Direct synthesis of poly(p-phenyleneethynylene)s from calcium carbide†
Nopparat
Thavornsin
a,
Mongkol
Sukwattanasinitt
b and
Sumrit
Wacharasindhu
*b
aProgram of Petrochemistry and Polymer Science, Faculty of Science, Chulalongkorn University, Bangkok 10330, Thailand. E-mail: nopparat.t@student.chula.ac.th
bNanotec-CU Center of Excellence on Food and Agriculture, Department of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok 10330, Thailand. E-mail: sumrit.w@chula.ac.th; Fax: +662 2187598; Tel: +662 2187634
First published on 12th August 2013
Abstract
An efficient method for the preparation of poly(p-phenyleneethynylene)s (PPEs) from direct coupling reactions between aryl diiodides and the inexpensive chemical feedstock calcium carbide is developed. A variety of PPEs can be prepared in high yields (71–93%) with the degree of polymerization between 36 and 128, offering fluorescence quantum efficiencies in the range of 0.34–0.71.
The Sonogashira reaction (Scheme 1) is among the most powerful tools for the construction of a C–C triple bond via Pd/Cu catalysed sp2–sp coupling reaction between aryl or alkenyl halides and terminal alkynes.1 Such reactions are essential for preparing acetylenic compounds that are important building blocks of natural products, pharmaceuticals and molecular materials.2 Due to environmental and economic pressures, over the past decade, development of this reaction has focused on new catalysts3 and new techniques such as microwave,4 ultrasonic irradiation5 or micro flow reactions.6 However, little investigation has been done on a more sustainable source of sp-carbon. Traditional synthetic methodology previously relied on the use of terminal alkynes such as substituted acetylenes, including expensive (trimethylsilyl)acetylene and acetylene gas,7 as sp-carbon providers. The former is a more favourable source due to its convenient handling but it is expensive and requires an extra deprotection step in some cases. The latter is appreciably more economical but its hazardous nature i.e. high flammability and explosiveness is prohibitive to broader adoption in routine laboratory experiments and industrial production. Therefore a better, safer and more economical source of acetylene is highly desirable for the Sonogashira reaction.
 | ||
| Scheme 1 Typical Sonogashira coupling reaction. | ||
Acetylene gas is produced by treating calcium carbide with water. The commercial process for making the calcium carbide is electric arch furnace of coke and lime. Since the first launch of a calcium carbide plant in 1894, it has become an important primary chemical feedstock for a broad range of commodity and specialty chemicals. With a constant low price and significant safety advantages over acetylene gas, if acetylenic derivatives can be synthesized directly from calcium carbide, such a process would offer significant cost and environmental advantages over existing reactions. The first direct use of calcium carbide in the Sonogashira reaction was reported in 2006 by Zhang,8 whereby symmetrical diarylethynes were synthesized in the presence of non-commercially available amino phosphine ligands. Subsequently, we reported the use of calcium carbide as a provider of sp-C in the synthesis of diarylethynes in the presence of a commercial Pd/Cu catalyst system.9 Recently, propagylamine derivatives have been prepared directly from calcium carbide via a Sonogashira type reaction.10 These examples demonstrated that calcium carbide could effectively serve as a C2 synthon in the synthesis of small molecular acetylenic derivatives,8–10 the transfer of this simple and economical source of acetylene into efficient synthesis of acetylenic containing polymer remains a challenge.
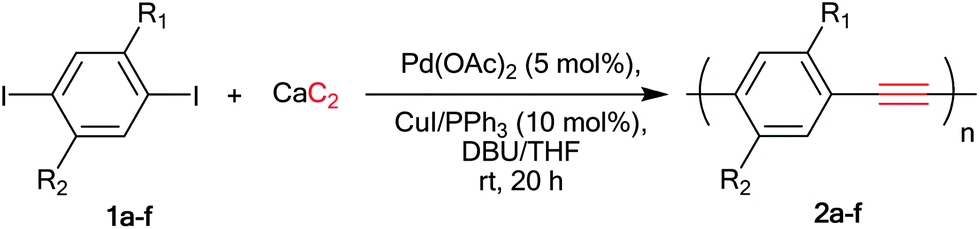
Poly(p-phenyleneethynylene)s (PPEs) are an attractive family of conjugated polymers in which aryl groups are fully π-conjugated by acetylene linkers. Such structural alignment extends electronic delocalization across the polymer backbone, providing properties such as charge mobility, electrical conductivity and photoluminescence.11 PPEs have found a large number of applications especially in the area of sensor,12 solar cell13 and display technologies.14 Typically, PPEs are prepared by polycondensation between aryl diiodide 1 and various acetylene sources through the Sonogashira coupling reaction as depicted in Scheme 2.
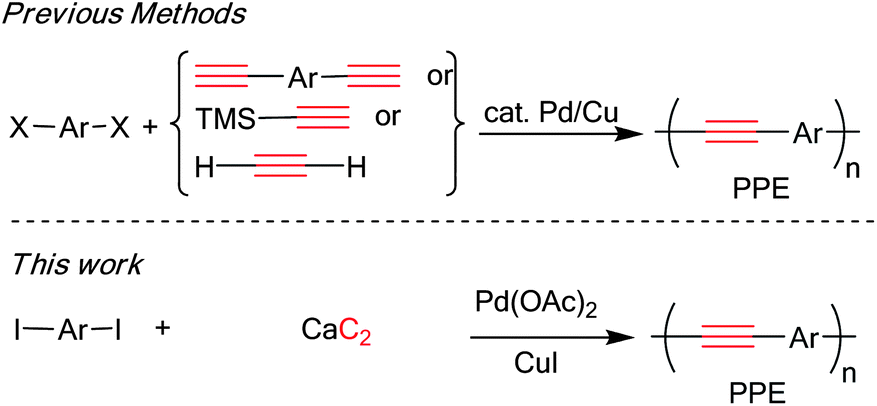 | ||
| Scheme 2 Synthetic method for PPEs via Sonogashira coupling. | ||
Diethynylarenes15 were first introduced as coupling partners and later on TMS-acetylene was used.2d,16 Later, the Bunz and Li groups reported the synthesis of PPEs directly from acetylene gas.17 This methodology gave high molecular weight homo-PPEs, considered to be economical due to the low price of acetylene gas. However, the success of this method relies heavily on close monitoring and regulating of stoichiometric acetylene gas, which require difficult equipment set up. We therefore investigated a direct synthesis of PPEs from calcium carbide which is less hazardous and provides a more economical carbon source than the acetylene gas. Herein, we would like to report that calcium carbide may be conveniently used in the condensation polymerization of diiodoarene to synthesize high molecular weight PPEs bearing various side chains (Scheme 2).
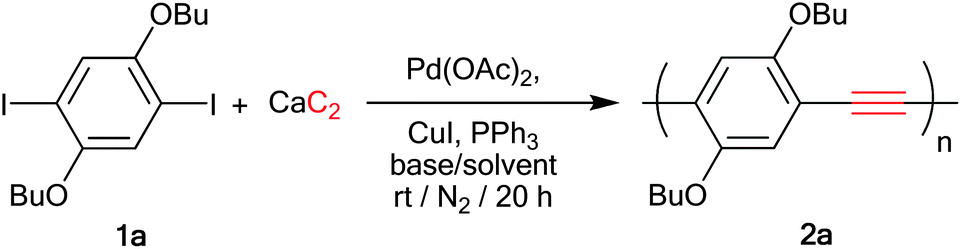
In our first model study, the polymerization was the coupling reaction between 1,4-dibutoxy-2,5-diiodobezene 1a and calcium carbide at room temperature in the presence of a Pd(OAc)2, CuI and PPh3 catalyst system. The resulting polymer was precipitated in excess methanol. The degree of polymerization was determined from the polymer solution in THF by gel permeation chromatography using standard polystyrene for universal calibration (Table 1). In the first attempt (entry 1), 1% of Pd(OAc)2 was used in a similar manner to the synthesis of the small molecules9 but the reaction gave only a low molecular weight oligomer, which was precipitated in methanol, along with the recovery of the starting diiodo compound. The polymerization was significantly improved by the increase of palladium amount to 5 mol%, which yielded 84% of a orange solid (entry 2) corresponding to the desired PPE (Mw = 11.7 kDa) without residual starting material 1a. Next, a set of experiments were carried out to determine the best solvent and base system (entries 2–7). The mixture of THF–DBU in a ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was found to be the best condition to produce the highest degree of polymerization (DPn = 36) and narrow polydispersity index (PDI) (Mw/Mn = 2.3) (entry 5). The slightly lower yield was mainly due to the loss of the high molecular weight portion, which was poorly soluble in CH2Cl2 solvent needed for the second precipitation.
:1 was found to be the best condition to produce the highest degree of polymerization (DPn = 36) and narrow polydispersity index (PDI) (Mw/Mn = 2.3) (entry 5). The slightly lower yield was mainly due to the loss of the high molecular weight portion, which was poorly soluble in CH2Cl2 solvent needed for the second precipitation.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Pd(OAc)2 (mol%) | Base/solvent | Yieldb (%) | M w (× 103) | DPnc | M w/Mnc |
| a Unless noted, CaC2 (6 equiv.), Pd(OAc)2, PPh3 (10 mol%), CuI (10 mol%) for 20 h. b Precipitation in CH3OH from CH2Cl2. c Determined with GPC using universal calibration with standard polystyrene. d TMS-acetylene were used instead of CaC2. | ||||||
| 1 | 1 | TEA/THF | N.A. | — | — | — |
| 2 | 5 | TEA/THF | 84 | 11.7 | 20 | 2.3 |
| 3 | 5 | TEA/MeCN | 90 | 6.8 | 13 | 2.1 |
| 4 | 5 | TEA/DMF | N.A. | — | — | — |
| 5 | 5 | DBU/THF | 71 | 20.2 | 36 | 2.3 |
| 6 | 5 | K2CO3/THF | N.A. | — | — | — |
| 7 | 5 | Cs2CO3/THF | N.A. | — | — | — |
| 8d | 5 | DBU/MeCN/THF | 100 | 9.6 | 17 | 2.3 |
The 1H and 13C NMR spectra of the orange solid product showed clean signals corresponding to PPE without end groups and diyne signals, suggesting a high molecular weight and defect free polymer (Fig. 1a and S13†). For comparison, the polymerization reaction between TMS-acetylene (entry 8) and 1a was tested using literature-reported conditions.16c The PPE obtained from these conditions also showed no diyne signal in 13C NMR (Fig. S19†) but displayed small peaks in the aromatic region suggesting the end group (Fig. 1b). The results agreed well with the GPC data showing a lower degree of polymerization (DPn = 17) comparing with the polymer obtained from our optimized calcium carbide method (entry 5). Both CaC2 and TMS-acetylene polymerization (entries 5 and 8) gave higher molecular weight and lower PDI than those from the conventional Pd-catalyzed coupling polymerization using the terminal alkynes.14g The high molecular weight and narrow PDI of PPEs have been attributed to the slow formation of the alkyne terminated monomers and oligomers that reduces the chance of their homocoupling.17b The homo-coupling side reaction causes the stoichiometric imbalance of the iodo and terminal alkyne groups that lead to low molecular weight and high PDI. The formation of the alkyne terminated monomers and oligomers in the CaC2 polymerization is governed by either the slow release of acetylene gas or calcium acetylide from CaC2. The release of these active species is self-controlled by HI, a by-product generated from the alkyne/iodocondensation,9 which is quite different from the direct use of acetylene gas that required very slow stirring.17b With the optimized conditions in hand, we then tested compatibility of the reaction with diiodobenzene containing different side chains. A series of diiodobenzenes (1b–f), prepared according to published procedures, were subjected to the Sonogashira coupling reaction with CaC2 under the optimized reaction conditions (Table 1, entry 5) and the results are summarized in Table 2. The dialkoxy-PPEs containing simple alkyl chains, such as 2b, 2c and 2d, were synthesized efficiently in good to excellent yields (Table 2, entries 2–4). Polymerization of 1c, bearing double ethylhexyloxy groups, gave the PPE with highest DPn = 128 and PDI = 2.0 when the reaction was carried out at room temperature for 20 h. This high degree of polymerization is attributed to the excellent solubility of the corresponding polymer in the reaction medium. This result suggested that the lower degree of polymerization of the other monomers was thus limited by the polymer solubility rather than the reaction efficiency. Interestingly, the polymerization condition is also compatible with the monomers containing multiple oxygenous substituents such as 1e and 1f (Table 2, entries 5 and 6). PPE 2e and 2f were prepared in 87 and 93% yields, respectively, as shown by the absence of end groups in the aromatic region in 1H NMR spectra (Fig. S17 and S18†), suggesting efficient polymerization of the monomers. The GPC measurement showed the degree of polymerization of 2eca. 51 while that of 2f could not be determined due to its poor solubility in THF. All PPEs were obtained as deep orange solids which typically gave blue to green emission in chloroform solution, except 2f which was soluble only in DMSO (Table S1†). The photophysical properties of the polymers including maximum absorption wavelength (λabs), maximum emission wavelength (λem), molar absorptivity (ε) and fluorescence quantum efficiency (Φf) were determined as presented in Table 2. Typically, each PPE exhibited a broad absorption band with the absorption maximum around 445 nm and showed the emission maximum around 475 nm, representing a rather small Stoke’s shift (Fig. S20†). These PPEs also gave relatively high quantum efficiencies (0.34–0.71). These photophysical parameters are consistent with the defect free π-conjugated PPEs reported with other polymerization methods,2d especially the high quantum efficiency.
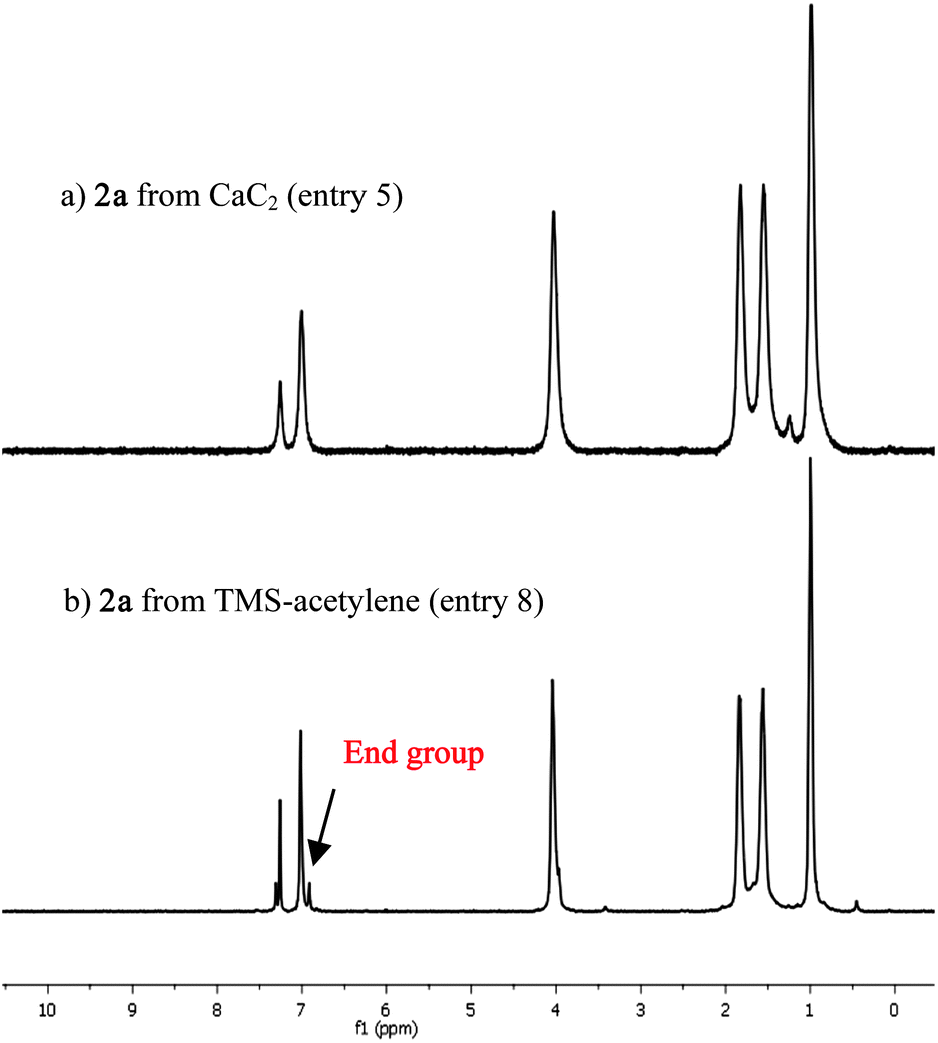 | ||
| Fig. 1 1H NMR spectrum of PPE 2a derived from: (a) CaC2; (b) TMS-acetylene. | ||
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Substituents | Yielda (%) | M w (× 103) | DPnb | M w/Mnb | λ abs/nm | λ em/nm | Logε |
Φ f |
| a Isolated yield by reprecipitation from CH3OH/CH2Cl2. b Determined by GPC using PS as reference. c Dissolved in CH2Cl2 and used quinine sulfate as reference standard. d Performed in DMSO. | |||||||||
| 1 | X: R1 = R2 = O(CH2)3CH3: 1a | 71 | 20.2 | 36 | 2.3 | 447 | 476 | 4.32 | 0.65 |
| 2 | X: R1 = R2 = O(CH2)7CH3: 1b | 78 | 40.9 | 48 | 2.4 | 448 | 476 | 4.30 | 0.66 |
| 3 | X: R1 = R2 = OCH2CH(CH2CH3)((CH2)3CH3): 1c | 83 | 91.3 | 12 | 2.0 | 48 | 479 | 4.41 | 0.71 |
| 4 | X: R1 = OMe, R2 = OCH2CH(CH2CH3)((CH2)3CH3): 1d | 93 | 26.9 | 47 | 2.2 | 450 | 478 | 4.02 | 0.67 |
| 5 | X: R1 = R2 = O(CH2)2O(CH2)2OCH3: 1e | 87 | 26.9 | 51 | 1.5 | 430 | 470 | 4.02 | 0.67 |
| 6 | X: R1 = R2 = O(CH2)3OH: 1f | 93 | N.A. | N.A. | N.A. | 445 | 481 | 4.11 | 0.34d |
In conclusion, this work demonstrates a novel and efficient polymerization of diiodobenzene derivatives to the corresponding defect free PPEs, directly from a low cost feedstock, calcium carbide, at ambient temperature. The polymerization method is compatible with various alkyl, ethylene glycol and aliphatic alcohol substituents. The extension of this polymerization method for preparation of PPEs with more functional pendant groups is in progress.
The authors wish to thank Prof. Pierre Dixneuf for discussions. This study is financially supported by the Thailand Research Fund (TRF-RSA5480004) and Nanotechnology Center (NANOTEC), NSTDA, Ministry of Science and Technology, Thailand, through its program of Center of Excellence Network. This work is part of the Project for Establishment of Comprehensive Center for Innovative Food, Health Products and Agriculture supported by the Thai Government Stimulus Package 2 (TKK2555, SP2), the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission (AM1006A-56) and the Ratchadaphiseksomphot Endowment Fund of Chulalongkorn University (RES560530126-AM).
Notes and references
- (a) K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef; for a review see: (b) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed; (c) H. Doucet and J. C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834–871 CrossRef CAS PubMed; (d) R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- (a) M. Yamashita, H. Horiguchi and K. Hirano, J. Org. Chem., 2009, 74, 7481–7488 CrossRef CAS PubMed; (b) C. J. Li, W. T. Salaven, IV, V. T. John and S. Banergee, Chem. Commun., 1997, 1569–1570 RSC; (c) M. Iyoda, A. Vorasingha and Y. Kuwatani, Tetrahedron Lett., 1998, 39, 4701–4704 CrossRef CAS; (d) U. H. F. Bunz, Macromol. Rapid Commun., 2009, 30, 772–805 CrossRef CAS PubMed.
- (a) M. B. Thathagar, J. Beckers and G. Rothenberg, Green Chem., 2004, 6, 215–218 RSC; (b) C. G. Frost and L. Mutton, Green Chem., 2010, 12, 1687–1703 RSC; (c) R. Thorwirth, A. Stolle and B. Ondruschka, Green Chem., 2010, 12, 985–991 RSC; (d) H. Doucet and J. C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834–871 CrossRef CAS PubMed; (e) S. Liu and J. Xiao, J. Mol. Catal. A: Chem., 2007, 270, 1–43 CrossRef CAS PubMed; (f) C. A. Fleckenstein and H. Plenio, Chem.–Eur. J., 2007, 13, 2701–2716 CrossRef CAS PubMed; (g) L. Yang, L. Zhao and C. J. Li, Chem. Commun., 2010, 46, 4184–4186 RSC; (h) R. Luque and D. J. MacQuarrie, Org. Biomol. Chem., 2009, 7, 1627–1632 RSC.
- (a) P. Appukkuttan, W. Dehaen and E. V. D. Eycken, Eur. J. Org. Chem., 2003, 4713–4716 CrossRef CAS; (b) H. Huang, H. Liu, H. Jiang and K. Chen, J. Org. Chem., 2008, 73, 6037–6040 CrossRef CAS PubMed; (c) K. M. Dawood, W. Solodenko and A. Kirschning, ARKIVOC, 2007, 5, 104–124 Search PubMed.
- (a) A. R. Gholap, K. Venkatesan, R. Pasricha, T. Daniel, R. J. Lahoti and K. V. Srinivasan, J. Org. Chem., 2005, 70, 4869–4872 CrossRef CAS PubMed; (b) S. S. Palimkar, P. H. Kumar, R. J. Lahoti and K. V. Srinivasan, Tetrahedron, 2006, 62, 5109–5115 CrossRef CAS PubMed.
- (a) L. M. Tan, Z. Y. Sem, W. Y. Chong, X. Liu, Hendra, W. L. Kwan and C. L. Lee, Org. Lett., 2013, 15, 65–67 CrossRef CAS PubMed; (b) R. Javaid, H. Kawanami, M. Chatterjee, T. Ishizaka, A. Suzuki and T. M. Suzuki, Chem. Eng. J., 2011, 167, 431–435 CrossRef CAS PubMed; (c) B. K. Singh, N. Kaval, S. Tomar, E. V. D. Eycken and V. S. Parmar, Org. Process Res. Dev., 2008, 12, 468–474 CrossRef CAS; (d) C. G. Frost and L. Mutton, Green Chem., 2010, 12, 1687–1703 RSC.
- (a) C. J. Li, D. Li and C. Costello, Org. Process Res. Dev., 1997, 1, 325–327 CrossRef CAS; (b) M. Pal and N. G. Kundu, J. Chem. Soc., Perkin Trans. 1, 1996, 449–451 RSC; (c) J. Moon, M. Jeong, H. Nam, J. Ju, J. H. Moon, H. M. Jung and S. Lee, Org. Lett., 2008, 10, 945–948 CrossRef CAS PubMed.
- W. Zhang, H. Wu, Z. Liu, P. Zhong, L. Zhang, X. Huang and J. Cheng, Chem. Commun., 2006, 4826–4828 RSC.
- P. Chuentragool, K. Vongnam, P. Rashatasakhon, M. Sukwattanasinitt and S. Wacharasindhu, Tetrahedron, 2011, 67, 8177–8182 CrossRef CAS PubMed.
- Z. Lin, D. Yu, Y. N. Sum and Y. Zhang, ChemSusChem, 2012, 5, 625–628 CrossRef CAS PubMed.
- (a) U. H. F. Bunz, Chem. Rev., 2000, 100, 1605–1644 CrossRef CAS PubMed; (b) U. H. F. Bunz, Adv. Polym. Sci., 2005, 177, 1–52 CrossRef CAS; (c) J. Zheng and T. M. Swager, Adv. Polym. Sci., 2005, 177, 151–179 CrossRef CAS; (d) G. Voskerician and C. Weder, Adv. Polym. Sci., 2005, 177, 209 CrossRef CAS.
- (a) T. S. Corbitt, L. Ding, E. Ji, L. K. Ista, K. Ogawa, G. P. Lopez, K. S. Schanze and D. G. Whitten, Photochem. Photobiol. Sci., 2009, 8, 998–1005 RSC; (b) E. L. Dane, S. B. King and T. M. Swager, J. Am. Chem. Soc., 2010, 132, 7758–7768 CrossRef CAS PubMed; (c) G. He, N. Yan, J. Yang, H. Wang, L. Ding, S. Yin and Y. Fang, Macromolecules, 2011, 44, 4759–4766 CrossRef CAS; (d) I. B. Kim, B. Erdogan, J. N. Wilson and U. H. F. Bunz, Chem.–Eur. J., 2004, 10, 6247–6254 CrossRef CAS PubMed; (e) R. L. Phillips, O. R. Miranda, D. E. Mortenson, C. Subramani, V. M. Rotello and U. H. F. Bunz, Soft Matter, 2009, 5, 607–612 RSC; (f) R. L. Phillips, O. R. Miranda, C. C. You, V. M. Rotello and U. H. F. Bunz, Angew. Chem., Int. Ed., 2008, 47, 2590–2594 CrossRef CAS PubMed; (g) S. W. Thomas, III, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef PubMed; (h) J. H. Wosnick, C. M. Mello and T. M. Swager, J. Am. Chem. Soc., 2005, 127, 3400–3405 CrossRef CAS PubMed; (i) X. Zhao, Y. Liu and K. S. Schanze, Chem. Commun., 2007, 2914–2916 RSC.
- (a) H. Hoppe, D. A. M. Egbe, D. Mühlbacher and N. S. Sariciftci, J. Mater. Chem., 2004, 14, 3462–3467 RSC; (b) J. K. Mwaura, X. Zhao, H. Jiang, K. S. Schanze and J. R. Reynolds, Chem. Mater., 2006, 18, 6109–6111 CrossRef CAS; (c) F. Silvestri and A. Marrocchi, Int. J. Mol. Sci., 2010, 11, 1471–1508 CrossRef CAS PubMed.
- (a) C. A. Breen, S. Rifai, V. Bulović and T. M. Swager, Nano Lett., 2005, 5, 1597–1601 CrossRef CAS PubMed; (b) A. Montali, P. Smith and C. Weder, Synth. Met., 1998, 97, 123–126 CrossRef CAS; (c) N. G. Pschirer, T. Miteva, U. Evans, R. S. Roberts, A. R. Marshall, D. Neher, M. L. Myrick and U. H. F. Bunz, Chem. Mater., 2001, 13, 2691–2696 CrossRef CAS; (d) C. Schmitz, P. Pösch, M. Thelakkat, H. W. Schmidt, A. Montali, K. Feldman, P. Smith and C. Weder, Adv. Funct. Mater., 2001, 11, 41–46 CrossRef CAS; (e) A. F. Thünemann and D. Ruppelt, Langmuir, 2001, 17, 5098–5102 CrossRef; (f) Y. Xu, P. R. Berger, J. N. Wilson and U. H. F. Bunz, Appl. Phys. Lett., 2004, 85, 4219–4221 CrossRef CAS; (g) J. Shinar, L. S. Swanson, F. Lu and Y. Ding, US Pat., 5 334 539, 1994.
- R. Giesa and R. C. Schulz, Makromol. Chem., 1990, 191, 857–867 CrossRef CAS.
- (a) M. Banno, T. Yamaguchi, K. Nagai, C. Kaiser, S. Hecht and E. Yashima, J. Am. Chem. Soc., 2012, 134, 8718–8728 CrossRef CAS PubMed; (b) A. Khan and S. Hecht, Chem. Commun., 2004, 300–301 RSC; (c) A. Khan, S. Müller and S. Hecht, Chem. Commun., 2005, 584–586 RSC.
- (a) C. J. Li, W. T. Slaven, IV, Y. P. Chen, V. T. John and S. H. Rachakonda, Chem. Commun., 1998, 1351–1352 RSC; (b) J. N. Wilson, S. M. Waybright, K. McAlpine and U. H. F. Bunz, Macromolecules, 2002, 35, 3799–3800 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data of 1a–f and PPE 2a–f. See DOI: 10.1039/c3py01068f |
| This journal is © The Royal Society of Chemistry 2014 |