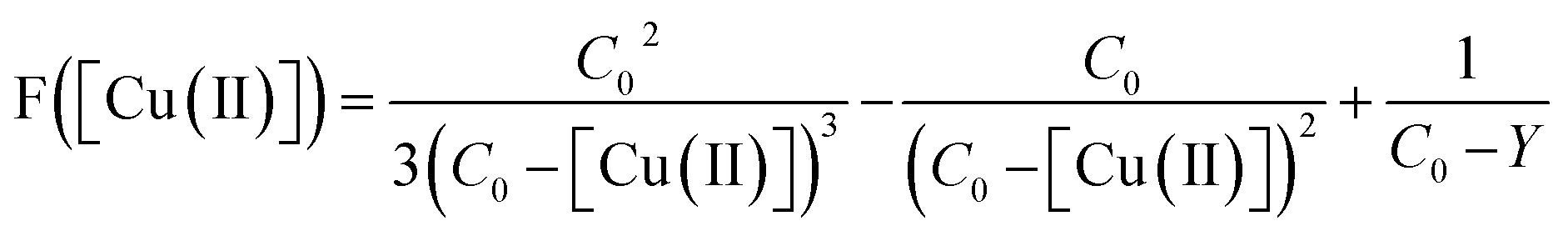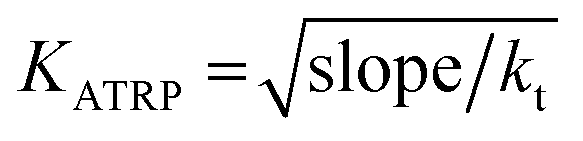DOI:
10.1039/C3PY01094E
(Paper)
Polym. Chem., 2014,
5, 524-534
The polymerisation of oligo(ethylene glycol methyl ether) methacrylate from a multifunctional poly(ethylene imine) derived amide: a stabiliser for the synthesis and dispersion of magnetite nanoparticles†
Received
13th August 2013
, Accepted 8th September 2013
First published on 10th September 2013
Abstract
A facile synthetic route to poly(ethylene imine)-graft-poly(oligo(ethylene glycol methyl ether)) (PEI-graft-POEGMA) functionalised superparamagnetic magnetite nanoparticles is described. The polymerisation of OEGMA from a model molecular amide demonstrated the feasibility of POEGMA synthesis under mild ATRP conditions (20 °C in ethanol) albeit with low initiator efficiencies. DFT studies suggest that the amide functionality is intrinsically of lower activity than ester functional monomers and initiators for atom transfer polymerisation (ATRP) as a consequence of higher bond dissociation energies and bond dissociation free energies (BDFE). However these studies further highlighted that use of an appropriate solvent could reduce the free energy of dissociation thereby reducing the relative difference in BDFE between the ester and amide groups. A commercial branched PEI sample was functionalised by reaction with 2-bromo-2-methylpropanoyl bromide giving an amide macroinitiator suitable for the atom transfer radical polymerisation (ATRP) of oligo(ethylene glycol methyl ether) methacrylate. The resulting PEI-graft-POEGMA copolymers were characterised by SEC, FT-IR and 1H and 13C NMR spectroscopy. PEI-graft-POEGMA coated magnetite nanoparticles were synthesised by a basic aqueous co-precipitation method and were characterised by transmission electron microscopy, thermogravimetric analysis and vibrating sample magnetometry and dynamic light scattering. These copolymer coated magnetite nanoparticles were demonstrated to be effectively stabilised in an aqueous medium. Overall the particle sizes and magnetic and physical properties of the coated samples were similar to those of uncoated samples.
Introduction
Magnetic iron oxide particles having sizes from few nanometers to micrometer scale are widely used in a number of biomedical applications such as drug delivery,1,2 magnetic resonance imaging3–5 and separation techniques.6 Of particular interest is magnetic drug targeting where magnetic particles and polymer coatings containing drugs are directed to tumour sites through the use of a directed external magnetic field gradient. By making the polymer coating thermoresponsive drug release can be triggered by local changes in temperature such as those induced in magnetite nanoparticles by an applied magnetic field. Thus thermoresponsive polymer–magnetic nanoparticles offer potential for simultaneous magnetically directed targeting, magnetically induced hyperthermic treatment and magnetically induced drug delivery.1,7,8
Magnetite (Fe3O4), which contains both Fe+2 and Fe+3 ions is the iron oxide type most used as the basis for magnetic nanoparticles due to its biocompatibility and excellent magnetic properties. If the grain size of magnetite particles is less than about 12 nm, then individual particles can have single magnetic domains leading to superparamagnetic behavior but suitable stabilization of the particles is needed in order to prevent aggregation, while keeping the superparamagnetic properties unchanged. Many synthesis methods have been reported on the aqueous synthesis of superparamagnetic magnetite nanoparticles of which chemical co-precipitation is the most frequently used owing to its simplicity. In this method Fe+2 and Fe+3 ions are mixed in a stoichiometric ratio of 1 to 2 in the presence of a base at high pH under inert conditions. The latter is to prevent oxidation of particles to maghemite which is also a superparamagnetic iron oxide but has a lower saturation magnetization value. The bare magnetite nanoparticles obtained are generally stabilized with surfactants or polymers by surface functionalization, preventing sedimentation and/or aggregation in solution.2 It has previously been shown that poly(ethylene imine) (PEI) can adsorb onto magnetite nanoparticles as a primary layer and a secondary layer of poly(ethylene oxide)-co-poly(glutamic acid) can give particles long term stability in physiological salt solution.9 Poly(ethylene glycol) (PEG) or poly(ethylene oxide) (PEO) has also been used by several groups to obtain stability in aqueous media.10–12 PEO possesses the advantage of being non-toxic and biocompatible and is widely utilized in biomedical applications. In contrast PEI has been investigated as an gene therapy carrier due to the high number of ammonium groups that are available to electrostatically interact with phosphate groups in DNA.13–15 Such polymeric ammonium groups are ideal cationic stabilisers for the magnetite water interface however PEI is relatively cytotoxic. In this study a PEI-graft-POEGMA polymer was designed as a stabilizer for magnetite to take advantage of the adsorption ability of the PEI backbone with the magnetite surface and the steric stabilisation from the comb-like of POEGMA chains.16 Copolymers formed from PEI and stimulus responsive polymers have been used to create systems which are ideal for controlled release.17,18 The PEI-graft-POEGMA copolymers presented in this research were specifically designed to take advantage of a number of design principles for nanoparticle stabilisation and biomedical application. The PEI backbone (of which a significant variety is commercially available) fulfils three functions, (1) to provide a number of amine groups that can directly interact with the iron oxide surface, (2) to provide amine groups amenable to functionalisation via simple and well-developed amide synthesis techniques, (3) provide the ability to fine tune stabiliser properties through the use of PEI of varying molecular architectures (branched and linear) and various molecular weights. For this initial study a low molecular weight branched PEI sample was chosen. Recent research reported on improved nanoparticle stability through the use of dendritic stabilisers relative to linear analogues.19 Whilst dendritic architectures are attractive for numerous applications the synthesis of such compounds is multi-step, time-consuming and costly. In contrast branched and hyperbranched architectures offer many of the advantages at a fraction of the cost and synthetic effort. The POEGMA polymer component is a bottle-brush type polymer with a polymethacrylic backbone with side chains of short chains of ethylene oxide. POEGMA has shown significant promise for bioconjugate systems due to its water solubility, protein and cell resistance and thermoresponsive properties.20–23 Thermoresponsive polymer coatings on magnetic nanoparticles to date have typically been introduced after particle synthesis by graft polymerisation or by seeded precipitation polymerisation and the most commonly employed thermoresponsive polymers to date has been poly(n-iso-propylacrylamide) and POEGMA.7,18,21,22,24–34 POEGMA is a poly methacrylate with graft oligo(ethylene oxide) side chains that has many of the properties of PEO whilst remaining amorphous and whose LCST can be fine tuned by varying side-chain length, end-groups and co-polymerising with other monomers.
PEI-graft-POEGMA based materials which can have their thermoresponsive behaviour tuned by the copolymerisation of 2-(2-methoxyethoxy)ethyl methacrylate (MEO2MA) and OEGMA have previously been synthesised by polymerising the copolymer first, then coupling it to the PEI core.35 Whilst this technique produced desirable materials (for gene delivery), it relied on a multiple stage synthesis using harsh conditions. Atom transfer radical polymerisation (ATRP) has been shown to be a robust and versatile technique for the creation of a wide range of polymers in relatively mild conditions.36–41 In this paper we will demonstrate that (i) OEGMA can be polymerised using a molecular amide initiator, (ii) the low initiator efficiency of amides in ATRP is a consequence of higher bond dissociation energies of the C–Br bond in amides, (iii) PEI can be modified to give a macro-initiator for ATRP by the reaction of amines within the PEI with an ATRP initiating moiety and (iv) the polymerisation of OEGMA from this modified PEI initiator is possible. This approach allows for the immediate and direct incorporation of the thermoresponsive functionality into the actual stabiliser structure with no need for post-functionalisation of the particles. Furthermore we will demonstrate the application of the resultant copolymer as a stabiliser for the synthesis of aqueous dispersed magnetite nanoparticles to obtain stable magnetic nano-materials in aqueous suspension.
Experimental
Materials
Ferric chloride (FeCl3) and ferrous sulfate heptahydrate (FeSO4·7H2O) were purchased from Riedel-deHaen and sodium hydroxide (NaOH) was purchased from J.T. Baker. All were analytical grade and used without further purification. Oligo(ethylene glycol methyl ether) methacrylate (Mn ≈ 360, Sigma-Aldrich), triethylamine (≥99%, Sigma-Aldrich), copper(I) bromide (98%, Sigma-Aldrich), 2-bromo-2-methylpropanoyl bromide (98%, Sigma-Aldrich), sodium bicarbonate (analytical reagent grade, Fisher Scientific) aluminium oxide (activated, neutral, for column chromatography 50–200 μm, Acros Organics) magnesium sulfate (97%, anhydrous, Acros Organics) methanol (analytical reagent grade, Fisher Scientific), tetrahydrofuran (analytical reagent grade, Fisher Scientific) and ethanol (analytical reagent, Fisher Scientific) were purchased and used without further purification. Dichloromethane (analytical reagent grade) was purchased from Fisher Scientific and immediately before use was dried and distilled over calcium hydride.
Characterization
1H and 13C NMR spectra of the polymers were recorded using a JEOL ECS spectrometer (300 MHz) at 25 °C in solutions of deuterated chloroform (CDCl3), d4-methanol or d8-ethanol. Molecular weight parameters were recorded by size exclusion chromatography (SEC) of THF solutions using two 5 μm mixed C PLgel columns at 40 °C and a Shodex RI-101 refractive index detector. Both SEC systems were calibrated using poly(methyl methacrylate) standards.
Particle sizes, distributions and morphologies of the nanoparticles were analyzed by FEI Tecnai G2 Sphera transmission electron microscope (TEM) operating at 200 kV by drying 30 μl of samples on carbon coated 200 mesh copper grids. Phase identification of synthesized nanoparticles was obtained by Rigaku X-ray diffractometer (XRD) by scanning 2-theta range of 20° to 70° at room temperature with 0.02 theta increments per 10 s. Magnetic properties of both bare and PEI-b-POEGMA coated particles were analyzed by vibrating sample magnetometer at dry state and room temperature.
DLS measurements were performed on the PEI-graft-POEGMA stabilised nanoparticle dispersions using a Malvern Zetasizer Nano ZS with Dispersion Technology Software (DTS) version 5.0 software. All measurements of 10 scans were repeated three times and the average at each temperature reported.
Syntheses
Poly(ethylene imine)-graft-(2-bromo-2-methyl)propanamide (macro-initiator).
PEI Mw 600 (5 g, 0.0083 mol) and triethylamine (1.5 ml, 0.011 mol) were dissolved in 200 ml of dichloromethane and placed into an ice bath at 0 °C and left stirring. 2-Bromo-2-methylpropanoyl bromide (15 g, 0.0653 mol), pre dissolved in 100 ml of dichloromethane, was added dropwise to this mixture. The resulting solution was left stirring at 0 °C for 3 hours and then stirring at room temperature for a further 18 hours. The solution was filtered to remove solids and then evaporated to leave a yellow viscous oil which was redissolved in 40 ml of dichloromethane. This was washed five times against a 10% saturated solution of sodium bicarbonate then left stirring over night with 3 g of activated charcoal and 5 g of anhydrous magnesium sulphate. Finally this was filtered again to remove solids before being dried under vacuum prior to NMR analysis. 1H NMR (CDCl3, 293 K, 400 MHz) 4.1 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–O–CH2, 3.8–3.3 O–CH2–CH2–O, 3.1 CH2–O–CH3, 2.0–1.7 CH2–C–CH3, 1.1–0.8 CH2–C–CH3.
O–O–CH2, 3.8–3.3 O–CH2–CH2–O, 3.1 CH2–O–CH3, 2.0–1.7 CH2–C–CH3, 1.1–0.8 CH2–C–CH3.
Poly(ethylene imine)-graft-poly(oligo(ethylene oxide) methyl ether methacrylate) (PEI-graft-POEGMA).
A typical synthesis was as follows: A Schlenk tube containing OEGMA (Mn 300, 5 g, 18.1 mmol), CuCl (0.036 g, 0.362 mmol), 4,4′-dinonyl-2,2′-bipyridine (dNBpy) (0.2959 g, 0.724 mmol) and ethanol (14 ml) was sealed and degassed with nitrogen for 45 minutes. PEI-initiator in ethanol (0.1 g ml−1) was injected via gastight syringe and then left stirring at room temperature under nitrogen for 48 hours. At timed intervals 1 ml samples were removed via syringe, exposed to air and then passed through a short alumina column to remove the catalytic system then diluted in THF for SEC and CDCl3 for NMR. The polymer was isolated by dropwise addition of the THF solution to an excess of cold, stirred hexane. The product precipitated as a green viscous liquid and was collected by centrifuge prior to drying overnight under vacuum at 35 °C before SEC and NMR analysis. 1H NMR (CDCl3, 293 K, 400 MHz) 4.1 CO–O–CH2, 3.8–3.3 O–CH2–CH2–O, 3.1 CH2–O–CH3, 2.0–1.7 CH2–C–CH3, 1.1–0.8 CH2–C–CH3.
Magnetite nanoparticles.
Typically FeCl3 (0.141 g) and FeSO4·7H2O (0.121 g) were dissolved in distilled water (40 ml) which was deaerated by bubbling with nitrogen for 30 minutes to remove any dissolved oxygen. The solution was then stirred for 15 minutes for complete mixing under nitrogen gas. A solution of PEI-graft-POEGMA (P1) (38 mg) in aqueous NaOH (0.257 g in 10 ml) was added to the iron salts solution rapidly and solution changes color from orange to black immediately. The resulting solution was then stirred at room temperature for 30 minutes. The particles were collected with a handheld magnet and centrifuged 2 times after washing with water. The final precipitate was then dried in a vacuum oven overnight at 60 °C. The magnetite nanoparticles without stabilizer were prepared in an identical procedure but without PEI-graft-POEGMA.
Modelling.
Initial molecular conformations were assessed and minimised using the semi-empirical PM6 method through the CS MOPAC interface in ChemBio3D Ultra version 12.0.2 (Cambridgesoft).42 All density functional theory (DFT) calculations were run using the GAMESS-US code version 11 (R1).43 All minimum energy confirmations and frequencies were determined at the B3-LYP/6-31+G(d) level of theory at 298.15 K.44,45 Single-point energy calculations were conducted with the BMK,46 M06-2X47 and B2GP-PLYP48 functionals using the augmented triple-ξ Dunning aug-cc-pVTZ basis set49 and unrestricted wave functions. DFT-D3 dispersion corrections were utilised in all cases;50,51 with additional parameters for the D3 corrections taken from the literature.52,53 The Cartesian coordinates of the minimised conformations and typical input parameters can be found in the electronic supplementary information (ESI). Calculations in solvents were performed using the conductor-like polarisable continuum model (C-PCM) combined with the universal solvation model (SMD) of Truhlar et al.54
Results and discussion
The polymerisation of OEGMA using an amide initiator
The difficulties associated with the use of amide initiators in ATRP are well known.55–61 Low initiator efficiencies resulting in higher than predicted molecular weight parameters and slow polymerizations with variable rates, have been the most common problems. Thus prior to utilizing a polyfunctional amide macroinitiator we investigated the activity of a secondary amide initiator, 2-bromo-2-methyl-N-propylpropanamide (PBrPA), in relation to the commonly employed EBriB ester based initiator (Scheme 2).
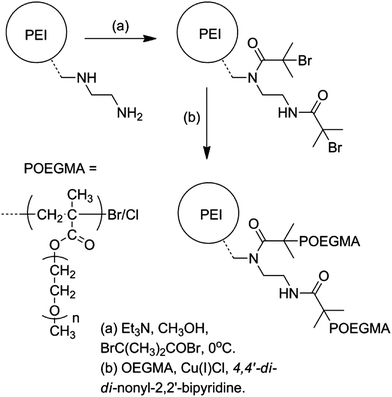 |
| | Scheme 1 Outline synthesis of PEI-graft-POEGMA. | |
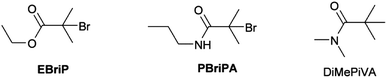 |
| | Scheme 2 Structures of initiators used in the ATRP of OEGMA. | |
2,2′-dinonyl-4,4′-dipyridine was used as the copper ligand and ethanol as the solvent with all reactions carried out at 25 °C. Representative results are given in Table 1. In agreement with previous reports of ATRP using amide ligands, the use of Cu(I)Br catalyst led to poorly controlled reactions with low conversions; replacing the Cu(I)Br with Cu(I)Cl led to relatively narrower polydispersity products but with poor initiation efficiencies. The effect of the Cu(I)Cl is to reduce the relative rate of propagation (of the methacrylate) to initiation (of the amide initiator). Increasing the total amount of solvent gave products with Mw/Mn < 1.3 and initiator efficiencies of around 0.7 to 0.8.
Table 1 Summary of the synthesis and molecular weight parameters for the ATRP of the EBriB and PBriBA initiated polymerisations of oligo(ethylene oxide methyl ether methacrylate) at 25 °C in ethanol (solvent![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :monomer 2:1) using a dinonyl-bipyridine ligand
:monomer 2:1) using a dinonyl-bipyridine ligand
| I |
[M]:[I] |
X |
t (h) |
M
n(exp)
|
M
n(th)
|
M
w/Mn |
Conv. (%) |
I
eff
|
|
Solvent:monomer = 3:1.
|
| EBriB |
50 |
Br |
24 |
11900 |
12150 |
1.31 |
81 |
1.02 |
| EBriB |
100 |
Br |
24 |
23950 |
25200 |
1.29 |
84 |
1.05 |
| PBriBA |
50 |
Br |
34 |
2750 |
4050 |
1.30 |
27 |
1.47 |
| PBriBA |
50 |
Br |
42 |
4300 |
3150 |
1.66 |
21 |
1.33 |
| EBriB |
50 |
Cl |
24 |
11100 |
11400 |
1.26 |
76 |
1.03 |
| PBriBA |
50 |
Cl |
20 |
19850 |
6000 |
1.31 |
40 |
0.30 |
| PBriBA |
50 |
Cl |
28 |
20100 |
7950 |
1.33 |
53 |
0.39 |
| PBriBA |
50 |
Cl |
36 |
18650 |
8900 |
1.18 |
59 |
0.48 |
| PBriBA |
50 |
Cl |
48 |
21750 |
11050 |
1.23 |
74 |
0.51 |
| EBriBa |
50 |
Cl |
20 |
12600 |
12000 |
1.17 |
80 |
0.95 |
| EBriBa |
50 |
Cl |
67 |
14900 |
13950 |
1.30 |
93 |
0.94 |
| PBriBAa |
50 |
Cl |
48 |
13650 |
9400 |
1.26 |
63 |
0.69 |
| PBriBAa |
50 |
Cl |
66 |
12400 |
9800 |
1.13 |
65 |
0.79 |
Calculations of bond dissociation energies and free energies
The cause of the poor initiator efficiency has previously been ascribed to rapid polymerisation of the initiator (typically a secondary amide) in comparison to the monomer (typically methacrylate monomers),56 decomposition of the initiator,59 loss of catalyst activity and/or complexation of the amide initiator with the copper/ligand catalyst system.60,62 With a view to understanding these results quantum chemical calculations were performed on some model systems to investigate the activity of secondary and tertiary amide initiators in relation to the commonly employed EBriB ester based initiator; the EBriB acting as a model initiator to compare initiation efficiencies and also as a model for the bromo-capped methacrylate polymer chain ends.
DFT calculations were carried out on methyl 2-bromo-2-methylpropanoate (MBriP), N-methyl 2-bromo-2-methylpropanamide (MBriPA), N,N-dimethyl 2-bromo-2-methylpropanamide (MBriPA2), methyl 2-chloro-2-methylpropanoate (MCliP) and N-methyl 2-chloro-2-methylpropanamide (MCliPA) (Scheme 3). Geometries were optimised using the B3LYP functional with the 6-31+Gd basis set previously employed for studies on ATRP initiators.63–66 Since the B3LYP functional is known to give very inaccurate values for thermochemical calculations and in particular free bond dissociation energies, further functionals were employed for free energy calculations.67,68 For the single point energy calculations we used the BMK and M06-2X functionals both of which have been reported to give good results in bond dissociation energy calculations46,47,68–72 and the double-hybrid functional B2G-PLYP functional which has also been shown to perform well for thermochemical calculations.48,52,73 In all cases Grimme's D3 dispersion energy correction was employed, which has been shown to improve BDE (and thermochemical) values for most functionals50,52,74 and the aug-cc-pVTZ basis set was used.49 The reference values for ΔH and ΔG for the dissociation of the C–Br bond were taken from the previous work of Coote et al.65 The results are summarised in Table 2.
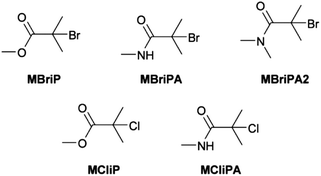 |
| | Scheme 3 Model structures used in DFT calculations. | |
Table 2 Summary of the results from the DFT calculations calculated at 298.15 K in the gas phase. All values in kJ mol−1
| Cmpd |
R–Br → R˙ + Br˙ |
|
|
B3-LYPa |
BMKb |
M06-2Xb |
B2G-PLYPb |
Litc |
|
6-31+G(d).
aug-cc-pVTZ-D3.
Calculated at the G3(MP2)-RAD level of theory at 298K in the gas phase; taken from ref. 65.
|
| MBriB |
ΔG |
185.0 |
217.8 |
206.5 |
207.7 |
221.2 |
| ΔH |
230.1 |
263.0 |
251.6 |
252.8 |
258.5 |
| MBriBA |
ΔG |
208.9 |
239.8 |
226.4 |
228.7 |
NA |
| ΔH |
251.0 |
282.0 |
268.6 |
270.8 |
NA |
| ΔGG |
|
−23.9 |
−22.0 |
−19.9 |
−21.0 |
|
| MBriBA2 |
ΔG |
195.4 |
228.3 |
216.1 |
216.2 |
NA |
| ΔH |
238.8 |
271.8 |
259.6 |
259.7 |
NA |
| ΔGG |
|
−10.4 |
−10.5 |
−9.6 |
−8.5 |
|
| MCliB |
ΔG |
225.9 |
276.0 |
265.6 |
254.2 |
278.5 |
| ΔH |
271.7 |
321.7 |
311.3 |
299.9 |
315.3 |
| MCliBA |
ΔG |
254.0 |
301.2 |
289.9 |
279.6 |
NA |
| ΔH |
296.8 |
343.9 |
332.6 |
322.3 |
NA |
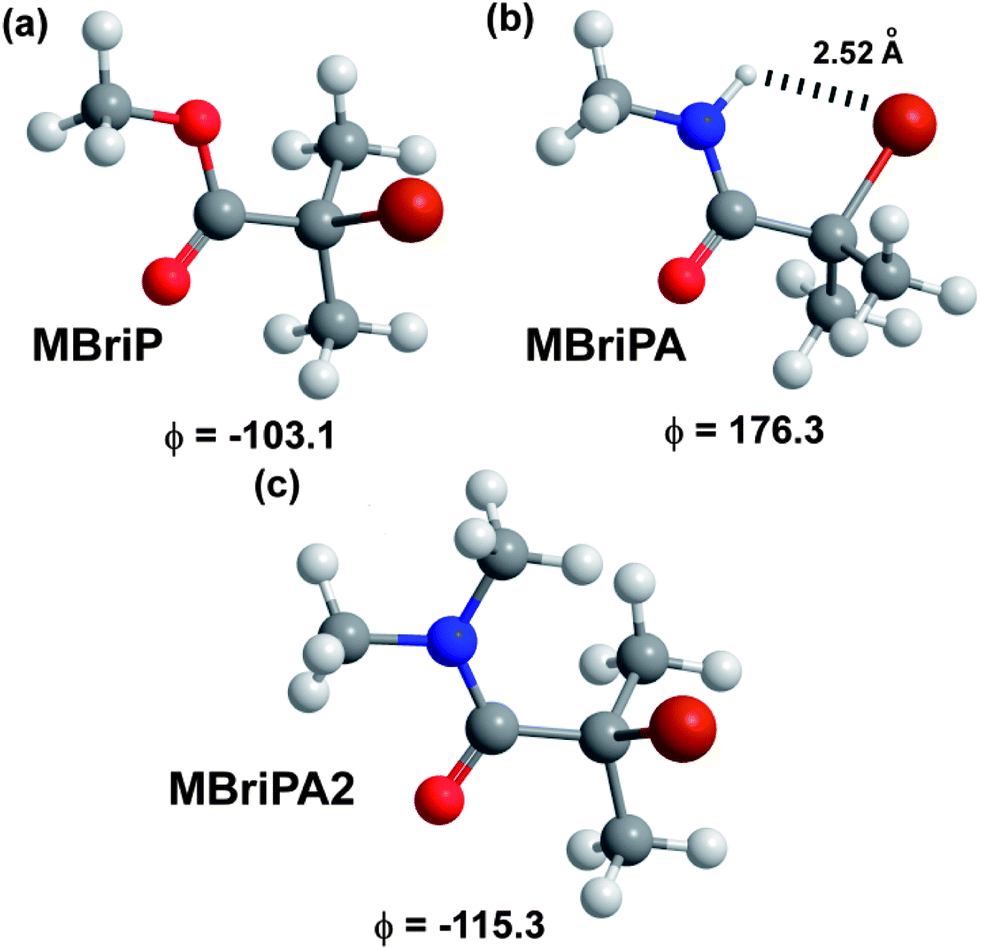
The closest ΔG and ΔH values to those in the literature were obtained for the BMK functional (UHF) confirming that this functional is the low cost method of choice for BDE calculations. Irrespective of the absolute values, of particular interest are the relative values of the bond dissociation free energies between the ester and amide initiators. Remarkably consistent values were obtained (excluding B3LYP) with an average value for the BDFE of −21.7 kJ mol−1 with a mean absolute deviation of 1.25 kJ mol−1. BDFE values for ATRP initiators have been demonstrated to be the major determinant for the equilibrium constants for activation of the initiators by the Cu catalysts. All else being equal (under identical reaction conditions with identical reagents) the relative BDFE values can be used to gauge the relative reactivity of the initiator species. In this case a −21.7 kJ mol−1 free energy difference would correspond to the ester being approximately 6335 times (KATRP(MBriPA)/KATRP(MBriP) = 0.000158) more active than the amide. In contrast MBriPA2 with two methyl groups on the amide nitrogen, gave an average −9.53 kJ mol−1 free energy difference would correspond to the ester only being approximately 47 times (KATRP(MBriPA2)/KATRP(MBriP) = 0.0214) more active than the amide. It has been previously reported that the experimental KATRP values for the secondary ATRP initiators ethylbromopropanoate and 2-bromo-N,N-diethylpropanamidewere 0.30 and 0.044 respectively giving only a ≈7 fold difference in activity.75 Thus for MBriP (and by default EBriB) the relative differences in reactivity mean that the polymerization should not proceed in a controlled manner if at all; such a low initiation activity, coupled with a methacrylate monomer, would lead to very slow polymerisation and very poor initiator efficiency. The actual lower activity of the amides is not surprising due to the amides generally having lower radical stabilisation energies than the equivalent esters.64 What is surprising is the calculated relative magnitude of difference in ATRP activity between and MBriP and between MBriPA and MBripA2. One possible reason presents itself whilst considering the minimal energy conformations of the three molecules which are illustrated in Fig. 1. The MBriP and MBriPA2 C–Br bond angles to the plane of the CO2 and CON are circa 77° and 65° respectively; for MBriPA however this angle is circa 4°. This appears to be a result of the presence of a intramolecular H–Br hydrogen bond. Such bonds have been seen for many α-Br aromatic amides.76,77 This H-bond has the apparent effect of strengthening the C–Br and increasing its BDE; entropic effects do not appear to play a role.
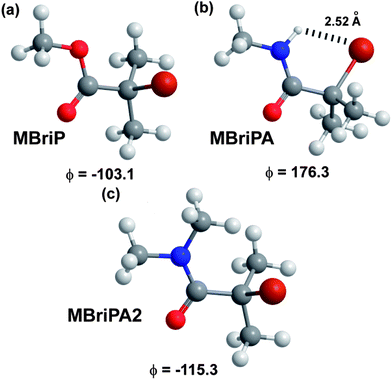 |
| | Fig. 1 Optimised minimum energy conformations (B3LYP/6-31+G(d)) with OC–C–Br dihedral angles obtained for model initiators. | |
To determine the experimental relative KATRP values for the two initiators EBriB and PBriBA used in the polymerisation of OEGMA, we followed the method described by Matyjaszewski et al. whereby a function of the Cu(II) concentration (eqn (1)) was measured against time.78,79 The increase in Cu(II) arises from the persistent radical effect and the slope of the plot of F([Cu(II)]) versus time gives a value for KATRP according to eqn (2).
| | 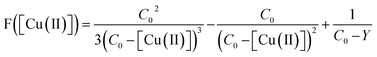 | (1) |
| | 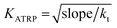 | (2) |
Thus degassed and sealed ethanol solutions of initiator:Cu(I)Cl:bipyridine (1:1:2) were prepared, the degassed initiators (EBriB or PBriBA) were introduced by syringe and the absorption at 740 nm was monitored with time using a UV-vis spectrometer. The extinction coefficient taken as a reference to determine the concentration of the Cu(II) was that of the bipyridine Cu(II)Cl2 complex. Given that we used a mixed halide system in the ATRP process (a Br initiator with Cu(I)Cl) the assumption of a constant extinction coefficient for the Cu(II) complex may be invalid (since we used Cu(II)Cl2 as the baseline measurement for 100% Cu(II)) and thus the values obtained cannot be taken as absolute. However the relative values should be significant. Measurements gave KATRP of 8.32 × 10−6 for the EBriB and 5.37 × 10−7 for PBriBA indicating that the activity of the ester is 15.5 times greater than that of the amide (plots of F[Cu(II)] versus time can be found in ESI). This is a remarkably different relative reactivity than that calculated from DFT. We surmised he most likely reason for this disparity was differing solvent effects on the two initiators. To ascertain the effect of the solvent on the reaction we carried out further calculations at the BMK/aug-cc-pVTZ level using the SMD solvent model of Truhlar.54 Xylene was chosen as a non-polar solvent with minimal H-bond properties and ethanol as the reaction solvent. The results of the calculations of ΔG and ΔH are illustrated graphically in Fig. 2 and values for −ΔGG and relative KATRP values (K/K0) are given in Table 3. A significant drop in the enthalpies and free energies of the bond dissociation is observed on going from the gas phase to a xylene solvent for the MBriP and the MBriPA initiators whereas the MBriPA2 actually decreases in ΔH but increases slightly in ΔG.
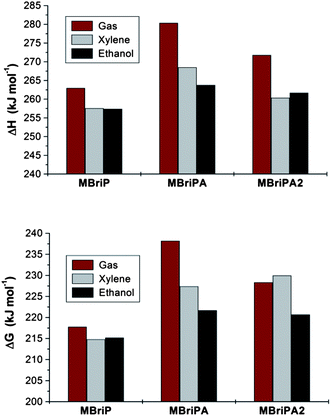 |
| | Fig. 2 Bond dissociation energies and free energies for model initiators in solvents calculated at the BMK/aug-cc-pVTZ//B3LYP/6-31+G(d) level of theory. | |
Table 3 Calculated (BMK/aug-cc-PVTZ) relative differences in BDFEs and relative KATRP values for the amide initiators from the ester initiator
| Cmpd |
|
Gas |
Xylene |
Ethanol |
|
ΔΔGG = difference between ΔG for compound relative to ΔG for ester (MBriP).
K/K0 = ratio of KATRP for compound to KATRP for ester (MBriP).
|
| MBriP |
ΔΔGa |
0 |
0 |
0 |
|
K/K0b |
1 |
1 |
1 |
| MBriPA |
ΔΔGa |
−20.4 |
−12.6 |
−6.6 |
|
K/K0b |
0.000264 |
0.00626 |
0.0710 |
| MBriPA2 |
ΔΔGa |
−10.5 |
−15.2 |
−5.5 |
|
K/K0b |
0.0144 |
0.00216 |
0.107 |
For the ester however there is no significant difference in these values between xylene and ethanol. The amides though show further significant drops in ΔG and ΔH in ethanol. Thus the MBriPA and MBriPA2 initiators are circa 14 and 9 less reactive than the ester. The former value is in excellent agreement with the measured experimental relative reactivity for the PrBriPA initiator (K/K0 = 0.0646, circa 15.5 times less reactive than the ester).
Thus whilst solvation in a polar protic solvent dramatically increases the rate of activation of the amide structures for ATRP it is still less than that of the ester. Significant solvent effects have previously been observed (DMF vs. DMSO).59,80 Complexation of the Cu catalyst by the amide group(s) has also been put forward as an explanation for the low activity of amide initiators though experimental results are contradictory on this point55,58 and no conclusive evidence for complexation has been put forward to the best of our knowledge. To confirm that complexation of the amide with the copper complex was not significant we conducted UV-vis spectroscopic studies of the Cu(I)Cl, Cu(II)Cl2 and Cu(II)Br2 in the presence of an analogue of the amide initiator (N-methylpivalamide, Scheme 1) and noted no significant differences in the UV spectra (see ESI†) that might be indicative of amide binding to Cu. This supports Adams et al. conclusion that amide complexation is not significant;58 complexation of copper by amides and peptides is rare without deprotonation of the nitrogen or a free acid function or histidine being present.81–84
The ATRP of methacrylates from amide initiators should always lead to poor initiator efficiencies, broader than optimal polydispersities and inconstant radical concentrations as a consequence of the more rapid propagation of the ester relative to initiation of the amide.
This supports the experimental data of Adams et al. who detected unreacted amide initiators even at high conversions.58This difference in reactivity between the amide initiator and the propagating methacrylate species, can in itself explain most experimental results observed for amide initiators used in standard ATRP conditions to date. The difference in activity can be ameliorated to a certain extent by appropriate choice of solvent. This is not to say that termination of amide activity (such as observed by Habraken et al.)59 does not take place but we believe it to be a consequence of the slower overall reaction times for amide initiated reactions.
We further note that given the high BDE of the MCliBA (Table 2), if any deactivation of an amide initiator by Cu(II) chloride complexes takes place, then for all intents and purposes this deactivates the initiator to any significant reactions for the lifetime of the polymerisation.
Synthesis of PEI-macroinitiator
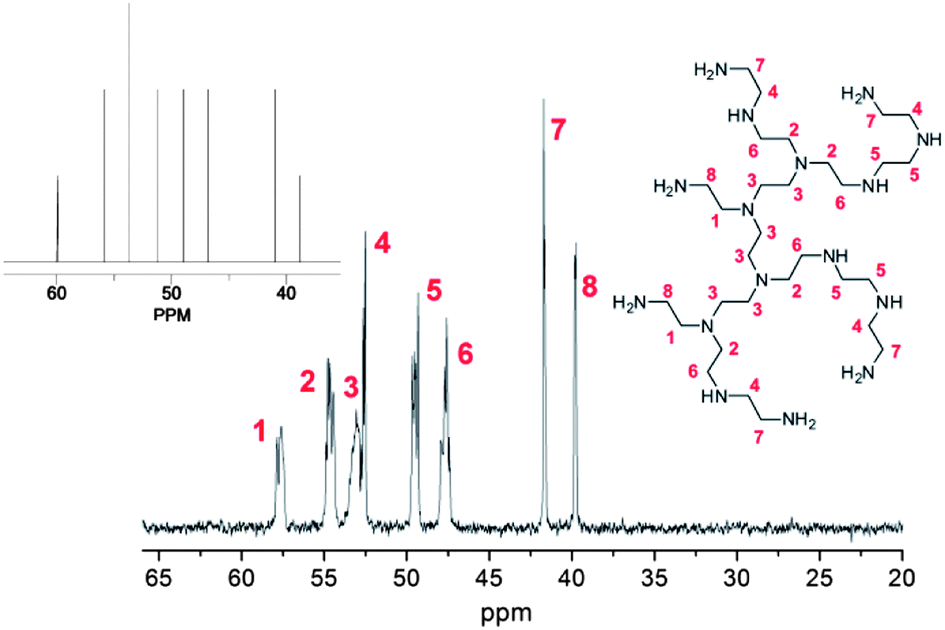
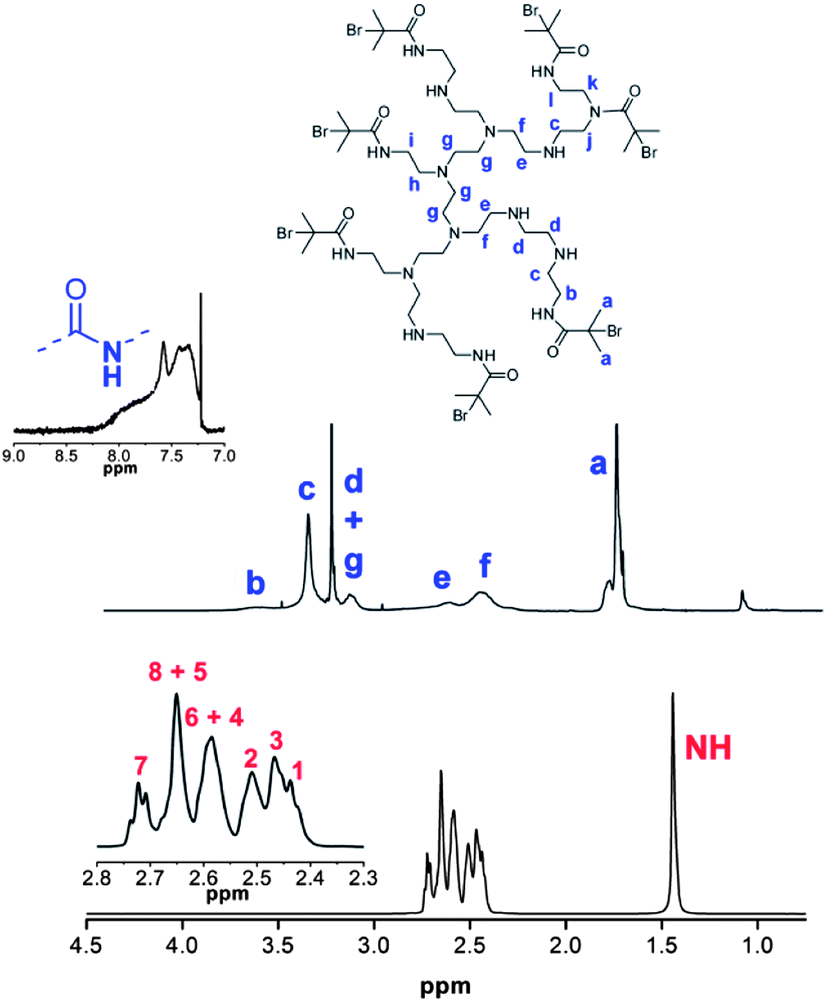
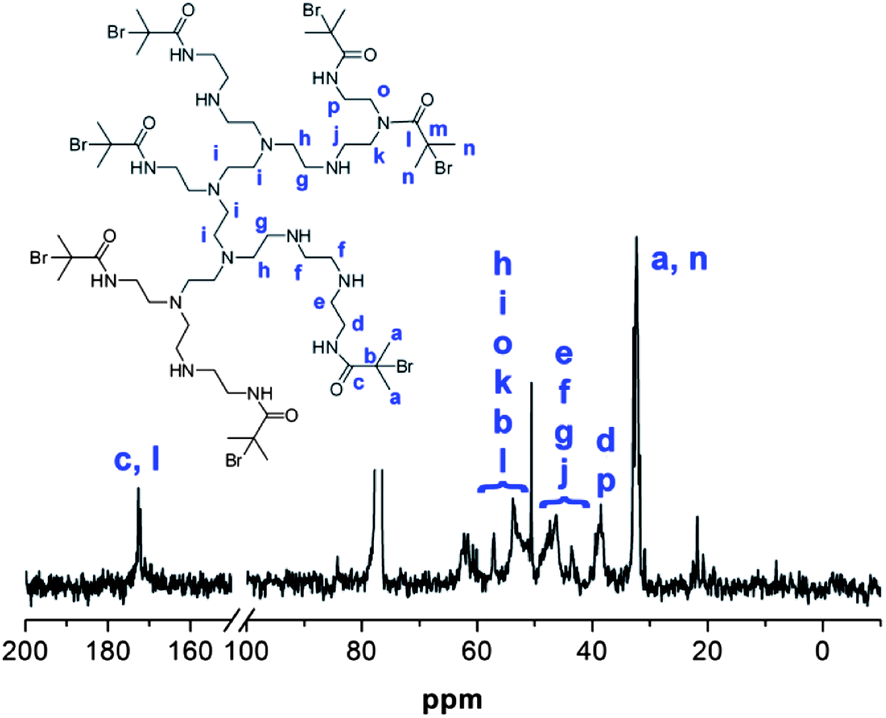
Whilst the manufacturer supplies an estimated ratio 25:50:25 for primary, secondary and tertiary amines respectively in the branched PEI used in this study, analysis of the structure, using previously reported 13C assignments,85 gave a percentage ratio of 39:38:23 for the 1°, 2° and 3° amine groups respectively (Fig. 3). This approximates to six 1°, six 2° and four 3° amines for a molecule of PEI with an Mn ≈ 600. The structure for PEI shown in Fig. 3 is used as a guideline only to illustrate the closest structure that can be constructed using this average as a basis. Using two dimensional NMR techniques (ESI) it was possible to assign the 1H NMR spectrum of PEI as shown in Fig. 4. The reaction of 2-bromo-2-methylpropanoyl bromide with low molecular weight PEI sample (Mn = 600) produced a PEI macro-initiator (PEI-Br) suitable for the ATRP of OEGMA (Scheme 1). Fig. 4 shows the 1H NMR spectra of PEI and PEI-Br for comparison and Fig. 5 the 13C spectrum of the PEI-Br. From integration of the 1H NMR spectrum the number of amide groups per PEI molecule was calculated to be 6.6 (based on the ratio of 1°, 2° and 3° amines given above and an Mn of 600). Due to the complexity of the 1H and 13C spectra the location of these sites within a PEI molecule can only be estimated. However the complete disappearance of the signals from carbons 7 and 8 in the 13C NMR (compare Fig. 3 and 5) suggest that all primary amines have effectively reacted and bear an amide function. From the number of initiating sites calculated to be present (6.6), it is clear that a proportion of secondary amines must also have reacted, which is supported by a shift in the signal for carbon 4 (adjacent to a secondary amine and carbon 7).
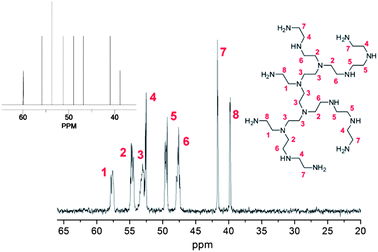 |
| | Fig. 3
13C NMR spectrum of PEI (Mn = 600) with assignments. Inset shows predicted spectrum for structure shown. | |
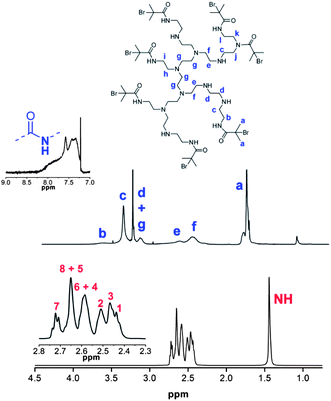 |
| | Fig. 4
1H NMR spectra and assignments of PEI (bottom) and PEI-Br (top). Assignments for PEI correspond to structure shown in Fig. 1. | |
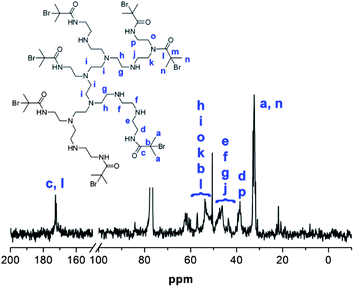 |
| | Fig. 5
13C NMR spectrum of PEI-Br with assignments. | |
Synthesis of PEI-graft-POEGMA
As previously noted the ATRP of OEGMA using the PBriBA initiator was successful, leading to controlled polymers with a range of molecular weights albeit with lower than desired initiation efficiencies (Table 4). In contrast the PEI-Br macro-initiator contains two distinct initiator functions (a primary and a secondary amide) which as demonstrated above, are expected to have different KATRP values, both of which are expected to be of lower value than the propagating ester unit. Despite this, by using similar reaction conditions to those previously used the PEI macro-initiator gave graft polymers with polydispersities under 1.4 (Table 4, Fig. 6). Generally the initiation efficiencies were low (0.22–0.63) except for sample P5 which was allowed to go to 99% conversion over an extended reaction time. Thus the results from these syntheses were variable but did allow the synthesis of graft copolymers with relatively narrow molecular eights.
Table 4 Summary of the synthesis and molecular weight parameters for the ATRP of one amide initiated (Am1) and five PEI-graft-POEGMA copolymers (PI–V) at 20 °C in ethanol
| ID |
[M]/[I] |
M
n,theo
|
SEC Mnb |
SECTripleMnc |
t (h) |
Convd (%) |
M
n/Mwb |
|
M
n,theo = [M]/[I] × Mn(0) × % conversion.
SEC Mn from RI response c against PMMA standards.
SEC Mn from triple-detection SEC.
Conversion from 1H NMR.
|
| P1 |
50 |
— |
15150 |
— |
|
— |
1.23 |
| P2 |
50 |
13700 |
27350 |
26500 |
|
83 |
1.40 |
| P3 |
50 |
10400 |
47200 |
— |
|
63 |
1.40 |
| P4 |
100 |
19500 |
30850 |
28400 |
|
59 |
1.25 |
| P5 |
100 |
32700 |
32150 |
— |
|
99 |
1.27 |
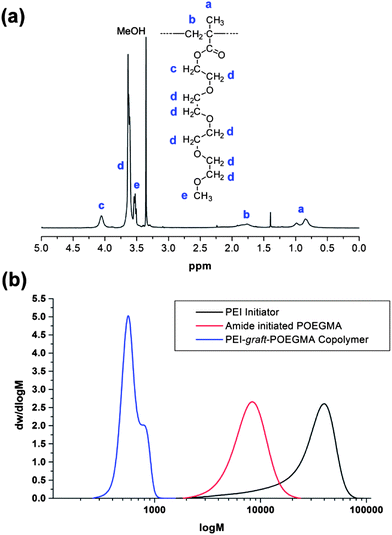 |
| | Fig. 6 (a) 1H NMR spectrum of PEI-graft-POEGMA (P1) sample. (b) SEC molecular weight distributions of PEI-Br, amide initiated POEGMA (Am1) and PEI-graft-POEGMA (P1). | |
Synthesis and characterisation of PEI-graft-POEGMA stabilised magnetic nanoparticles
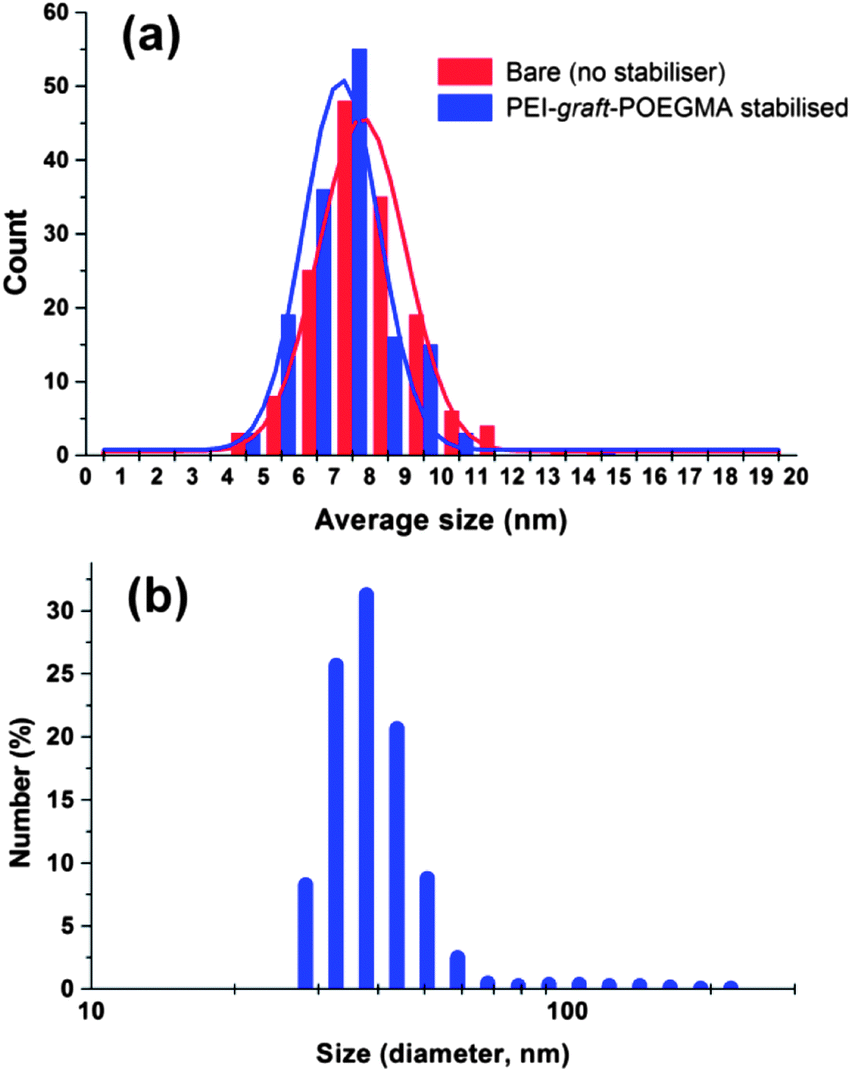
The synthesis of superparamagnetic magnetite nanoparticles by chemical coprecipitation method is known to yield crystalline random oriented particles with various morphologies, having an average size in between 6 and 10 nm with a fairly broad size distribution.86 The morphology of the particles prepared in the presence of PEI-graft-POEGMA (P1) was analyzed by transmission electron microscopy (Fig. 7) while size and size distribution analyses were performed by measuring 150 individual particles and dynamic light scattering (DLS) (Fig. 8). The presence of the polymer did not affect the size and size distribution significantly. As seen from Fig. 6, the average size of the bare particles was determined to be 7.9 nm while particles coated with PEI-graft-POEGMA (P1) had an average size of 7.4 nm. The size distribution of both samples were almost the same with standard deviations of 1.35 nm and 1.50 nm for PEI-graft-POEGMA (P1) coated and bare particles respectively. DLS analysis of a 1 wt% dispersion of the stabilised particles at 20.1 °C gave an n-average particle diameter of 48.3 nm with a standard deviation of 8.58 nm. This particle diameter is considerably larger than that observed by TEM (7.4 ± 1.5 nm) and indicates that the particles form clusters in solution as is well documented.87–91
 |
| | Fig. 7 TEM Micrograph of PEI-graft-POEGMA (P1) coated superparamagnetic iron oxide nanoparticles. | |
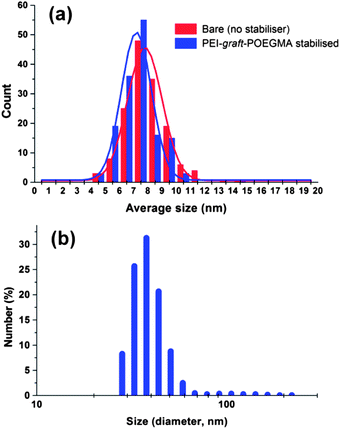 |
| | Fig. 8 Particle size distribution of (a) iron oxide nanoparticles and (b) PEI-graft-POEGMA coated superparamagnetic iron oxide nanoparticles based on DLS measurements. | |
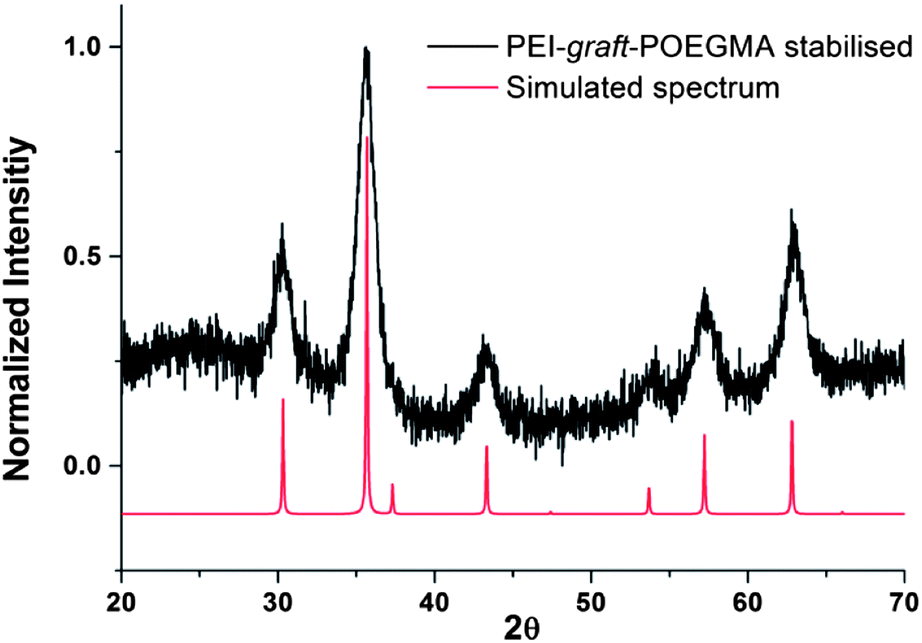
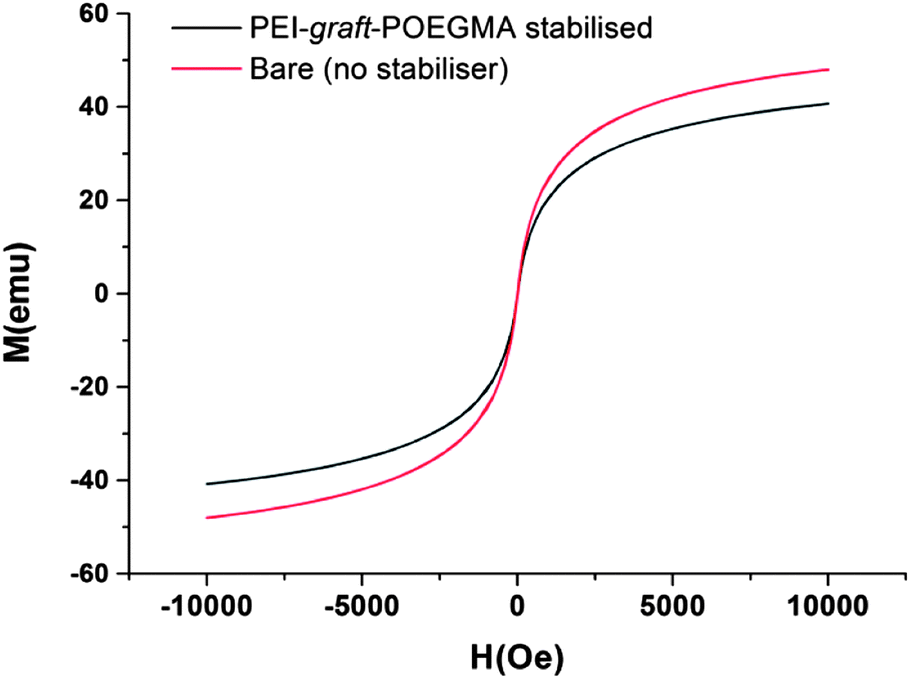
As can be seen from Fig. 7, the particles had shown no specific shape but irregular morphologies no different from the unstabilised bare samples. High magnification imaging showed cubic, octahedral and spherical morphologies as is often obtained from chemical co-precipitation. X-ray diffraction analysis of the nanoparticles synthesized in the presence of PEI-graft-POEGMA, given in Fig. 9, shows the characteristic reflections of peaks assigned to magnetite and/or maghemite without any impurities of other iron oxide phases. High resolution TEM shows the lattice fringes with a d-spacing of 0.254 nm which can be indexed to 311 plane of magnetite, showing the crystalline nature of the product. Selected area diffraction pattern of the particles showed the existence of randomly oriented small crystals of the magnetite phase (ESI†). As was expected for the co-precipitation method there was no difference in the morphology or diffraction pattern between the particles prepared with and without PEI-graft-PEOGMA (ESI†). The magnetic properties of saturation magnetization, coercivity, and remanence of the PEI-graft-POEGMA coated and bare nano-particles were analyzed by vibrating sample magnetometry. Fig. 10 shows the corresponding hysteresis loop with no coercivity or remanence indicating that both samples have a superparamagnetic nature. The saturation magnetization of particles coated with PEI-graft-POEGMA was found to be 40.7 emu g−1 while for bare particles it is 48 emu g−1 which is less than bulk magnetite. The lowering of magnetization can be attributed to the presence of non-magnetic PEI-graft-POEGMA coating as the crystal structure, morphology and average size of the nanoparticles remain unchanged.92 Thermogravimetric analysis (TGA) (Fig. 11) showed 6% weight loss for uncoated superparamagnetic iron oxide nanoparticles due to the release of absorbed water from the nanoparticle surfaces. On the other hand PEI-graft-POEGMA coated iron oxide nanoparticles shows a further loss of 8.5% demonstrating the existence of a degradable organic component (the PEI-graft-POEGMA coating) on the magnetic iron oxide nanoparticles.
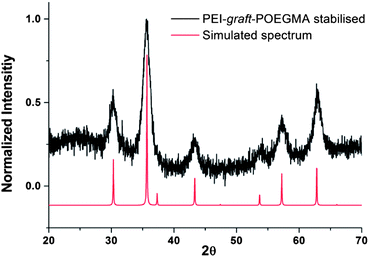 |
| | Fig. 9 X-ray diffraction pattern for PEI-graft-POEGMA coated magnetite nanoparticles compared simulated pattern of magnetite. nanoparticles. | |
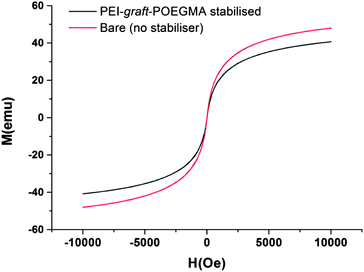 |
| | Fig. 10 Magnetization curves of bare and PEI-graft-POEGMA (P1) coated iron oxide nanoparticles. | |
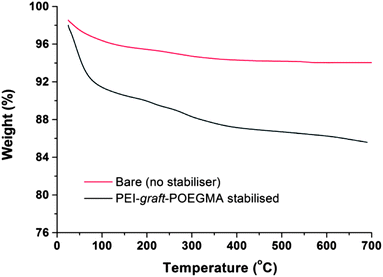 |
| | Fig. 11 TGA curves of bare and PEI-graft-POEGMA (P1) coated iron oxide nanoparticles. | |
The superparamagnetic magnetite nanoparticles prepared in the presence of PEI-graft-POEGMA (P1) were dispersed in distilled water by sonication after the synthesis. Although some settling occurred over time, the dispersed particles largely remained stable in suspension over days (ESI†). It should be noted that the bare particles without any surface modifications aggregated and precipitated in a matter of minutes.
Conclusions
In summary we have developed a facile synthetic route to poly(ethylene imine)-graft-poly(oligo(ethylene glycol methyl ether)) (PEI-graft-POEGMA) functionalised superparamagnetic magnetite nanoparticles. The synthesis of a poly-amide ATRP macroinitiator from a commercial branched PEI sample suitable for the polymerisation of oligo(ethylene glycol methyl ether) methacrylate allows for the ready functionalisation of PEI. Whilst only OEGMA has been utilised as a monomer in this study we envisage that this synthetic route opens the door to various possible functionalisations of PEI with various polymers. The utility of the PEI-graft-POEGMA synthesised was demonstrated by its ready use as a stabiliser in the preparation of superparamagnetic magnetite nanoparticles which retained all of the characteristics of nanoparticles prepared in the absence of stabiliser. Both the PEI-graft-POEGMA and the PEI-POEGMA stabilised nanoparticles are expected to display lower critical solution temperatures and the thermoresponsive properties will be reported in a forthcoming publication.
Notes and references
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed
 .
.
- M. Chanana, Z. Mao and D. Wang, J. Biomed. Nanotechnol., 2009, 5, 652–668 CrossRef CAS PubMed .
- Z. R. Stephen, F. M. Kievit and M. Zhang, Mater. Today, 2011, 14, 330–338 CrossRef CAS .
- C. Alexiou, R. J. Schmid, R. Jurgons, M. Kremer, G. Wanner, C. Bergemann, E. Huenges, T. Nawroth, W. Arnold and F. G. Parak, Eur. Biophys. J., 2006, 35, 446–450 CrossRef CAS PubMed .
- F. Y. Cheng, C. H. Su, Y. S. Yang, C. S. Yeh, C. Y. Tsai, C. L. Wu, M. T. Wu and D. B. Shieh, Biomaterials, 2005, 26, 729–738 CrossRef CAS PubMed .
- S. Bucak, S. Sharpe, S. Kuhn and T. A. Hatton, Biotechnol. Prog., 2011, 27, 744–750 CrossRef CAS PubMed .
- S. Purushotham, P. E. J. Chang, H. Rumpel, I. H. C. Kee, R. T. H. Ng, P. K. H. Chow, C. K. Tan and R. V. Ramanujan, Nanotechnology, 2009, 20, 305101 CrossRef CAS PubMed .
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167 CrossRef CAS .
- A. F. Thünemann, D. Schütt, L. Kaufner, U. Pison and H. Möhwald, Langmuir, 2006, 22, 2351–2357 CrossRef PubMed .
- X. Liu, Y. Guan, Z. Ma and H. Liu, Langmuir, 2004, 20, 10278–10282 CrossRef CAS PubMed .
- L. Harris, J. Goff, A. Carmichael, J. Riffle, J. Harburn, T. G. S. Pierre and M. Saunders, Chem. Mater., 2003, 15, 1367–1377 CrossRef CAS .
- A. Mukhopadhyay, N. Joshi, K. Chattopadhyay and G. De, ACS Appl. Mater. Interfaces, 2011, 4, 142–149 Search PubMed .
- H. K. Nguyen, P. Lemieux, S. V. Vinogradov, C. L. Gebhart, N. Guerin, G. Paradis, T. K. Bronich, V. Y. Alakhov and A. V. Kabanov, Gene Ther., 2000, 7, 126–138 CrossRef CAS PubMed .
- O. Boussif, F. Lezoualch, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS .
- M. Turk, S. Dincer, I. S. Yulug and E. Piskin, J. Controlled Release, 2004, 96, 325–340 CrossRef CAS PubMed .
- C. Flesch, Y. Unterfinger, E. Bourgeat-Lami, E. Duguet, C. Delaite and P. Dumas, Macromol. Rapid Commun., 2005, 26, 1494–1498 CrossRef CAS .
- C. Y. Quan, H. Wei, Y. X. Sun, S. X. Cheng, K. Shen, Z. W. Gu, X. Z. Zhang and R. X. Zhuo, J. Nanosci. Nanotechnol., 2008, 8, 2377–2384 CrossRef CAS PubMed .
- H. Cheng, J. L. Zhu, Y. X. Sun, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, Bioconjugate Chem., 2008, 19, 1368–1374 CrossRef CAS PubMed .
- T. Gillich, C. Acikgöz, L. Isa, A. D. Schlüter, N. D. Spencer and M. Textor, ACS Nano, 2012, 7, 316–329 CrossRef PubMed .
- D. C. Popescu, R. Lems, N. A. A. Rossi, C. T. Yeoh, J. Loos, S. J. Holder, C. V. C. Bouten and N. A. J. M. Sommerdijk, Adv. Mater., 2005, 17, 2324–2329 CrossRef CAS .
- J. F. Lutz, Adv. Mater., 2011, 23, 2237–2243 CrossRef CAS .
- J. F. Lutz, O. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed .
- Z. Zhang, M. Zhang, S. F. Chen, T. A. Horbetta, B. D. Ratner and S. Y. Jiang, Biomaterials, 2008, 29, 4285–4291 CrossRef CAS PubMed .
- S. R. Deka, A. Quarta, R. Di Corato, A. Riedinger, R. Cingolani and T. Pellegrino, Nanoscale, 2011, 3, 619–629 RSC .
- J. Guo, W. Yang, Y. Deng, C. Wang and S. Fu, Small, 2005, 1, 737–743 CrossRef CAS PubMed .
- Y.-H. Lien, T.-M. Wu, J.-H. Wu and J.-W. Liao, J. Nanopart. Res., 2011, 13, 5065–5075 CrossRef CAS .
- R. Narain, M. Gonzales, A. S. Hoffman, P. S. Stayton and K. M. Krishnan, Langmuir, 2007, 23, 6299–6304 CrossRef CAS PubMed .
- M. Turk, S. Dincer and E. Piskin, J. Tissue Eng. Regener. Med., 2007, 1, 377–388 CrossRef CAS PubMed .
- H. Malonne, F. Eeckman, D. Fontaine, A. Otto, L. D. Vos, A. Moës, J. Fontaine and K. Amighi, Eur. J. Pharm. Biopharm., 2005, 61, 188–194 CrossRef CAS PubMed .
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS .
- Z. Hu, T. Cai and C. Chi, Soft Matter, 2010, 6, 2115–2123 RSC .
- J. F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS .
- S. J. Holder, G. G. Durand, C. T. Yeoh, E. Illi, N. J. Hardy and T. H. Richardson, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7739–7756 CrossRef CAS .
- S. J. Holder, N. A. Rossi, C.-T. Yeoh, G. G. Durand, M. J. Boerakker and N. A. Sommerdijk, J. Mater. Chem., 2003, 13, 2771–2778 RSC .
- R. Zhang, Y. Wang, F. S. Du, Y. L. Wang, Y. X. Tan, S. P. Ji and Z. C. Li, Macromol. Biosci., 2011, 11, 1393–1406 CrossRef CAS PubMed .
- D. J. Siegwart, J. K. Oh and K. Matyjaszewski, Prog. Polym. Sci., 2012, 37, 18–37 CrossRef CAS PubMed .
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 CrossRef CAS PubMed .
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS PubMed .
- J. S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7901–7910 CrossRef CAS .
- H. Bergenudd, G. Coullerez, M. Jonsson and E. Malmstrom, Macromolecules, 2009, 42, 3302–3308 CrossRef CAS .
- X. S. Wang, S. F. Lascelles, R. A. Jackson and S. P. Armes, Chem. Commun., 1999, 1817–1818 RSC .
- J. J. Stewart, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS PubMed .
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen and S. Su, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS .
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS .
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS .
- A. D. Boese and J. M. L. Martin, J. Chem. Phys., 2004, 121, 3405–3416 CrossRef CAS PubMed .
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed .
- A. Karton, A. Tarnopolsky, J.-F. o. Lamère, G. C. Schatz and J. M. L. Martin, J. Phys. Chem. A, 2008, 112, 12868–12886 CrossRef CAS PubMed .
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS .
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154119 CrossRef PubMed .
- R. Peverati and K. K. Baldridge, J. Chem. Theory Comput., 2008, 4, 2030–2048 CrossRef CAS .
- L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC .
- L. Goerigk and S. Grimme, J. Chem. Theory Comput., 2011, 7, 291–309 CrossRef CAS .
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed .
- H. Rettig, E. Krause and H. G. Börner, Macromol. Rapid Commun., 2004, 25, 1251–1256 CrossRef CAS .
- A. Limer and D. M. Haddleton, Macromolecules, 2006, 39, 1353–1358 CrossRef CAS .
- P. M. Wright, G. Mantovani and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7376–7385 CrossRef CAS .
- D. J. Adams and I. Young, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6082–6090 CrossRef CAS .
- G. J. Habraken, C. E. Koning and A. Heise, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6883–6893 CrossRef CAS .
- K.-Y. Baek, M. Kamigaito and M. Sawamoto, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1937–1944 CrossRef CAS .
- Y. Mei, K. L. Beers, H. C. M. Byrd, D. L. VanderHart and N. R. Washburn, J. Am. Chem. Soc., 2004, 126, 3472–3476 CrossRef CAS PubMed .
- M. Senoo, Y. Kotani, M. Kamigaito and M. Sawamoto, Macromolecules, 1999, 32, 8005–8009 CrossRef CAS .
- A. A. Isse, A. Gennaro, C. Y. Lin, J. L. Hodgson, M. L. Coote and T. Guliashvili, J. Am. Chem. Soc., 2011, 133, 6254–6264 CrossRef CAS PubMed .
- M. L. Coote, C. Y. Lin, A. L. Beckwith and A. A. Zavitsas, Phys. Chem. Chem. Phys., 2010, 12, 9597–9610 RSC .
- C. Y. Lin, M. L. Coote, A. Gennaro and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 12762–12774 CrossRef CAS PubMed .
- W. Tang, Y. Kwak, W. Braunecker, N. V. Tsarevsky, M. L. Coote and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 10702–10713 CrossRef CAS PubMed .
- H. Kruse, L. Goerigk and S. Grimme, J. Org. Chem., 2012, 77, 10824–10834 CrossRef CAS PubMed .
- E. I. Izgorodina, M. L. Coote and L. Radom, J. Phys. Chem. A, 2005, 109, 7558–7566 CrossRef CAS PubMed .
- A. S. Menon, G. P. Wood, D. Moran and L. Radom, J. Phys. Chem. A, 2007, 111, 13638–13644 CrossRef CAS PubMed .
- I. Y. Zhang, J. Wu, Y. Luo and X. Xu, J. Chem. Theory Comput., 2010, 6, 1462–1469 CrossRef CAS .
- R. J. O'Reilly, A. Karton and L. Radom, Int. J. Quantum Chem., 2012, 112, 1862–1878 CrossRef CAS .
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2008, 112, 1095–1099 CrossRef CAS PubMed .
- A. Tarnopolsky, A. Karton, R. Sertchook, D. Vuzman and J. M. L. Martin, J. Phys. Chem. A, 2007, 112, 3–8 CrossRef PubMed .
- B. Chan and L. Radom, J. Phys. Chem. A, 2012, 116, 4975–4986 CrossRef CAS PubMed .
- W. Tang and K. Matyjaszewski, Macromolecules, 2007, 40, 1858–1863 CrossRef CAS .
- D. Wang, J. Wang, D. Zhang and Z. Li, Sci. China: Chem., 2012, 55, 2018–2026 CrossRef CAS .
- Y.-Y. Zhu, L. Jiang and Z.-T. Li, CrystEngComm, 2009, 11, 235–238 RSC .
- W. Tang, N. V. Tsarevsky and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 1598–1604 CrossRef CAS PubMed .
- W. A. Braunecker, N. V. Tsarevsky, A. Gennaro and K. Matyjaszewski, Macromolecules, 2009, 42, 6348–6360 CrossRef CAS .
- S. Steig, F. Cornelius, P. Witte, B. B. P. Staal, C. E. Koning, A. Heise and H. Menzel, Chem. Commun., 2005, 5420–5422 RSC .
- H. Sigel and R. B. Martin, Chem. Rev., 1982, 82, 385–426 CrossRef CAS .
- V. Jószai, Z. Nagy, K. Ősz, D. Sanna, G. Di Natale, D. La Mendola, G. Pappalardo, E. Rizzarelli and I. Sóvágó, J. Inorg. Biochem., 2006, 100, 1399–1409 CrossRef PubMed .
- H. Sigel, Inorg. Chem., 1975, 14, 1535–1540 CrossRef CAS .
- B. Bóka, A. Myari, I. Sóvágó and N. Hadjiliadis, J. Inorg. Biochem., 2004, 98, 113–122 CrossRef PubMed .
- A. von Harpe, H. Petersen, Y. X. Li and T. Kissel, J. Controlled Release, 2000, 69, 309–322 CrossRef CAS .
- A. H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed .
- R. Sondjaja, T. Alan Hatton and M. K. C. Tam, J. Magn. Magn. Mater., 2009, 321, 2393–2397 CrossRef CAS PubMed .
- A. Ditsch, P. E. Laibinis, D. I. C. Wang and T. A. Hatton, Langmuir, 2005, 21, 6006–6018 CrossRef CAS PubMed .
- J. F. Berret, N. Schonbeck, F. Gazeau, D. El Kharrat, O. Sandre, A. Vacher and M. Airiau, J. Am. Chem. Soc., 2006, 128, 1755–1761 CrossRef CAS PubMed .
- J. H. Park, G. von Maltzahn, L. Zhang, M. P. Schwartz, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Adv. Mater., 2008, 20, 1630–1635 CrossRef CAS PubMed .
- S. A. Corr, S. J. Byrne, R. Tekoriute, C. J. Meledandri, D. F. Brougham, M. Lynch, C. Kerskens and L. O'Dwyer, J. Am. Chem. Soc., 2008, 130, 4214–4215 CrossRef CAS PubMed .
- Y. Yuan, D. Rende, C. L. Altan, S. Bucak, R. Ozisik and D. A. Borca-Tasciuc, Langmuir, 2012, 28, 13051–13059 CrossRef CAS PubMed .
Footnotes |
| † Electronic supplementary information (ESI) available: Plots of Cu(II) function with time; UV-vis absorption spectra of Cu(I)Cl, Cu(II)Cl2 and Cu(II)Br2 solutions with DiMePiVA; X-ray diffraction pattern of PEI-graft-POEGMA stabilised nanoparticles; high magnification TEM images of magnetite nanoparticles; pictures of particle suspensions; X-ray diffraction pattern and TEM of bare (unstabilised) nanoparticles 2D 1H–13C and 1H–15N NMR spectra of PEI; inputs for DFT calculations; Cartesian co-ordinates of energy minimised conformations of initiators. See DOI: 10.1039/c3py01094e |
| ‡ These authors made equal contributions to the research described in this manuscript. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a