 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCationic polythiophenes as responsive DNA-binding polymers†
Analyn C.
Carreon
abc,
Webster L.
Santos
b,
John B.
Matson
*b and
Regina C.
So
*a
aDepartment of Chemistry, Ateneo de Manila University, Schmitt Hall, Loyola Heights, Katipunan, Quezon City 1108, Philippines. E-mail: rso@ateneo.edu; Fax: +63 2 4261323; Tel: +63 2 4266001 ext. 5620
bDepartment of Chemistry, Virginia Tech, 800 West Campus Drive, Blacksburg, VA 24061, USA. E-mail: jbmatson@vt.edu; Fax: +1 540 2313255; Tel: +1 540 2313329
cDepartment of Chemistry, Xavier University – Ateneo de Cagayan University, Corrales Avenue, Cagayan de Oro City 9000, Philippines. E-mail: acarreon@xu.edu.ph; Fax: +63 8822 723116 2029; Tel: +63 88 8583116 ext. 3110
First published on 23rd September 2013
Abstract
A new water soluble cationic polythiophene derivative, poly(N,N,N-trimethyl-3-(2-(thiophen-3-yl)acetamido)propan-1-aminium iodide), was synthesized via two consecutive post-polymerization functionalizations of poly(methyl 2-(thiophen-3-yl)acetate). This conjugated polymer binds DNA at N/P = 5 and forms polyplexes at N/P = 10. Its potential use as a theranostic gene delivery vehicle is investigated here.
Amongst the heavy interest in developing gene delivery vectors over the past decade, a new class of gene delivery agents with combined imaging capacity has recently begun to take hold.1,2 These theranostic gene delivery vehicles aim to offer both treatment and diagnostic capabilities within the same particle, potentially improving treatments by enabling synchronous delivery and monitoring of therapeutic genes. Most theranostic agents for gene delivery rely on magnetic nanoparticles,3 quantum dots,4 nanogold,5 carbon nanoparticles,6 or other inorganic nanomaterials as the imaging component. In these materials, a cationic coating of passivating ligands or polymers is typically incorporated into the formulation to enable DNA binding. While inorganic-based gene materials are typically very photostable and versatile, questions of toxicity may limit their potential applications. An alternative strategy is to use the cationic, DNA-binding component as the imaging agent itself, thereby simplifying the construct to only two components.7
Conjugated polymers are well known fluorescent materials that are under intense study in a variety of applications, including sensors,8 displays,9 and photovoltaics.10 Beyond these traditional applications, the increased quantum yields and photostability of conjugated polymers compared with small molecule fluorophores11 make them attractive as potential imaging components in theranostic drug and gene delivery agents.12 Despite their ideal photophysical properties and generally low toxicity,13 conjugated polymers are underexplored as theranostic gene delivery vectors. Only a few reports are available on two-component gene delivery agents using conjugated polymers.14–16 In these studies, poly(phenylene ethynylene),14,15 and poly(fluorenylene phenylene)16 were found to be effective gene transfection agents. We turned here to one of the simplest conjugated polymers, polythiophene, to evaluate its potential to form discrete polyplexes when bound to DNA and lay the foundation for its use as a theranostic gene delivery vector.
Polythiophene is an especially intriguing candidate for theranostic gene delivery due to its ability to change its emission profile in response to changes in a variety of stimuli, including temperature (thermochromism),17,18 solvent (solvatochromism),18 surfactants (surfactochromism),19,20 irradiation (photochromism),21 and the presence of certain other chemical species (affinitychromism).22–25 These properties may open opportunities not only to track DNA delivery but also to specifically trigger DNA delivery or to monitor environmental changes that affect polyplex stability. We report here a straightforward synthetic route to cationic polythiophene and demonstrate its ability to form polyplexes with plasmid DNA.
Scheme 1 describes the overall route of the synthesis of polymer 4. First, thiophene 3-acetic acid (3TAA) was esterified with methanol and catalytic H2SO4 to yield thiophene 1 according to a literature procedure.26 Oxidative polymerization of monomer 1 was carried out in CHCl3 with FeCl3 to generate polymer 2, which was isolated by precipitation into acidic methanol to afford polymer 2 as a brown powder.26 Polymerization was monitored by the disappearance of proton peaks at the 2 and 5 positions of the thiophene ring using 1H NMR (see ESI† for details). Three peaks were observed at the 4 position (7.14, 7.17, 7.21 ppm) corresponding to the four triad configurations (HT–HT, TT–HT, HT–HH, TT–HH) of monosubstituted polythiophenes.27 This lack of regioregularity is typical of polythiophenes prepared by oxidative routes.28
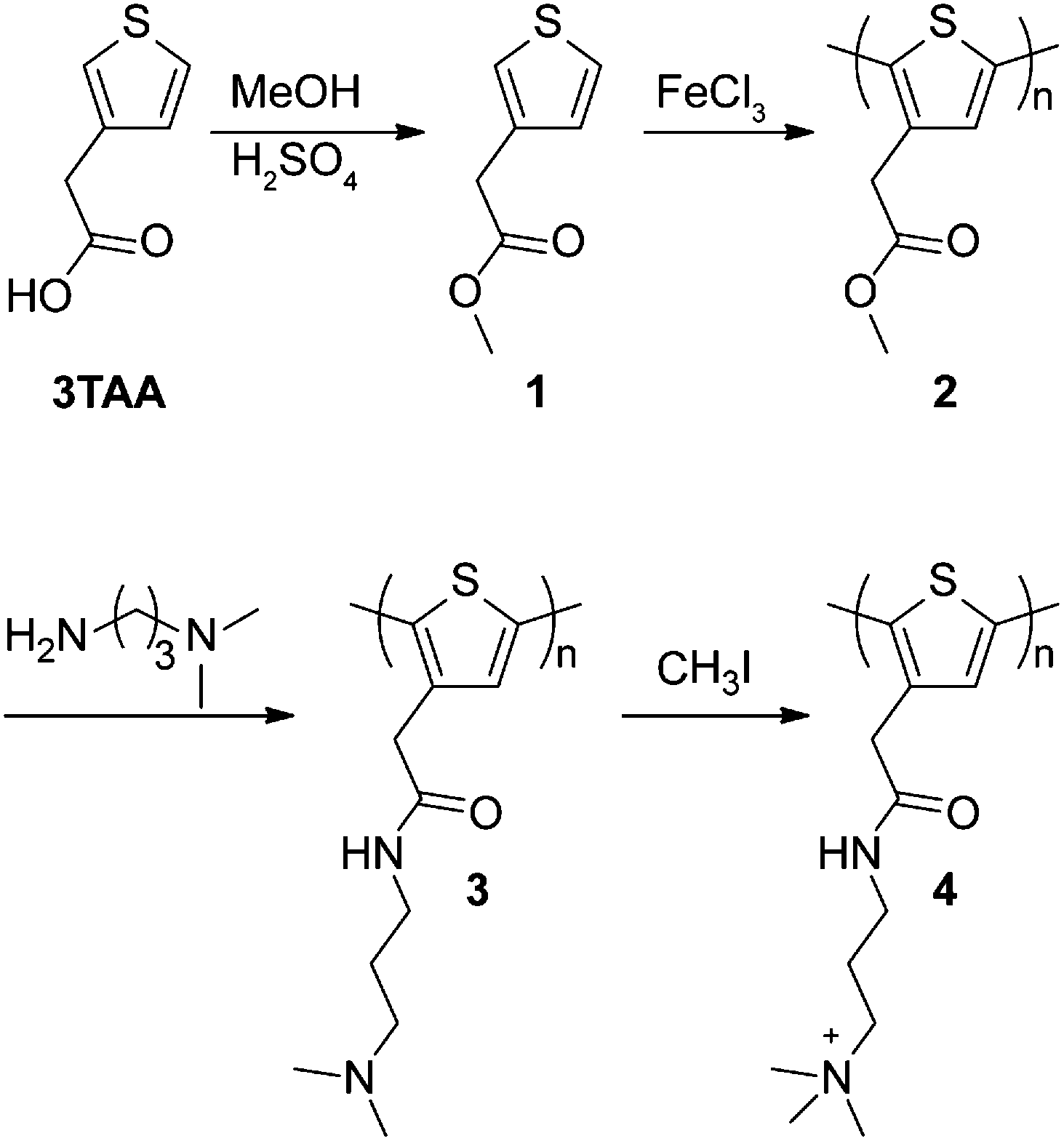 | ||
| Scheme 1 Synthesis of cationic polythiophenes 3 and 4 through post-polymerization functionalization of neutral polythiophene 2. | ||
To increase the stability of polymer side chains, polythiophenes 3 and 4, wherein the ester group was replaced with an amide linker, were synthesized by consecutive post-polymerization reactions on polymer 2. NMR spectra for polymers 2, 3, and 4 are included in the ESI.† Conversion of the side chain ester to an amide was accomplished by reaction of 3-(dimethylamino)-1-propylamine with polymer 2 to yield tertiary amine-containing polythiophene 3. Reaction progress was monitored by following the colour change from brown to red during the reaction. Polymer 3 was isolated by precipitation and recovered by filtration as an orange solid. 1H and 13C NMR confirmed >90% conversion to the desired tertiary amine. IR spectroscopy also confirmed conversion of the ester to the desired amide as the carbonyl peak shifted from 1730 cm−1 to 1644 cm−1 (Fig. 1). The presence of an amide N–H bend at 1550 cm−1 is also observed in polymer 3. A small peak remaining at 1730 cm−1 indicates that the remaining impurity is the methyl ester from polymer 2. This conclusion is confirmed by 13C NMR (Fig. S7, ESI†).
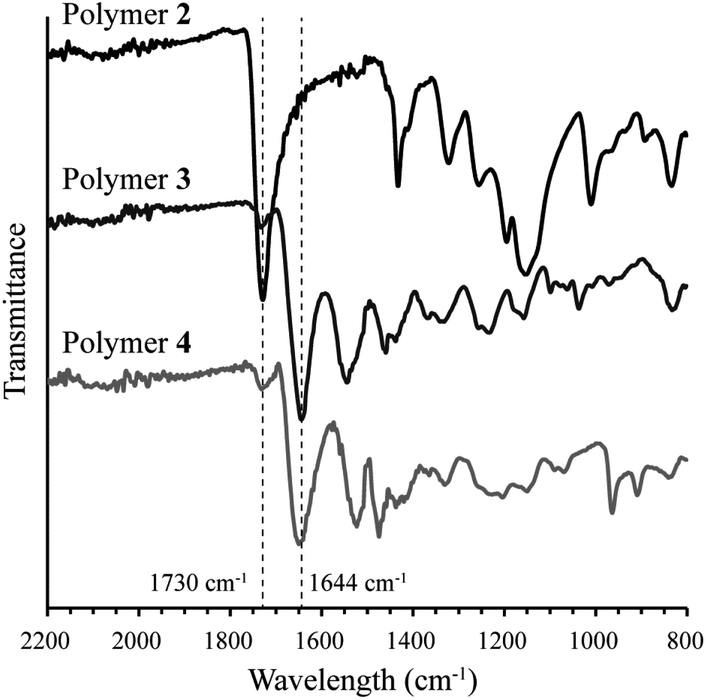 | ||
| Fig. 1 FT-IR spectra of polymers 2, 3 and 4 (attenuated total reflectance). The y-axis of each spectrum was adjusted for clarity. The dashed lines highlight the loss of the methyl ester at 1730 cm−1 and the appearance of the amide at 1644 cm−1 in going from polymer 2 to 3 and upon further derivatization to polymer 4. | ||
Polymer 4 was synthesized by hypermethylation of polymer 3 with CH3I (10 equiv.) in MeOH/Et2O at room temperature. The product precipitated as the reaction progressed and was obtained as a dark brown solid. Integration of the 1H NMR spectrum showed 93% conversion to the quaternized product (Fig. S8, ESI†). Complete conversion was likely limited by precipitation of the product from the reaction medium. The IR spectrum of polymer 4 is similar to polymer 3, with a slight shift of carbonyl stretch to 1655 cm−1 and a shift of the amide N–H bend to 1529 cm−1 (Fig. 1). The peak at 1731 cm−1 appears to be a remnant from polymer 2.
Over multiple trials, polymer 2 was obtained with Mn values ranging from 4100–7100 g mol−1 (corresponding to degrees of polymerization from 26 to 46) with polydispersity values ranging from 2.12–2.53 (Table S1, ESI†) as determined by gel permeation chromatography (GPC) (Fig. S9, ESI†). Polymers 3 and 4 were found to be soluble in water as well as DMF and DMSO. However, dynamic light scattering (DLS) measurements showed that polymers 3 and 4 were prone to aggregation in these solvents and several solvent mixtures, so their molecular weights could not be verified.
MALDI-TOF mass spectrometry was also found to be unsuitable for the characterization of polymers 3 and 4, which is consistent with previous reports.22,29 The thermal properties of polymers were also investigated (Fig. S10, ESI†). All were found to be stable up to 200 °C, with polymers 3 and 4 showing slight mass loss before 200 °C, likely corresponding to the loss of water.26
Polymers containing both tertiary and quaternary amines are suitable for DNA binding because of favourable electrostatic interactions and have been studied as gene delivery vehicles.30 As such, both polymers 3 and 4 were investigated for DNA binding. Polymer 4 bears a permanent positive charge as a result of the quaternary amines, giving it the potential advantage of making stable polyplexes across a broad pH range. On the other hand, polymer 3, which contains tertiary amines, is sensitive to solution pH—it could potentially be deprotonated to disassemble polyplexes at high pH. The pKa value of polymer 3 was determined to be 8.3 by potentiometric titration (Fig. S11, ESI†). DNA binding assays of polymers 3 and 4 were carried out at various N/P ratios using a fixed amount of plasmid DNA (pUC19) (Fig. 2). It was found that both polymers 3 and 4 can bind pUC19 at minimum N/P ratios of 1.5 and 5, respectively. Both values are low compared with other conjugated polymer gene delivery vehicles,14–16 indicating that a high percentage of the amines are available for DNA binding. The lower N/P ratio required for full DNA binding in polymer 3versus polymer 4 is surprising, as polymers containing quaternary amines typically bind DNA with equal or better efficiency than analogues with tertiary amines.31 We speculate that the less efficient binding of DNA in polymer 4versus polymer 3 may be a result of a stiffer polythiophene backbone induced by quaternization, which may both increase charge repulsion between repeating units and decrease attractive interactions by removing the hydrogen bonding capacity of the amine unit. Previous studies have found that extended chain conformations can hinder DNA binding.32
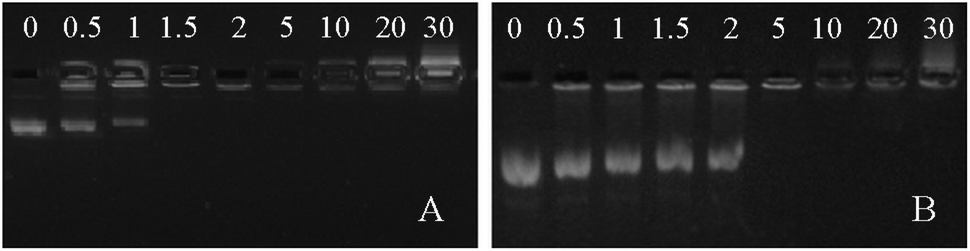 | ||
| Fig. 2 DNA binding assays of polymers 3 (A) and 4 (B) in 2% agarose gel with N/P ratio from 0.5 to 30. | ||
The polyplex size of polymer 4 bound to pUC19 was studied using DLS at N/P ratios of 5, 10, 20 and 30 (Fig. 3). Large aggregates were observed at N/P = 5, indicating either polyplex aggregation or poor formation of polyplexes. At N/P = 10, the average particle diameter was 173.0 nm while the diameter decreased to 144.1 nm and 99.9 nm at N/P = 20 and 30, respectively. These results are consistent with previous observations showing that as the concentration of the cations increases, polyplex size decreases.33 Polyplex stability was measured in buffer by DLS over a period of 4 h, with the data showing stable polyplex size over this period (Fig. S12, ESI†). The ability of polymer 4 to form polyplexes with plasmid DNA demonstrates its potential as a nonviral vector. All of the polyplexes obtained showed average diameters below 200 nm in size, similar to polyplexes made from poly(2-(dimethylamino)ethyl methacrylate) (pDMAEMA), a common polycation-based gene delivery vector.34 Zeta potential measurements showed positive surface charge for polymer 4 at all three N/P ratios (Fig. 3D).
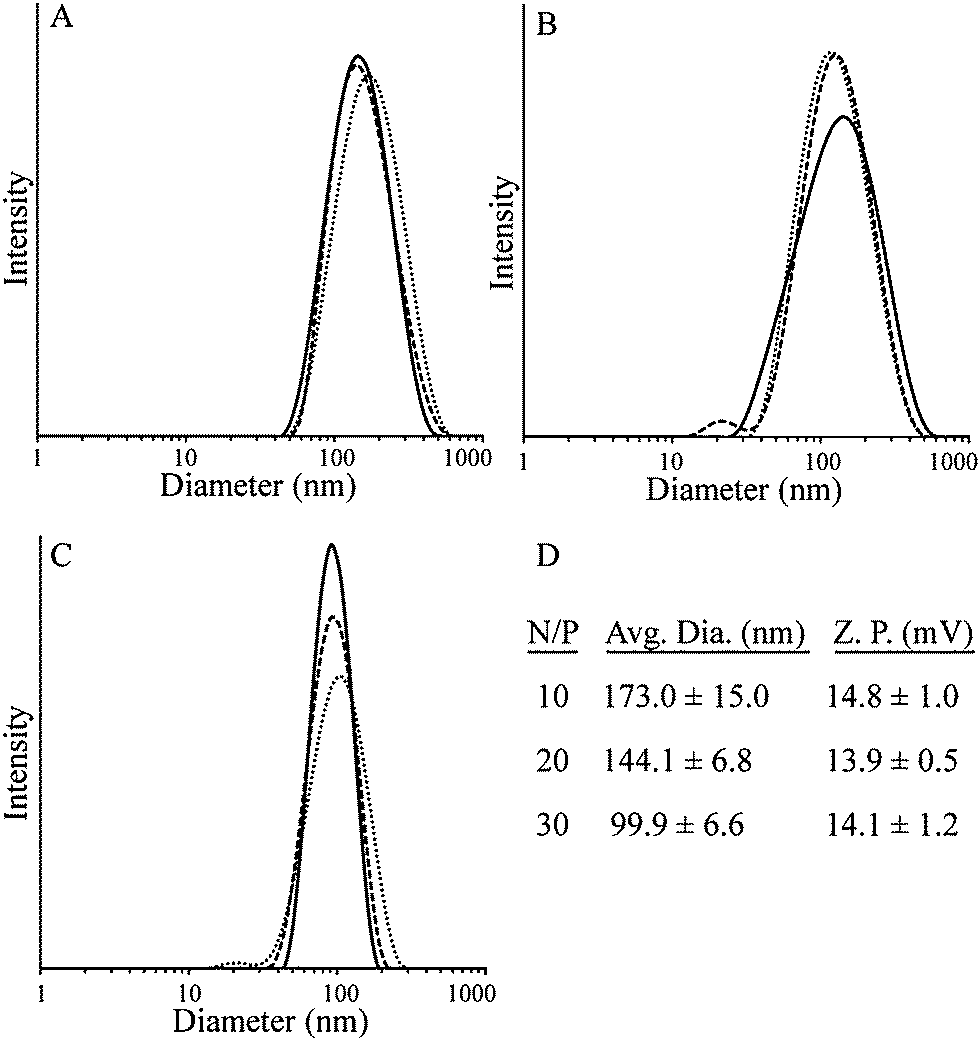 | ||
| Fig. 3 DLS and zeta potential data of polymer 4 with pUC19 plasmid DNA at N/P ratios of 10 (A), 20 (B) and 30 (C) with average diameter values (D). DLS data shown are the results of three trials for each N/P ratio. Zeta potential data represent the average and standard deviation of at least three measurements at each N/P ratio. | ||
We also investigated the fluorescence properties of polymer 4 to evaluate its potential as an imaging agent (Fig. 4). The peak emission in aqueous buffer of free polymer 4 was found to be 413 nm. Upon addition of plasmid DNA (N/P ratio of 5), the peak emission wavelength shifted to 419 nm. This redshift has been previously observed in imidazolium-containing polythiophenes and is likely due to changes in the conformational structure of the cationic polythiophene as it binds to DNA.35 The noticeable albeit small change in emission peak is exciting because of possible future applications as cationic conjugated polymer gene delivery vehicles. For example, temporal and spatial subcellular localization of DNA release from a polyplex could be detected using this strategy. Detection of DNA unpackaging has been studied in complex theranostic systems, such as those using quantum dots,36 but not to our knowledge in simple, two-component polyplexes. Understanding the factors that affect dissociation of polyplexes inside the cell is an important part of making more effective gene delivery vehicles, and imaging agents capable of reflecting whether DNA is bound or unbound may facilitate studies in this area.7 Additionally, polythiophenes inherently possess or can be modified to display a variety of properties that merit their further exploration as theranostic gene delivery vehicles, including fluorescence amplification,37 light responsiveness,21 and sensitivity to surfactants,20 temperature,17,38 solvents,18 and light.21 Moreover, cationic polythiophenes have shown minimal toxicity in several cell lines, further adding to their potential as theranostic gene delivery agents.29
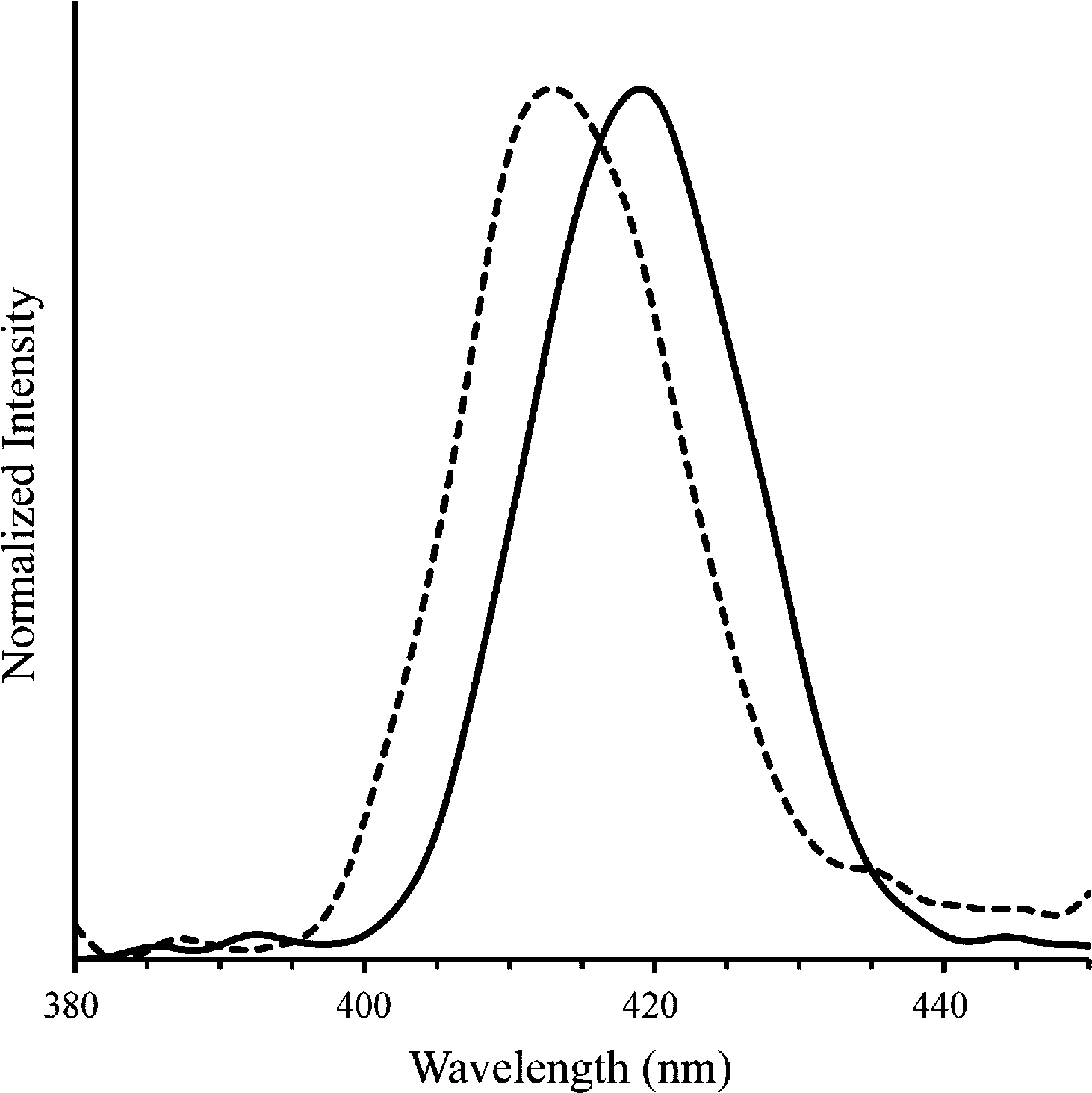 | ||
| Fig. 4 Emission spectra of polymer 4 alone (dashed line; λmax = 413 nm) and with plasmid DNA (solid line; λmax = 419 nm) in TBE buffer at N/P = 5 (λex at absorbance max, 404 and 410 nm, respectively). | ||
Conclusions
Cationic polythiophene derivatives were successfully synthesized using consecutive post-polymerization reactions. Both polymers were found to form polyplexes with pUC19 plasmid DNA at low N/P ratios. In the case of polymer 4, which contains quaternary amines, polyplexes with hydrodynamic diameters of less than 200 nm were formed at N/P ratios of 10 and above. Fluorescence spectroscopy showed a slight but noticeable redshift in the peak emission of polymer 4 when plasmid DNA was added. The described polyplexes are a simple, two-component system that adds to the growing number of theranostic gene delivery agents. The change in peak emission upon binding of DNA demonstrates how future designs with larger emission shifts might expand the possibilities of theranostic particles. Particles with large shifts between bound and unbound DNA may enable researchers not only to visualize polyplexes, but also to provide spatiotemporal information on polyplex dissociation. Other photophysical properties of polythiophene, including thermochromism, affinitychromism, and others, may make cationic polythiophenes a valuable new tool in theranostic gene delivery.Acknowledgements
This work was supported by a PhD Sandwich Program through the Commission on Higher Education, Philippines. The authors thank Prof. Tim Long, Alison Schultz, and Sean Hemp for assistance with GPC and DLS. We gratefully acknowledge Wenyu Zhang for the plasmid DNA and Molly Congdon for FT-IR assistance.Notes and references
- J. Xie, S. Lee and X. Y. Chen, Adv. Drug Delivery Rev., 2010, 62, 1064–1079 CrossRef CAS PubMed.
- J. M. Knipe, J. T. Peters and N. A. Peppas, Nano Today, 2013, 8, 21–38 CrossRef CAS PubMed.
- D. Yoo, J. H. Lee, T. H. Shin and J. Cheon, Acc. Chem. Res., 2011, 44, 863–874 CrossRef CAS PubMed.
- Y. P. Ho and K. W. Leong, Nanoscale, 2010, 2, 60–68 RSC.
- E. C. Dreaden, A. M. Alkilany, X. H. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- X. Q. Zhang, M. Chen, R. Lam, X. Y. Xu, E. Osawa and D. Ho, ACS Nano, 2009, 3, 2609–2616 CrossRef CAS PubMed.
- T. Krasia-Christoforou and T. K. Georgiou, J. Mater. Chem. B, 2013, 1, 3002–3025 RSC.
- H. N. Kim, Z. Q. Guo, W. H. Zhu, J. Yoon and H. Tian, Chem. Soc. Rev., 2011, 40, 79–93 RSC.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS PubMed.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- Z. Y. Tian, J. B. Yu, C. F. Wu, C. Szymanski and J. McNeill, Nanoscale, 2010, 2, 1999–2011 RSC.
- G. M. Yang, F. T. Lv, B. Wang, L. B. Liu, Q. Yang and S. Wang, Macromol. Biosci., 2012, 12, 1600–1614 CrossRef CAS PubMed.
- K. Y. Pu and B. Liu, Adv. Funct. Mater., 2011, 21, 3408–3423 CrossRef CAS.
- A. T. Silva, N. Alien, C. M. Ye, J. Verchot and J. H. Moon, BMC Plant Biol., 2010, 10, 291 CrossRef CAS PubMed.
- J. H. Moon, E. Mendez, Y. Kim and A. Kaur, Chem. Commun., 2011, 47, 8370–8372 RSC.
- X. L. Feng, Y. L. Tang, X. R. Duan, L. B. Liu and S. Wang, J. Mater. Chem., 2010, 20, 1312–1316 RSC.
- J. Wang, Q. Zhang, K. J. Tan, Y. F. Long, J. Ling and C. Z. Huang, J. Phys. Chem. B, 2011, 115, 1693–1697 CrossRef CAS PubMed.
- T. Shiraki, A. Dawn, Y. Tsuchiya and S. Shinkai, J. Am. Chem. Soc., 2010, 132, 13928–13935 CrossRef CAS PubMed.
- L. Wang, Q. Feng, X. Wang, M. Pei and G. Zhang, New J. Chem., 2012, 36, 1897–1901 RSC.
- M. Knaapila, R. C. Evans, V. M. Garamus, L. s. Almásy, N. m. K. Székely, A. Gutacker, U. Scherf and H. D. Burrows, Langmuir, 2010, 26, 15634–15643 CrossRef CAS PubMed.
- G. Yang, H. Yuan, C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 2334–2337 CAS.
- H.-A. Ho, M. Boissinot, M. G. Bergeron, G. Corbeil, K. Doré, D. Boudreau and M. Leclerc, Angew. Chem., Int. Ed., 2002, 41, 1548–1551 CrossRef CAS.
- K. Doré, S. Dubus, H.-A. Ho, I. Lévesque, M. Brunette, G. Corbeil, M. Boissinot, G. Boivin, M. G. Bergeron, D. Boudreau and M. Leclerc, J. Am. Chem. Soc., 2004, 126, 4240–4244 CrossRef PubMed.
- M. Béra-Abérem, H.-A. Ho and M. Leclerc, Tetrahedron, 2004, 60, 11169–11173 CrossRef PubMed.
- H.-A. Ho and M. Leclerc, J. Am. Chem. Soc., 2004, 126, 1384–1387 CrossRef CAS PubMed.
- M. Aydın, Z. Durmus, H. Kavas, B. Esat, H. Sözeri, A. Baykal, F. Yılmaz and M. S. Toprak, Polyhedron, 2011, 30, 1120–1126 CrossRef PubMed.
- S. Sengmany, C. Ceballos, R. Belhadj, C. Cachet-Vivier, E. L. Gall, B. Brissault, J. Penelle and E. Léonel, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 900–911 CrossRef CAS.
- Y. Wang, B. L. Lucht and W. B. Euler, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2002, 43, 1160–1160 CAS.
- L. B. Liu, M. H. Yu, X. R. Duan and S. Wang, J. Mater. Chem., 2010, 20, 6942–6947 RSC.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS PubMed.
- M. A. Wolfert, P. R. Dash, O. Nazarova, D. Oupicky, L. W. Seymour, S. Smart, J. Strohalm and K. Ulbrich, Bioconjugate Chem., 1999, 10, 993–1004 CrossRef CAS PubMed.
- N. A. Jones, I. R. C. Hill, S. Stolnik, F. Bignotti, S. S. Davis and M. C. Garnett, Biochim. Biophys. Acta, Gene Struct. Expression, 2000, 1517, 1–18 CrossRef CAS.
- S. De Smedt, J. Demeester and W. Hennink, Pharm. Res., 2000, 17, 113–126 CrossRef CAS.
- P. van de Wetering, J. Y. Cherng, H. Talsma, D. J. A. Crommelin and W. E. Hennink, J. Controlled Release, 1998, 53, 145–153 CrossRef CAS.
- B. Liu and G. C. Bazan, Chem. Mater., 2004, 16, 4467–4476 CrossRef CAS.
- H. H. Chen, Y. P. Ho, X. Jiang, H. Q. Mao, T. H. Wang and K. W. Leong, Mol. Ther., 2008, 16, 324–332 CrossRef CAS PubMed.
- H. A. Ho, K. Doré, M. Boissinot, M. G. Bergeron, R. M. Tanguay, D. Boudreau and M. Leclerc, J. Am. Chem. Soc., 2005, 127, 12673–12676 CrossRef CAS PubMed.
- S. D. Jung, D. H. Hwang, T. Zyung, W. H. Kim, K. G. Chittibabu and S. K. Tripathy, Synth. Met., 1998, 98, 107–111 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, GPC and NMR spectra, and TGA thermograms. See DOI: 10.1039/c3py01069d |
| This journal is © The Royal Society of Chemistry 2014 |