 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multifunctional lipid-coated polymer nanogels crosslinked by photo-triggered Michael-type addition†
Yingkai
Liang
a and
Kristi L.
Kiick
*abc
aDepartment of Materials Science and Engineering, University of Delaware, 201 DuPont Hall, Newark, DE 19716, USA. E-mail: kiick@udel.edu; Tel: +1-302-831-0201
bBiomedical Engineering, University of Delaware, Newark, DE 19716, USA
cDelaware Biotechnology Institute, 15 Innovation Way, Newark, DE 19716, USA
First published on 21st October 2013
Abstract
Novel, multifunctional lipid-coated polymer nanogels crosslinked by photo-triggered Michael-type addition were prepared via the use of liposome templates. The photo-sensitive gelation and temporal control of the crosslinking reaction were confirmed by oscillatory rheology experiments of bulk hydrogels, and the production of nanogels was confirmed via dynamic light scattering and transmission electron microscopy. The surface functionality of the lipid-coated nanogels was demonstrated by surface modification with a reactive fluorescent dye. These multifunctional lipid-coated nanogels, given the mild preparation conditions, ease of size control, and versatility of the chemical linkages used as cross-links, have significant potential for use in polymeric nanoparticulate drug delivery systems.
Introduction
Nanotechnology has had a significant impact on the development of nanoscale vehicles and devices in the field of drug delivery and tissue engineering,1–5 and many types of nanoparticles have been developed over the past few decades, including inorganic nanoparticles, lipid-based carriers, and polymeric nanostructures (e.g., polymer–drug conjugates and block copolymer micelles).6,7 Although metallic nanoparticles provide opportunities for magnetic resonance imaging (MRI)8 or photothermal therapy,9 they are not biodegradable and many are not small enough to be cleared from circulation easily, which may lead to long-term toxicity due to potential accumulation in the body. Polymeric nanoparticulate systems have attracted much attention as potential drug carriers due to enhanced therapeutic efficacy that is imparted by improvements in the solubility of hydrophobic drugs, extension of circulation half-life, reduction of immunogenicity, and sustained drug release.10,11 In addition, nanoparticles that are responsive to external stimuli such as pH,12 light,13,14 temperature,15,16 magnetic fields,17 and ultrasound18 have also been developed to achieve on-demand/triggered drug release.19 Particularly, nanoparticles are “small” enough (10–150 nm) to passively accumulate in specific tissues (e.g., tumors) through the enhanced permeability and retention (EPR) effect and sufficiently “large” (high surface area-to-volume ratios) for surface functionalization with specific ligands to actively target the site of disease.7,20–23Many polymeric nanoparticles are based on non-covalent assembly, which may render their structure unstable and lead to subsequent loss of drug when circulating in large volumes of biological fluids.24–26 To address these concerns, crosslinked polymer nanogels with improved stability have been developed.27–32 Composed of highly crosslinked hydrophilic polymer chains, nanogels maintain structural integrity even under dilute conditions. Due to their gel-like structures, nanogels have a high degree of porosity that allows efficient encapsulation of therapeutic molecules.33 In addition, the high water content and mechanical softness of nanogels may enhance their biocompatibility and positively impact cellular uptake and biodistribution.34–36 Nanogels have thus emerged as promising materials for the delivery of a wide range of therapeutic molecules including chemotherapeutics,29,31 proteins,30,32 and siRNAs.33
A major obstacle in the preparation of nanogels is macrogelation, which ultimately promotes bulk gelation (to yield either a microgel or hydrogel).37 Many procedures have been employed to avoid macrogelation during crosslinking, including microemulsion/inverse microemulsion polymerization methods.38 In these approaches, each droplet of polymer solution serves as a nanoreactor, permitting isolated crosslinking of each droplet and the facile incorporation of bioactive molecules.39 However, the necessary use of organic solvents in the formation of the emulsions can be detrimental to the therapeutic cargo.40 Alternatively, preparation of nanogels from nanotemplates such as liposomes largely avoids the use of organic solvents during chemical crosslinking,41 as hydrogels are formed in the aqueous lumen of the liposome. In addition, the use of liposomes generally permits better control over the size of the resulting nanogels, given the uniformity of liposome populations and the simplicity of the manipulation of their size via extrusion methods.42
Although the preparation of nanogels using liposome-templated methods is a common approach,43–49 most reported strategies are based on radical polymerization of vinyl monomers/methacrylated macromers or ionic crosslinking of alginate, which exerts less control of network development and may result in heterogeneous network structures. While Michael-type addition reactions have been widely used as a crosslinking strategy for hydrogel preparation under mild, physiologically relevant conditions,50,51 they have not been commonly employed in the field of nanogel synthesis,52 owing to their spontaneous reaction kinetics.53 The application of photolabile protecting groups and photo-catalyzed chemistries provides spatial and temporal regulation of the physical and chemical properties of materials.54–57 The photocleavage reaction has been widely used in photodegradable crosslinkers,58–60 side chain functionalization of photoresponsive block copolymers,61–63 and photo-activated bioconjugates.64–67 Recently, photolabile chemistry has also been utilized as a strategy for light-triggered crosslinking of alginate hydrogels, which displayed improved mechanical properties and homogeneity.68 The application of these strategies to yield photolabile, protected precursors for Michael-type additions would permit the light-dependent production of homogeneous network structures with more precise control of the reaction, which would thus expand the utility of Michael-type additions in the synthesis of nanogels.
We report here the production of novel lipid-coated polymer nanogels, which are crosslinked by photo-triggered Michael-type additions of thiols and maleimides, via liposome-templated methods. Specifically, photolabile o-nitrobenzyl protecting groups have been employed to protect PEG–thiols and thus to permit triggering of nanogel formation upon photo-irradiation; the o-nitrobenzyl groups undergo photocleavage into o-nitrosobenzaldehyde upon irradiation with UV light, releasing free thiols for subsequent crosslinking reactions with maleimides.54,69 The photo-triggered chemistry provides temporal control of the reaction and avoids macroscale gelation during nanogel synthesis, while the lipid coating offers additional functional features to the nanogels by preventing burst release of therapeutic molecules and providing surface versatility. The photo-triggered gelation of bulk hydrogels was monitored via oscillatory rheology and the size distribution and colloidal stability of the nanogels were measured via dynamic light scattering and electrophoretic light scattering, respectively. The morphology and size of the nanogels were confirmed via transmission electron microscopy (TEM). The chemical reactivity of the nanogels was probed via their modification with a fluorescent dye. Owing to the ability to control degradation of the resulting polymeric networks, these nanogels have potential application as targeted drug-delivery vehicles and imaging probes.
Experimental section
Materials
Four-arm, thiol-functionalized PEG (PEG-SH, Mn 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) and four-arm, maleimide-functionalized PEG (PEG-Mal, Mn 10000 g mol−1) were purchased from JenKem Technology USA Inc. (Allen, TX, USA). 1-α-Phosphatidylcholine from egg (egg-PC), cholesterol, o-nitrobenzyl bromide, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl) and N-hydroxysulfosuccinimide (sulfo-NHS) were purchased from Sigma-Aldrich (St. Louis, MO, USA). DSPE-PEG2000 carboxylic acid was purchased from Nanocs Inc. (New York, NY, USA). 1-Aminomethylpyrene hydrochloride was purchased from AnaSpec Inc. (Fremont, CA, USA). All other reagents and materials were purchased from Fisher Scientific unless otherwise noted (Pittsburgh, PA, USA). 1H NMR spectra were acquired under standard quantitative conditions at ambient temperature on a Bruker AV400 NMR spectrometer (Billerica, MA, USA). All samples were dissolved in CDCl3.
000 g mol−1) and four-arm, maleimide-functionalized PEG (PEG-Mal, Mn 10000 g mol−1) were purchased from JenKem Technology USA Inc. (Allen, TX, USA). 1-α-Phosphatidylcholine from egg (egg-PC), cholesterol, o-nitrobenzyl bromide, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl) and N-hydroxysulfosuccinimide (sulfo-NHS) were purchased from Sigma-Aldrich (St. Louis, MO, USA). DSPE-PEG2000 carboxylic acid was purchased from Nanocs Inc. (New York, NY, USA). 1-Aminomethylpyrene hydrochloride was purchased from AnaSpec Inc. (Fremont, CA, USA). All other reagents and materials were purchased from Fisher Scientific unless otherwise noted (Pittsburgh, PA, USA). 1H NMR spectra were acquired under standard quantitative conditions at ambient temperature on a Bruker AV400 NMR spectrometer (Billerica, MA, USA). All samples were dissolved in CDCl3.
Synthesis of photolabile, protected PEG-SH
Four-arm PEG-SH, protected with o-nitrobenzyl groups, was synthesized via previously published methods, with some modifications.62,70 Four-arm, thiol-terminated PEG (150 mg or 0.015 mmol) was dissolved in 5 mL of DMF, and then neutralized with TEA (100 μL or 0.72 mmol). The solution was cooled down to 0 °C and a solution of 2-nitrobenzyl bromide (50 mg or 0.23 mmol in 5 mL acetone) was added dropwise over 15 min. The reaction mixture was stirred vigorously at 0 °C for 1 h and then at room temperature for 24 h. The resulting solution was dialyzed against DI water for 24 h (MWCO 1000) and then lyophilized to afford a white powder product. The functionality of the resulting polymers was determined via1H NMR (in CDCl3), with an average value of 71 ± 5%. 1H NMR (Fig. S1†) (400 MHz, CDCl3) δ 7.99 (d, 4H, Ar), 7.55 (dd, 8H, Ar), 7.45 (d, 4H, Ar), 4.17 (s, 8H, CH2), 3.90–3.40 (d, 900H, CH2CH2O), 2.64 (t, 8H, CH2).Bulk gel preparation via photo-triggered Michael-type addition and subsequent rheological analysis
The PEG hydrogel was prepared by the photo-triggered Michael-type addition of protected PEG-SH and PEG-Mal (at a final concentration of 5 wt% or 15 wt%). The solution was well mixed by vortexing and then was exposed for 2 hours to UV light (365 nm; Black-Ray® mercury lamp) at an intensity of 10 mW cm−2. Oscillatory rheology experiments conducted on a stress-controlled AR-G2 rheometer (TA Instruments, New Castle, DE, USA) were used to characterize the photo-triggered cross-linking and subsequent mechanical properties of the bulk PEG hydrogels. Experiments were conducted at room temperature using an 8 mm diameter, steel plate geometry with 100 μm gap distance, with oscillatory time, frequency and strain sweeps performed. Strain sweeps were performed on samples from 0.1% to a maximum strain of 1000% to determine the limit of the linear viscoelastic region. Dynamic oscillatory time sweeps were collected at angular frequencies of 6 rad s−1 and 1% strain chosen from the linear viscoelastic region. Rheological properties were examined by frequency sweep experiments (ω = 0.1 to 100 rad s−1) at a fixed strain amplitude of 1%. The protected PEG-SH and PEG-Mal precursors were dissolved in water separately with a concentration of 5 wt% or 15 wt%. The precursors were then combined, vortexed, and loaded onto the rheometer stage. Hydrogels were formed by exposing the solution to UV light continuously for 2 hours (365 nm at 10 mW cm−2; OmniCure® S2000, 200 W UV lamp with bandpass filters and high power fiber light guide) or periodically by shuttering the light (365 nm at 10 mW cm−2; light on for 5 min, light off for 20 min); the G′ and G′′ values were monitored throughout the crosslinking experiments.Synthesis of lipid-coated nanogels (NGs)
A chloroform solution (1 mL) with 5 mg egg-PC and 0.5 mg of cholesterol was placed in a round-bottom flask, with evaporation of the chloroform under nitrogen flushing to yield a thin film of the lipid that was then dried under vacuum overnight at room temperature to remove the organic solvent. A 15 wt% PEG solution was prepared by dissolving 75 mg PEG-Mal and 75 mg o-nitrobenzyl-protected PEG-SH in 1 mL DI water; this solution was used to hydrate to the lipid film, with ten alternating cycles of 30 s vortexing and 5 min quiescent incubation at room temperature.48 The solution was then sonicated for 30 min to completely solubilize the lipid. The resulting multilamellar liposomes were extruded 20 times through a 0.8 μm polycarbonate membrane (Whatman, Piscataway, NJ, USA), 20 times through a 0.2 μm membrane, and finally 21 times through a 0.1 μm membrane using an Avanti® Mini-Extruder (Avanti Polar Lipids Inc., Alabaster, AL, USA). The resulting unilamellar liposomes were then diluted 5 times (to avoid macroscale gelation upon UV irradiation). The solutions were irradiated under UV light (365 nm) for 2 hours with a light intensity of 10 mW cm−2. The nanogels were stored at 4 °C until further use.Surface functionalization of lipid-coated nanogels
Chloroform solution (1 mL) with 4 mg egg-PC, 1 mg DSPE-PEG2000-COOH and 0.5 mg cholesterol was placed in a round-bottom flask, with nitrogen flushing and drying to yield a thin film. Carboxylic acid-functionalized liposomes, and then lipid-coated nanogels (COOH-NGs), were produced as described above and stored at 4 °C until further use. A solution of COOH-NG (1 mL, 1.2 mg mL−1) was then added to EDC–sulfo-NHS solution (50 mM EDC and 25 mM sulfo-NHS dissolved in phosphate buffer pH = 7.2) to yield a total final volume of 2.5 mL. The solution was stirred for 45 min at 4 °C to activate the carboxylic acid groups and then 0.5 mg 1-aminomethylpyrene (dissolved in 50 μL DMSO) was added dropwise to achieve a final dye concentration of 0.2 mg mL−1. The solution was subsequently stirred for 4 hours at room temperature. The resulting dye-conjugated nanogels (dye-conjugated NGs, 2.5 mL) were then dialyzed against 2 L water overnight and washed 3 times with water at 4000 rpm for 20 min using Amicon Ultra-15 Centrifugal Filter Units (MWCO: 30 kDa, Millipore, Billerica, MA) to remove unconjugated dye molecules. The emission fluorescence spectrum of the dye-conjugated nanogels was measured from 350–600 nm with a slit width of 1.5 nm (after excitation at 333 nm with a slit width of 3 nm) using a HORIBA Jobin Yvon SPEX FluoroMax-4 spectrofluorometer. Microscopy images of the fluorescently labeled NGs were obtained via confocal laser scanning microscopy (CLSM, Zeiss, 510 NLO, Germany). A COOH-NG control was prepared under identical reaction conditions and procedures, without the coupling reagents, to confirm that non-reacted dye was removed under the reported conditions.Dynamic light scattering (DLS) and zeta potential analysis
The average size and size distribution of the nanogels were analyzed via dynamic light scattering (DLS) and the zeta potential determined via analysis of light scattering on a Malvern Zetasizer Nano ZS apparatus with Malvern Instruments DTS software (v.6.01) (Malvern Instruments, UK). The analyses were conducted on solutions of the nanogels in DI water and at 25 °C. For the stability measurement, analyses were conducted in PBS buffer at different temperatures. The mean hydrodynamic diameter of the nanogels (dh) was computed from the intensity of the scattered light using the Malvern software package based on the theory of Brownian motion and the Stokes–Einstein equation. The zeta potential of the nanogels was analyzed using the electrophoretic light scattering spectrophotometer of the instrument.Transmission electron microscopy (TEM) characterization of nanogel morphology
Transmission electron microscopy (TEM) images were acquired using a FEI Tecnai-12 microscope operating at an accelerating voltage of 120 kV. Images were collected on a Gatan CCD camera. TEM samples were prepared by applying a drop of nanogel solution (∼3 μL) onto a carbon-coated copper TEM grid (300 mesh size). The excess solution was wicked away after 1 min using a piece of filter paper. A droplet of 1% phosphotungstic acid aqueous solution (∼3 μL) was then added onto the grid and wicked away after 1 min using a piece of filter paper. The samples were dried at room temperature for 3 hours before imaging.Results and discussion
Photodeprotection and photo-triggered gelation
The photolabile, protected PEG-SH was synthesized as shown in Scheme 1. The thiols from the PEG chains were protected by the photolabile o-nitrobenzyl groups to yield o-nitrobenzyl thioether moieties. 1H NMR spectroscopy was used to confirm the functionality of the polymer protection by o-nitrobenzyl groups. The data shown in Fig. S1† indicate the presence of both aromatic protons (7.4–8.0 ppm) of the o-nitrobenzyl group, and the ethylene protons (3.4–3.9 ppm) of the polymer, which is consistent with results reported in similar photoprotection systems.65,69–71 Based on the integration of the aromatic and ethylene group protons, a functionality of 71 ± 5% was obtained; this functionality was consistently obtained across multiple syntheses. Upon UV irradiation, the o-nitrobenzyl groups undergo photocleavage into o-nitrosobenzaldehyde, releasing free thiols for subsequent in situ Michael-type additions.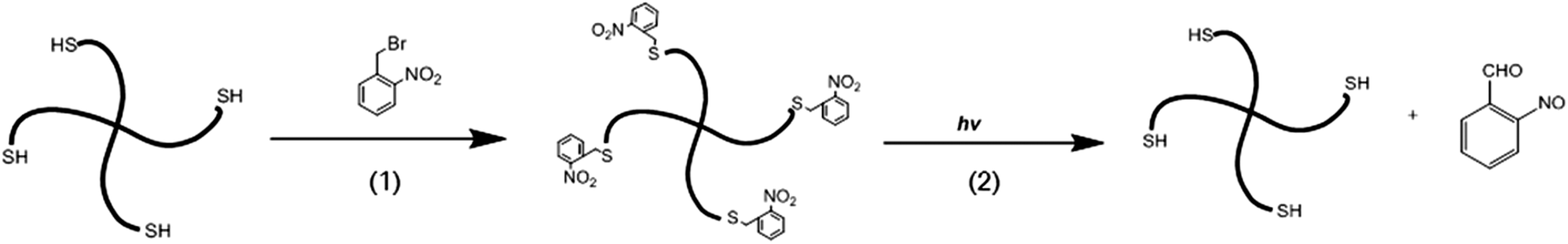 | ||
| Scheme 1 Schematic of the protection of 4-arm PEG-thiols with photolabile groups. (1) DMF, 24 h, R.T. (2) Water, 365 nm, 10 mW cm−2. | ||
The photo-triggered gelation experiments were carried out at ambient temperature by irradiation of the mixed solution of hydrogel precursors at a wavelength of 365 nm and an intensity of 10 mW cm−2 (Scheme 2). The hydrogel turned yellow after gelation, which may be attributed to the o-nitrosobenzaldehyde produced by the photocleavage.62 Hydrogel formation was confirmed via oscillatory rheology in situ during network development (Fig. 1). The data in Fig. 1 show that homogeneous solutions of protected PEG-SH and PEG-Mal in water did not exhibit any increase in the elastic modulus (G′) before UV irradiation, while the Michael-type addition between thiol and maleimide usually occurs within a few seconds under similar conditions.50,72 After 5 min irradiation, a rapid increase in G′ was observed, confirming gelation and supporting the expected photo-triggered crosslinking mechanism. The rheology results are also plotted in log scale as shown in Fig. S2,† to better show that the viscous modulus (G′′) remained essentially unchanged before UV irradiation and ultimately increased to approximately 100 Pa when irradiation was completed. The slow increase of G′ at later time points during the measurement is likely a result of some continued polymerization (owing to the slow deprotection kinetics) and in part due to the radical polymerization of the remaining PEG-Mal functional groups that have not yet reacted with thiol.73 Data presented in Fig. S3† illustrate that the G′ of a PEG-Mal control (lacking PEG-SH) began to increase after approximately 30 min, indicating the polymerization of PEG-Mal and suggesting that the increase in G′ of the PEG-Mal/PEG-SH bulk gel after 30 min irradiation may arise in part from this polymerization. In order to demonstrate that the crosslinking reaction between thiol and maleimide groups occurs mainly as a result of UV exposure, the polymerization was interrupted at approximately 15, 40 and 65 min, for a period of 20 min each time, simply by shuttering the UV light source. The data in Fig. 1b illustrate that little or no increase in elastic modulus was observed during the periods when there is no UV irradiation, indicating the temporal control of this light-dependent crosslinking strategy. Similar to other light-dependent thiol–ene polymerization strategies as those previously reported by Bowman, Anseth and co-workers,74 the photo-triggered Michael-type addition might also have significant potential in the preparation of biomimetic hydrogels. The temporal control achieved by this strategy would allow premixing and appropriate positioning (e.g., in a tissue defect) of hydrogel precursors prior to crosslinking. The spatial control enabled by this chemistry, while not investigated here, might extend the capability of Michael-type additions to create patterned devices, materials and structures.
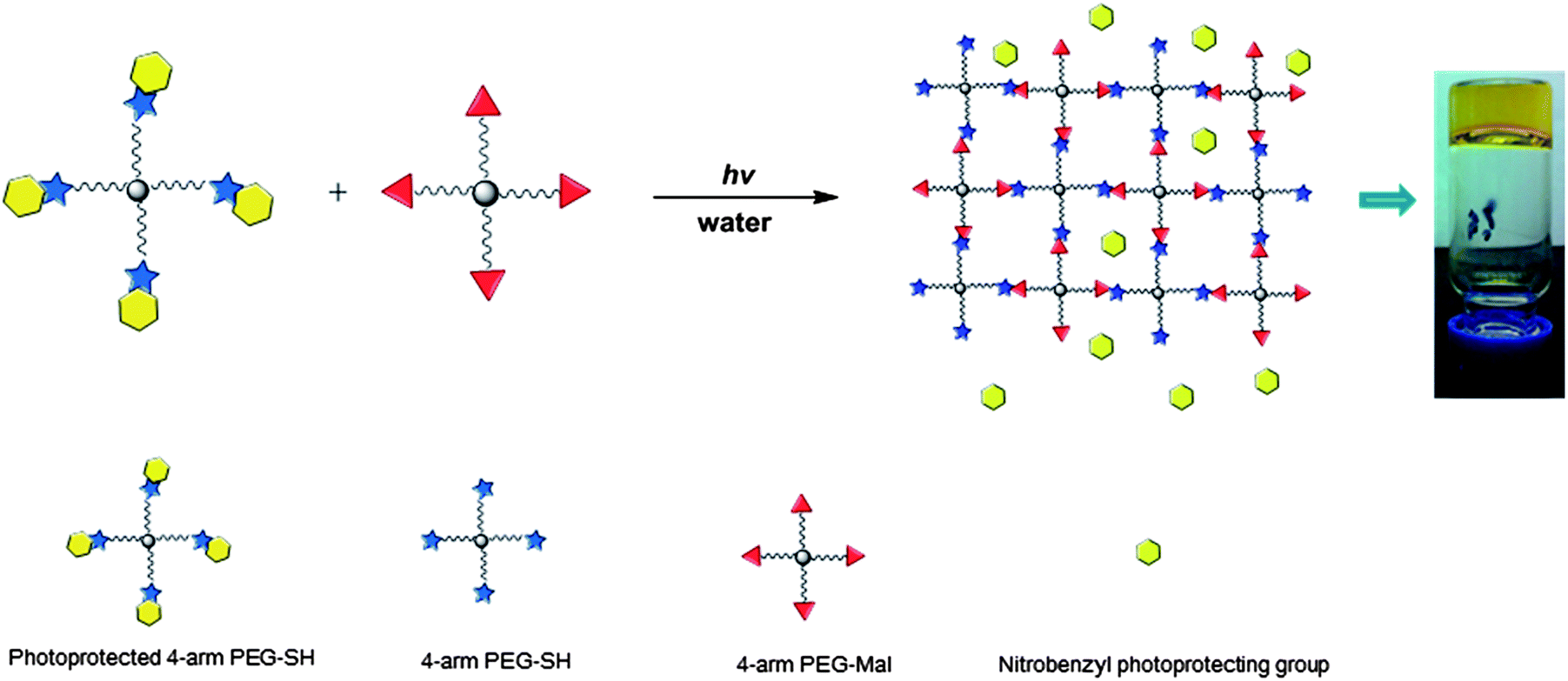 | ||
| Scheme 2 Schematic representation of the photo-triggered formation of PEG-based hydrogels via Michael-type addition. | ||
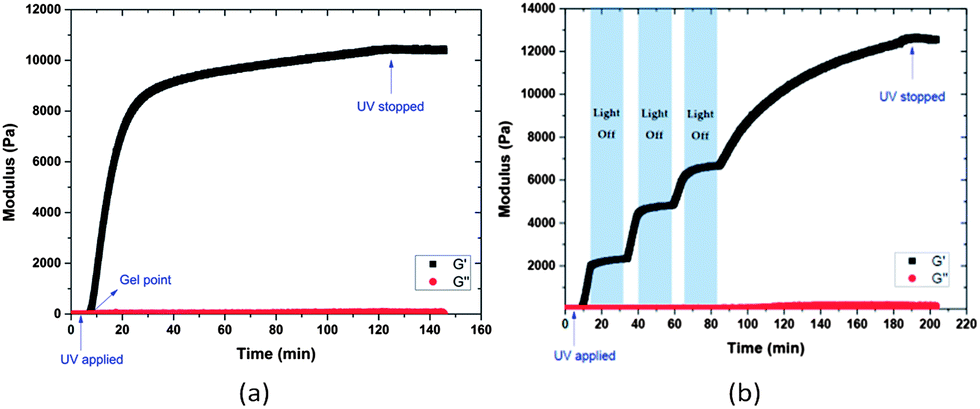 | ||
| Fig. 1 Oscillatory rheology time sweeps of 15 wt% PEG hydrogels formed by photo-triggered Michael-type addition with (a) continuous and (b) periodic irradiation (365 nm and 10 mW cm−2). | ||
It should be noted that the mechanical properties of these hydrogels can be readily tuned not only by varying the UV irradiation time, but also by changing the polymer concentration. As shown in Fig. S4,† an expected increase in elastic modulus was observed upon an increase in polymer concentration. Specifically, the measured G′ of the 5 wt% gels is 2 kPa, while that of the 15 wt% gels increased to approximately 12 kPa. Such changes in hydrogel properties with variation in crosslinking density have been regularly employed to modify the delivery properties of various hydrogels, which could be potentially applied to these nanogel systems.50,75
Synthesis and characterization of lipid-coated nanogels (NGs)
The lipid-coated PEG nanogels were prepared via a liposome-templated method as shown in Scheme 3. Upon hydration of a lipid thin film with the polymer solutions, multilamellar liposomes form with hydrogel precursors encapsulated in the aqueous lumen. By extrusion of the multilamellar liposome suspension through polycarbonate membranes of specific pore size, small unilamellar liposomes with well-defined size distribution can be prepared.44 The solutions of the unilamellar liposomes, prepared via these accepted strategies, were then diluted to prevent the potential macrogelation of any unencapsulated precursors outside the liposomes. After UV irradiation, precursors were crosslinked by photo-triggered Michael-type addition inside the lipid vesicles, forming lipid-coated nanogels.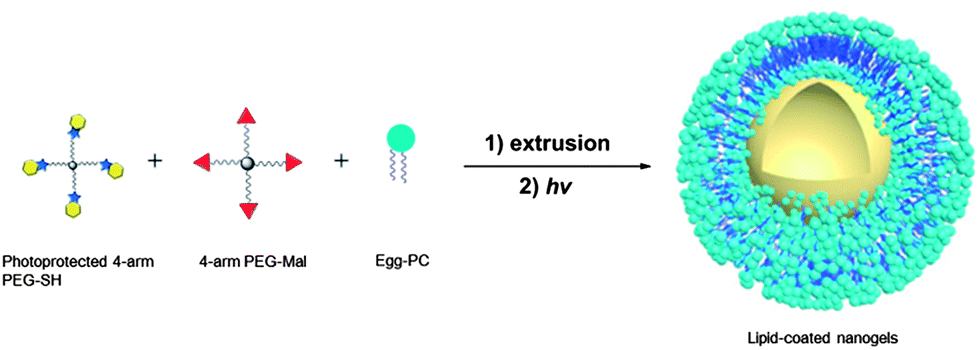 | ||
| Scheme 3 Schematic representation of the synthesis of PEG-based nanogels via liposome templates in aqueous solutions. (1) DI water, polycarbonate membrane; (2) 2 h, 365 nm, 10 mW cm−2. | ||
The hydrodynamic diameter and surface charge of the lipid-coated nanogels in aqueous solution were determined via dynamic light scattering (DLS) and zeta potential measurements, respectively. As shown in Fig. 2, the lipid-coated nanogels have an average diameter of 160 nm with a polydispersity index (PDI) of 0.25, which is very close to that of the liposomes prepared under the same conditions (154 nm, PDI 0.14). The similarity in size between the lipid-coated nanogels and liposomes confirmed the successful use of the template to form nanogels of prescribed size as expected based on the use of other liposome-templated nanogel systems.42,45 For example, work reported by An et al.42 demonstrated the size control and monodispersity of nanogels prepared from liposome templates by varying the polycarbonate membrane with specific pore sizes. The lipid-coated nanogels exhibited slightly negative zeta potentials (−7.1 mV), which may be attributed to the lipid layer since the surface charge of bare nanogels became neutral when the lipid layer was removed by detergents such as Triton X-100 (data not shown). Work reported by Wang et al.76 also indicated that conventional liposomes composed of egg-PC and cholesterol (2:1 molar ratio) demonstrate a zeta potential value of −2.7 mV (average diameter: 110 nm), which is in good agreement with our results. Retention of the lipid layer offers advantages for the subsequent use of these hydrogel nanoparticles; the slight negative charge introduced by the lipid layer should improve the stability of the nanogels in solution (and in circulation) by preventing self-aggregation and non-specific cellular uptake, based on electrostatic repulsion.77,78
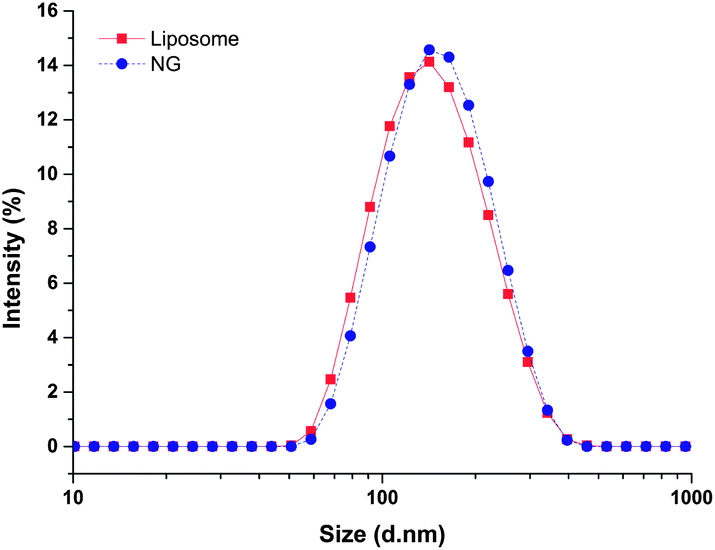 | ||
| Fig. 2 Dynamic light scattering analysis of the hydrodynamic size of lipid-coated nanogels and liposomes in water at room temperature. | ||
The morphology of the lipid-coated nanogels dried from aqueous solution was confirmed via transmission electron microscopy (TEM); representative data from these experiments are shown in Fig. 3. As illustrated in the images, the lipid-coated nanogels showed a reasonably well-defined spherical morphology. The average diameter of the nanogels (calculated from analysis via Image J of over 100 nanoparticles on the TEM images) is 110 ± 35 nm, which is in good agreement with results obtained from the DLS measurements presented in Fig. 2. The relatively smaller size observed from the TEM images compared with DLS results is likely due to the dehydration of the nanogels during the TEM sample preparation, which has been commonly observed.30,31,79
 | ||
| Fig. 3 TEM images of the lipid-coated nanogels dried from aqueous solution. | ||
The stability of these nanogels under physiologically relevant buffer conditions was evaluated via DLS at different temperatures. The data shown in Fig. S5† indicate that changes in hydrodynamic size of the nanogels upon temperature changes are almost negligible between 25 °C to 50 °C, suggesting their stability against aggregation at physiological temperatures. The nanogel solutions were also stable and showed no significant aggregation for several months when stored at 4 °C.
Surface functionalization of lipid-coated nanogels
Owing to their high surface area-to-volume ratio, the therapeutic effect of nanoparticles can be enhanced by surface modification with ligands that allow targeting of specific tissues and/or fluorescence imaging.7,80 To examine the potential suitability of these lipid-coated nanogels for such applications, carboxylic lipid-coated nanogels (COOH-NGs) composed of PEG based nanogels and a mixture of egg-PC and DSPE-PEG2000-COOH in a 4/1 ratio (w/w) were formulated. Poly(ethylene glycol) (PEG) on nanogel surfaces has been demonstrated to prolong circulation and improve evasion of immune clearance.81 A reactive fluorescent dye, 1-aminomethylpyrene, was then introduced onto the surface of the COOH-NGs via an EDC–sulfo-NHS coupling reaction to yield dye-conjugated nanogels (dye-conjugated NGs) (Scheme 4). A control experiment was performed on COOH-NGs under identical conditions and procedures, without the coupling reagents, to demonstrate that unconjugated pyrene can be removed under such conditions. The chemical conjugation of pyrene was confirmed by fluorescence spectroscopy; data from these experiments are presented in Fig. 4. As illustrated in the data, the COOH-NG control showed no obvious fluorescence emission from 425 nm to 525 nm, while the solution of dye-conjugated nanogels displayed the characteristic excimer emissions of pyrene molecules at approximately 450 nm and 490 nm.82–84 The fluorescence spectra of various nanogels were normalized to the o-nitrosobenzaldehyde peak at 360 nm (Fig. S6†), at which both the COOH-NGs and dye-conjugated NGs revealed strong fluorescence emission due to the o-nitrosobenzaldehyde residues encapsulated in the nanogel networks; the concentration of the o-nitrosobenzaldehyde is expected to be similar in the various nanogels.69 The emission from the dye-conjugated NGs closely matches the spectrum of pyrene dissolved in DMSO, suggesting that the chromophore remains intact after conjugation. These results illustrate that pyrene has been covalently conjugated to the surface of nanogels via EDC–sulfo-NHS coupling; the fact that no significant signal was observed in the same region from the COOH-NG control demonstrates that unbound pyrene molecules were removed under these experimental conditions.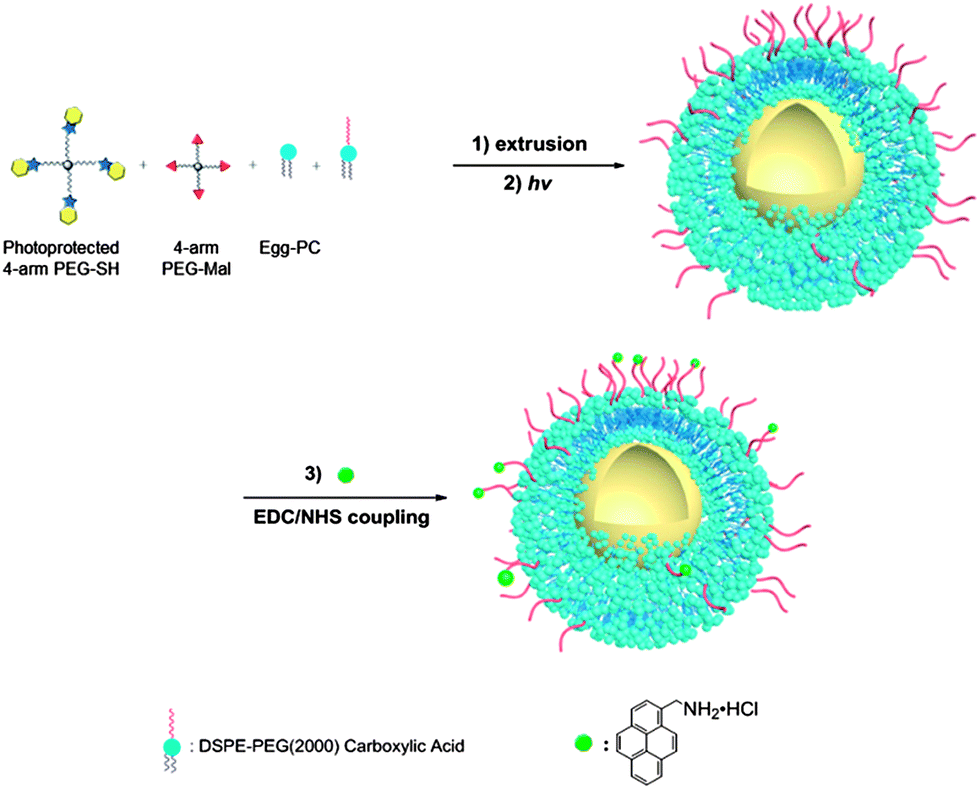 | ||
| Scheme 4 Schematic representation of the synthesis of carboxylic acid-functionalized, lipid-coated nanogels and the surface conjugation of a reactive fluorescent dye. (1) DI water, polycarbonate membrane; (2) 2 h, 365 nm, 10 mW cm−2; (3) EDC–sulfo-NHS solution, 1-aminomethylpyrene (dissolved in DMSO), pH = 7.2, 4 h, R.T. | ||
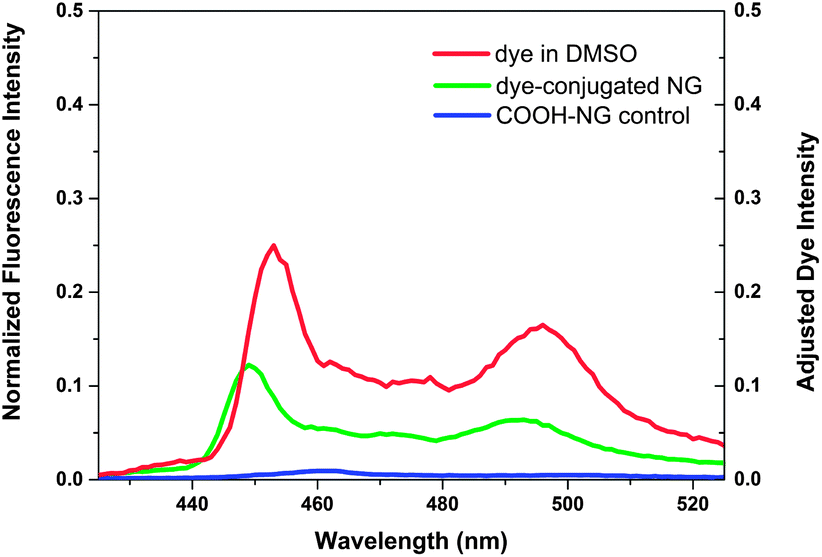 | ||
| Fig. 4 Fluorescence spectra of carboxylic acid-functionalized, lipid-coated nanogels and dye-conjugated nanogels in aqueous solution; λexc = 333 nm, 20 °C; spectra of nanogels were normalized to the o-nitrosobenzaldehyde peak at 360 nm, which should be of similar intensity for the various nanogels. | ||
The hydrodynamic diameter and surface charge of the COOH-NGs and dye-conjugated NGs in aqueous solution were also measured by dynamic light scattering (DLS) and electrophoretic light scattering, respectively, as shown in Fig. 5. The COOH-NGs have an average diameter of 156 nm with a PDI of 0.17, which closely matches that of the COOH-functionalized liposome (141 nm, PDI: 0.12), as expected. The size analysis of the dye-conjugated NGs revealed that they have an average diameter of 181 nm with a PDI of 0.23, illustrating the lack of significant change in hydrodynamic diameter after conjugation of the dye. The slight increase in size could be explained by slight swelling of the nanogels due to the retention of a small amount of DMSO from the reaction (Fig. 5a).31 As shown in Fig. 5b, the COOH-NGs exhibit a slightly greater negative surface charge (−16.3 mV) than the NGs, as expected owing to the presence of the carboxylic acid group. The negative zeta potential of dye-conjugated NGs (−15.8 mV) became only slightly less negative after the conjugation of pyrene, which is likely due to the relatively low coupling efficiency of EDC-mediated reactions at pH = 7.2.85
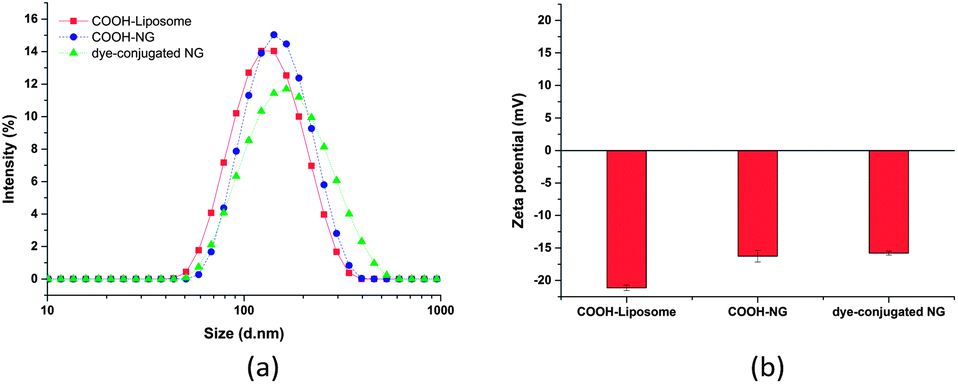 | ||
| Fig. 5 Dynamic light scattering and zeta potential analysis of functional lipid-coated nanogels. Size distribution (a) and surface charge density (b) of carboxylic acid-functionalized liposomes, carboxylic acid-functionalized lipid-coated nanogels and dye-conjugated nanogels in water at room temperature. | ||
The surface modification was also confirmed via fluorescence microscopy of the pyrene-modified NGs; a representative image from confocal laser scanning microscopy is shown in Fig. 6. As shown in the figure, the dye-conjugated nanogels appear as individual green dots on the dark background, while no green particles were observed in the fluorescence microscopy image of the COOH-NG control (Fig. S7†). Work recently reported by Singh et al.32 also confirmed the conjugation of carboxyflourescein-labeled model peptide to nanogels and the removal of unbound peptide via similar methods. Consistent with this report, these confocal microscopy data are consistent with the spectroscopy data and confirm the successful conjugation of pyrene. These results also suggest that the nanogels likely have the capacity to be functionalized with a broad range of biomolecules to enhance therapeutic efficacy.
 | ||
| Fig. 6 Fluorescence microscopy image of dye-conjugated nanogels in aqueous solution, characterized by confocal laser scanning microscopy (CLSM). | ||
In contrast to traditional radical-chain-growth photo-polymerization of polymers modified with acrylate or methacrylate moieties, formation of hydrogels via the step-growth mechanism of Michael-type addition may afford networks of greater homogeneity and improved mechanical integrity.86 Also, compared to step-growth thiol–ene chemistry74 and the recently developed photocaged amine-catalyzed thiol-Michael addition,56 the photolabile-protection strategy of thiol groups permits triggering of subsequent crosslinking reactions without the addition of any initiators or catalysts, and might thus positively affect the integrity and bioactivity of encapsulated therapeutic agents. In addition to the o-nitrobenzyl moiety employed here, similar photolabile-protection of thiols or amines could be achieved by coumarin and p-methoxyphenacyl moieties.66,67,71 Therefore, this photo-triggered Michael-type addition crosslinking strategy provides an initiator/catalyst-free photo-crosslinking method for polymer network formation with the associated advantages of homogeneity and spatiotemporal control, potentially diversifying the uses for the photo-mediated click reaction toolbox.
Compared with other existing nanogel systems, the lipid-coated nanogels synthesized from photo-triggered Michael-type addition in this report provide mild reaction conditions and control of the network development, offering new strategies in the design of nanogel materials. The surface charge and functionality introduced by the lipid coating may increase circulation stability and offer versatility of the nanogels in multiple applications, making them promising candidates as nanoparticle therapeutics.
As stimuli-triggered drug delivery systems are becoming more and more attractive, several chemically labile linkages have been employed in the synthesis of nanogels to allow on-demand or targeted release of therapeutic molecules.30–32,75,79 In particular, our group has exploited the reversibility of Michael-type reactions in the presence of glutathione (GSH), a thiol-containing tripeptide localized in cellular compartments or tumor microenvironments.87 Hydrogels prepared with arylthiol-modified PEGs based on thiol–maleimide Michael-type addition showed significantly more rapid degradation under conditions similar to those in intracellular compartments and tumor microenvironments (10 mM GSH) than under reducing conditions in blood circulation (10 μM GSH).72 Therefore, the incorporation of these GSH-responsive arylthiol–maleimide linkages in nanogel synthesis suggests potential opportunities in the development of nanoparticles for intracellular or targeted delivery of therapeutics with increased stability in blood circulation. Considering the hydrophilic nature of these nanogels, hydrophilic drugs and proteins could be incorporated into these systems via physical encapsulation or chemical conjugation. Our ongoing studies have also preliminarily shown that these nanogels have higher drug encapsulation efficiency compared to liposome carriers due to their crosslinked network structure. Investigations of such nanogel systems with stimuli responsiveness are underway and will be the subject of future reports.
Conclusions
Novel, well-defined lipid-coated polymer nanogels were successfully synthesized via photo-triggered Michael-type addition using liposome templates. The nanotemplate method offers facile control of the size of the nanogels and mild synthetic conditions. The photo-triggered crosslinking and temporal control achieved by these chemical strategies, confirmed by oscillatory rheology experiments, permit the synthesis of these nanogel templates and suggest opportunities for expanding the tool-box for photo-triggered Michael-type additions and patterning bulk gels via these methods. Through surface modification with functional ligands such as targeting moieties and cell penetration peptides, these nanogels provide a promising platform for nanoparticle therapeutics.Acknowledgements
This work was supported by the University of Delaware. The contents of the manuscript are the sole responsibility of the authors and do not necessarily reflect the official views of the University of Delaware. The authors acknowledge Dr Linqing Li for assistance with confocal imaging, Tianzhi Luo for assistance with TEM imaging and Yingchao Chen for discussion of schematic designs.Notes and references
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS PubMed.
- J. J. Shi, A. R. Votruba, O. C. Farokhzad and R. Langer, Nano Lett., 2010, 10, 3223–3230 CrossRef CAS PubMed.
- K. Riehemann, S. W. Schneider, T. A. Luger, B. Godin, M. Ferrari and H. Fuchs, Angew. Chem., 2009, 48, 872–897 CAS.
- J. J. Shi, Z. Y. Xiao, N. Kamaly and O. C. Farokhzad, Acc. Chem. Res., 2011, 44, 1123–1134 CrossRef CAS PubMed.
- T. Dvir, B. P. Timko, D. S. Kohane and R. Langer, Nat. Nanotechnol., 2011, 6, 13–22 CrossRef CAS PubMed.
- T. Lammers, W. E. Hennink and G. Storm, Br. J. Cancer, 2008, 99, 392–397 CrossRef CAS PubMed.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- J. H. Gao, H. W. Gu and B. Xu, Acc. Chem. Res., 2009, 42, 1097–1107 CrossRef CAS PubMed.
- C. J. Murphy, A. M. Gole, J. W. Stone, P. N. Sisco, A. M. Alkilany, E. C. Goldsmith and S. C. Baxter, Acc. Chem. Res., 2008, 41, 1721–1730 CrossRef CAS PubMed.
- R. Gref, Y. Minamitake, M. T. Peracchia, V. Trubetskoy, V. Torchilin and R. Langer, Science, 1994, 263, 1600–1603 CAS.
- R. Langer, Nature, 1998, 392, 5–10 CAS.
- W. W. Gao, J. M. Chan and O. C. Farokhzad, Mol. Pharmaceutics, 2010, 7, 1913–1920 CrossRef CAS PubMed.
- B. Yan, J.-C. Boyer, N. R. Branda and Y. Zhao, J. Am. Chem. Soc., 2011, 133, 19714–19717 CrossRef CAS PubMed.
- R. Tong, H. D. Hemmati, R. Langer and D. S. Kohane, J. Am. Chem. Soc., 2012, 134, 8848–8855 CrossRef CAS PubMed.
- H. Wei, X.-Z. Zhang, Y. Zhou, S.-X. Cheng and R.-X. Zhuo, Biomaterials, 2006, 27, 2028–2034 CrossRef CAS PubMed.
- M. Nakayama, T. Okano, T. Miyazaki, F. Kohori, K. Sakai and M. Yokoyama, J. Controlled Release, 2006, 115, 46–56 CrossRef CAS PubMed.
- F. de Cogan, A. Booth, J. E. Gough and S. J. Webb, Soft Matter, 2013, 9, 2245–2253 RSC.
- Z.-G. Gao, H. D. Fain and N. Rapoport, J. Controlled Release, 2005, 102, 203–222 CrossRef CAS PubMed.
- B. P. Timko, T. Dvir and D. S. Kohane, Adv. Mater., 2010, 22, 4925–4943 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS.
- J. D. Byrne, T. Betancourt and L. Brannon-Peppas, Adv. Drug Delivery Rev., 2008, 60, 1615–1626 CrossRef CAS PubMed.
- Y. H. Bae and H. Yin, J. Controlled Release, 2008, 131, 2–4 CrossRef CAS PubMed.
- S. Kim, Y. Z. Shi, J. Y. Kim, K. Park and J. X. Cheng, Expert Opin. Drug Delivery, 2010, 7, 49–62 CrossRef CAS PubMed.
- S. Jiwpanich, J. H. Ryu, S. Bickerton and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 10683–10685 CrossRef CAS PubMed.
- J.-H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227–17235 CrossRef CAS PubMed.
- D. A. Heller, Y. Levi, J. M. Pelet, J. C. Doloff, J. Wallas, G. W. Pratt, S. Jiang, G. Sahay, A. Schroeder, J. E. Schroeder, Y. Chyan, C. Zurenko, W. Querbes, M. Manzano, D. S. Kohane, R. Langer and D. G. Anderson, Adv. Mater., 2013, 25, 1449–1454 CrossRef CAS PubMed.
- Y. Chen, P. A. Wilbon, J. H. Zhou, M. Nagarkatti, C. P. Wang, F. X. Chu and C. B. Tang, Chem. Commun., 2013, 49, 297–299 RSC.
- M. A. Azagarsamy, D. L. Alge, S. J. Radhakrishnan, M. W. Tibbitt and K. S. Anseth, Biomacromolecules, 2012, 13, 2219–2224 CrossRef CAS PubMed.
- M. Q. Li, Z. H. Tang, H. Sun, J. X. Ding, W. T. Song and X. S. Chen, Polym. Chem., 2013, 4, 1199–1207 RSC.
- S. Singh, F. Topuz, K. Hahn, K. Albrecht and J. Groll, Angew. Chem., Int. Ed., 2013, 52, 3000–3003 CrossRef CAS PubMed.
- M. H. Smith and L. A. Lyon, Acc. Chem. Res., 2012, 45, 985–993 CrossRef CAS PubMed.
- K. A. Beningo and Y. L. Wang, J. Cell Sci., 2002, 115, 849–856 CAS.
- X. Banquy, F. Suarez, A. Argaw, J.-M. Rabanel, P. Grutter, J.-F. Bouchard, P. Hildgen and S. Giasson, Soft Matter, 2009, 5, 3984–3991 RSC.
- T. J. Merkel, S. W. Jones, K. P. Herlihy, F. R. Kersey, A. R. Shields, M. Napier, J. C. Luft, H. L. Wu, W. C. Zamboni, A. Z. Wang, J. E. Bear and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 586–591 CrossRef CAS PubMed.
- M. M. Yallapu, M. Jaggi and S. C. Chauhan, Drug Discovery Today, 2011, 16, 457–463 CrossRef CAS PubMed.
- S. Nayak and L. A. Lyon, Angew. Chem., Int. Ed., 2005, 44, 7686–7708 CrossRef CAS PubMed.
- D. Klinger and K. Landfester, Soft Matter, 2011, 7, 1426–1440 RSC.
- K. Raemdonck, J. Demeester and S. De Smedt, Soft Matter, 2009, 5, 707–715 RSC.
- Y. Sasaki and K. Akiyoshi, Chem. Rec., 2010, 10, 366–376 CAS.
- S. Y. An, M. P. N. Bui, Y. J. Nam, K. N. Han, C. A. Li, J. Choo, E. K. Lee, S. Katoh, Y. Kumada and G. H. Seong, J. Colloid Interface Sci., 2009, 331, 98–103 CrossRef CAS PubMed.
- S. Kazakov, M. Kaholek, I. Teraoka and K. Levon, Macromolecules, 2002, 35, 1911–1920 CrossRef CAS.
- S. Kazakov, M. Kaholek, D. Kudasheva, I. Teraoka, M. K. Cowman and K. Levon, Langmuir, 2003, 19, 8086–8093 CrossRef CAS.
- T. G. Van Thienen, B. Lucas, F. M. Flesch, C. F. van Nostrum, J. Demeester and S. C. De Smedt, Macromolecules, 2005, 38, 8503–8511 CrossRef CAS.
- J. P. Schillemans, F. M. Flesch, W. E. Hennink and C. F. van Nostrum, Macromolecules, 2006, 39, 5885–5890 CrossRef CAS.
- T. G. Van Thienen, K. Raemdonck, J. Demeester and S. C. De Smedt, Langmuir, 2007, 23, 9794–9801 CrossRef CAS PubMed.
- J. Park, S. H. Wrzesinski, E. Stern, M. Look, J. Criscione, R. Ragheb, S. M. Jay, S. L. Demento, A. Agawu, P. L. Limon, A. F. Ferrandino, D. Gonzalez, A. Habermann, R. A. Flavell and T. M. Fahmy, Nat. Mater., 2012, 11, 895–905 CrossRef CAS PubMed.
- J. S. Hong, W. N. Vreeland, S. H. DePaoli Lacerda, L. E. Locascio, M. Gaitan and S. R. Raghavan, Langmuir, 2008, 24, 4092–4096 CrossRef CAS PubMed.
- T. Nie, A. Baldwin, N. Yamaguchi and K. L. Kiick, J. Controlled Release, 2007, 122, 287–296 CrossRef CAS PubMed.
- T. Nie, R. E. Akins and K. L. Kiick, Acta Biomater., 2009, 5, 865–875 CrossRef CAS PubMed.
- E. A. Scott, M. D. Nichols, L. H. Cordova, B. J. George, Y. S. Jun and D. L. Elbert, Biomaterials, 2008, 29, 4481–4493 CrossRef CAS PubMed.
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 448–477 CrossRef CAS PubMed.
- H. Zhao, E. S. Sterner, E. B. Coughlin and P. Theato, Macromolecules, 2012, 45, 1723–1736 CrossRef CAS.
- B. J. Adzima, Y. H. Tao, C. J. Kloxin, C. A. DeForest, K. S. Anseth and C. N. Bowman, Nat. Chem., 2011, 3, 256–259 CrossRef CAS PubMed.
- W. X. Xi, M. Krieger, C. J. Kloxin and C. N. Bowman, Chem. Commun., 2013, 49, 4504–4506 RSC.
- S. W. Thomas, Macromol. Chem. Phys., 2012, 213, 2443–2449 CrossRef CAS.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- A. M. Kloxin, M. W. Tibbitt and K. S. Anseth, Nat. Protoc., 2010, 5, 1867–1887 CrossRef CAS PubMed.
- A. M. Kloxin, M. W. Tibbitt, A. M. Kasko, J. A. Fairbairn and K. S. Anseth, Adv. Mater., 2010, 22, 61–66 CrossRef CAS PubMed.
- J. Q. Jiang, X. Tong, D. Morris and Y. Zhao, Macromolecules, 2006, 39, 4633–4640 CrossRef CAS.
- G. Liu and C. M. Dong, Biomacromolecules, 2012, 13, 1573–1583 CrossRef CAS PubMed.
- X. Jiang, C. A. Lavender, J. W. Woodcock and B. Zhao, Macromolecules, 2008, 41, 2632–2643 CrossRef CAS.
- Y. Luo and M. S. Shoichet, Nat. Mater., 2004, 3, 249–253 CrossRef CAS PubMed.
- Y. Luo and M. S. Shoichet, Biomacromolecules, 2004, 5, 2315–2323 CrossRef CAS PubMed.
- J. H. Wosnick and M. S. Shoichet, Chem. Mater., 2008, 20, 55–60 CrossRef CAS.
- R. G. Wylie and M. S. Shoichet, J. Mater. Chem., 2008, 18, 2716–2721 RSC.
- J. Cui, M. Wang, Y. Zheng, G. M. Rodríguez Muñiz and A. del Campo, Biomacromolecules, 2013, 14, 1251–1256 CrossRef CAS PubMed.
- G. Delaittre, T. Pauloehrl, M. Bastmeyer and C. Barner-Kowollik, Macromolecules, 2012, 45, 1792–1802 CrossRef CAS.
- T. Pauloehrl, G. Delaittre, M. Bastmeyer and C. Barner-Kowollik, Polym. Chem., 2012, 3, 1740–1749 RSC.
- R. M. Hensarling, E. A. Hoff, A. P. LeBlanc, W. Guo, S. B. Rahane and D. L. Patton, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1079–1090 CrossRef CAS.
- A. D. Baldwin and K. L. Kiick, Polym. Chem., 2013, 4, 133–143 RSC.
- L.-T. Ng, S. Jönsson, S. Swami and K. Lindgren, Polym. Int., 2002, 51, 1398–1403 CrossRef CAS.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS.
- W. Chen, M. Zheng, F. H. Meng, R. Cheng, C. Deng, F. J. Jan and Z. Y. Zhong, Biomacromolecules, 2013, 14, 1214–1222 CrossRef CAS PubMed.
- X. Y. Wang, T. Ishida, M. Ichihara and H. Kiwada, J. Controlled Release, 2005, 104, 91–102 CrossRef CAS PubMed.
- S. Tatur, M. Maccarini, R. Barker, A. Nelson and G. Fragneto, Langmuir, 2013, 29, 6606–6614 CrossRef CAS PubMed.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657–3666 CrossRef CAS PubMed.
- J. X. Ding, F. H. Shi, C. S. Xiao, L. Lin, L. Chen, C. L. He, X. L. Zhuang and X. S. Chen, Polym. Chem., 2011, 2, 2857–2864 RSC.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS PubMed.
- C. Huang, K. G. Neoh, L. Wang, E.-T. Kang and B. Shuter, J. Mater. Chem., 2010, 20, 8512–8520 RSC.
- D. A. Ossipov, X. Yang, O. Varghese, S. Kootala and J. Hilborn, Chem. Commun., 2010, 46, 8368–8370 RSC.
- M.-C. Li, R.-M. Ho and Y.-D. Lee, J. Mater. Chem., 2011, 21, 2451–2454 RSC.
- J.-S. Yang, C.-S. Lin and C.-Y. Hwang, Org. Lett., 2001, 3, 889–892 CrossRef CAS PubMed.
- U. Freudenberg, A. Hermann, P. B. Welzel, K. Stirl, S. C. Schwarz, M. Grimmer, A. Zieris, W. Panyanuwat, S. Zschoche, D. Meinhold, A. Storch and C. Werner, Biomaterials, 2009, 30, 5049–5060 CrossRef CAS PubMed.
- M. W. Tibbitt, A. M. Kloxin, L. A. Sawicki and K. S. Anseth, Macromolecules, 2013, 46, 2785–2792 CrossRef CAS.
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectrum of four-arm nitrobenzyl protected PEG-SH; photorheology of PEG-SH/Mal hydrogels and PEG-Mal control; storage moduli of PEG-SH/Mal hydrogels at different concentrations; DLS of lipid-coated nanogels in PBS at different temperatures; full fluorescence spectra of various nanogels; fluorescence microscopy image of the COOH-NG control. See DOI: 10.1039/c3py01269g |
| This journal is © The Royal Society of Chemistry 2014 |