
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The structure and UV spectroscopy of benzene–water (Bz–W6) clusters using time-dependent density functional theory†
Divya
Sharma
and
Martin J.
Paterson
*
Institute of Chemical Sciences, School of Engineering and Physical Sciences, Heriot Watt University, Edinburgh, EH14 4AS, UK. E-mail: m.j.paterson@hw.ac.uk
First published on 1st September 2014
Abstract
DFT and MP2 calculations are performed to obtain optimized ground state geometries and binding energies of the cage and the prism conformers of water W6 clusters and Bz–W6 clusters using the aug-cc-pVDZ basis set. The cage conformer of Bz–W6 system is found to be more stable than prism conformer for all range of DFT functionals and MP2. Time dependent-DFT is then used to study UV spectroscopy of Bz, water W6 clusters and Bz–W6 clusters at both the MP2 and wB97XD optimized ground state geometries using the B3LYP, CAM-B3LYP and M06-2X functionals with 6-31++G(d,p) and aug-cc-pVTZ basis sets. Our results predict minor differences in the UV spectroscopy of cage and prism conformers W6 and Bz–W6 clusters that may be observable with high-resolution spectroscopy. The M06-2X and CAM-B3LYP functionals perform consistently with each other. Benzene-mediated excitations of the water W6 cluster towards longer wavelengths above 170 nm are noticed in both the cage and prism geometries of Bz–W6. Benzene is found to be influenced after interacting with the cage and prism W6 geometries, and is seen to undergo a red shift in the main π→π* electronic transition, in which the degeneracy is slightly broken. Charge transfer (CT) states and diffuse Rydberg-type states are also found to play an important role in the spectroscopy of such systems.
Introduction
Water clusters are of fundamental importance in many areas of chemistry, and are also a useful model system to understand the relation of the properties of the gas phase clusters to the condensed phase, ice, and liquid water. The structures and properties of water Wn clusters have been studied extensively both theoretically and experimentally because of their importance in many physical, chemical and biological fields, such as in the understanding of cloud and ice formation, bio-chemical processes, etc.1–11 The interactions of aromatic molecules with water solvents, and the role of these weak interactions to determine the physical and chemical properties of such systems is an area of particular interest for many theoretical and experimental studies. In this context, the simplest system of this type, benzene–water complexes, considering benzene as a prototype of intermolecular interactions involving aromatic systems, has been studied extensively.1,12–23 The study of complexes of benzene (Bz) with water clusters is also of considerable astrophysical importance and can replicate polycyclic aromatics-ice systems to gain better understanding on the interactions of polycyclic aromatic hydrocarbons (PAHs), which may account for up to 20% of galactic carbon,24,25 with water being the most abundant molecule in icy grain mantles.26 Benzene has been detected in the proto-planetary nebula CRL 61827,28 and is amongst the list of known interstellar molecules. Benzene may be thought of as a prototypical PAH compound that plays an important role as an intermediate in the formation of PAHs from acetylene,28–31 and water clusters can be a good representation of interstellar ice surfaces. It has been observed that UV irradiation of water ice containing PAHs may play an important role in the formation of complex organic species such as alcohols, quinones and ethers.32 Photo-desorption is an important process to account for the high gas phase abundances of water under astrophysically relevant conditions.33,34 Benzene and its derivatives, also being environmental pollutants35,36 and the constituents of many organic products, e.g. solvents, perfumes, etc. can be efficiently excited electronically by UV radiation present in the environment. Therefore, the computational study of the ground and excited states of the complexes of benzene with water clusters is of possible environmental as well as astrophysical relevance.Numerous theoretical and experimental studies have been performed on the ground state properties such as binding energies and IR spectra of Bz–Wn clusters, and also on non-covalent interactions such as hydrogen bonding interactions that dominate such systems.1,6,11,17–20,37–41 Zwier and their co-workers15,18–20,22,23,39 have undertaken detailed experimental studies on Bz–Wn clusters using resonant ion-dip spectroscopy (RIDIRS) and resonant two-photon ionization (R2PI) techniques. It was observed that the water O–H stretch spectra depends on the size of cluster and is sensitive to the number, strength and the type of hydrogen bonds in which it participates while C–H stretch in benzene was found insensitive to the size of water cluster with n ≤ 7.20 For n = 3–5, the water cluster was considered to bind to benzene primarily through single water molecule on or near benzene six-fold axis and the O–H spectra was consist of free O–H stretches, π H-bonded O–H stretches and the single donor O–H stretches. The significant spectral signature was observed at n ≥ 6 in O–H stretch spectra, showing new transitions that are associated with the double-donor O–H stretches, which exist, in the more compact large Bz–Wn clusters. Experimental evidence predicted the change in shape of cluster from cyclic to non-cyclic at n = 6 such that Bz–W6 cluster was found likely to exist as a cage shaped arrangement of six water molecules giving the lowest energy structure.
A detailed computational study on geometries, binding energies and infrared (IR) spectra of Bz–Wn (n = 1–10) clusters has been carried out by Prakash et al.17 using hybrid meta DFT based M05-2X method with the 6-31+G** basis set. Using Bader's theory of atoms in molecule (AIM) approach, it is found that the nonconventional H-bonding interactions, such as O–H⋯π interactions are present in all these clusters with additional contributions from C–H⋯O and lone pair (lp)⋯π interactions, leads to overall stability of these clusters. It is also noted that π H-bonded O–H stretching vibrational frequencies are red shifted in all the clusters. The inverted book conformer of water hexamer is found to have highest binding energy in compared to all other water clusters.17 Slipchenko et al.42 investigated the structures and bonding in water Wn (n = 1–8)-Bzn (n = 1–2) complexes computationally using an effective fragment potential (EFP) method. It is predicted that benzene can act both as an H-bond donor and acceptor in the water-benzene complexes and all of the larger water-benzene complexes are dominated by H-bonding interactions i.e., O–H⋯π and C–H⋯O interactions. The interactions between such systems are considered complicated due to the existence of various possible structures that depend on the number and type of hydrogen bonds involved in these complexes as the benzene–water interactions are weaker than the water–water interactions and the very small energy differences between different isomers are involved in such complexes.
Excited state properties of such systems have received very little attention as of yet. Computational studies on low lying excited states of Bz–Wn (n = 1–6) complexes have been performed by Upadhyay et al.43 using configuration interaction method involving all the singly excited configurations (CIS). The binding energies of these clusters were also calculated and complex of water dimer with benzene (Bz–W2) is found to be the most stable among various complexes, with a high stability of Bz–W6 complex as well. It is found that the whole Bz–Wn complex as well as the water cluster Wn undergoes an expansion in size following excitation to lowest singlet excited states. The small blue shifts in the excitation energies are observed in the electronic absorption spectra in going from Bz to higher complexes (Bz–Wn (n = 4–6)). Photon-stimulated desorption in an astrophysical context has not been studied extensively until recently. A few experiments have been performed on photo processing of laboratory models of Bz-ice complexes by UV radiation.44–46 Experimental study44 on photo-processes in model interstellar ices by modeling multilayer films of benzene & water deposited on a sapphire substrate at a temperature of around 80 K has investigated three distinct photo-desorption mechanisms in such systems: (i) direct adsorbate-mediated desorption of benzene; (ii) indirect adsorbate-mediated desorption of water; (iii) substrate-mediated desorption of both benzene and water. The translational temperature of both desorbed species i.e., benzene and water molecule is found to be more than the ambient temperature of the complex system. Recent experimental study on photon- and electron-induced desorption from laboratory models of interstellar ice grains by mimicking the conditions found in dense interstellar clouds has been performed by Thrower et al.46 The desorption cross sections and first order rate coefficients for the desorption processes in benzene–water (Bz–Wn) complexes have been obtained. It is observed that photon absorption by benzene can make H2O desorption possible at wavelengths where photon-absorption cross-section for H2O is negligible.
The electronically excited states play an important role in the photochemistry and electronic spectroscopy and are the subject of our investigations detailed below. The main aim of the present study is to investigate the spectroscopy and photochemistry of interstellar ice analogs i.e., Bz–W6 cluster quantum mechanically to provide insight into elementary process involved in their processing, and to study important electronic transitions involved in such systems using time dependent DFT with range of well developed DFT functionals for response theory.
It has been observed from earlier studies that the water hexamer (H2O)6 cluster is the smallest water cluster that allows non-cyclic structures and a more three dimensional structure, it can be considered as the building block of many ice forms.1,3 We chose a water hexamer (W6) cluster since this is frequently taken as a benchmark system for many computational chemistry studies.1–3,47–49 The hydrogen-bond arrangements of these six water molecules in the cluster determine the shape and stability of the cluster. A few low-lying iso-energetic conformers of the water hexamer cluster are ring, book, cage and prism with 6, 7, 8 and 9 number of hydrogen bonds, respectively. There have been uncertainties in literature on predicting the exact order in energetics of very close lying water hexamer conformers. Previous theoretical investigations have found the cage shaped water W6 cluster, with four dangling hydrogen atoms (non-hydrogen bonded atoms) and eight hydrogen bonds, to be the most stable one giving the minimum energy, among the other low lying conformers i.e., prism, book, ring and chair forms.2,5,11,49 DFT-D calculations on the water hexamer has predicted the energetic order prism < cage < book < chair.50,51 Most recent computational studies have predicted prism, cage and book conformers as the three lowest energy conformers with ZPE correction, with prism as the lowest energy structure, and cage conformer is found to be very closer to prism in energy.9,47 A very recent theoretical study on the water hexamer using full dimensional Diffusion Quantum Monte-Carlo simulations has predicted the presence of cage and prism conformers at low temperatures,10 and in agreement with the experimental measurements of broadband rotational spectra of the water hexamer formed in supersonic expansion.48 Experimental study by Pate et al. has also established the cage conformer of water hexamer as the global minimum energy structure.48 Taking account of both theoretical9 and experimental48 evidences of existence of cage and prism conformers at low temperatures, that are relevant to astrophysical interstellar conditions, and are also established as lowest energy conformers of water hexamer by very recent studies,9,47,48 we have chosen both the cage and prism structures as model systems in our present study.
The benzene (Bz) molecule is then brought closer to the water W6 cluster and it binds to the water cluster through hydrogen bonded interactions and thus form Bz–W6 complex system. The benzene molecule binds to the water cluster mainly through O–H⋯π hydrogen bonds where one of the free dangling hydrogen atom of the water cluster points toward the π electron cloud of the benzene ring.17,40,42 Small contributions from C–H⋯O and lone pair (lp)⋯π hydrogen bonded interactions also stabilize the Bz-(H2O)6 cluster.17 Both cage and prism geometries of water W6 cluster interact with Bz to give Bz–W6 cluster and are predicted to retain their cage and prism structures in Bz–W6 cluster too.
Results and discussion
Ground state structures
The ground state geometries of both the cage and prism conformers of W6 clusters and Bz–W6 clusters were optimized using density functional theory with Truhlar's meta hybrid functional M05-2X functional and the 6-31+G(d,p) basis set. M05-2X & M06-2X are non-local functionals with double the amount of non-local (Hartree–Fock) exchange, and are found to perform better than standard hybrid functionals for systems involving non-covalent interactions, and modeling electronic excitation energies to both valence and Rydberg states.52,53In order to properly investigate any long range correction due to dispersion effects in such complex systems, the obtained optimized ground state geometries were further re-optimized using the following range of hybrid functionals LC-wPBE, wB97X, wB97XD, B2PLYP, B2PLYPD, in addition to second order Moller–Plesset perturbation theory (MP2), and employing the augmented correlation-consistent polarized-valence double zeta basis set (aug-cc-pvDZ). Long range corrected functionals such as LC-wPBE, CAM-B3LYP, wB97X, wB97XD functionals account the non-Coulomb part of exchange functionals, which typically dies off too rapidly and gives inaccurate results at large distances, where wB97XD functional also includes an empirical atom-atom dispersion corrections.54–56Fig. 1(a) and (b) shows the MP2/aug-cc-pVDZ optimized geometries of cage and prism form of Bz–W6 cluster, respectively.
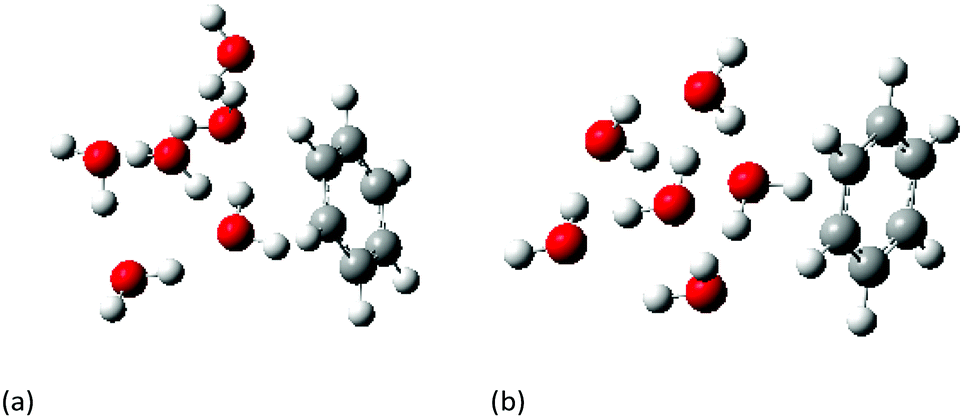 | ||
| Fig. 1 MP2/aug-cc-pVDZ optimized geometries of C6H6−(H2O)6 clusters, (a) cage form (b) prism form. | ||
The binding energy (BE) of the Bz–W6 cluster is calculated using
| |BE| = (EBz–W6− (EW6 + EBz)) | (1) |
| Prism | LC-wPBE | wB97X | wB97XD | MP2 | B2PLYP | B2PLYPD |
|---|---|---|---|---|---|---|
| E Bz–W6 (au) | −690.5773 | −690.8295 | −690.7965 | −689.197 | −690.377 | −690.3948 |
| E W6 (au) | −458.4780 | −458.6150 | −458.5969 | −457.6430 | −458.351 | −458.3600 |
| E Bz (au) | −232.0941 | −232.2051 | −232.1888 | −231.540 | −232.019 | −232.0236 |
| BE (kcal mol−1) | 3.24 (2.47) | 5.84 (4.76) | 6.80 (5.65) | 9.04 (4.35) | 3.93 (2.54) | 7.03 (4.79) |
| ZPE corrected | ||||||
| E Bz–W6 (au) | −690.3237 | −690.5751 | −690.5415 | −688.946 | −690.126 | −690.1432 |
| E W6 (au) | −458.3271 | −458.4630 | −458.4445 | −457.492 | −458.201 | −458.2097 |
| E Bz (au) | −231.9923 | 232.10382 | −232.0878 | −231.440 | −231.919 | −231.9237 |
| BE (kcal mol−1) (with ZPE) | 2.69 | 5.20 | 5.76 | 8.08 | 3.20 | 6.12 |
| BE (kcal mol−1) from previous computational studies: 4.63a, 4.70b, 5.0d | ||||||
| Cage | LC-wPBE | wB97X | wB97XD | MP2 | B2PLYP | B2PLYPD |
|---|---|---|---|---|---|---|
| a Taken from ref. 17. Method: M05-2X/6-31+G** and ZPE correction. b Taken from ref. 49. Method: MP2/6-31+G** with 50% BSSE correction. c Taken from ref. 43. Method: B3LYP/6-31+G** and MP2 single point. d Taken from ref. 17. Method: M05-2X/6-31+G** and BSSE correction. | ||||||
| E Bz–W6 (au) | −690.5781 | −690.8295 | −690.7971 | −689.198 | −690.378 | −690.3957 |
| E W6 (au) | −458.4780 | −458.6141 | −458.5966 | −457.642 | −458.351 | −458.3590 |
| E Bz (au) | −232.0942 | −232.2051 | −232.1888 | −231.540 | −232.019 | −232.024 |
| BE (kcal mol−1) | 3.69 (2.67) | 6.41 (5.41) | 7.35 (6.14) | 9.91 (5.18) | 4.68 (2.94) | 7.88 (5.58) |
| ZPE corrected | ||||||
| E Bz–W6 (au) | −690.3245 | −690.5754 | −690.541 | −688.947 | −690.127 | −690.1439 |
| E W6 (au) | −458.3275 | −458.4626 | −458.444 | −457.492 | −458.201 | −458.209 |
| E Bz (au) | −231.9923 | 232.1038 | −232.087 | −231.440 | −231.919 | −231.923 |
| BE (kcal mol−1) (with ZPE) | 2.91 | 5.66 | 6.19 | 8.73 | 3.74 | 6.76 |
| BE (kcal mol−1) from previous computational studies: 5.65a, 4.94b, 5.69c, 6.62d | ||||||
In order to get correct energetics of both the cage and prism forms of Bz-W6 clusters, basis set superposition error (BSSE) is also calculated using the counterpoise (CP) method and corrected binding energies are also listed in Table 1. Bindings energies are reduced noticeably after applying BSSE correction and it ranges from 2.67–6.14 kcal mol−1 for the cage conformer and 2.47–5.65 kcal mol−1 for the prism conformer.
Fig. 2(a) and 2(b) depict the variation of BE's of the prism and cage forms of Bz–W6 clusters versus different DFT hybrid functionals and MP2 with ZPE and BSSE correction, respectively. It is clearly seen from these results that the cage conformer is more stable than the prism conformer, for all of the various functionals employed, as the cage form is found to have higher binding energy (BE) than the prism form, which is also consistent with previous computational studies.17,40,49 The magnitude of BE's of Bz–W6 cluster (with ZPE correction) for both the cage and prism forms with different computational methods vary in the following order: LC-wPBE < B2PLYP < wB97X < wB97XD < B2PLYPD < MP2. With ZPE correction, Bz–W6 cluster is found to be most stable at the MP2 level with the highest BE, and least stable at the LC-wPBE level of calculation. However with BSSE correction, the ‘wB97XD’ functional gives higher binding energies than MP2, that clearly indicates that BSSE error is higher for wavefunction methods and energies are overestimated by MP2 level of calculations as shown in Fig. 3(a) and 3(b) for both prism and cage conformers, respectively. The MP2 method is used extensively in computational chemistry and is considered reliable for ground state geometry optimizations and to estimate the energies of the loosely bound hydrogen bonded and dispersion bound complexes by accounting the electron-correlation effects including dispersion.57 Taking into account the reliability and success of MP2 method in the past, and highest BE values with ZPE correction for our Bz–W6 clusters, we have used MP2 optimized ground state geometries for main discussion on TD-DFT studies.
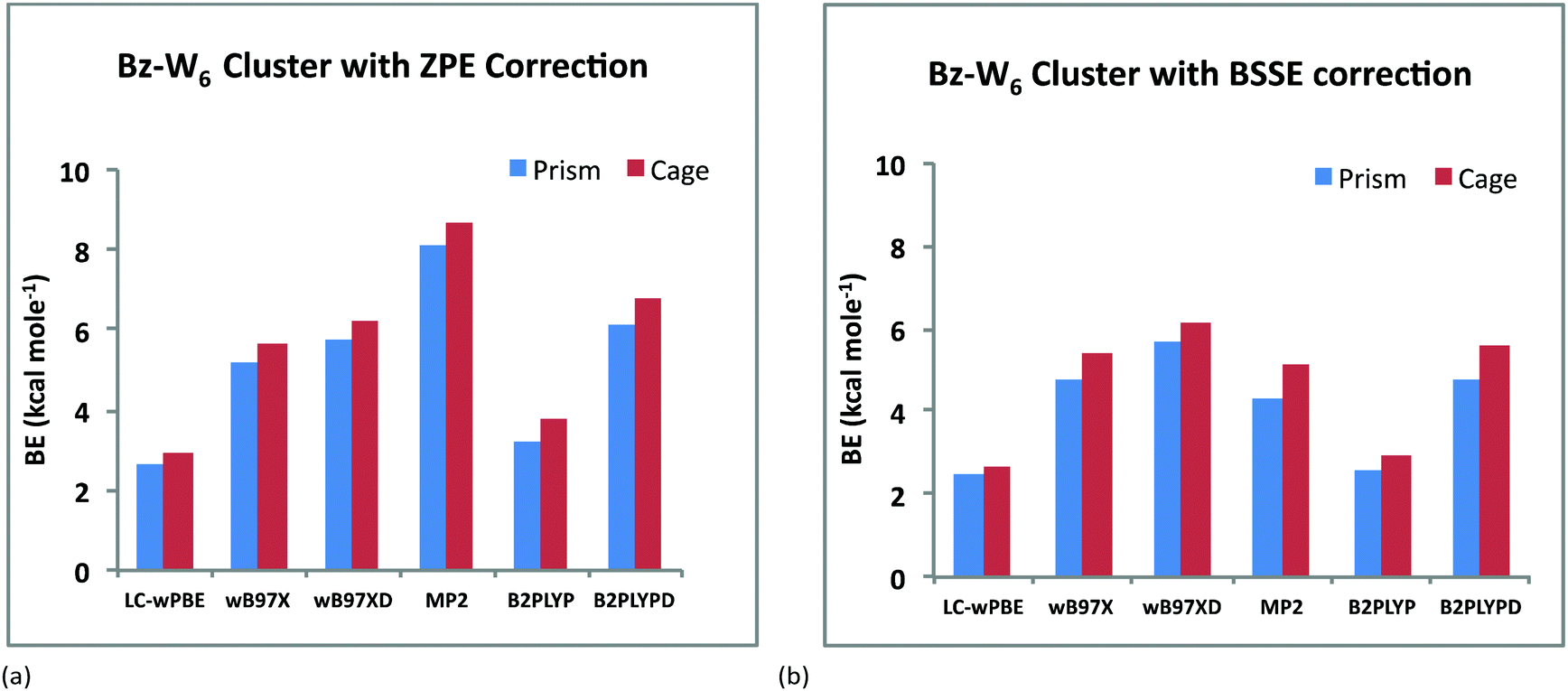 | ||
| Fig. 2 Variation of BE's (kcal mol−1) of Bz–W6 clusters versus different DFT hybrid functionals and MP2 method for both prism and cage forms: (a) with ZPE correction (b) with BSSE correction. | ||
 | ||
| Fig. 3 Variation of BE's (kcal mol−1) (with and without BSSE correction) of Bz–W6 clusters versus different DFT hybrid functionals and MP2 method (a) prism conformer (b) cage conformer. | ||
Electronic excitations in Bz–W6 clusters
In recent years, TD-DFT linear response theory58–61 has become the most widely used electronic structure method for calculating vertical electronic excitation energies. These TD-DFT calculations provide the Ultraviolet (UV) absorption spectra giving the information about the excitation wavelength and oscillator strength (f) of each excited state.The oscillator strength can be written as,62
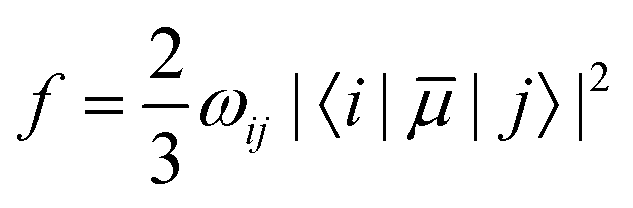 | (2) |
![[small mu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0cd.gif) |j〉|2 is the associated transition dipole moment.
|j〉|2 is the associated transition dipole moment.
We have performed time-dependent DFT calculations on both MP2/aug-cc-pVDZ and wB97XD/aug-cc-pVDZ optimized ground state geometries of Bz, W6 and Bz–W6 clusters using three different functionals i.e., B3LYP, CAM-B3LYP and M06-2X with the 6-31++G(d,p) basis set.
UV spectrum of benzene shows three well-known absorption bands at 4.9, 6.20, and 6.94 eV related to three electronic excitations from the ground state to the excited states with symmetries 1B2U, 1B1U, and 1E1U, respectively.63 The calculated valence π→π* excitation energies (experimental values in parenthesis) using M06-2X functional are 1B2U: 5.47(4.963,64), 1B1U: 6.27(6.20,636.1964), and 1E1U: 6.98(6.94,636.9664) eV, respectively. Our results show good agreement with the experiments. 1B1U and 1E1U excited states are computed with an accuracy of about 0.07 eV, while overestimated by about 0.5 eV for 1B2U. CAM-B3LYP and B3LYP functional also compare well with the experiments and values are listed in Tables 2 and 3.
| DFT functional | E (eV) | λ (nm) | Oscillator strength (f) | Electronic transition |
|---|---|---|---|---|
| Previous work:a Exptl/taken from ref. 63.b Exptl/taken from ref. 64.c Exptl/taken from ref. 65.d Exptl/taken from ref. 66. | ||||
| M06-2X | 5.45 (5.47, 4.9a,b) | 227.6 (226.8) | 0.0001 (0.0000) | π→π* |
| 6.24 (6.27, 6.20a,c, 6.19b) | 198.6 (197.5) | 0.00014 (0.0000) | π→π* | |
| 6.88 (6.99, 6.94a, 6.96b) | 180.2 (177.3) | 0.3967 (0.6084, 1.25d) | π→π* | |
| 6.87 | 180.4 | 0.2920 | π→π* | |
| 6.99 | 177.4 | 0.1871 | π→π* | |
| 7.02 | 176.6 | 0.0882 | Bz CT state | |
| CAM-B3LYP | 5.38 (5.40, 4.9a,b) | 230.3 (229.8) | 0.0002 (0.0000) | π→π* |
| 6.08 (6.10, 6.20a,c, 6.19b) | 204.0 (203.3) | 0.0006 (0.0000) | π→π* | |
| 6.88 (6.98, 6.94a, 6.96b) | 180.2 (177.7) | 0.4353 (0.6301, 1.25d) | π→π* | |
| 6.90 | 179.7 | 0.4323 | π→π* | |
| 7.09 | 174.9 | 0.1133 | Bz diffuse state | |
| 7.89 | 157.1 | 0.0851 | σ→π* | |
| 7.88 | 157.4 | 0.0312 | Bz CT state | |
| B3LYP | 5.28 (5.30, 4.9a,b) | 234.7 (233.9) | 0.0002 (0.0000) | π→π* |
| 5.96 (5.98, 6.20a,c, 6.19b) | 208.1 (207.4) | 0.0003 (0.0000) | π→π* | |
| 6.71 (6.86, 6.94a, 6.96b) | 184.7 (180.7) | 0.1747 (0.6064, 1.25d) | π→π* | |
| 6.75 | 183.6 | 0.1719 | π→π* | |
| 6.80 | 182.4 | 0.1622 | W-CT state | |
| 6.94 | 178.7 | 0.1056 | Bz CT state | |
| 7.13 | 174.0 | 0.0527 | W-CT state | |
| 6.51 | 190.4 | 0.0489 | Bz diffuse state | |
| DFT functional | E (eV) | λ (nm) | Oscillator strength (f) | Electronic transition |
|---|---|---|---|---|
| Previous work:a Exptl/taken from ref. 63.b Exptl/taken from ref. 64.c Exptl/taken from ref. 65.d Exptl/taken from ref. 66. Lowest π→π* singlet vertical excitation energy (eV) in Bz–W6 cage cluster = 6.17e and f = 0.004e, eCIS/taken from ref. 43. | ||||
| M06-2X | 5.46 (5.47, 4.9a,b) | 226.9 (226.8) | 0.0000 (0.0000) | π→π* |
| 6.26 (6.27, 6.20a,c, 6.19b) | 198.0 (197.5) | 0.0000 (0.0000) | π→π* | |
| 6.90 (6.99, 6.94a, 6.96b) | 179.7 (177.3) | 0.4475 (0.6084, 1.25d) | π→π* | |
| 6.93 | 178.9 | 0.4107 | π→π* | |
| 6.99 | 177.5 | 0.052 | Bz CT state | |
| CAM-B3LYP | 5.40 (5.40, 4.9a,b) | 229.6 (229.8) | 0.0000 (0.0000) | π→π* |
| 6.10 (6.10, 6.20a,c, 6.19b) | 203.4 (203.3) | 0.0000 (0.0000) | π→π* | |
| 6.90 (6.98, 6.94a, 6.96b) | 179.6 (177.7) | 0.4723 (0.6301, 1.25d) | π→π* | |
| 6.94 | 178.7 | 0.4633 | π→π* | |
| 7.71 | 160.7 | 0.0807 | W-Rydberg state | |
| 7.12 | 174.1 | 0.0636 | Bz CT state | |
| B3LYP | 5.30 (5.30, 4.9a,b) | 233.8 (233.9) | 0.0000 (0.0000) | π→π* |
| 5.97 (5.98, 6.20a,c, 6.19b) | 207.7 (207.4) | 0.0000 (0.0000) | π→π* | |
| 6.80 (6.86, 6.94a, 6.96b) | 182.2 (180.7) | 0.1980 (0.6064, 1.25d) | π→π* | |
| 6.77 | 183.2 | 0.1917 | π→π* | |
| 6.76 | 183.4 | 0.1701 | Bz diffuse state | |
| 6.86 | 180.7 | 0.1157 | W-Rydberg state | |
| 6.89 | 180.0 | 0.1101 | Bz CT state | |
| 6.72 | 184.5 | 0.1039 | Bz diffuse state | |
For Bz–W6 prism shaped geometry, the UV spectra results obtained from TD-DFT calculations on MP2 optimized ground state Bz–W6 prism shaped cluster, using all three functionals are presented in Fig. 4(a). Fig. 4(a) shows the diverse spectra for three different functionals where B3LYP functional generates the strongest peak at around 182 nm, with other less intense peaks within the range 174–190 nm, while CAM-B3LYP functional predicts transitions over the spectral range 157–180 nm, with strongest one centered around 180 nm and less intense peaks at around 157 nm and 175 nm. M06-2X and CAM-B3LYP functionals are consistent to generate the strongest intensity peak at 180 nm. It is also noted that B3LYP predicts few less intense peaks above 180 nm. In order to investigate the effect of Bz interaction with isolated water cluster W6 and also to investigate how it affects excitation features in Bz–W6 cluster, it is important to compare the UV spectra of Bz–W6 cluster with UV spectra of isolated water cluster W6 and benzene Bz. The performances of all three functionals i.e., B3LYP, CAM-B3LYP and M06-2X are also tested on prism-shaped W6 & Bz–W6 clusters and Bz molecule (see Fig. 4(b)–4(d)).
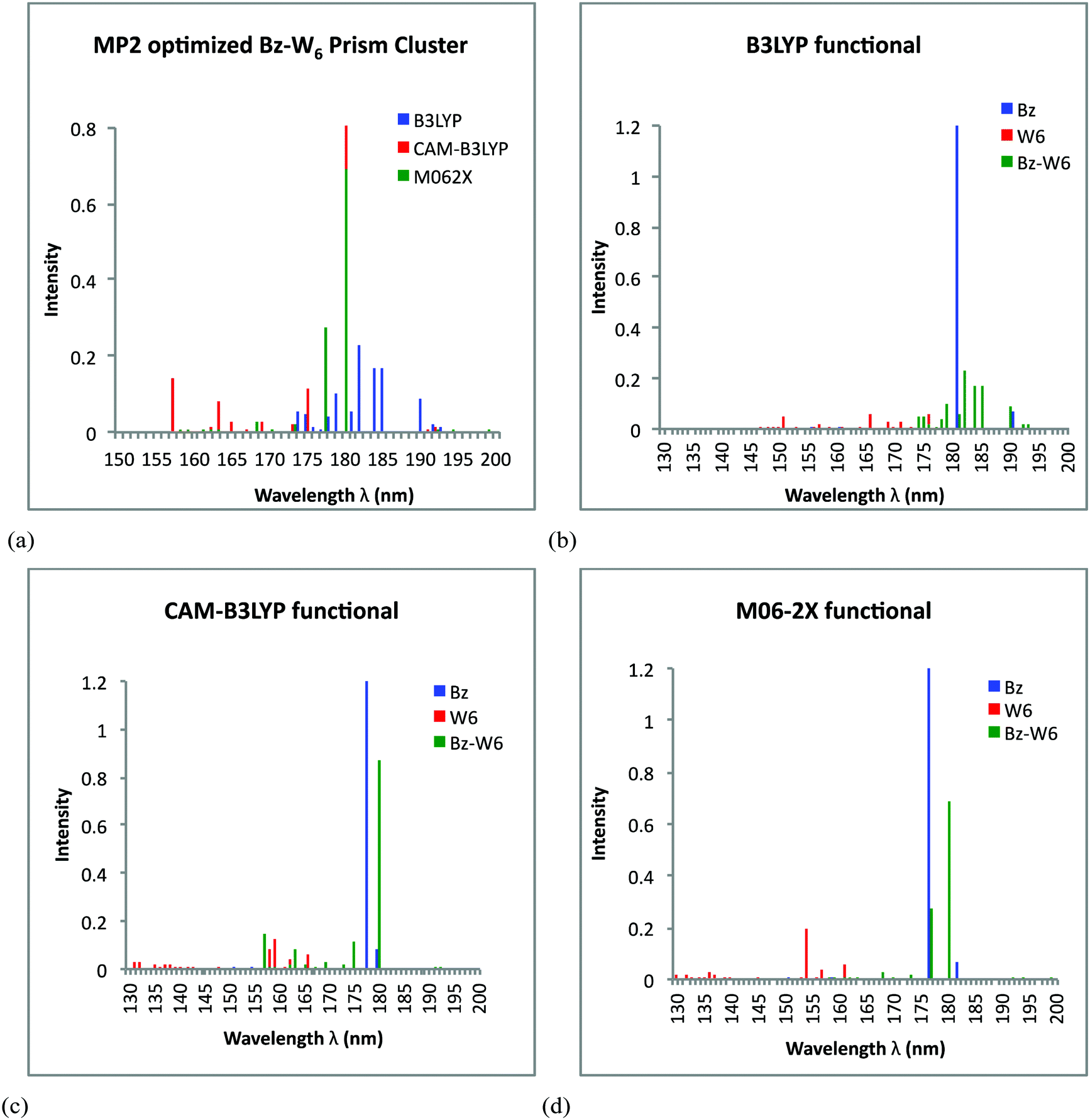 | ||
| Fig. 4 Simulated UV spectra obtained from TD-DFT calculations on MP2 optimized ground state geometries of prism shaped water W6 cluster, Bz–W6 cluster and Bz. (a) Comparison of B3LYP, CAM-B3LYP and M06-2X functionals performance on Bz–W6 prism cluster. (b) Performance of B3LYP functional. (c) Performance of CAM-B3LYP functional. (d) Performance of M06-2X functional. | ||
It is also important to investigate the nature of these transitions and to understand whether Bz or W6 cluster excitations, plays the dominant role in Bz–W6 clusters. So, we have also analyzed the orbital transformations for those excitations that are associated with the high oscillator strengths or high intensity peaks found in the prism Bz–W6 spectra for all three functionals, and are presented in Table 2.
Fig. 4(b) clearly indicates different excitation features in Bz–W6 prism cluster with respect to excitation features in isolated W6 prism cluster and Bz molecule given by B3LYP functional. The excitations in Bz–W6 cluster are shifted towards longer wavelengths (above 174 nm extended up to 193 nm) with respect to W6 cluster where electronic excitations are dominant only in shorter wavelength region i.e., below 176 nm. The Bz excitation feature at around 181 nm is of very high intensity, with another less intense peak at 190 nm. In Bz–W6 prism cluster, it is interesting to see that one of the strong intensity peak at around 182 nm is due to a charge transfer feature of the W6 cluster to Bz system. Another strong peaks at around 184 nm and 185 nm is identified as the π→π* electronic transition of benzene. Few weak excitations showing partial charge transfer from benzene to W6 moiety and Bz locally diffuse excitations are also seen at wavelengths around 179 nm and 190 nm, respectively. It is interesting to note that presence of Bz enhances the excitations in W6 cluster towards longer wavelengths above 176 nm, not present in isolated W6 cluster. The intensity of peaks associated with water excitations in Bz–W6 prism cluster are also found to be higher than in isolated water W6 cluster.
Fig. 4(c) shows that the CAM-B3LYP functional predicts a few excitations below 170 nm region in Bz–W6 cluster which are in close proximity to excitations in W6 cluster. Similarly peaks observed at longer wavelengths at around 175 nm and 180 nm in the Bz–W6 cluster (completely absent in W6 cluster) are found to be closer to Bz excitation feature at 178 nm. Using the CAM-B3LYP functional, it is found that the strongest peak at around 180 nm is the π→π* electronic transition of benzene. A second intense peak at 157 nm is predicted due to the combined influence of σ→π* transition from σ bonding orbital to π anti-bonding orbital of the benzene at 157.1 nm and the benzene charge transfer (CT) state at 157.4 nm, while third peak at around 175 nm corresponds to locally diffuse charge state associated with Bz excitation. We can also see that Bz excitations in Bz–W6 prism cluster are red-shifted as compared to isolated Bz spectra. Thus, it is realized that Bz excitations are also influenced by presence of W6 cluster around it.
It is seen from Fig. 4(d) that Bz–W6 spectra are red shifted with respect to W6 spectra, using M06-2X functional. Since, excitations observed in Bz system are above 175 nm wavelength region and therefore more likely to influence excitations at longer wavelengths in Bz–W6 cluster rather than W6 cluster. The M06-2X functional predicts the π→π* transition feature of benzene at around 180 nm for the strongest peak, while another strong peak at around 177 nm is due to combined effect of π→π* transition feature of benzene at 177.36 nm, and benzene charge transfer (CT) state at 176.56 nm, in which charge from Bz ring is transferred to W6 cluster in prism shaped Bz–W6 complex system.
It is found that most of the peak excitations in Bz–W6 prism cluster are influenced by Bz excitations as compared to water W6 excitations. It is also noted that intensity of the strongest peak is much higher in Bz and Bz–W6 cluster than those in W6 cluster, which hold for all three functionals.
Among various long range corrected functionals, wB97XD functional is found to be one of the most promising DFT functional used for systems involving general non-covalent interactions.67 It is already mentioned above that wB97XD provides highest binding energies among all DFT functionals on Bz–W6 clusters with BSSE correction, and it is same for both cage and prism conformers (see Table 1 and Fig. 3). Therefore, TD-DFT calculations on wB97XD-optimized ground state geometries of Bz–W6 clusters with three functionals i.e., B3LYP, CAM-B3LYP and M06-2X are also performed.
Similar UV spectral characteristics are obtained from TD-DFT calculations on the wB97XD optimized ground state Bz–W6 prism shaped cluster, where UV spectra undergo a red shift in going from the CAM-B3LYP to the B3LYP functional (see ESI: Fig. S1 and Table S1†). Again we clearly see that the strongest intensity peak associated with the benzene π→π* transition undergoes a small red shift in going from Bz to Bz–W6 cluster for both CAM-B3LYP and M06-2X functionals, and the magnitude of shift is slightly larger for MP2 optimized geometries than wB97XD optimized geometries. The intensities are also found to be much stronger in Bz–W6 cluster than those in W6 cluster. It is again noted that presence of Bz enhances the excitations in W6 cluster towards wavelengths above 170 nm. The strong intensity π→π* transition features of Bz has shown small blue shift of order of around 2–3 nm in wB97XD optimized ground state geometries of Bz–W6 cluster with respect to MP2 geometries.
Now, focusing on cage form of Bz–W6 geometry, UV spectra results obtained from TD-DFT calculations on the MP2 optimized Bz–W6 cage cluster using all three B3LYP, CAM-B3LYP and M06-2X functional are shown in Fig. 5(a). The important electronic transitions corresponding to high oscillator strengths or strong intensity peaks in UV spectra of cage shaped Bz–W6 cluster for all three functionals, are presented in Table 3.
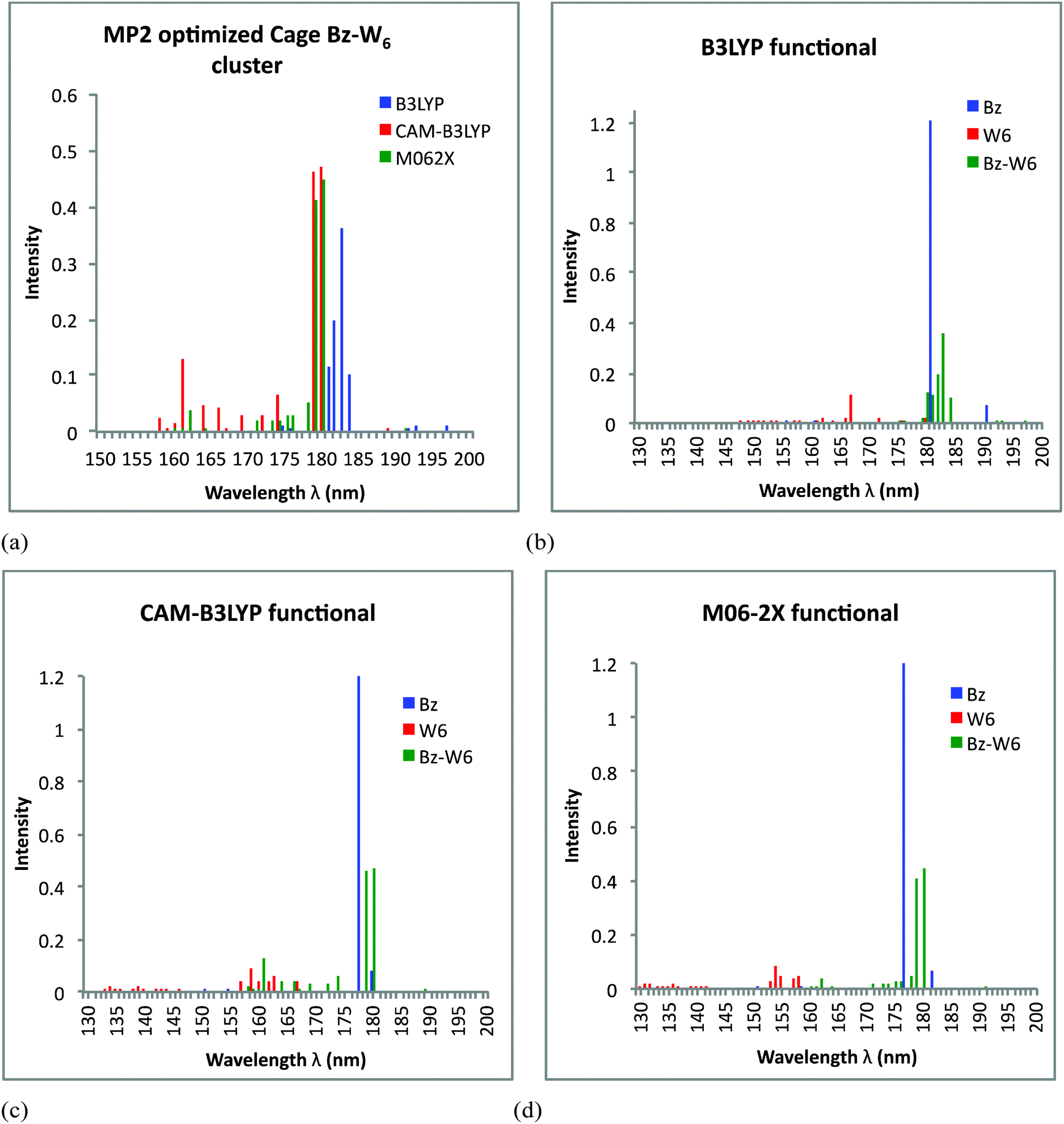 | ||
| Fig. 5 Simulated UV spectra obtained from TDDFT calculations on MP2 optimized ground state geometries of cage shaped water W6 cluster, Bz–W6 cluster and Bz. (a) Comparison of B3LYP, CAM-B3LYP and M062X functional performance on Bz–W6 prism cluster. (b) Performance of B3LYP functional. (c) Performance of CAM-B3LYP functional. (d) Performance of M06-2X functional. | ||
Fig. 5(a) shows that in Bz–W6 cage cluster, electronic excitations predicted by B3LYP functional are red shifted with respect to excitations in M06-2X and CAM-B3LYP level of calculations. Both CAM-B3LYP and M06-2X functionals are consistent to find strong intensity peaks very close to each other at wavelength around 179–180 nm. However, a few less intense peaks at shorter wavelengths below 174 nm are also observed in CAM-B3LYP level of calculations.
Fig. 5(b) shows that excitations in Bz–W6 cage cluster are shifted towards longer wavelengths (region above 180 nm) with respect to W6 cluster using B3LYP functional. It is interesting to notice that excitations in isolated water W6 cluster above 170 nm wavelength range are rarely observed, however the B3LYP functional generates some weaker transitions at around 180–181 nm. It is found that strongest peaks generated at around 183 nm in UV spectra corresponds to contributions from π→π* transition of benzene, and from a locally diffuse state due to Bz excitation. The another strong peak at around 182 nm is associated with π→π* transition of benzene while other peaks at around 180 nm and 181 nm correspond to Bz charge transfer excitation and to local Rydberg type state associated with W6 cluster excitation, respectively. It is also again evident from spectra that intensity of water excitations increases in Bz–W6 cluster as compared to the isolated water W6 cluster.
Fig. 5(c) indicates that excitations found in Bz–W6 cage cluster do not overlap with the peaks found in isolated W6 cage cluster for CAM-B3LYP functional. However, some weak excitation peaks at shorter wavelengths below 170 nm lie close to the W6 cluster excitation region and some of the strong peaks at longer wavelengths (above 170 nm) lie close to Bz strongest intensity peak centered at around 178 nm. The analysis of these excitations has shown that with CAM-B3LYP, two close lying peaks with strong intensities at around 179 nm and 180 nm are associated with π→π* transition of benzene. The third peak at around 161 nm corresponds to locally diffuse Rydberg state due to W6 cluster excitation while another peak at around 174 nm seems like Bz charge transfer state.
From Fig. 5(d) we see that peaks in Bz–W6 cluster are red shifted (above 170 nm) with respect to W6 cluster with W6 excitations lies below 161 nm as given by M06-2X functional. The M06-2X functional accounts for the π→π* transition of benzene at around 180 nm and 179 nm which corresponds to strong intensities too, while another weak excitation at around 178 nm seems to be due to a benzene charge transfer excitation.
Benzene is found to dominate all the excitations in the Bz–W6 cage cluster with M06-2X. It is again noted that once again the intensities of peaks are much stronger in Bz–W6 cage cluster than isolated water cage W6 cluster.
The UV spectra results obtained from TD-DFT calculations on the wB97XD optimized ground state Bz–W6 cage shaped cluster, using all three functionals are generally consistent with the one obtained on MP2 optimized geometries. Most of the strong intensity peaks are found around the wavelength range 175–180 nm for all three hybrid functionals (see ESI: Fig. S2 and Table S2†). UV spectra obtained by M06-2X is quite similar to those predicted by CAM-B3LYP functional. B3LYP again shows transitions towards longer wavelengths with respect to transitions given by CAM-B3LYP and M06-2X functionals.
It is interesting to see that the peaks are slightly red-shifted for TD-DFT calculations on MP2 optimized geometries as compared to wB97XD optimized ground state geometries for both prism and cage Bz–W6 clusters (see ESI: Fig. S3 and S4†), which is true for all three functionals. The nature of the transitions in UV spectra for both MP2 and wB97XD optimized geometries are generally quite consistent.
We again note that water excitations are observed in Bz–W6 cage cluster at longer wavelengths, which are completely absent in isolated cage W6 system. The intensities of water excitations are also stronger in Bz–W6 cluster as compared to isolated W6 cluster. Again we clearly see that the benzene π→π* transition undergoes a small red shift in Bz–W6 cluster with respect to isolated Bz. The intensities are also found to be much stronger in Bz–W6 cluster than those in W6 cluster.
It is worth mentioning that the lowest valence transitions of Bz from ground to excited states 11B2U and 11B1U are strictly dipole forbidden on symmetry grounds. The low lying vertical π→π* excited states in Bz–W6 prism clusters show small red shift in excitation wavelengths, and oscillator strengths become slightly allowed as compared to dipole forbidden 11B2U and 11B1U excited states in Bz i.e., (Bz(f = 0.0000)→Bz–W6(f = 0.0001 − 0.0006)), indicating slight allowed character in restricted lowest energy π→π* electronic transitions due to the presence of prism W6 conformer around Bz. However, no such difference has been noticed in cage conformer of Bz–W6 cluster.
In UV spectra of both Bz–W6 prism and cage clusters, the strong intensity π→π* electronic transitions (bright states) with high oscillator strengths are red-shifted towards longer wavelengths with respect to corresponding dipole allowed degenerate 1E1U excited states of isolated Bz. The magnitude of red-shifts are about 3–4 nm in Bz–W6 prism clusters, while Bz–W6 cage clusters give red-shifts of about 2–3 nm. The degeneracies of these states are only slightly broken by about 1.0 nm in both Bz–W6 cage and prism clusters. It is seen that cage Bz–W6 cluster shows higher individual transition oscillator strengths than prism Bz–W6 cluster (see Tables 2 and 3). However due to overlapping of very close lying peaks in prism Bz–W6 cluster, the intensity of the strongest peak is larger in prism case than in cage one for CAM-B3LYP and M06-2X functionals, as degeneracy of strongest intensity π→π* electronic transitions is broken slightly lesser in prism cluster than in cage cluster. The oscillator strengths of these bright π→π* electronic states are lower in Bz–W6 clusters as compared to isolated Bz, illustrating the effect of water cluster around Bz, and existence of other close lying new Bz charge transfer and diffuse states, which also increase the intensities of water excitations and shift such water excitations towards longer wavelengths as also seen in recent experimental studies.44–46 It is clear from above results and discussion that the benzene interaction with W6 cluster plays a significant role in giving new excitation features in UV spectra of Bz–W6 clusters.
In order to calibrate basis set effects on the UV spectra, we have also performed TD-DFT calculations using Dunning's correlation-consistent aug-cc-pVTZ basis set on MP2 optimized ground state geometries of Bz, W6 and Bz–W6 clusters. For all three employed functionals, the peak electronic excitations show the small red-shift of around 1–2 nm for both isolated Bz and Bz–W6 clusters relative to those electronic excitations generated using 6-31++G(d,p) basis set, while small blue shift of around 1 nm is seen in W6 clusters, and that holds for both prism and cage conformers. The nature of peak excitations associated with higher oscillator strengths in Bz–W6 clusters (for prism and cage conformers) is generally quite consistent for both aug-cc-pVTZ and 6-31++G(d,p) calculations (see ESI: Tables S3 and S4†).
Conclusions
TD-DFT calculations have been performed on both MP2 and wB97XD optimized geometries for Bz, W6 and Bz–W6 clusters. We observe minor differences in the UV spectroscopy of cage and prism conformers of W6 and Bz–W6 clusters. Our results and discussions also show that TD-DFT calculations are functional-dependent, with CAM-B3LYP and M06-2X generally showing good agreement. We have calculated some interesting features for water W6 cluster excitations at longer wavelengths above 170 nm in both Bz–W6 cage and prism geometries which are absent in isolated water W6 cluster, indicating benzene-mediated excitations in the W6 cluster as observed in recent experimental studies.44–46 The intensities of W6 excitations are also found to increase in Bz–W6 cluster relative to those in isolated W6 clusters. It is predicted that for both MP2 and wB97XD optimized geometries, benzene π→π* transition undergoes small red shift in Bz–W6 cluster with respect to isolated benzene, for both cage and prism geometries and that the degeneracy of this transition is very slightly broken compared to the isolated gas-phase benzene. Our results also predict that charge transfer (CT) states and locally excited diffuse states play an important role in such systems due to complicated nature of benzene–water (π⋯HO) hydrogen bonding interactions prevailing in such systems, including conventional π→π* type interactions between benzene and electrostatic interactions between hydrogen bonded water W6 cluster.Acknowledgements
Thanks are given to Prof. Martin McCoustra for helpful discussion. DS acknowledges the LASSIE FP7 Marie Curie Initial Training Network (ITN) for funding, while both DS and MJP acknowledge support from European Research Council (ERC grant number 25899).References
- K. Kim, K. D. Jordan and T. S. Zwier, Low-Energy Structures and Vibrational Frequencies of the Water Hexamer – Comparison with Benzene-(H2O)(6), J. Am. Chem. Soc., 1994, 116, 11568–11569 CrossRef CAS.
- M. D. Tissandier, S. J. Singer and J. V. Coe, Enumeration and evaluation of the water hexamer cage structure, J. Phys. Chem. A, 2000, 104, 752–757 CrossRef CAS.
- K. Liu, M. G. Brown, C. Carter, R. J. Saykally, J. K. Gregory and D. C. Clary, Characterization of a cage form of the water hexamer, Nature, 1996, 381, 501–503 CrossRef CAS.
- S. Maheshwary, N. Patel, N. Sathyamurthy, A. D. Kulkarni and S. R. Gadre, Structure and stability of water clusters (H2O)(n), n=8–20: An ab initio investigation, J. Phys. Chem. A, 2001, 105, 10525–10537 CrossRef CAS.
- D. M. Upadhyay, M. K. Shukla and P. C. Mishra, An ab initio study of water clusters in gas phase and bulk aqueous media: (H2O)(n), n=1–12, Int. J. Quantum Chem., 2001, 81, 90–104 CrossRef CAS.
- A. Lenz and L. Ojamae, A theoretical study of water clusters: the relation between hydrogen-bond topology and interaction energy from quantum-chemical computations for clusters with up to 22 molecules, Phys. Chem. Chem. Phys., 2005, 7, 1905–1911 RSC.
- A. Lenz and L. Ojamae, Theoretical IR spectra for water clusters (H2O)(n) (n=6–22, 28, 30) and identification of spectral contributions from different H-bond conformations in gaseous and liquid water, J. Phys. Chem. A, 2006, 110, 13388–13393 CrossRef CAS PubMed.
- A. Lenz and L. Ojamae, On the stability of dense versus cage-shaped water clusters: Quantum-chemical investigations of zero-point energies, free energies, basis-set effects and IR spectra of (H2O)(12) and (H2O)(20), Chem. Phys. Lett., 2006, 418, 361–367 CrossRef CAS PubMed.
- D. M. Bates and G. S. Tschumper, CCSD(T) complete basis set limit relative energies for low-lying water hexamer structures, J. Phys. Chem. A, 2009, 113, 3555–3559 CrossRef CAS PubMed.
- Y. Wang, V. Babin, J. M. Bowman and F. Paesani, The water hexamer: cage, prism, or both. Full dimensional quantum simulations say both, J. Am. Chem. Soc., 2012, 134, 11116–11119 CrossRef CAS PubMed.
- Y. I. Neela, A. S. Mahadevi and G. N. Sastry, Hydrogen Bonding in Water Clusters and Their Ionized Counterparts, J. Phys. Chem. B, 2010, 114, 17162–17171 CrossRef CAS PubMed.
- M. Baron and V. J. Kowalewski, The liquid water-benzene system, J. Phys. Chem. A, 2006, 110, 7122–7129 CrossRef CAS PubMed.
- D. Feller, Strength of the benzene-water hydrogen bond, J. Phys. Chem. A, 1999, 103, 7558–7561 CrossRef CAS.
- S. Y. Fredericks, J. M. Pedulla, K. D. Jordan and T. S. Zwier, OH stretch IR spectra of (H2O)(3) and benzene-(H2O)(3), Theor. Chem. Acc., 1997, 96, 51–55 CrossRef CAS.
- A. J. Gotch and T. S. Zwier, Multiphoton Ionization Studies of Clusters of Immiscible Liquids 0.1. C6h6-(H2o)N, N=1,2, J. Chem. Phys., 1992, 96, 3388–3401 CrossRef CAS PubMed.
- P. Jedlovszky, A. Kereszturi and G. Horvai, Orientational order of the water molecules at the vicinity of the water-benzene interface in a broad range of thermodynamic states, as seen from Monte Carlo simulations, Faraday Discuss., 2005, 129, 35–46 RSC.
- M. Prakash, K. G. Samy and V. Subramanian, Benzene-Water (BZW(n) (n=1–10)) Clusters, J. Phys. Chem. A, 2009, 113, 13845–13852 CrossRef CAS PubMed.
- R. N. Pribble, A. W. Garrett, K. Haber and T. S. Zwier, Resonant Ion-Dip Infrared-Spectroscopy of Benzene-H2o and Benzene-Hod, J. Chem. Phys., 1995, 103, 531–544 CrossRef CAS PubMed.
- R. N. Pribble and T. S. Zwier, Size-Specific Infrared-Spectra of Benzene-(H2o)(N) Clusters (N=1 through 7) – Evidence for Noncyclic (H2o)(N) Structures, Science, 1994, 265, 75–79 CAS.
- R. N. Pribble and T. S. Zwier, Probing Hydrogen-Bonding in Benzene-(Water)(N) Clusters Using Resonant Ion-Dip Ir Spectroscopy, Faraday Discuss., 1994, 97, 229–241 RSC.
- P. Tarakeshwar, H. S. Choi, S. J. Lee, J. Y. Lee, K. S. Kim, T. K. Ha, J. H. Jang, J. G. Lee and H. Lee, A theoretical investigation of the nature of the pi-H interaction in ethene-H2O, benzene-H2O, and benzene-(H2O)(2), J. Chem. Phys., 1999, 111, 5838–5850 CrossRef CAS PubMed.
- C. J. Gruenloh, J. R. Carney, C. A. Arrington, T. S. Zwier, S. Y. Fredericks and K. D. Jordan, Infrared spectrum of a molecular ice cube: The S-4 and D-2d water octamers in benzene-(water)(8), Science, 1997, 276, 1678–1681 CrossRef CAS.
- C. J. Gruenloh, J. R. Carney, F. C. Hagemeister, C. A. Arrington, T. S. Zwier, S. Y. Fredericks, J. T. Wood and K. D. Jordan, Resonant ion-dip infrared spectroscopy of the S-4 and D-2d wafer octamers in benzene-(water)(8) and benzene(2)-(water)(8), J. Chem. Phys., 1998, 109, 6601–6614 CrossRef CAS PubMed.
- L. J. Allamandola, Interstellar PAHs: Yesterday, today, and tomorrow, Abstr. Pap. Am. Chem. Soc., 2006, 231 Search PubMed.
- L. J. Allamandola, D. M. Hudgins, C. W. Bauschlicher and S. R. Langhoff, Carbon chain abundance in the diffuse interstellar medium, Astron. Astrophys., 1999, 352, 659–664 CAS.
- L. J. Allamandola, M. P. Bernstein, S. A. Sandford and R. L. Walker, Evolution of interstellar ices, Space. Sci. Rev., 1999, 90, 219–232 CrossRef CAS.
- J. Cernicharo, A. M. Heras, A. G. G. M. Tielens, J. R. Pardo, F. Herpin, M. Guelin and L. B. F. M. Waters, Infrared Space Observatory's discovery of C-4H2, C-6H2, and benzene in CRL 618, Astrophys. J., 2001, 546, L123–L126 CrossRef CAS.
- P. M. Woods, T. J. Millar and A. A. Zijlstra, The synthesis of benzene in the proto-planetary nebula CRL 618, Astrophys. J., 2002, 574, L167–L170 CrossRef CAS.
- I. Cherchneff, J. R. Barker and A. G. G. M. Tielens, Polycyclic Aromatic Hydrocarbon Formation in Carbon-Rich Stellar Envelopes, Astrophys. J., 1992, 401, 269–287 CrossRef CAS PubMed.
- P. M. Woods and K. Willacy, Benzene formation in the inner regions of protostellar disks, Astrophys. J., 2007, 655, L49–L52 CrossRef CAS.
- M. Frenklach and E. D. Feigelson, Formation of Polycyclic Aromatic-Hydrocarbons in Circumstellar Envelopes, Astrophys. J., 1989, 341, 372–384 CrossRef CAS PubMed.
- M. P. Bernstein, S. A. Sandford, L. J. Allamandola, J. S. Gillette, S. J. Clemett and R. N. Zare, UV irradiation of polycyclic aromatic hydrocarbons in ices: Production of alcohols, quinones, and ethers, Science, 1999, 283, 1135–1138 CrossRef CAS.
- C. Dominik, C. Ceccarelli, D. Hollenbach and M. Kaufman, Gas-phase water in the surface layer of protoplanetary disks, Astrophys. J., 2005, 635, L85–L88 CrossRef CAS.
- K. Willacy and D. A. Williams, Desorption Processes in Molecular Clouds – Quasi-Steady-State Chemistry, Mon. Not. R. Astron. Soc., 1993, 260, 635–642 CrossRef CAS PubMed.
- S. V. S. Rana and Y. Verma, Biochemical toxicity of benzene, J. Environ. Biol., 2005, 26, 157–168 CAS.
- R. Duarte-Davidson, C. Courage, L. Rushton and L. Levy, Benzene in the environment: an assessment of the potential risks to the health of the population, Occup. Environ. Med., 2001, 58, 2–13 CrossRef CAS.
- J. Ma, D. Alfe, A. Michaelides and E. Wang, The water-benzene interaction: Insight from electronic structure theories, J. Chem. Phys., 2009, 130 CAS.
- R. N. Pribble and T. S. Zwier, Size-Specific Infrared Spectra of Benzene-(H2O)n Clusters (n=1 through 7): Evidence for Noncyclic (H2O)n Structures, Science, 1994, 265, 75–79 CAS.
- A. W. Garrett and T. S. Zwier, Multiphoton Ionization Studies of Clusters of Immiscible Liquids 0.2. C6h6-(H2o)N, N=3–8 and (C6h6)2-(H2o)1,2, J. Chem. Phys., 1992, 96, 3402–3410 CrossRef CAS PubMed.
- S. J. Kim, H. I. Seo and B. H. Boo, Theoretical investigations for the molecular structures and binding energies for C(6)H(6)(H(2)O)(n), (n=1–7) complexes, Mol. Phys., 2009, 107, 1261–1270 CrossRef CAS.
- M. Alberti, N. F. Lago and F. Pirani, Benzene water interaction: From gaseous dimers to solvated aggregates, Chem. Phys., 2012, 399, 232–239 CrossRef CAS PubMed.
- L. V. Slipchenko and M. S. Gordon, Water-Benzene Interactions: An Effective Fragment Potential and Correlated Quantum Chemistry Study, J. Phys. Chem. A, 2009, 113, 2092–2102 CrossRef CAS PubMed.
- D. M. Upadhyay and P. C. Mishra, Binding of benzene with water clusters (H2O)(n), n=1–6, in the ground and lowest singlet excited states, J. Mol. Struct. (THEOCHEM), 2002, 584, 113–133 CrossRef CAS.
- J. D. Thrower, M. P. Collings, M. R. S. McCoustra, D. J. Burke, W. A. Brown, A. Dawes, P. D. Holtom, P. Kendall, N. J. Mason, F. Jamme, H. J. Fraser, I. P. Clark and A. W. Parker, Surface science investigations of photoprocesses in model interstellar ices, J. Vac. Sci. Technol., A, 2008, 26, 919–924 CAS.
- J. D. Thrower, D. J. Burke, M. P. Collings, A. Dawes, P. D. Holtom, F. Jamme, P. Kendall, W. A. Brown, I. P. Clark, H. J. Fraser, M. R. S. McCoustra, N. J. Mason and A. W. Parker, Desorption of hot molecules from photon irradiated interstellar ices, Astrophys. J., 2008, 673, 1233–1239 CrossRef CAS PubMed.
- J. D. Thrower, A. G. M. Abdulgalil, M. P. Collings, M. R. S. McCoustra, D. J. Burke, W. A. Brown, A. Dawes, P. J. Holtom, P. Kendall, N. J. Mason, F. Jamme, H. J. Fraser and F. J. M. Rutten, Photon- and electron-stimulated desorption from laboratory models of interstellar ice grains, J. Vac. Sci. Technol., A, 2010, 28, 799–806 CAS.
- B. Temelso, K. A. Archer and G. C. Shields, Benchmark Structures and Binding Energies of Small Water Clusters with Anharmonicity Corrections, J. Phys. Chem. A, 2011, 115, 12034–12046 CrossRef CAS PubMed.
- C. Perez, M. T. Muckle, D. P. Zaleski, N. A. Seifert, B. Temelso, G. C. Shields, Z. Kisiel and B. H. Pate, Structures of cage, prism, and book isomers of water hexamer from broadband rotational spectroscopy, Science, 2012, 336, 897–901 CrossRef CAS PubMed.
- J. Y. Lee, J. Kim, H. M. Lee, P. Tarakeshwar and K. S. Kim, Structures, vibrational frequencies, and infrared spectra of the hexa-hydrated benzene clusters, J. Chem. Phys., 2000, 113, 6160–6168 CrossRef CAS PubMed.
- A. K. Kelkkanen, B. I. Lundqvist and J. K. Norskov, Density functional for van der Waals forces accounts for hydrogen bond in benchmark set of water hexamers, J. Chem. Phys., 2009, 131, 046102 CrossRef PubMed.
- B. Santra, A. Michaelides, M. Fuchs, A. Tkatchenko, C. Filippi and M. Scheffler, On the accuracy of density-functional theory exchange-correlation functionals for H bonds in small water clusters. II. The water hexamer and van der Waals interactions, J. Chem. Phys., 2008, 129, 194111 CrossRef PubMed.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions, J. Chem. Theory Comput., 2006, 2, 364–382 CrossRef.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- J. D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS PubMed.
- Y. Tawada, T. Tsuneda, S. Yanagisawa, T. Yanai and K. Hirao, A long-range-corrected time-dependent density functional theory, J. Chem. Phys., 2004, 120, 8425–8433 CrossRef CAS PubMed.
- K. E. Riley and P. Hobza, Assessment of the MP2 method, along with several basis sets, for the computation of interaction energies of biologically relevant hydrogen bonded and dispersion bound complexes, J. Phys. Chem. A, 2007, 111, 8257–8263 CrossRef CAS PubMed.
- E. Runge and E. K. U. Gross, Density-Functional Theory for Time-Dependent Systems, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- K. Burke, J. Werschnik and E. K. U. Gross, Time-dependent density functional theory: Past, present, and future, J. Chem. Phys., 2005, 123 Search PubMed.
- M. A. L. Marques and E. K. U. Gross, Time-dependent density functional theory, Annu. Rev. Phys. Chem., 2004, 55, 427–455 CrossRef CAS PubMed.
- H. H. Telle, R. J. Donovan and A. Gonzalez Urena, Laser chemistry: spectroscopy, dynamics and applications, John Wiley & Sons, Chichester, 2007 Search PubMed.
- A. Hiraya and K. Shobatake, Direct Absorption-Spectra of Jet-Cooled Benzene in 130–260-Nm, J. Chem. Phys., 1991, 94, 7700–7706 CrossRef CAS PubMed.
- E. Pantos, J. Philis and A. Bolovinos, Extinction Coefficient of Benzene Vapor in Region 4.6 to 36 Ev, J. Mol. Spectrosc., 1978, 72, 36–43 CrossRef CAS.
- E. Lassettre, A. Skerbele, M. A. Dillon and K. J. Ross, High-Resolution Study of Electron-Impact Spectra at Kinetic Energies between 33 and 100 Ev and Scattering Angles to 16 Degrees, J. Chem. Phys., 1968, 48, 5066–5096 CrossRef CAS PubMed.
- M. W. Williams, R. A. Macrae, R. N. Hamm and E. T. Arakawa, Collective Oscillations in Pure Liquid Benzene, Phys. Rev. Lett., 1969, 22, 1088–1091 CrossRef CAS.
- L. Goerigk and S. Grimme, A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: UV spectra (Fig. S1 and S2) and important electronic transitions corresponding to higher oscillator strengths for wB97XD optimized Bz–W6 prism and cage shaped clusters (Tables S1 and S2); comparison of UV spectra obtained from TD-DFT calculations on MP2 and wB97XD optimized ground state geometries of prism and cage shaped Bz–W6 cluster (Fig. S3 and S4); electronic transitions obtained using B3LYP, CAM-B3LYP and M06-2X hybrid functionals with aug-cc-pVTZ basis set on MP2 optimized Bz–W6 prism and cage shaped clusters (Tables S3 and S4). See DOI: 10.1039/c4pp00211c |
| This journal is © The Royal Society of Chemistry and Owner Societies 2014 |