentrations indicating solubilisation of pyrene in the hydrophobic microdomains of the aggregates. The critical aggregate concentration (CAC) of polymers was measured by the standard procedure from emission spectra of pyrene from the plot of I383/I373versus the logarithm of the copolymer concentration, which is shown for CP2 in Fig. S9B (see Fig. S10† for CD2). The CAC values of 3.3, 3.2 and 2.3 mg L−1 were determined for CP2, CP3 and CP4, respectively. Among the three copolymers from the P(MAECA-co-PEGMA) series, aggregation tendency gradually improved with increasing MAECA contents in the polymer main chain. Interestingly, CP1 was unable to increase the fluorescence intensity of pyrene with increasing polymer concentration, indicating the lack of aggregation to form hydrophobic microdomains due to the overall high hydrophilicity of CP1. In the P(MAECA-co-DMAEMA) series, CD1 and CD2 gave CAC values of 41 and 16 mg L−1, respectively. Here too, aggregation probability increases with increasing MAECA components in the copolymer. Note that the CAC values are much smaller than those of low molecular weight surfactants and comparable with other polymer micelle-like aggregates.Furthermore, self-assembly behaviour of CP2, CP4 and CD2 was investigated by 1H NMR spectroscopy in two different solvents, D2O and DMSO-d6. The DMSO-d6 is a good solvent for both MAECA and PEGMA/DMAEMA units in the copolymer, whereas D2O does not solubilise the hydrophobic MAECA moiety. The 1H NMR spectra of CP2 (Fig. 5A), CP4 (Fig. S11†) and CD2 (Fig. 5B) in DMSO-d6 show all characteristic peaks from both the co-monomers, while the resonance signal in D2O disappeared at 0.60 ppm from the methyl group of hydrophobic cholate units. These results revealed that CP2, CP4 and CD2 formed higher order structure through the arrangement of hydrophilic and hydrophobic components in the copolymers in aqueous media. In the aggregates the hydrophilic units were uncovered to the outer surface and hydrophobic parts were favoured to the core, and as a result we did not observe the 0.60 ppm peak from hydrophobic cholate moieties in the core in the NMR spectra of CP2, CP4 and CD2 in D2O.
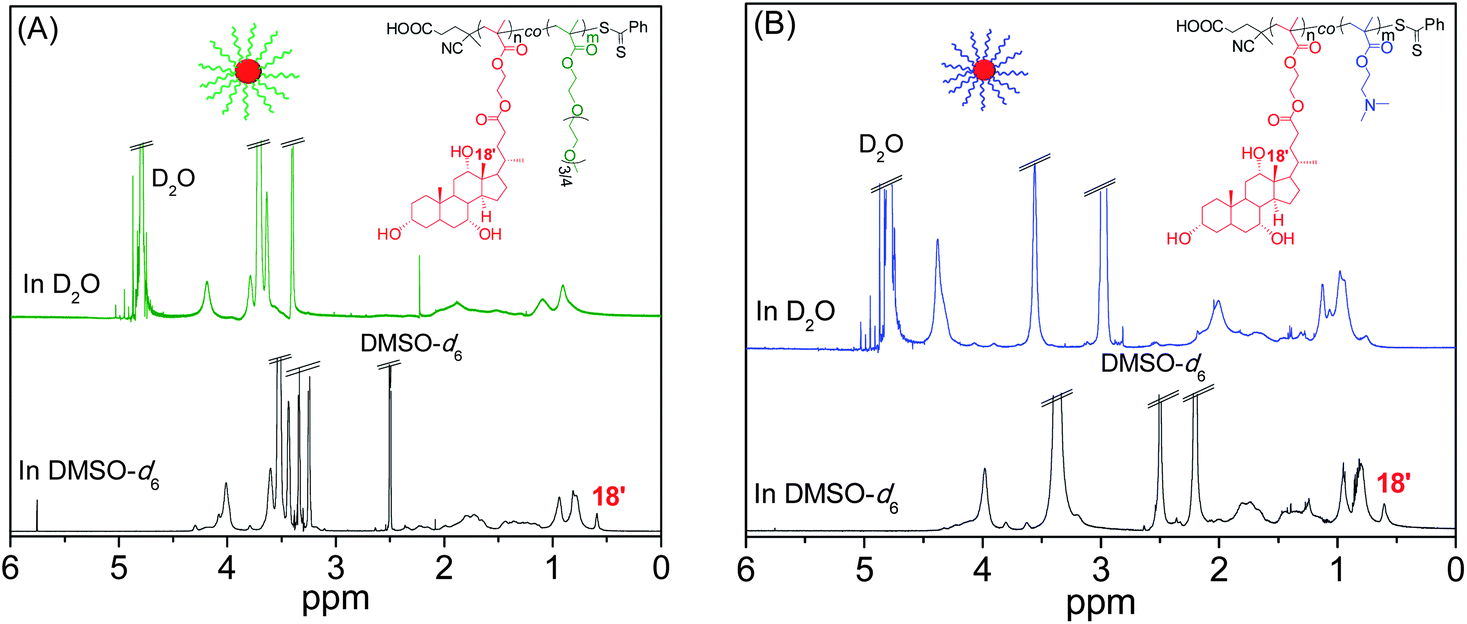 | ||
| Fig. 5 1H NMR spectra of (A) CP2 and (B) CD2 in DMSO-d6 and D2O at 25 °C. | ||
In order to study the aggregation behaviour in aqueous solution, the hydrodynamic diameter (Dh) of various copolymers was determined by DLS at 25 °C with the sample concentration of 1 mg mL−1 (w/v). CP2 (2.8 and 95 nm), CP3 (3.0 and 95 nm), and CP4 (4.6 and 96 nm) displayed two peaks, one at around 3–5 nm and another peak at approximately 95 nm (Fig. 6A). The smaller size in the range of 3 to 5 nm might be attributed to the size of a single chain conformation. Larger size in the range of 95–96 nm suggests formation of aggregated particles. CP1 showed only one peak in Fig. 6A with a Dh value of 2.7 nm, confirming its unimeric structure with insufficient amount of hydrophobic units in the polymer chain for self-assembly. This result confirmed the above pyrene encapsulation studies, where CP1 was unable to increase the fluorescence intensity of pyrene with increasing polymer concentration, due to the lack of self-assembly formation capabilities. Similarly, we measured Dh values for CD1 at pH 7 and CD2 at pH 5 (Fig. 6B). Both CD1 and CD2 showed single size distribution with Dh values of ∼100 nm, indicating their self-assembly in water to yield multi-molecular aggregates.
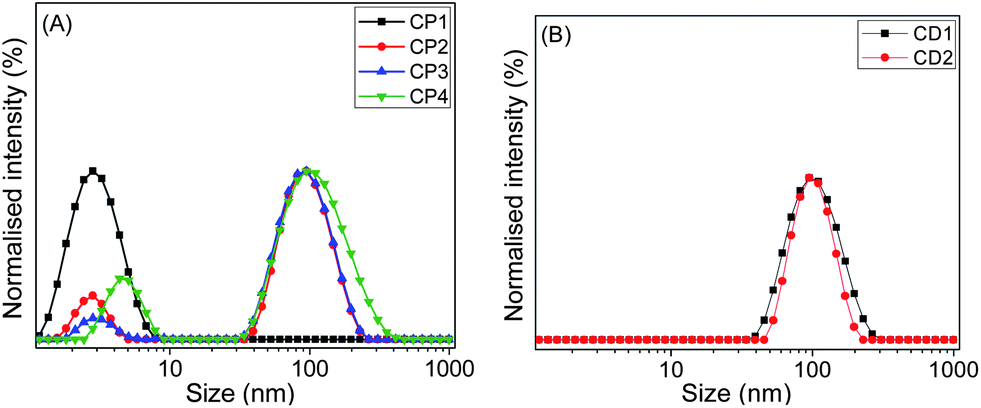 | ||
| Fig. 6 DLS size distributions of (A) CP1, CP2, CP3 and CP4, and (B) CD1 and CD2 in aqueous medium at 25 °C (concentration: 1 mg mL−1). | ||
To further characterize the aggregates and obtain direct visualization of their size and morphology, we investigated thin films of the random copolymers by imaging techniques such as AFM and SEM. The morphology of CP2, CP3 and CP4 copolymers was found to be spherical, with an average size of 78, 71 and 76 nm, respectively (Fig. 7). These copolymers formed micellar structures in water due to the aggregation of the hydrophobic cholate component towards the core and hydrophilic PEGMA units constitute the corona of the micelles. The AFM result for CP4 was also supported by SEM analysis, where we observed ∼78 nm size with spherical structure (Fig. S12†). Similarly, CD1 gave globular shape in the SEM image with a diameter of ∼82 nm (Fig. S12†). Note that sizes estimated from the AFM/SEM study are slightly lower than DLS data as reported elsewhere.40 Also, the observed diameters by DLS and AFM analyses are much higher than the contour length of the corresponding copolymers, indicating some another sort of self-assemblies.41
 | ||
| Fig. 7 AFM images of (A) CP2, (B) CP3 and (C) CP4. Copolymer solutions (0.2 mg mL−1) were deposited onto cover-slips. | ||
Nile red encapsulation using copolymer aggregates
The encapsulation of nile red, a model hydrophobic dye, by various water soluble copolymers was spectroscopically investigated. Typically, 10 mM nile red stock solution was prepared in THF, and 100 μL aliquots were added to 4 mL of the polymer solutions in deionised water (1.0 mg mL−1, w/v). The solution was stirred for 24 h at room temperature to eliminate THF, and then it was filtered through a 0.45 μm syringe filter to remove insoluble nile red.42 At this point, the aqueous solution of CP1 was colourless, which further demonstrates the inability of CP1 to self-assemble into higher order structure. However, CP2, CP3 and CP4 (Fig. 8A) solutions were pink coloured, which were studied by UV-vis and fluorescence spectroscopy.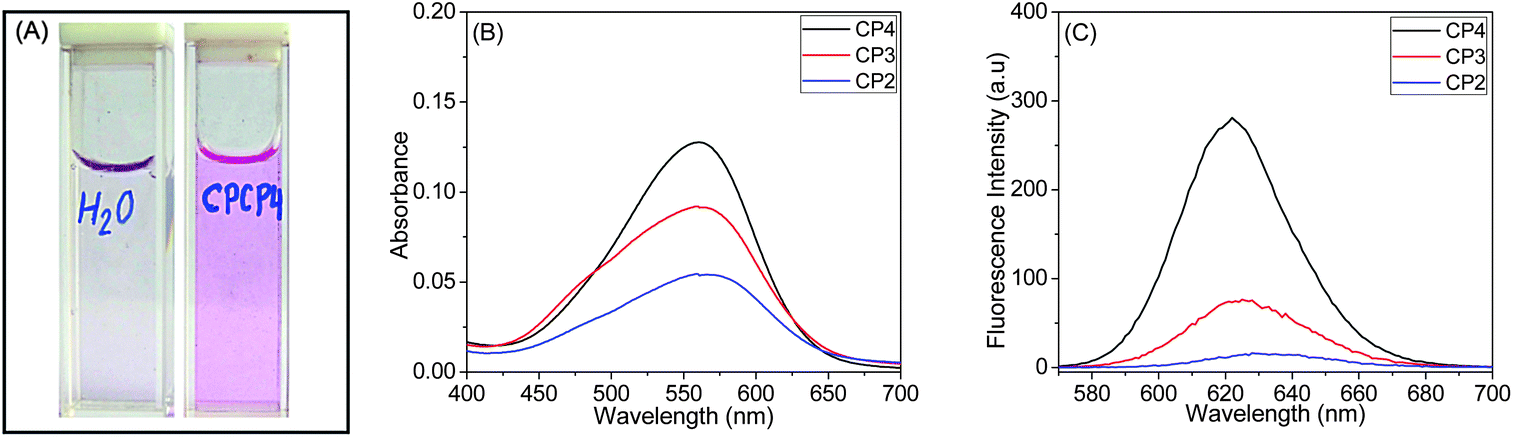 | ||
| Fig. 8 (A) CP4 (CPCP4 in the picture) loaded with nile red. (B) UV-vis and (C) corresponding fluorescence emission (λex = 550 nm) spectra of nile red solubilised by CP2, CP3 and CP4. | ||
Fig. 8B shows UV-vis spectra of nile red loaded CP2, CP3 and CP4, where we observed λmax values of 568, 563 and 560 nm for CP2, CP3 and CP4, respectively. Also, absorbance of copolymers gradually increased with increasing MAECA content in the copolymer. The fluorescence emission spectra in Fig. 8C suggest relatively low fluorescence intensity for CP2 (λmax = 628 nm) and CP3 (λmax = 625 nm) compared to CP4 with a λmax of 622 nm. The fluorescence intensity of CP4 is roughly 10 times higher than CP3. These results indicate that CP4 has more hydrophobic environment due to the presence of more number of MAECA units. Since the P(MAECA-co-PEGMA) copolymer series is thermoresponsive and the P(MAECA-co-DMAEMA) copolymers are both thermo- and pH-sensitive, external stimuli such as thermo- and pH-induced controlled release of hydrophobic molecules (e.g., nile red and doxorubicin) from the polymeric micelles are under investigation.43
Conclusions
RAFT polymerization has been used to design bile acid containing thermo-, and dual thermo- and pH-responsive random copolymers. The random distribution of co-monomers in the copolymer chains was confirmed by the reactivity ratios of two monomers for the two systems (MAECA with PEGMA and DMAEMA), and further supported by a single Tg value of copolymers which varies smoothly with composition between the Tg values of the respective homopolymers. The thermal stability of copolymers increased with increasing MAECA contents in copolymer. The LCST can easily be tuned for the P(MAECA-co-PEGMA) series by varying the co-monomer composition of copolymers. For P(MAECA-co-DMAEMA) copolymers, the LCST can be adjusted between 31 and 73 °C by changing the pH and co-monomer contents in the copolymer. 1H NMR and fluorescence spectroscopy indicated self-assembly from random copolymers in water to micellar structures with a spherical shape, which were characterized by AFM and SEM studies, while aggregation was dependent on hydrophilic PEGMA or DMAEMA to hydrophobic MAECA contents in the copolymers. Cholic acid based copolymers were capable of encapsulating hydrophobic nile red molecules in aqueous solutions. Further studies are in progress to achieve bile acid containing random copolymer architectures with high drug loading capacity, and controlled release capability at different pHs and temperatures.Acknowledgements
The authors thank the Indian Institute of Science Education and Research-Kolkata for providing research facilities. S. Pal is grateful to CSIR, Government of India for his fellowship.Notes and references
- E. Sievänen, Molecules, 2007, 12, 1859–1889 CrossRef PubMed.
- T. B. N. Satyanarayana, U. Maitra and A. J. Savyasachi, Eur. J. Org. Chem., 2012, 3658–3664 CrossRef CAS.
- C. Corpechot, F. Carrat, A. M. Bonnand, R. E. Poupon and P. Poupon, Hepatology, 2000, 32, 1196–1199 CrossRef CAS PubMed.
- J. Tamminen and E. Kolehmainen, Molecules, 2001, 6, 21–46 CrossRef CAS.
- P. Babu, N. M. Sangeetha and U. Maitra, Macromol. Symp., 2006, 241, 60–67 CrossRef CAS.
- X. X. Zhu and M. Nichifor, Acc. Chem. Res., 2002, 35, 539–546 CrossRef CAS PubMed.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- P. V. Mendonça, A. C. Serra, C. L. Silva, S. Simões and J. F. J. Coelho, Prog. Polym. Sci., 2013, 38, 445–461 CrossRef PubMed.
- J. Hao, H. Li and X. X. Zhu, Biomacromolecules, 2006, 7, 995–998 CrossRef CAS PubMed.
- J.-P. Xu, J. Ji, W.-D. Chen and J.-C. Shen, Macromol. Biosci., 2005, 5, 164–171 CrossRef CAS PubMed.
- Y. Shao, C. Lavigueur and X. X. Zhu, Macromolecules, 2012, 45, 1924–1930 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS.
- S. Boissé, J. Rieger, A. Di-Cicco, P.-A. Albouy, C. Bui, M.-H. Li and B. Charleux, Macromolecules, 2009, 42, 8688–8696 CrossRef.
- (a) A. Benrebouh, D. Avoce and X. X. Zhu, Polymer, 2001, 42, 4031–4038 CrossRef CAS; (b) H. Liu, D. Avoce, Z. Song and X. X. Zhu, Macromol. Rapid Commun., 2001, 22, 675–680 CrossRef CAS; (c) D. Avoce, H. Y. Liu and X. X. Zhu, Polymer, 2003, 44, 1081–1087 CrossRef CAS.
- Nonappa and U. Maitra, Eur. J. Org. Chem., 2007, 3331–3336 CrossRef CAS.
- K. Dan, N. Bose and S. Ghosh, Chem. Commun., 2011, 47, 12491–12493 RSC.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS PubMed.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Advances in RAFT polymerization, 2005, vol. 46, pp. 8458–8468 Search PubMed.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- Y. K. Chong, J. Krstina, T. P. T. Le, G. Moad, A. Postma, E. Rizzardo and S. H. Thang, Macromolecules, 2003, 36, 2256–2272 CrossRef CAS.
- T. Shimoboji, Z. L. Ding, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 2002, 13, 915–919 CrossRef CAS PubMed.
- S. V. Aathimanikandan, E. N. Savariar and S. Thayumanavan, J. Am. Chem. Soc., 2005, 127, 14922–14929 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- K. Bauri, S. Pant, S. G. Roy and P. De, Polym. Chem., 2013, 4, 4052 RSC.
- C. Zhu, S. Jung, S. Luo, F. Meng, X. Zhu, T. G. Park and Z. Zhong, Biomaterials, 2010, 31, 2408–2416 CrossRef CAS PubMed.
- T. Kelen and F. J. Tüdös, J. Macromol. Sci., Part A: Pure Appl. Chem., 1981, 16, 1283–1297 CrossRef.
- R. Hoogenboom, M. W. M. Fijten, S. Wijnans, A. M. J. van der Berg, H. M. L. Thijs and U. S. Schubert, J. Comb. Chem., 2006, 8, 145–148 CrossRef CAS PubMed.
- G. Odian, Principles of Polymerization, Wiley India, New Delhi, 4th edn, 2008, p. 472 Search PubMed.
- A. Krebs, B. Bruchmann, A. Müller-Cristadoro, R. Al-Hellani and A. D. Schlüter, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1372–1377 CrossRef CAS.
- S. G. Roy, K. Bauri, S. Pal, A. Goswami, G. Madras and P. De, Polym. Int., 2013, 62, 463–473 CrossRef CAS.
- Y. Tong, L. Chen, X. He and Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4341–4360 CrossRef CAS.
- Z.-J. Song, H. Li and X. X. Zhu, Chin. J. Polym. Sci., 2003, 21, 521–526 CAS.
- T. G. Fox, Bull. Am. Phys. Soc., 1956, 1, 123–128 CAS.
- T. Ishizone, A. Seki, M. Hagiwara, S. Han, H. Yokoyama, A. Oyane, A. Deffieux and S. Carlotti, Macromolecules, 2008, 41, 2963–2967 CrossRef CAS.
- Z. M. O. Rzaev, S. Dinçer and E. Piskin, Prog. Polym. Sci., 2007, 32, 534–595 CrossRef CAS PubMed.
- J. Park, M. Moon, M. Seo, H. Choi and S. Y. Kim, Macromolecules, 2010, 43, 8304–8313 CrossRef CAS.
- D. Fournier, R. Hoogenboom, H. M. L. Thijs, R. M. Paulus and U. S. Schubert, Macromolecules, 2007, 40, 915–920 CrossRef CAS.
- J. F. Gohy, S. Creutz, M. Garcia, B. Mahltig, M. Stamm and R. Jerome, Macromolecules, 2001, 33, 6378 CrossRef.
- W.-J. Song, J.-Z. Du, N.-J. Liu, S. Dou, J. Cheng and J. Wang, Macromolecules, 2008, 41, 6935–6941 CrossRef CAS.
- E. Rakhmatullina, T. Braun, M. Chami, V. Malinova and W. Meier, Langmuir, 2007, 23, 12371–12379 CrossRef CAS PubMed.
- S. Bhatia, A. Mohr, D. Mathur, V. S. Parmar, R. Haag and A. K. Prasad, Biomacromolecules, 2011, 12, 3487–3498 CrossRef CAS PubMed.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The synthesis protocol for MAECA, NMR characterization of monomers, homopolymers and various copolymers, FT-IR and ESI-MS spectra, GPC RI traces of various polymers, DSC and TGA thermograms of P(MAECA-co-DMAEMA) copolymers, phase transitions of CD2 at different pHs, fluorescence spectra and corresponding CAC determination plots, SEM images, and solubility studies of both the copolymer series. See DOI: 10.1039/c3py01317k |
| This journal is © The Royal Society of Chemistry 2014 |