
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbohydrate-based N-heterocyclic carbenes for enantioselective catalysis†
Alexander S.
Henderson
,
John F.
Bower
* and
M. Carmen
Galan
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: john.bower@bris.ac.uk; m.c.galan@bris.ac.uk
First published on 30th September 2014
Abstract
Versatile syntheses of C2-linked and C2-symmetric carbohydrate-based imidazol(in)ium salts from functionalised amino-carbohydrate derivatives are reported. The novel NHCs were ligated to [Rh(COD)Cl]2 and evaluated in Rh-catalysed asymmetric hydrosilylation of ketones with good yields and promising enantioselectivities.
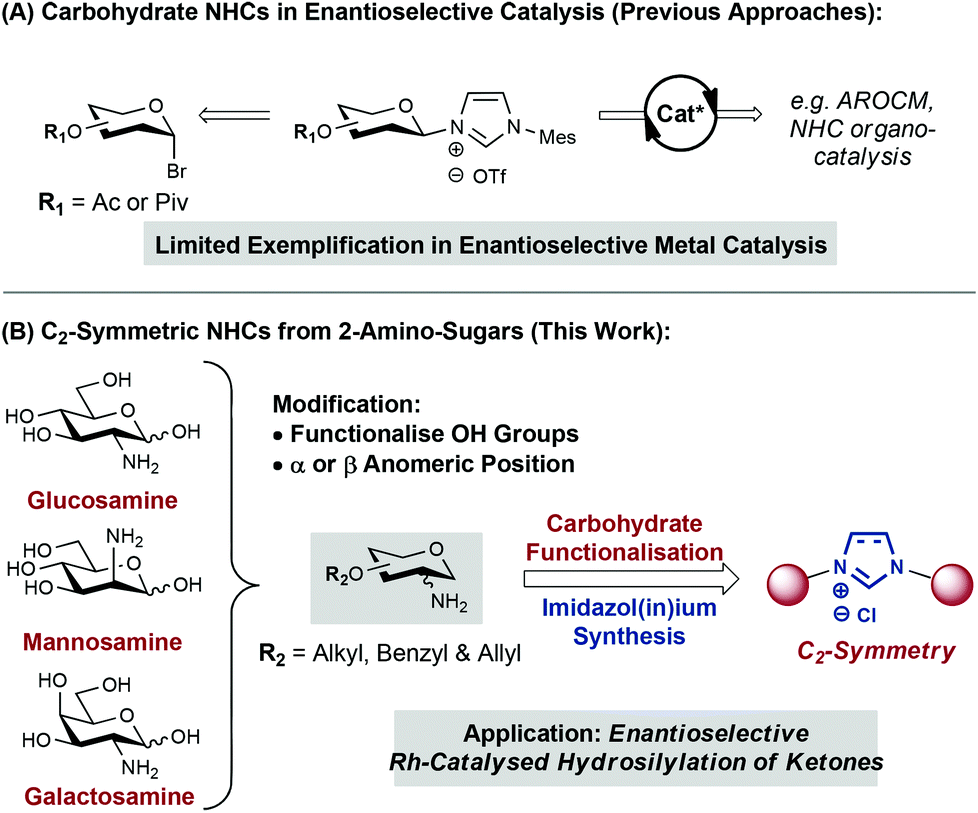
N-heterocyclic carbenes (NHCs) have been extensively exploited over the last few decades as ligands in transition-metal catalysis.1,2 However, the use of chiral NHCs in enantioselective catalysis remains underdeveloped.2 A key challenge resides in the development of systems that are able to relay efficiently ligand chirality to the coordination sphere of the metal centre. Most efforts in this area have been devoted to modification of the NHC backbone or the use of chiral motifs (e.g. functionalised arenes, amino acids) as N-substituents.1–3 Complementary methodologies that enable the incorporation of cheap and diversifiable chiral building blocks onto NHC scaffolds will likely accelerate the development of efficient ligand systems.1d
Carbohydrates are one of the most diverse and important classes of biomolecule. Nature provides in carbohydrates a toolkit of well-defined chirality that is primed for modification. It is not surprising then, that carbohydrate scaffolds have been employed successfully as ligands for enantioselective transition-metal catalysis.2,4 Within this area, the design of NHC-based systems has received relatively little attention (Fig. 1A).5 Anomeric reactivity has been exploited to append the NHC unit (via nitrogen) to C1 of the carbohydrate.5a,c,d,f,g Related C3- and C6-linked monosaccharide systems have also been disclosed.5b,e,h In most cases, application to enantioselective transition-metal catalysis has not been pursued.5a–c,e–g A C1-linked carbohydrate-functionalized Ru catalyst was evaluated in asymmetric ring-opening cross-metathesis (AROCM) but high yields could only be achieved with modest enantioselectivities (up to 26 ee).5d More recently, elegant work by Sollogoub and co-workers has demonstrated that C6-linked NHC-capped cyclodextrins provide chiral “cavities” that mediate enantioselective gold-catalysed alkene cyclopropanation in up to 59% ee.5h Nevertheless, applications of carbohydrate-based NHCs to enantioselective transition-metal catalysis remain underexplored.5d
 | ||
| Fig. 1 Carbohydrate NHCs in asymmetric catalysis. | ||
As part of our ongoing interest in imidazolium-linked sugar building blocks for oligosaccharide synthesis,6 we became interested in their application as carbene ligands for catalysis. Herein we report flexible synthetic entries to a series of C2-linked and C2-symmetric carbohydrate-based NHCs (Fig. 1B). As a proof of concept, we demonstrate the complexation of these to afford a series of neutral Rh(I) catalysts, that show promising activity for enantioselective ketone hydrosilylation.
Previous reports have linked carbohydrates to the imidazolium core via the C1, C3 or C6 positions.5 To prepare moderately rigid NHC complexes that might allow the chirality of the glycan to be propagated to the substrate during catalysis, attachment via C6 was deemed as suboptimal. Anomeric (C1) attachment was also discounted to avoid problems associated with diastereocontrol at this centre and the stability of the eventual NHC. Therefore, and given the availability of C2 amino-carbohydrates, we have targeted a series C2-linked imidazol(in)ium salts (Fig. 1B).
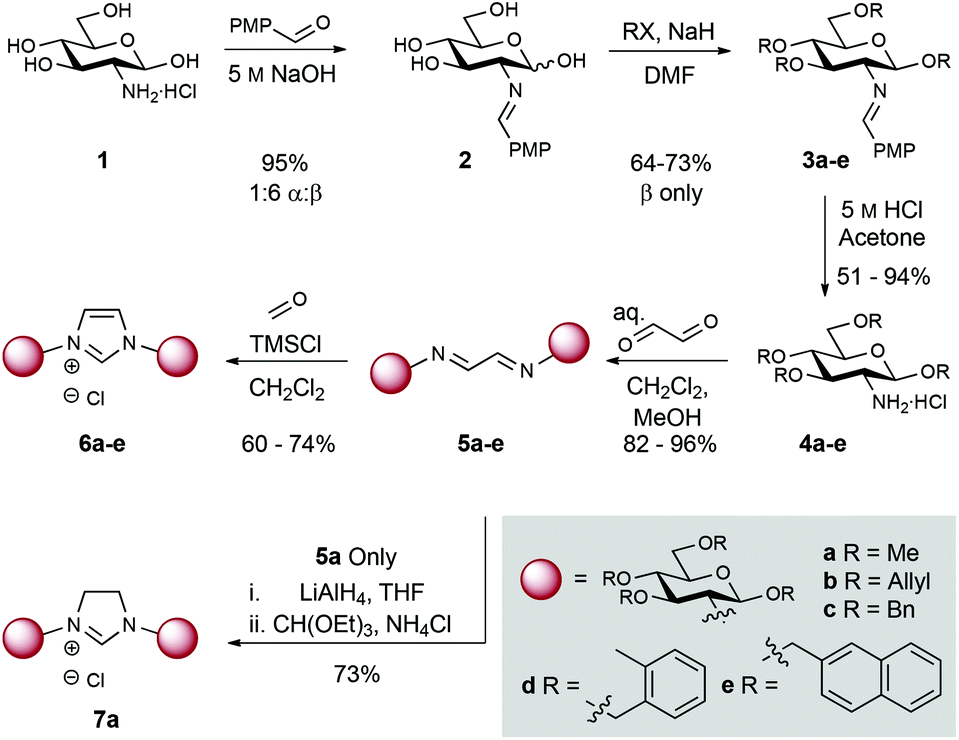
Commercially available D-glucosamine hydrochloride, which bears an equatorial amine at C2, was initially chosen for this study (Scheme 1). Treatment of amine 1 with p-anisaldehyde under basic conditions provided imine 2 in 95% yield (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 α:β mixture). Alkylation of the remaining hydroxyl groups with either methyl iodide, allyl bromide, benzyl bromide, 1-(bromomethyl)-2-methylbenzene, or 2-(bromomethyl)naphthalene in the presence of NaH gave compounds 3a–e in 64–73% yield. Imine hydrolysis (5 M HCl) afforded differentially protected amino building blocks 4a–e (51–94% yield). Bidirectional condensation with glyoxal then generated bis-iminoethylidene derivatives 5a–e in 82–96% yield. Ring-closure to the corresponding imidazolium chlorides 6a–e was achieved in 60–74% yield using TMSCl and paraformaldehyde.71H NMR data unambiguously confirmed the formation of the C2 derived glucoside-imidazolium structures (singlet at δ: 12.31–11.96 ppm corresponding to the C2H imidazolium, and carbohydrate anomeric signals (d, J1,2 = 8.0–8.5 Hz at δ: 6.40–5.52 ppm). Additionally, glucosamine derived imidazolinium 7a was prepared by reduction of 5a to the corresponding diamine (LiAlH4), and subsequent condensation with triethyl orthoformate. This allowed access to a comparable imidazolinium ligand.
:6 α:β mixture). Alkylation of the remaining hydroxyl groups with either methyl iodide, allyl bromide, benzyl bromide, 1-(bromomethyl)-2-methylbenzene, or 2-(bromomethyl)naphthalene in the presence of NaH gave compounds 3a–e in 64–73% yield. Imine hydrolysis (5 M HCl) afforded differentially protected amino building blocks 4a–e (51–94% yield). Bidirectional condensation with glyoxal then generated bis-iminoethylidene derivatives 5a–e in 82–96% yield. Ring-closure to the corresponding imidazolium chlorides 6a–e was achieved in 60–74% yield using TMSCl and paraformaldehyde.71H NMR data unambiguously confirmed the formation of the C2 derived glucoside-imidazolium structures (singlet at δ: 12.31–11.96 ppm corresponding to the C2H imidazolium, and carbohydrate anomeric signals (d, J1,2 = 8.0–8.5 Hz at δ: 6.40–5.52 ppm). Additionally, glucosamine derived imidazolinium 7a was prepared by reduction of 5a to the corresponding diamine (LiAlH4), and subsequent condensation with triethyl orthoformate. This allowed access to a comparable imidazolinium ligand.
 | ||
| Scheme 1 Synthesis of β-glucosamine based NHC·HCl salts. | ||
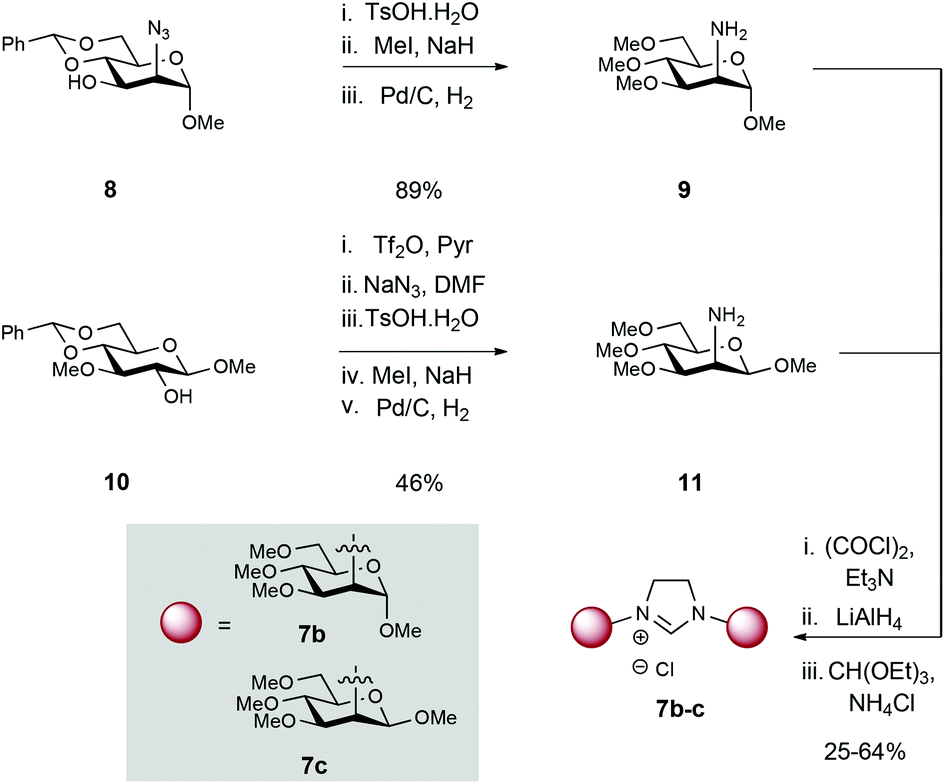
To study the effects of sugar ring substituent configuration (C2 axial vs. equatorial) on overall catalytic efficiency and selectivity, mannosamine scaffolds were also targeted (Scheme 2). Azido-containing mannopyranoside 8,8 which can be prepared in 3 steps from commercial 1-O-methyl-α-D-mannopyranoside, was subjected to acetal hydrolysis (TsOH), followed by permethylation with methyl iodide and NaH. Subsequent Pd-catalysed hydrogenation of the azide furnished amine 9 in 89% yield over 3 steps.
 | ||
| Scheme 2 Synthesis of mannosamine based NHC·HCl salts. | ||
Attempts to condense 9 following the same conditions as described for the glucose series (Scheme 1), proceeded with low efficiency, probably due to decreased reactivity and steric hindrance of the axial amine (vs. equatorial amine as in 4a–e).9 Consequently, we elected to explore conversion to the corresponding imidazolinium salts. To this end, mannosamine derivative 9 was reacted with oxalyl chloride to yield the corresponding bis-amide. Carbonyl reduction with LiAlH4 followed by thermal condensation/cyclization with CH(OEt)3 in the presence of NH4Cl afforded imidazolinium 7b.
β-D-Mannosamine 11 was obtained from known β-D-glucoside 10 (Scheme 2),10 which is accessible from commercial 1,2:5,6-di-O-isopropylidene-α-D-glucofuranose (see ESI† for details). Triflation of the C2 OH was followed by displacement with azide (NaN3) to install the synthetically challenging β-D-mannosamine scaffold. Sequential acetal hydrolysis, permethylation and azide reduction then afforded amine 11 in a 46% over 5 steps. In an identical fashion to 9, the imidazolinium moiety was constructed by formation of bis-amide, reduction to the secondary amine and then thermal cyclisation with HC(OEt)3 and NH4Cl. 1H NMR data confirmed the formation of the glycan-imidazolinium structures (singlet at δ: 9.96–9.69 ppm corresponding to the C2H imidazolinium and carbohydrate (H-1) signals (d, J1,2 = 1.5 (7b) and 2.0 (7c) Hz) at δ: 5.12 (7b) and 4.47 (7c) ppm).
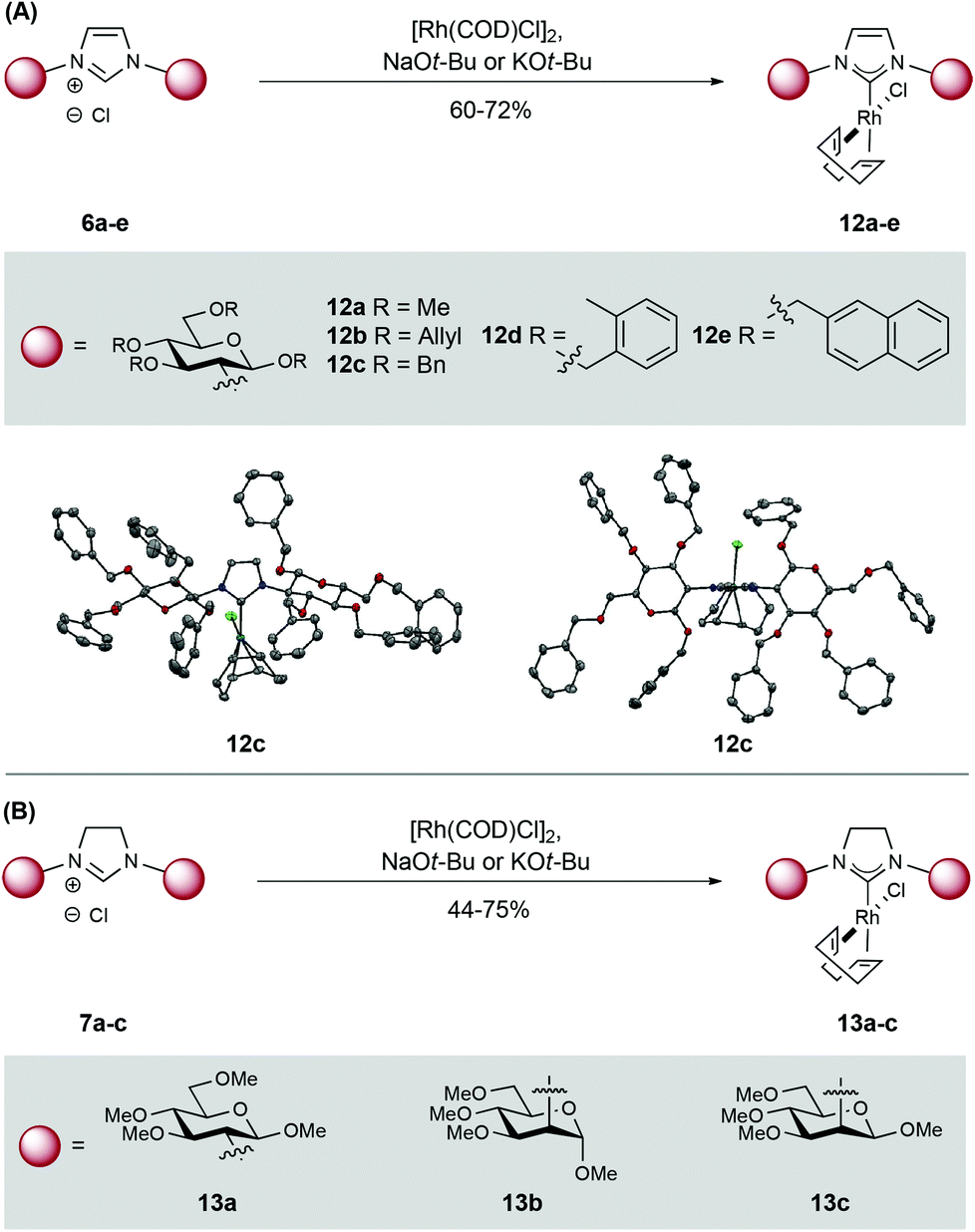
To demonstrate utility, ligation of the carbenes derived from imidazol(in)iums 6a–e and 7a–c to Rh(I) was pursued. Pleasingly, base-promoted complexation (NaOt-Bu or KOt-Bu) to [Rh(COD)Cl]2 proceeded smoothly and the target Rh-NHCs 12a–e and 13a–c were isolated in moderate to good yield. These complexes were purified by flash column chromatography and show good stability in the solid state (Scheme 3).11 Complex 12c was characterized by X-ray diffraction (Scheme 3A) and this revealed an N–C–N angle of 103.2° and a C–Rh bond length of 2.03 Å; these values are in line with other reported Rh-NHC complexes.12
 | ||
| Scheme 3 Complexation of NHC ligands to [Rh(COD)Cl]2. | ||
As a benchmark reaction, we investigated the application of [Rh(NHC)]-based catalysts 12–13 to enantioselective ketone hydrosilylation, a process that is sensitive to the electronic and steric demands of the substrate.13 Complex 12a catalysed the 1,2-additon of diphenylsilane to acetophenone 14a14 and, following acid promoted (HCl) cleavage of the silyl ether, alcohol 15a was isolated in 62% yield and 80:20 er (Table 1, entry 1). Hydrolysis under basic conditions (K2CO3, MeOH) provided an increased yield of 15a but no change in er (entry 2).15 Complex 13a, which is the imidazolinium analogue of 12a, provided similar levels of enantioselectivity (80:20 R:S), (entry 3). Allylated and benzylated complexes 12b–e, which possess bulkier modifying groups on the carbohydrate unit compared to 12a, gave lower levels of enantiocontrol (entries 5–7).16 Changing the configuration of the C2 amine in the glycan from equatorial (glucos-) to axial (mannos-) (13b/c) had a detrimental effect in both yield and enantioselectivity (entries 9 and 10). Interestingly, a switch in preference from R to S was observed for the formation of 15a when changing from an α to a β configuration at C1 (13bvs.13c). These results highlight the importance of substituent configuration and size on the carbohydrate scaffold and show that these factors can affect the enantioselectivity of the reaction.
|
|
|||
|---|---|---|---|
| Entry | Complex | Yielda (%) | er (R:S)b |
|
a Isolated yields.
b
R:S ratios were determined by chiral HPLC using the corresponding racemate as a standard.
c Optimised silyl ether cleavage conditions: K2CO3, MeOH, 2 h.
|
|||
| 1 | 12a | 62 | 80:20 |
| 2 | 12a | 89c | 81:19 |
| 3 | 13a | 85c | 80:20 |
| 4 | 12b | 76 | 65:35 |
| 5 | 12c | 19 | 67:33 |
| 6 | 12d | 29 | 68:32 |
| 7 | 12e | 23 | 60:40 |
| 8 | 13b | 56c | 56:44 |
| 9 | 13c | 51c | 43:57 |
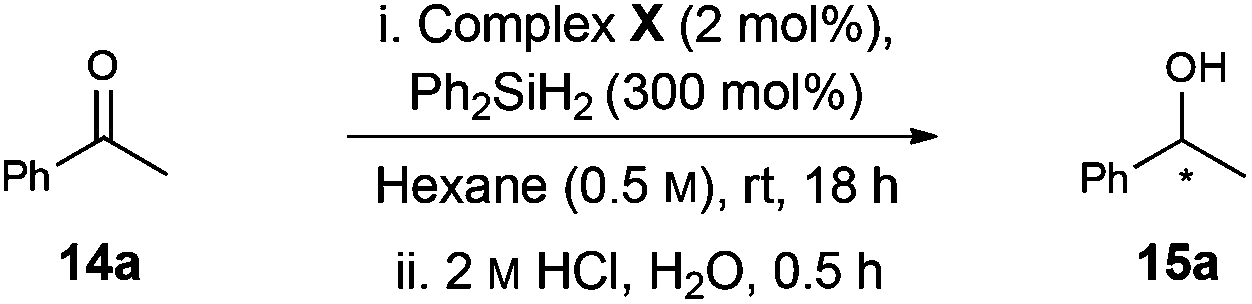
To explore scope further, hydrosilylation of structurally diverse ketones 14b–d was explored using complex 12a (Scheme 4). Pleasingly, reaction yields were uniformly high (84–96% yield) and the products were formed with similar levels of enantioselectivity (71:29–75:25; R:S) across the range of alkyl–alkyl (15b), aryl–alkyl (15c), and bicyclic (15d) ketone motifs. Evidently further optimisation is required, but these results demonstrate that the carbohydrate-based NHC ligand of 12a is effective at relaying chiral information to the “active” coordination sites of the Rh-complex.
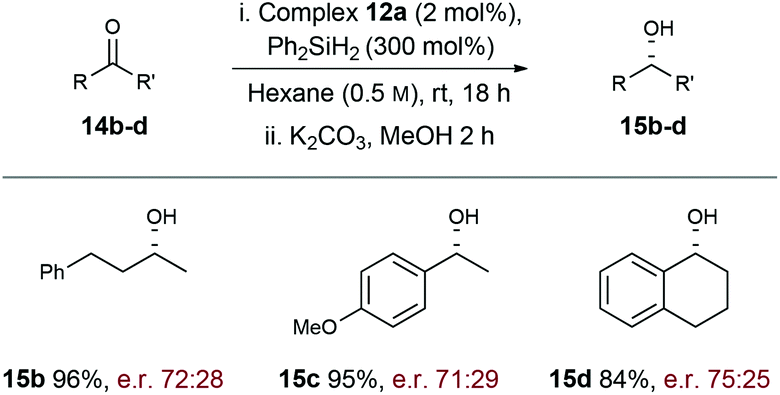 | ||
| Scheme 4 Exploration of ketone scope with complex 12a. | ||
In conclusion, we outline flexible routes to a family of novel C2-linked and C2-symmetric carbohydrate-based NHCs. Suitable selection and modification of the carbohydrate unit is readily achieved and this provides “tunable” access to a diverse range of derivatives. The corresponding Rh(I)-complexes are accessed easily and display promising enantioselectivities in ketone hydrosilylation. Overall, the results described here highlight the potential of this family of simple and modifiable carbohydrate derived NHCs as ligands for enantioselective transition metal catalysis. The development and application of related classes of chiral NHC will be reported in due course.
Acknowledgements
We thank the EPSRC Bristol Chemical Synthesis CDT (ASH), EPSRC (EP/J007455/1) and the Royal Society (JFB) and the EPSRC (EP/J002542/1 and EP/L001926/1) (MCG). Dr M. Haddow (University of Bristol) is thanked for XRDS of 12c.Notes and references
- (a) W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162 CrossRef CAS; (b) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef PubMed; (c) L. A. Schaper, S. J. Hock, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2013, 52, 270 CrossRef CAS PubMed; (d) L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. César, Chem. Rev., 2011, 111, 2705 CrossRef CAS PubMed.
- (a) V. César, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619 RSC; (b) F. Wang, L. J. Liu, W. Wang, S. Li and M. Shi, Coord. Chem. Rev., 2012, 256, 804 CrossRef CAS PubMed.
- (a) A. Schumacher, M. Bernasconi and A. Pfaltz, Angew. Chem., Int. Ed., 2013, 52, 7422 CrossRef CAS PubMed; (b) A. Monney and M. Albrecht, Coord. Chem. Rev., 2013, 257, 2420 CrossRef CAS PubMed; (c) F. Glorius, G. Altenhoff, R. Goddard and C. Lehmann, Chem. Commun., 2002, 2704 RSC; (d) D. M. Lindsay and D. McArthur, Chem. Commun., 2010, 46, 2474 RSC; (e) Y. Zhao and S. R. Gilbertson, Org. Lett., 2014, 16, 1033 CrossRef CAS PubMed; (f) K. C. Nicolaou and H. J. Mitchell, Angew. Chem., Int. Ed., 2001, 40, 1576 CrossRef CAS.
- (a) M. M. K. Boysen, Chem. – Eur. J., 2007, 13, 8648 CrossRef CAS PubMed; (b) S. Castillón, C. Claver and Y. Díaz, Chem. Soc. Rev., 2005, 34, 702 RSC; (c) M. Diéguez, O. Pàmies and C. Claver, Chem. Rev., 2004, 104, 3189 CrossRef PubMed; (d) B. Gyurcsik and L. Nagy, Coord. Chem. Rev., 2000, 203, 81 CrossRef CAS; (e) D. Steinborn and H. Junicke, Chem. Rev., 2000, 100, 4283 CrossRef CAS PubMed.
- (a) F. Tewes, A. Schlecker, K. Harms and F. Glorius, J. Organomet. Chem., 2007, 692, 4593 CrossRef CAS PubMed; (b) J. C. Shi, N. Lei, Q. Tong, Y. Peng, J. Wei and L. Jia, Eur. J. Inorg. Chem., 2007, 2221 CrossRef CAS; (c) T. Nishioka, T. Shibata and I. Kinoshita, Organometallics, 2007, 26, 1126 CrossRef CAS; (d) B. K. Keitz and R. H. Grubbs, Organometallics, 2010, 29, 403 CrossRef CAS; (e) C. C. Yang, P. S. Lin, F. C. Liu and I. J. B. Lin, Organometallics, 2010, 29, 5959 CrossRef CAS; (f) T. Shibata, H. Hashimoto, I. Kinoshita, S. Yano and T. Nishioka, Dalton Trans., 2011, 40, 4826 RSC; (g) T. Shibata, S. Ito, M. Doe, R. Tanaka, H. Hashimoto, I. Kinoshita, S. Yano and T. Nishioka, Dalton Trans., 2011, 40, 6778 RSC; (h) M. Guitet, P. Zhang, F. Marcelo, C. Tugny, J. Jimnez-Barbero, O. Buriez, C. Amatore, V. Mouriès-Mansuy, J. P. Goddard, L. Fensterbank, Y. Zhang, S. Roland, M. Ménand and M. Sollogoub, Angew. Chem., Int. Ed., 2013, 52, 7213 CAS.
- (a) M. C. Galan, A. T. Tran and C. Bernard, Chem. Commun., 2010, 46, 8968 RSC; (b) A. T. Tran, R. Burden, D. T. Racys and M. C. Galan, Chem. Commun., 2011, 47, 4526 RSC; (c) M. C. Galan, A. T. Tran, K. Bromfield, S. Rabbani and B. Ernst, Org. Biomol. Chem., 2012, 10, 7091 RSC; (d) M. C. Galan, R. A. Jones and A. T. Tran, Carbohydr. Res., 2013, 375, 35 CrossRef CAS PubMed.
- L. Hintermann, Beilstein J. Org. Chem., 2007, 3, 22 CrossRef PubMed.
- A. Popelová, K. Kefurt, M. Hlaváčková and J. Moravcová, Carbohydr. Res., 2005, 340, 161 CrossRef PubMed.
- A. Burkhardt, H. Görls and W. Plass, Carbohydr. Res., 2008, 343, 1266 CrossRef CAS PubMed.
- C. M. Nycholat and D. R. Bundle, Carbohydr. Res., 2009, 344, 1397 CrossRef CAS PubMed.
- The Rh(I)-complexes described here were stored under air and no deterioration in catalytic activity was observed over 6 months.
- (a) P. A. Evans, E. W. Baum, A. N. Fazal and M. Pink, Chem. Commun., 2005, 63 RSC; (b) W. Duan, Y. Ma, F. He, L. Zhao, J. Chen and C. Song, Tetrahedron: Asymmetry, 2013, 24, 241 CrossRef CAS PubMed.
- (a) Review: K. Riener, M. P. Högerl, P. Gigler and F. E. Kühn, ACS Catal., 2012, 2, 613 CrossRef CAS; (b) Mechanistic study: N. Schneider, M. Finger, C. Haferkemper, S. Bellemin-Laponnaz, P. Hofmann and L. H. Gade, Angew. Chem., Int. Ed., 2009, 48, 1609 CrossRef CAS PubMed; (c) W. L. Duan, M. Shi and G. B. Rong, Chem. Commun., 2003, 2916 RSC; (d) L. H. Gade, V. César and S. Bellemin-Laponnaz, Angew. Chem., Int. Ed., 2004, 43, 1014 CrossRef CAS PubMed; (e) A. Albright and R. E. Gawley, J. Am. Chem. Soc., 2011, 133, 19680 CrossRef CAS PubMed.
- The use of hexane as solvent led to greatest levels of asymmetric induction for the reduction of 14a to 15a. See ESI† for further catalytic data with the use of other common solvents.
- 1H NMR of crude material (Table 1, entry 1) showed full conversion of 14a to a mixture of silylated alcohol (∼90%) and silyl enol ether byproduct (∼10%). De-silylation using aq. HCl was inefficient, while the use of K2CO3 in MeOH allowed the isolation of 15a in a yield that reflects the efficiency of the reduction step.
- During catalytic runs with 12c–e (entries 4–6), catalyst precipitation was noted. HRMS of these precipitates was consistent with partial O-debenzylation occurring. We postulate the reductive cleavage of the benzyl ethers and concomitant formation of the alcohol, results in a less soluble catalyst and/or leads to deactivation.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and data, NMR spectra and X-ray data for 12c. CCDC 1017544. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob02056a |
| This journal is © The Royal Society of Chemistry 2014 |