
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A facile approach to tryptophan derivatives for the total synthesis of argyrin analogues†
Chou-Hsiung
Chen‡
,
Sivaneswary
Genapathy‡
,
Peter M.
Fischer
and
Weng C.
Chan
*
School of Pharmacy, Centre for Biomolecular Sciences, University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: weng.chan@nottingham.ac.uk; Tel: +44(0)-115-9515080
First published on 22nd October 2014
Abstract
A facile route has been established for the synthesis of indole-substituted (S)-tryptophans from corresponding indoles, which utilizes a chiral auxiliary-facilitated Strecker amino acid synthesis strategy. The chiral auxiliary reagents evaluated were (S)-methylbenzylamine and related derivatives. To illustrate the robustness of the method, eight optically pure (S)-tryptophan analogues were synthesized, which were subsequently used for the convergent synthesis of a potent antibacterial agent, argyrin A and its analogues.
Tryptophan and its derivatives are key biosynthetic precursors in many nonribosomal peptide antibiotics. These peptidic compounds typically display a wide spectrum of biological activities. Specifically, the argyrins, a family of eight 24-membered macrocyclic peptides isolated from the marine myxobacterium Archangium gephyra,1,2 have been associated with antibacterial,2–4 antiinflammatory2 and antiproteasomal5,6 activities. All biologically active members of the family contain an unusual (S)-4-methoxy-tryptophan residue.1–6 Argyrin A and B display potent antibacterial activity against the opportunistic human pathogen Pseudomonas aeruginosa,1,2,7,8 which is the result of functional inhibition of elongation factor G (EF-G).7,8 Utilizing the nomenclature recently introduced by Villar et al.,9 argyrin B7 appeared to engage in a compact binding mode with EF-G, in which ‘peripheral atoms’ and ‘substituent atoms’ make extensive contacts with EF-G. This binding mode reveals intimate polar and nonpolar interactions between EF-G and the 4-methoxy-tryptophan side-chain (substituent atoms) of argyrin B.
While the (S)-4-methoxy-tryptophan residue in argyrin A has been identified as crucial for its antiproteasomal activity in human cancer cell lines,6 the structure–activity relationships (SARs) of this residue that govern the peptide antibacterial property remain unexplored. Therefore a variety of (S)-Trp analogues were prepared in order to investigate the role of this indolyl amino acid residue in antibacterial activity.
Current methods employed for the synthesis of enantiomerically pure (S)-Trp analogues rely on either enzymatic or chemical approaches. The enzymatic approach involves a final-step resolution of the racemates using N-acylase enzyme, which affords a mixture of the desired product and the unprocessed (R)-N-acetyl-tryptophan.10 Chemical approaches in the past have exploited various chiral auxiliaries to access enantiomerically pure (S)-Trp analogues. For example, Ma et al.11 utilize a Schöllkopf chiral auxiliary whilst Buelow et al.6 employ a DuanPhos ligand. We herein report our progress towards the development of a flexible and operationally simple synthetic route to a raft of (S)-Trp analogues. Our strategy exploits a previously reported asymmetric Strecker synthesis of aliphatic α-aminonitriles that employs chiral auxiliary reagents, such as (S)-α-methylbenzylamine, which in turn delivers aliphatic (S)-amino acids.12 The new (S)-Trp analogues were then used for the total chemical synthesis of argyrin A and related analogues. In contrast to previously reported solution-based fragment condensation approaches,10a,13 we also report for the first time a robust Fmoc/tBu solid-phase peptide synthesis (SPPS) strategy for the stepwise assembly of the octapeptide.
In our appraisal of the α-substituted benzylamine-facilitated Strecker synthesis of new (S)-Trp analogues (Scheme 1), several key challenges were addressed, including the stability of intermediates, the availability of chiral auxiliary reagents that are inexpensive and are tolerant to a wide range of substrates, and the stereoselectivity of the condensation reaction.
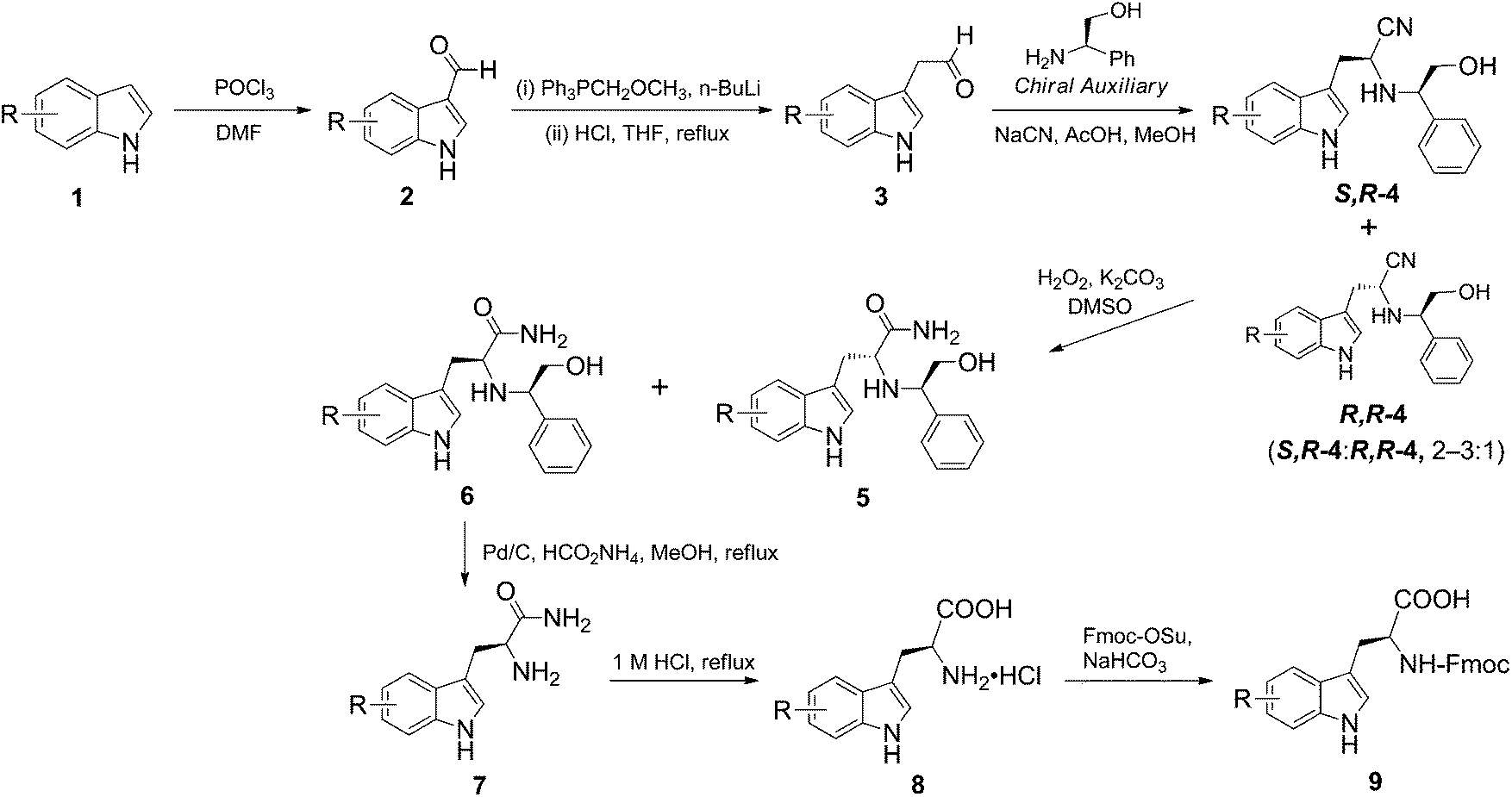 | ||
| Scheme 1 Synthesis of tryptophan analogues. | ||
Thus, a variety of commercially available indoles 1 were subjected to Vilsmeier formylation to give indole-3-carbaldehydes 2, which were then homologated via a Wittig reaction to afford the 2-(indol-3-yl)acetaldehydes 3. In the latter transformation, efficiency of the acid hydrolysis of the enol ether intermediates was found to be dependent on the electronic nature of substituents in the indole ring. Regardless of the position, indoles with an electron-donating substituent (2e–h) were more labile to acid-mediated decomposition than their counterparts with an electron-withdrawing substituent (2b–d) (Table 1). Moreover, the 2-(indol-3-yl)acetaldehydes 3e–h were typically found to be unstable on storage. Hence, the two-step homologation reaction was carefully monitored by TLC to avoid decomposition and the reaction products were immediately subjected to Strecker condensation to yield the α-aminonitriles 4.
| Compound 3 | R = | HCl (M) | Reaction time (h) | Crude yield (%) |
|---|---|---|---|---|
| a | H | 0.4 | 2.5 | 69 |
| b | 5-Br | 0.4 | 2 | 74 |
| c | 5-Cl | 0.4 | 2 | 74 |
| d | 6-F | 0.4 | 2 | 73 |
| e | 7-Et | 0.4 | 1.5 | 65 |
| f | 5-Me | 0.4 | 1.5 | 60 |
| g | 5-MeO | 0.1 | 1.5 | 55 |
| h | 4-MeO | 0.1 | 1.5 | 47 |
For the crucial three-component Strecker condensation reaction, we started our study by evaluating the effectiveness of the chiral auxiliary agent (R)-2-phenylglycinol 1014 in comparison to the standard reagent (S)-α-methylbenzylamine 11 (Fig. 1).12 Using 2-(indol-3-yl)acetaldehyde 3a as the model substrate, initial condensation of 10 with the protonated aldehyde 3a generates an imine intermediate which in turn is attacked by a cyanide anion to yield the α-aminonitrile 4a.12 As anticipated, the nucleophilic nitrile addition occurs at the more favourable, less shielded diastereotopic re-face to afford the (S,R)-diastereomer 4a as the predominant product. Similarly, (S)-α-methylbenzylamine 11 gave the re-addition product, a (S,S) variant of 4a. Both the chiral auxiliary agents 10 and 11 gave the products 4 with a favourable dr 2–3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which was determined by RP-HPLC analysis; a dr in this range was also observed for all indole variants (a–h) examined in this study.
:1, which was determined by RP-HPLC analysis; a dr in this range was also observed for all indole variants (a–h) examined in this study.
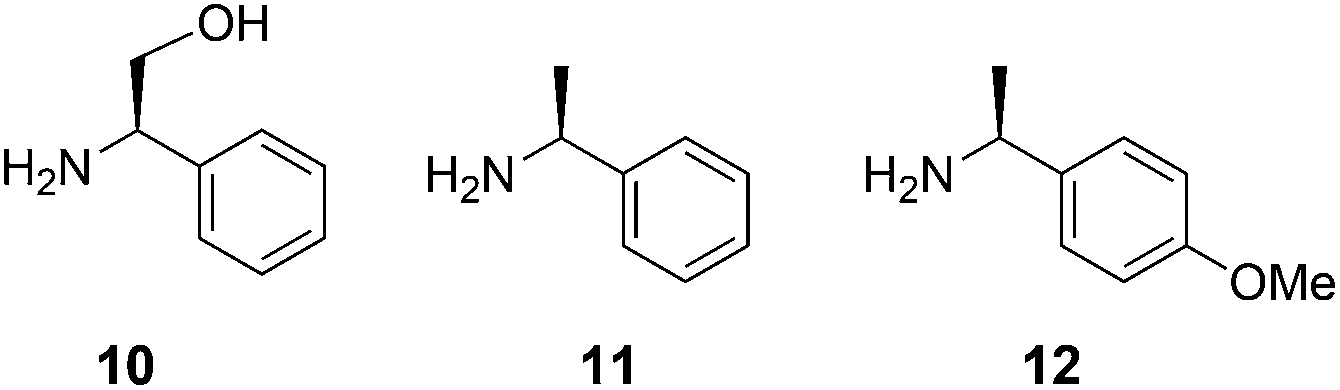 | ||
| Fig. 1 The chiral auxiliaries amine reagents used in the Strecker synthesis strategy. | ||
For the next transformation step, initial attempts to hydrolyse the α-aminonitriles 4 directly to α-amino acids proved to be difficult. Typical acid- or base-catalysed hydrolytic methods required concentrated acid or strong alkali at high temperature,15 which in our case resulted in an intractable mixture of decomposition products. Hence, we opted for an indirect route, by converting the α-aminonitriles 4 to α-amino amides 5 and 6, which was achieved by employing 30% aqueous H2O2–DMSO–K2CO3.16,17 The additive DMSO significantly accelerated the reaction under mild basic conditions. In fact, the H2O2-enabled oxidation of α-aminonitriles was highly efficient and gave exclusively the α-amino amide diastereomers 5 and 6 in good yields. At this stage, we found that the α-amino amides 5 and 6 were readily and efficiently separated by preparative silica gel flash-chromatography, to deliver the required diastereomerically pure compounds 6. It is worth noting that, when (S)-α-methylbenzylamine 11 was utilized, we were unable to achieve reliable separation of the corresponding α-(methylbenzylamino) amides by column chromatography.
Since the S-isomer is the required enantiomer, compounds 6 were used for subsequent investigations. We next established that an unmasking step, i.e. the debenzylation step, followed by mild acid hydrolysis was the most robust procedure to finally furnish the (S)-Trp analogues 8. It has been previously reported that the methyl group in the α-position could prevent N-debenzylation under standard hydrogenolysis conditions.18 Although the use of the more active Pearlman's catalyst Pd(OH)2/C to remove the N-α-methylbenzyl group was considered, it poses a risk of reducing the indole system.19–21 To avoid this, we employed catalytic phase transfer hydrogenolysis to promote the N-debenzylation of compounds 6.22 Accordingly, 10% Pd/C in the presence of excess ammonium formate in MeOH,23 was used to afford specifically 7a and 7d–h.
However, hydrogenolysis of the halogen-substituted compounds 6b and 6c led to concomitant dehalogenation, resulting in 7a as the only isolated product. Selective hydrogenolytic debenzylation in the presence of bromo- and chloro-substituted phenyls remain poorly explored. Li et al. reported the use of chloride salt in the selective hydrogenolysis of O-benzyl groups in the presence of aryl chloride.24 Unfortunately, using similar conditions, we observed dehalogenation of our substrates 6b and 6c.
To overcome the above problem, we investigated the use of an alternative chiral auxiliary reagent, 4-methoxy-α-methylbenzylamine (PMB) 12, to generate the α-amino amides 13 and 14. Various oxidative, reductive and acidolytic methods could subsequently be used to remove the PMB group.25 Our initial attempts of oxidative removal using CAN or DDQ26 were unsuccessful. Gratifyingly, treatment of 13 and 14 with TFA under reflux enabled efficient acidolytic removal of PMB to yield 7b and 7c, respectively (Scheme 2).
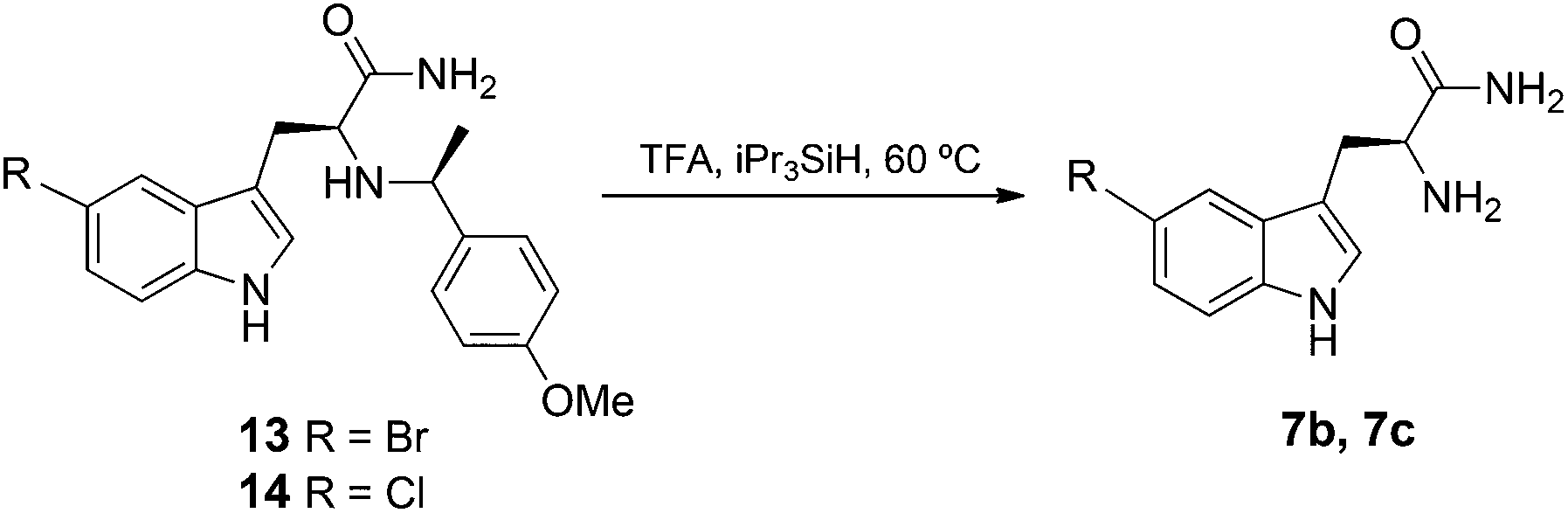 | ||
| Scheme 2 The acidolytic removal of N-PMB group. | ||
The unmasked α-amino amides 7a–h were subsequently subjected to mild hydrolysis using 1 M aqueous HCl under reflux condition for 5 h. This afforded the α-amino acids 8 in good-to-excellent yields (>80%). Since these new tryptophan analogues are required for peptide synthesis, without further purification, compounds 8 were Fmoc-protected using standard procedures to afford 9. In addition to using 1H NMR to establish purity, their absolute configuration was established. Specifically, the observed [α]24D −29 and −23 for 9a and 9g, respectively, are comparable to the values reported in the literature, [α]25D −29.5 (c 1, DMF)27 and −24.5 (c 1, MeOH).10b The outlined synthetic routes therefore delivered a raft of enantiomerically pure (S)-Fmoc-tryptophan analogues.
The Fmoc-protected amino acids 9 were then used for the total chemical synthesis of argyrin A and analogues thereof. This entailed stepwise assembly of the peptide sequence using Fmoc/tBu solid-phase peptide synthesis (SPPS) followed by acidolytic (TFA–CH2Cl2–H2O–iPr3SiH) release of the linear peptide and subsequent head-to-tail macrocyclization (Scheme 3). In this context, the (S)-Fmoc-β-phenylseleno cysteine 15 was used as a masked precursor for the dehydroalanine residue, and the thiazole dipeptide28 was N-Boc protected (16) (see ESI†). Thus, the synthesized linear peptides 18 were subjected to PyBOP-mediated macrocyclization to yield 19, followed by an oxidation–elimination reaction to afford the desired argyrin A 20h and related analogues 20a–g. The purity of the cyclic peptides were established by 1H NMR and reversed-phase HPLC.
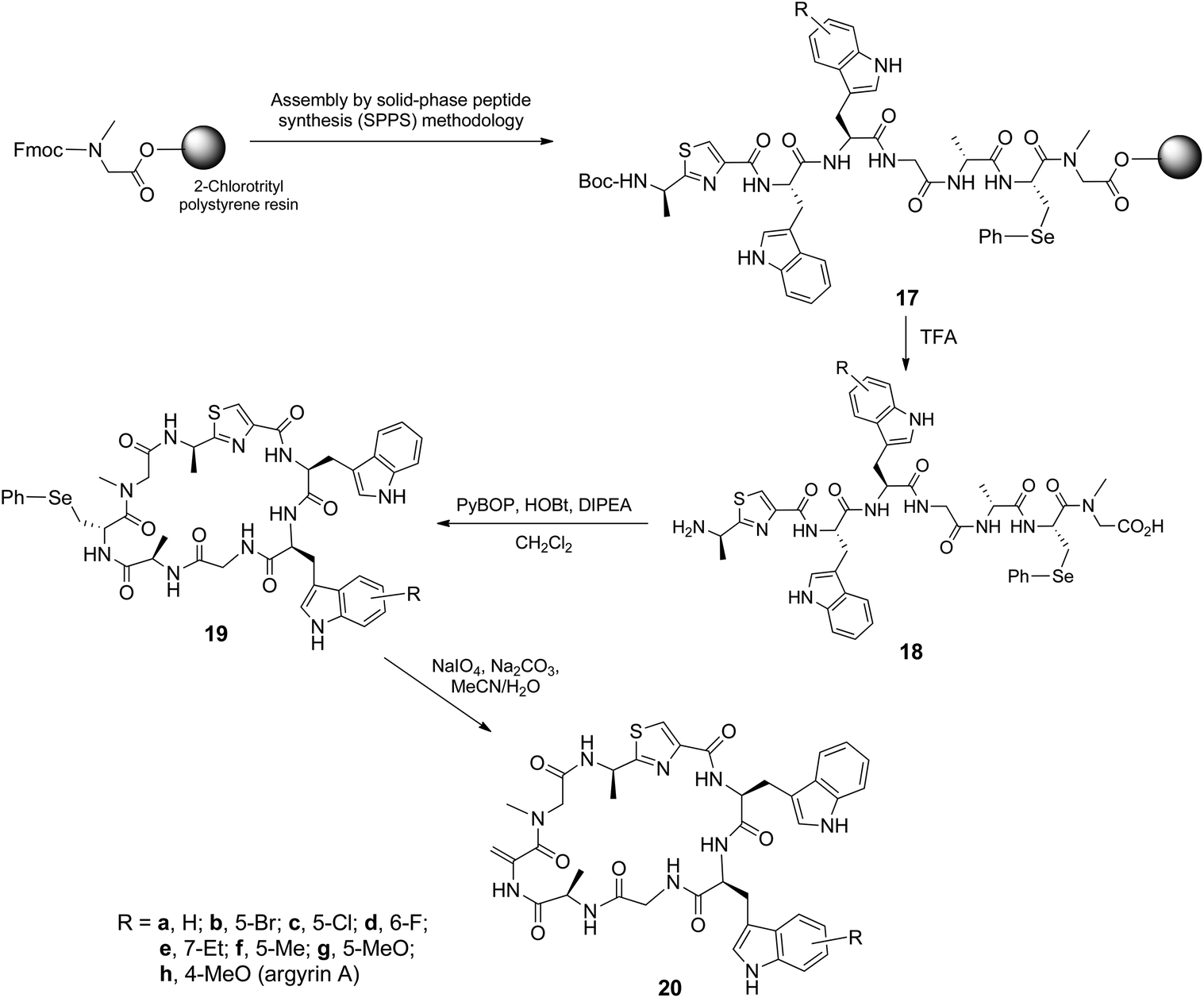 | ||
| Scheme 3 Fmoc/tBu solid-phase synthesis followed by macrocyclization to afford argyrin A and its analogues. | ||
The antibacterial activity of a selection of argyrin analogues was evaluated against two Gram-negative bacteria, Pseudomonas aeruginosa PAO1 and Proteus mirabilis Hauser 1885 (ESI Table S1†). While the activity of argyrin A 20h against P. aeruginosa PAO1 (MIC50 = 19.8 ± 1.6 μM) is comparable to a reported value8 (Fig. 2), we were pleasantly surprised that the argyrin analogue 20g, comprised of a (S)-5-methoxy-tryptophan residue, showed MIC50 of 90–100 μM against both bacteria. In contrast, the (S)-5-chloro- and (S)-5-methyl-tryptophan containing analogues 20c and 20f were essentially inactive. The experimental complex structure between bacterial EF-G and argyrin B shows a H-bond from Ser417 to the O of the 4-methoxy group in the Trp residue.7 Preliminary inspection of the complex structure suggests that either the H-bond is maintained but weaker due to an increased distance, or the 5-OMe is capable of H-bond interaction with Tyr683. However, argyrin analogues lacking a H-bond acceptor in a suitable position (20c and 20f) were inactive.
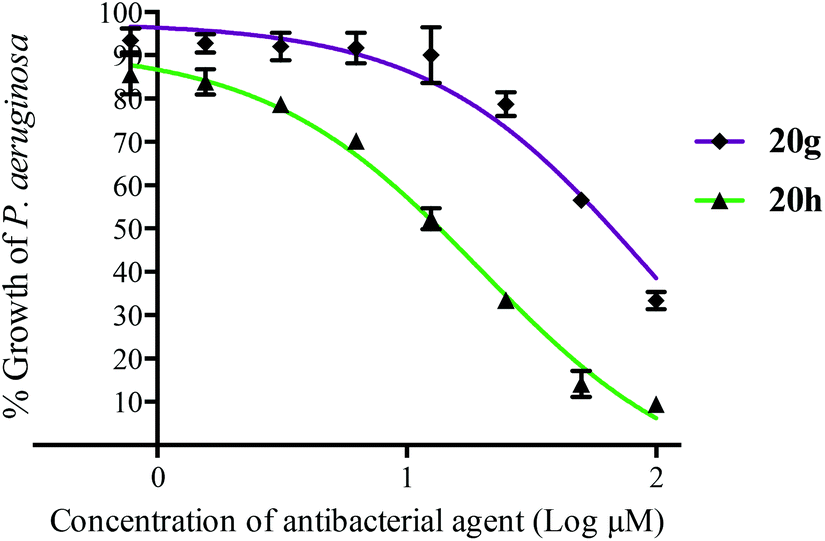 | ||
| Fig. 2 The effects of argyrin A 20h and analogue 20g on the growth of amoxicillin-resistant P. aeruginosa PAO1 in Muller–Hinton broth at 13 h. | ||
Conclusions
A facile approach for the synthesis of enantiomerically pure (S)-tryptophan with halo-, methoxy- and alkyl-substitution has been established using inexpensive (S)-methylbenzylamine-based chiral auxiliary reagents. Using our range of new (S)-Trp analogues, we have also discovered that the antimicrobial activity of argyrin A, although dependent on the (S)-4-methoxy-tryptophan residue, tolerates a (S)-5-methoxy-tryptophan residue. We anticipate this will provide a useful platform for the design and synthesis of more potent and species-specific antibacterial agent.Acknowledgements
We would like to acknowledge financial support from CS Bio Company Inc., USA (to C.H.C.) and Vice-Chancellor's Scholarship for Research Excellence, University of Nottingham (to S.G.).Notes and references
- E. Selva, L. Gastaldo, G. S. Saddler, G. Toppo, P. Ferrari, G. Carniti and B. P. Goldstein, J. Antibiot., 1996, 49, 145–149 CrossRef CAS.
- F. Sasse, H. Steinmetz, T. Schupp, F. Petersen, K. Memmert, H. Hofmann, C. Heusser, V. Brinkmann, P. von Matt, G. Hofle and H. Reichenbach, J. Antibiot., 2002, 55, 543–551 CrossRef CAS.
- P. Ferrari, K. Vekey, M. Galimberti, G. G. Gallo, E. Selva and L. F. Zerilli, J. Antibiot., 1996, 49, 150–154 CrossRef CAS.
- L. Vollbrecht, H. Steinmetz and G. Hofle, J. Antibiot., 2002, 55, 715–721 CrossRef CAS.
- I. Nickeleit, S. Zender, F. Sasse, R. Geffers, G. Brandes, I. Soerensen, H. Steinmetz, S. Kubicka, T. Carlomagno, D. Menche, I. Guetgemann, J. Buer, A. Gossler, M. P. Manns, M. Kalesse, R. Frank and N. P. Malek, Cancer Cell, 2008, 14, 23–35 CrossRef CAS PubMed.
- L. Buelow, I. Nickeleit, A.-K. Girbig, T. Brodmann, A. Rentsch, U. Eggert, F. Sasse, H. Steinmetz, R. Frank, T. Carlomagno, N. P. Malek and M. Kalesse, ChemMedChem, 2010, 5, 832–836 CrossRef CAS PubMed.
- B. Nyfeler, D. Hoepfner, D. Palestrant, C. A. Kirby, L. Whitehead, R. Yu, G. Deng, R. E. Caughlan, A. L. Woods, A. K. Jones, S. W. Barnes, J. R. Walker, S. Gaulis, E. Hauy, S. M. Brachmann, P. Krastel, C. Studer, R. Riedl, D. Estoppey, T. Aust, N. R. Movva, Z. Wang, M. Salcius, G. A. Michaud, G. McAllister, L. O. Murphy, J. A. Tallarico, C. J. Wilson and C. R. Dean, PLoS One, 2012, 7, e42657 CAS.
- P. Bielecki, P. Lukat, K. Huesecken, A. Doetsch, A. H. Steinmetz, R. W. Hartmann, R. Mueller and S. Haeussler, ChemBioChem, 2012, 13, 2339–2345 CrossRef CAS PubMed.
- E. A. Villar, D. Beglov, S. Chennamadhavuni, J. A. Porco Jr., D. Kozakov, S. Vajda and A. Whitty, Nat. Chem. Biol., 2014, 10, 723–731 CrossRef CAS PubMed.
- (a) S. V. Ley, A. Priour and C. Heusser, Org. Lett., 2002, 4, 711–714 CrossRef CAS PubMed; (b) G. Blaser, J. M. Sanderson, A. S. Batsanov and J. A. K. Howard, Tetrahedron Lett., 2008, 49, 2795–2798 CrossRef CAS PubMed; (c) R. J. M. Goss and P. L. A. Newill, Chem. Commun., 2006, 4924–4925 RSC.
- J. Ma, W. Yin, H. Zhou, X. Liao and J. M. Cook, J. Org. Chem., 2009, 74, 264–273 CrossRef CAS PubMed.
- K. Harada, Nature, 1963, 200, 1201 CrossRef CAS.
- W. Wu, Z. Li, G. Zhou and S. Jiang, Tetrahedron Lett., 2011, 52, 2488–2491 CrossRef CAS PubMed.
- R. H. Dave and B. D. Hosangadi, Tetrahedron, 1999, 55, 11295–11308 CrossRef CAS.
- A. Arasappan, S. Venkatraman, A. I. Padilla, W. L. Wu, T. Meng, Y. Jin, J. Wong, A. Prongay, V. Girijavallabhan and F. G. Njoroge, Tetrahedron Lett., 2007, 48, 6343–6347 CrossRef CAS PubMed.
- A. R. Katritzky, B. Pilarski and L. Urogdi, Synthesis, 1989, 949–950 CrossRef CAS.
- E. Vedejs and C. Kongkittingam, J. Org. Chem., 2001, 66, 7355–7364 CrossRef CAS PubMed.
- R. Baltzly and P. B. Russell, J. Am. Chem. Soc., 1953, 75, 5598–5602 CrossRef CAS.
- R. C. Bernotas and R. V. Cube, Synth. Commun., 1990, 20, 1209–1212 CrossRef CAS.
- M. Truong, F. Lecornue and A. Fadel, Tetrahedron: Asymmetry, 2003, 14, 1063–1072 CrossRef CAS.
- D. Hazelard, A. Fadel and C. Girard, Tetrahedron: Asymmetry, 2006, 17, 1457–1464 CrossRef CAS PubMed.
- G. Brieger and T. J. Nestrick, Chem. Rev., 1974, 74, 567–580 CrossRef CAS.
- S. Ram and L. D. Spicer, Tetrahedron Lett., 1987, 28, 515–516 CrossRef CAS.
- J. Li, S. Wang, G. A. Crispino, K. Tenhuisen, A. Singh and J. A. Grosso, Tetrahedron Lett., 2003, 44, 4041–4043 CrossRef CAS.
- P. J. Kocienski, Protecting groups, Georg Thieme Verlag, New York, 2005 Search PubMed.
- S. D. Bull, S. G. Davies, G. Fenton, A. W. Mulvaney, R. S. Prasad and A. D. Smith, J. Chem. Soc., Perkin Trans. 1, 2000, 3765–3774 RSC.
- S. Kokinaki, L. Leondiadis and N. Ferderigos, Org. Lett., 2005, 7, 1723–1724 CrossRef CAS PubMed.
- S. V. Ley and A. Priour, Eur. J. Org. Chem., 2002, 3995–4004 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures for the chemical synthesis and characterization of Trp analogues and their synthetic intermediates, as well as argyrin analogues and their antimicrobial tests. See DOI: 10.1039/c4ob02107j |
| ‡ C.H.C. and S.G. contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |