
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improved synthesis of the super antioxidant, ergothioneine, and its biosynthetic pathway intermediates†
Peguy Lutete
Khonde
and
Anwar
Jardine
*
Department of Chemistry, University of Cape Town, Cape Town, South Africa. E-mail: anwar.jardine@uct.ac.za
First published on 20th October 2014
Abstract
Ergothioneine and mycothiol are low molecular mass redox protective thiols present in actinomycetes, in particular mycobacteria. We report the improved chemical synthesis of ergothioneine (ESH) and biosynthetic pathway intermediates using either histidine or ESH as the starting material. The detailed mechanism of ESH biosynthesis has not yet been completely elucidated and substrates for enzymes in the pathway will provide valuable tools to aid this study. Particularly interesting is the PLP dependent β-lyase, EgtE, of mycobacteria, having the capability of cleaving the substrate, S-(β-amino-β-carboxyethyl)ergothioneine sulfoxide, to provide ESH. A synthetic route toward ESH pathway intermediates also allowed the preparation of stable isotopically labelled hercynine-d3 which was enzymatically transformed into ESH-d3. The deuterated ergothioneine biosynthetic pathway metabolites are valuable tools for future studies.
Many Gram positive bacteria, such as Mycobacterium tuberculosis, lack the redox protective molecule, glutathione, and instead produce mycothiol and ergothioneine (ESH) as their principal low molecular mass thiols.1,2 ESH is a thiohistidine betaine derivative with a thiol group at the C2 atom (ε-position) of the imidazole ring (Scheme 1). Present knowledge indicates that ESH may play a critical role in the in vivo and in vitro survival of mycobacteria.3 Recently, it was found that ESH is actively secreted into culture media by Mycobacterium smegmatis.4
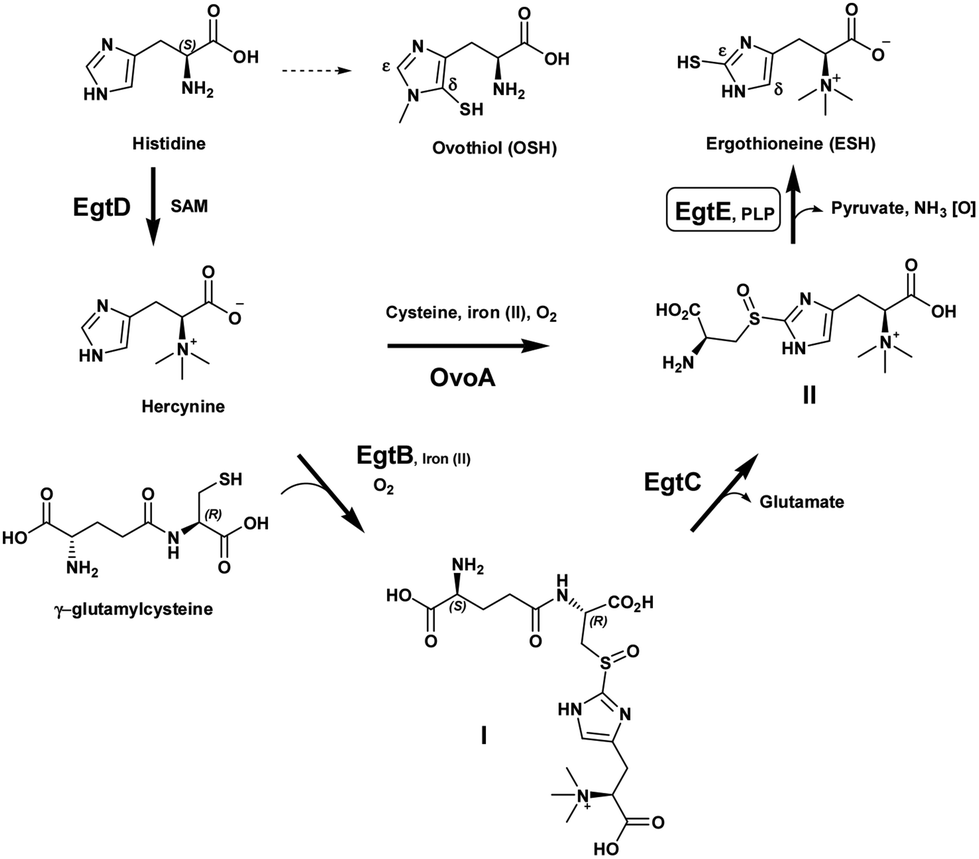 | ||
| Scheme 1 ESH biosynthesis. | ||
A structural variant of ESH, ovothiol A, also serves as an anti-oxidant albeit in sea urchin eggs as well as in the pathogens, Leishmania major and Trypanosoma cruzi.5
Humans do not synthesize ESH, but possess an active transport system, a cation transporter (OCTN1) with high specificity for its uptake from dietary sources.6 The exact function of ESH in humans is currently in the spotlight, in particular its potent antioxidant activity.7 Recent commercial interest in ESH as a super anti-oxidant molecule has added an even greater value to improve the synthetic process development of this molecule.
In 1956, Heath et al. elucidated ESH biosynthesis in Claviceps purpurea. They demonstrated that histidine or a compound closely related to histidine might be a precursor of ESH; subsequent publications disclosed the biosynthetic assembly of ESH utilizing organisms such as Neurospora crassa and Mycobacterium smegmatis with the aid of radio isotopic labelling (14C and 35S).8–10
Melville et al. further established the participation of the S-(β-amino-β-carboxyethyl)ergothioneine sulfoxide as an intermediate in ESH synthesis by incubation of hercynine in cell-free extracts of Neurospora crassa in the presence of O2 and Fe2+.11
It has now been established that ESH is synthesized by the sequential action of five enzymes, encoded by the genes egtA, egtB, egtC, egtD and egtE (Scheme 1).12 EgtA is considered to be a γ-glutamyl cysteine ligase and catalyzes the formation of γ-glutamylcysteine. Histidine is methylated by an S-adenosylmethionine (SAM) dependent methyl transferase, EgtD, to give the trimethyl ammonium betaine, hercynine. Hercynine is then converted into S-(β-amino-β-carboxyethyl)ergothioneine sulfoxide (II) via an iron(II)-dependent oxidase (EgtB) which requires oxygen and γ-glutamylcysteine to produce γ-glutamylcysteinylhercynine (I). The exact nature of the latter transformation, in particular the sulfoxide formation, is still under investigation. Subsequently, a putative class-II glutamine amidotransamidase, EgtC, mediates the hydrolysis of the N-terminus glutamic acid, providing S-(β-amino-β-carboxyethyl)ergothioneine sulfoxide (II). Finally, EgtE, a pyridoxal 5-phosphate (PLP)-dependent β-lyase, gives the final product, ESH.
Recently, the research focus with regard to these mercaptohistidines has shed light on the mechanism of C–S bond formation at the δ- or ε-positions of the imidazole ring.13 OvoA is an iron(II) dependent sulfoxide synthase which catalyzes the first step in ovothiol A synthesis and is a homolog of EgtB. Interestingly, the substrate specificity of EgtB vs. OvoA in achieving C–S bond formation differs significantly. OvoA is very selective towards its sulfur donor substrate and only accepts L-cysteine while it prefers histidine as the co-substrate. However, EgtB requires γ-glutamyl-L-cysteine as the sulfur donor. Furthermore, it is selective toward α-N,N,N-methylation on the histidine, i.e. hercynine as the co-substrate. Surprisingly, OvoA switches its sulfurization pattern on the histidine ring from the δ-carbon to the ε-carbon depending on the level of α-N-methylation.14 Thus, OvoA converts hercynine directly into S-(β-amino-β-carboxyethyl)ergothioneine sulfoxide (II) and produces a minor amount of the δ-sulfoxide (ovothiol substitution pattern) when α-N,N-dimethyl histidine is used as the co-substrate (Scheme 1).
While the enzymes EgtB and EgtC have been expressed in a functional form, EgtE is still elusive and none of the enzymes have been thoroughly studied due to the lack of readily available substrate intermediates.
Here we report the improved synthesis of the ESH and its biosynthetic precursor, S-(β-amino-β-carboxyethyl)ergothioneine sulfoxide (II). The latter sulfoxide is the substrate for the mycobacterial enzyme, EgtE. However, the absolute chirality of the sulfoxide is not known for the natural substrate or the synthetic one. Prior synthesis of intermediate (II) was reported in 1974 but was elaborate and irreproducible and resulted in a low overall yield of 8.5%.15 The authors reported only the position of the aromatic proton resonance and no further structural confirmation. An optical rotation, [α]D +74.4 (c = 0.5, H2O), was reported which did not reconcile with the authentic natural product [α]D +9.1 (c = 0.5, H2O). However, the m.p. of both natural and synthetic products was recorded as 188–190 °C. Nonetheless, it was claimed that synthetic S-(β-amino-β-carboxyethyl)ergothioneine sulfoxide (II) was extensively cleaved to ESH using crude cell free extracts of Neurospora crassa.
A large number of naturally occurring sulfoxides have known absolute configurations and in some instances, the differences in the biological activity of both diastereomers have been determined.16S-substituted cysteine sulfoxides occur almost exclusively in the RcSs configuration in nature. It is therefore possible that the isolated S-(β-amino-β-carboxyethyl)ergothioneine sulfoxide (II) has the RcSs configuration. However, the latter sulfoxide's absolute chirality and its importance are yet to be established.
Two different routes to the target compound, S-(β-amino-β-carboxyethyl)ergothioneine sulfoxide (II), were considered. In this approach, retrosynthetic cleavage of S-(β-amino-β-carboxyethyl)ergothioneine sulfoxide (II) gave the β-chloro-alanine methyl ester and ESH (Scheme 2 and S1†). Thus, S-alkylation of a protected chloromethyl alanine ester (2), derived from serine, provided the core structure (3). The resulting sulfide (3) was oxidised using either mCPBA or H2O2.
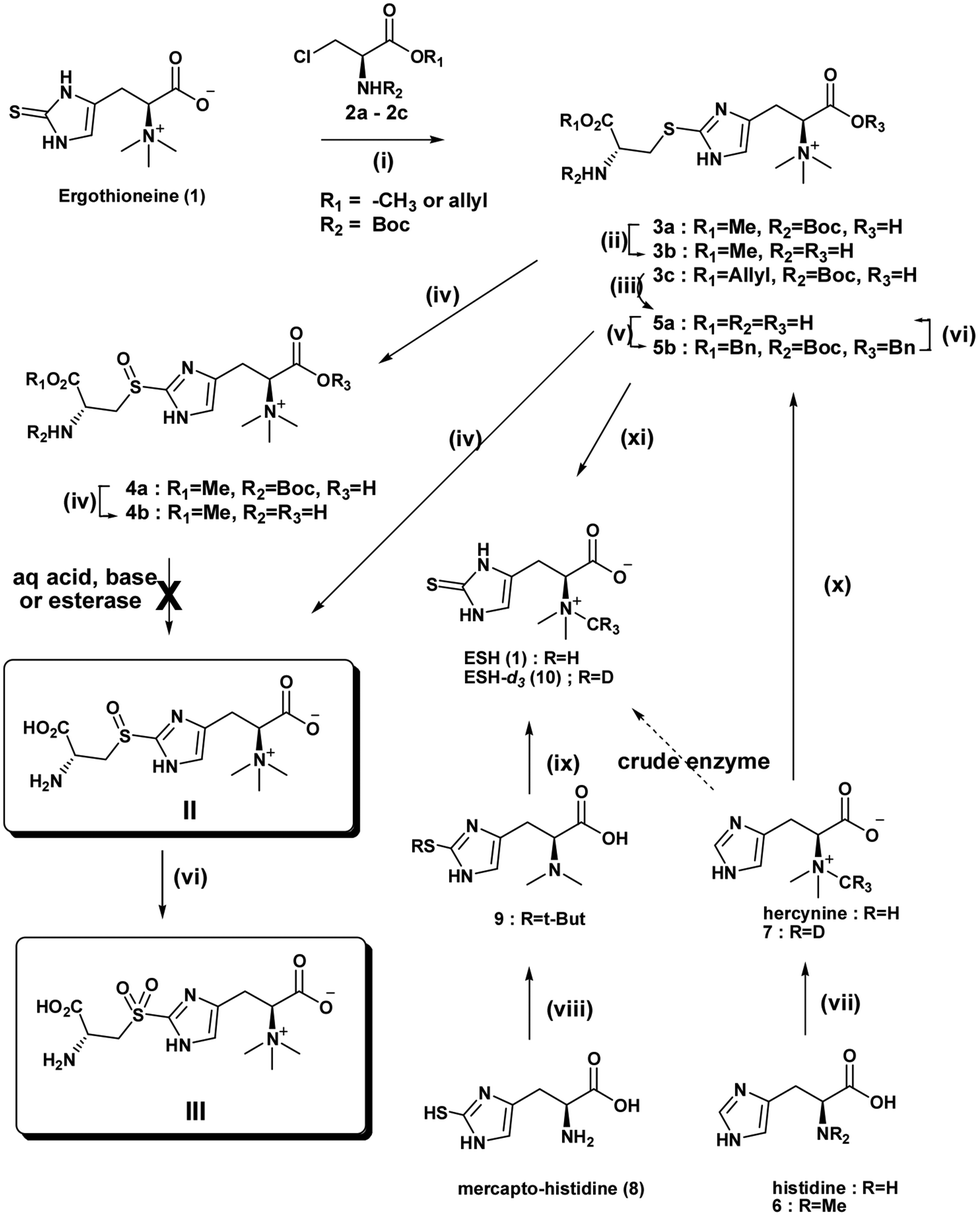 | ||
Scheme 2 Reagents and conditions: (i) Et3N–H2O, 5 h at 30 °C (3a); (ii) N-Boc deprotection: TFA, DCM–H2O, 0–5 °C (3b), (4b) or (II); (iii) (a) RhCl(PPh3)3, EtOH–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) reflux, (b) TFA, DCM–H2O, 0–5 °C (5a); (iv) mCPBA, H2O–DCM 1:1, 5 h at 25 °C (4a) or (II); (v) (a) Boc2O–NaOH, H2O–CH3CN, rt, overnight, (b) BnBr–DMF, rt, overnight; (vi) Pd/C, 3 eq. TFA, H2 50 psi, rt, 12 h; (vii) H2O2, 3 h at rt (III); (viii) (a) CH2O, sodium triacetoxyborohydride–CH3CN, 18–24 h at rt (6), (b) NH4OH, CH3I or CD3I–MeOH, 24 h at rt (7); (ix) (a) t-BuOH–HCl, refluxed for 3–4 h, S-t-butyl mercaptohistidine, (b) CH2O, sodium triacetoxyborohydride–THF, 6–8 h at 10 °C (9); (x) (a) NH4OH, CD3I–MeOH, 24 h, rt, (b) HCl, 2-mercaptopropionic acid refluxed for 21 h (quantitative); (xi) Br2, cysteine HCl, H2O, 1 h at 0 °C (5a); (xii) 3-mercaptopropionic acid, HCl–H2O, 19 h reflux. :1) reflux, (b) TFA, DCM–H2O, 0–5 °C (5a); (iv) mCPBA, H2O–DCM 1:1, 5 h at 25 °C (4a) or (II); (v) (a) Boc2O–NaOH, H2O–CH3CN, rt, overnight, (b) BnBr–DMF, rt, overnight; (vi) Pd/C, 3 eq. TFA, H2 50 psi, rt, 12 h; (vii) H2O2, 3 h at rt (III); (viii) (a) CH2O, sodium triacetoxyborohydride–CH3CN, 18–24 h at rt (6), (b) NH4OH, CH3I or CD3I–MeOH, 24 h at rt (7); (ix) (a) t-BuOH–HCl, refluxed for 3–4 h, S-t-butyl mercaptohistidine, (b) CH2O, sodium triacetoxyborohydride–THF, 6–8 h at 10 °C (9); (x) (a) NH4OH, CD3I–MeOH, 24 h, rt, (b) HCl, 2-mercaptopropionic acid refluxed for 21 h (quantitative); (xi) Br2, cysteine HCl, H2O, 1 h at 0 °C (5a); (xii) 3-mercaptopropionic acid, HCl–H2O, 19 h reflux. | ||
Ishikawa et al. suggested that the reaction may possibly occur via the formation of the cyclic ethylenimine carboxylic acid intermediate produced by an intramolecular SN2 reaction of the β-chloroalanine (2), followed by the ring opening induced by nucleophilic attack of the sulfur atom of ESH, giving the major product N-Boc methyl ester (3a).
Sulfoxidation reaction conditions with H2O2 previously investigated by Ishikawa et al. led to an overoxidation to the sulfone and no analytical evidence was provided.11 In order to prevent overoxidation of the sulfoxide, mCPBA was used. Its milder nature and potential for controlled sulfoxidation compared to hydrogen peroxide is advantageous. The sulfide methyl ester (3a) was subjected to S-oxidation using one equivalent of mCPBA to afford the sulfoxide methyl ester (4a). The synthetic product is most likely a mixture of RcSs and RcRs diastereomers. The lowest steric energy conformations (total energy 34.37 kcal mol−1) of the sulfide methyl ester (3a) indicated potential face selectivity toward sulfoxidation, which could lead predominantly to the SR diastereoisomer sulfoxide derivative (S1.3 & S1.4†). 1H NMR spectra of the sulfoxide methyl ester (4b) displayed evidence of diastereoselectivity (ca. 3:1 ratio) (Fig. S1.4.2 and S1.4.3†). However, only a crystal structure, supported by CD (circular dichroism) spectra, will help establish the absolute configuration of the major chiral sulfoxide and also that of the natural sulfoxide (II). Deliberate oxidation of sulfide (3b), (4b) or sulfoxide (5a) to the sulfone (III) was achieved with excess oxidant.
Finally, attempted global deprotection of the Boc group and hydrolysis of the methyl ester under aqueous acidic conditions gave only the methyl ester sulfide (3b) or methyl ester sulfoxide (4b) from 3a and 4a respectively. Unsuccessful acid, base or esterase mediated ester hydrolysis obligated reconsideration of the synthetic route. Stable amino acid methyl esters have been reported before.17,18 An allyl ester derivative of β-chloroserine provided the N-Boc allyl ester sulfide (3c) after S-alkylation. Sulfoxidation followed by mild RhCl(PPh3)3 catalysed allyl ester cleavage19 and acid mediated Boc protecting group removal gave the target S-(β-amino-β-carboxyethyl)ergothioneine sulfoxide (II) in a moderate overall yield of 63%.
With the second retrosynthetic approach, cleavage of the histidine moiety of S-(β-amino-β-carboxyethyl)ergothioneine sulfoxide (3) gave cysteine and bromohercynine derivative (Scheme 2). This route has received the most attention as it provides the required sulfurization of histidine to provide the commercially important ESH. S-(β-amino-β-carboxyethyl)ergothioneine sulfide (5a) was synthesized in one pot using a slightly modified Erdelmeier method (Scheme 2).20 However, a large quantity of salt by-products had to be removed which hampered purification. Furthermore, the treatment of the S-(β-amino-β-carboxyethyl)ergothioneine sulfide (5a) with 3-mercaptopropionic acid at 90 °C for 18 h gave ESH. The conversion of the sulfide (5a) to the bis-benzyloxy N-Boc protected ester (5b) allowed organic extraction and removal of salts to give a clean benzyl ester (5b). Global deprotection of the N-Boc benzyl ester (5b) was achieved by hydrogenation (Pd/C) in the presence of TFA under 50 psi hydrogen pressure to give pure S-(β-amino-β-carboxyethyl)ergothioneine sulfide (5a). Biphasic sulfoxidation of the sulfide (5a) with mCPBA in a DCM–water mixture gave the S-(β-amino-β-carboxyethyl)ergothioneine sulfoxide (II) in a low overall yield of 36%.
The diastereomers of all S-substituted cysteine sulfoxides exhibit 1H NMR spectra with a characteristic ABX pattern for the S(O)CH2CH(NH2) methylene protons. 1H NMR spectra of the S-(β-amino-β-carboxyethyl)ergothioneine sulfide (5a), sulfoxide (II) and sulfone (III) were consistent with the reported structures. Diagnostic ABX coupling patterns were observed for the α- and β-protons, thus giving rise to sets of doublets at about 4.0–4.5 ppm and 3.3–3.6 ppm respectively. Coupling constants JAX, JBX and JAB were approx. 10, 5 and 14 Hz respectively. 1H NMR COSY spectra as well as 13C NMR aided in complete assignment of the structure.
Improved synthesis of ESH
Here we report the improved synthesis of the ESH via the biosynthetic precursor, S-(β-amino-β-carboxyethyl)ergothioneine sulfide. The latter sulfide and its sulfoxide (II) are substrates for the mycobacterial enzyme, EgtE, that produces ESH. Our process starts with a commercially available N-benzyl protected histidine rather than the unprotected form (key difference). We discovered that the bromination of the N-benzyl protected hercynine intermediate was achieved with N-bromosuccinimide (NBS) to give a N-debenzylated 5-bromohercynine derivative using DMF as solvent in 90% yield. The advantage of the latter intermediate is that subsequent process steps are almost quantitative, relatively simple, all at room temperature, shortened and allow an overall synthesis yield of 70%. Thus, our process is at least 2 times better in overall yield than any prior patented or published process.20 The high yields of the intermediate products also allow viable isotopic labelling steps to be performed. Isotopes are usually very expensive and are advantaged by high yield conversions. The final step involves a biomimetic pyridoxal phosphate (PLP) mediated cleavage of the sulfide or sulfoxide substrates with crude enzymatic extracts of M. smegmatis to give ESH (Scheme 3).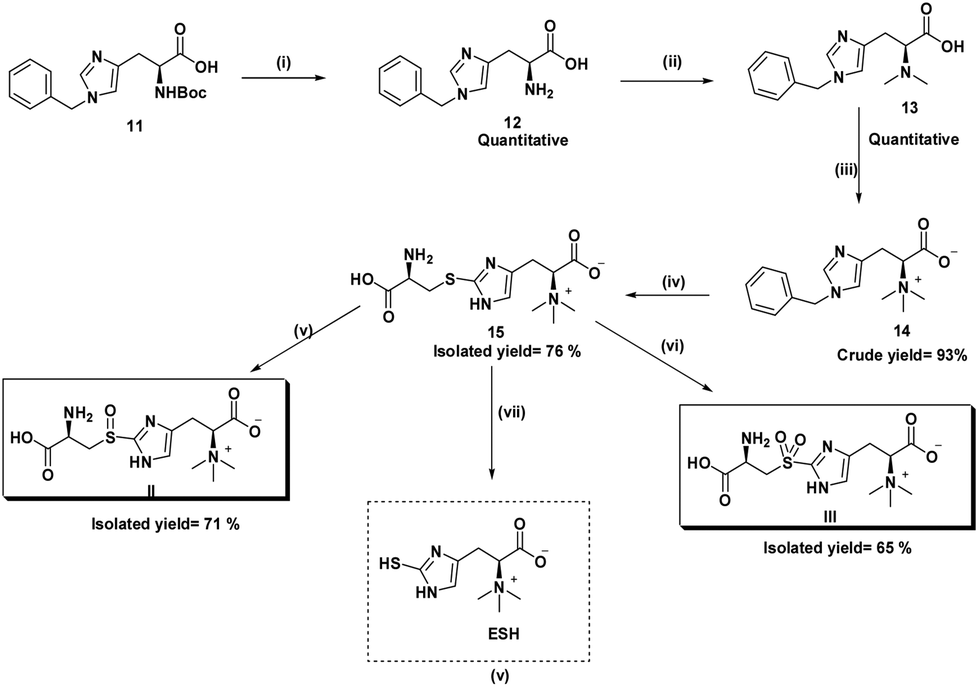 | ||
| Scheme 3 Improved synthesis of sulfoxide (II), sulfone (III) and ESH: (i) TFA, DCM, rt, overnight; (ii) CH2O, NaBH(OAc)3, CH3CN, 24 h at rt; (iii) MeI, THF, 24 h at rt; (iv) (a) NBS (2.5 eq.), DMF, dark at rt, (b) L-cysteine (2.5 eq.), DMF, 24 h at rt; (v) p-toluene sulfonic acid (cat), H2O2 (2.4 eq.); (vi) boric acid (cat), H2O2 (4.8 eq.); (vii) C–S enzyme lyase or PLP non-enzymatic cleavage. | ||
Previously, the biosynthesis pathway of ESH was elucidated utilising radiolabeled intermediates. Advances in synthetic procedures have now made it possible to synthesize the same intermediates incorporating stable isotopes. These intermediates are valuable internal standards in the quantitation of pathway metabolites during external stimuli or drug treatment. Here we also report the synthesis of ESH-d3 (10) and demonstrate its biosynthesis from deuterated hercynine.
Hercynine-d3 (7) was synthesized in a two-step reaction starting with the commercially available L-histidine (Scheme 2). The first step involved reductive amination using aqueous formaldehyde and sodium triacetoxyborohydride to give N,N-dimethyl histidine (6). The second step involved the quaternarization of the crude N,N-dimethyl histidine (6) using methyl-d3 iodide under basic conditions to give the hercynine-d3 (7). Characteristic HRMS peaks indicating M + 3 for hercynine-d3 (7) were obtained (S3.2.1†).
ESH-d3 (10) was synthesized in two sequential reaction steps starting with the S-tert-butyl protected 2-mercaptohistidine (9), derived from mercaptohistidine (8).21 Selective N-methylation with methyl-d3 iodide, followed by S-tert-butyl deprotection using 2-mercaptopropionic acid (tert-butyl scavenger) in HCl gave ESH-d3 (10). Characteristic HRMS peaks indicating M + 3 for the ESH-d3 (10) were obtained (S3.2.2†).
We compared the enzymatic and non-enzymatic PLP mediated synthesis of ESH from the synthetic sulfoxide (II). To this end, crude M. smegmatis cell free extracts were isolated from cultures grown and harvested at the late exponential phase, characterised by high enzymatic activity.10
The crude enzymatic transformation of the ESH biosynthetic pathway precursors, including sulfide and sulfoxide variants, was evaluated by the concomitant production of ESH in excess of basal levels as determined by LCMS. ESH precursor metabolites hercynine-d3 (7), hercynyl cysteine methyl ester sulfoxide (4b), (β-amino-β-carboxyethyl)ergothioneine sulfide (15) and (β-amino-β-carboxyethyl)ergothioneine sulfoxide (II) were incubated with the crude cell free extract at 37° C, pH = 7.4 for 1 day, and analyzed by LCMS.
The control reaction containing only the crude M. smegmatis cell free extract was also treated under the same conditions as the four metabolites. The concentration of ESH thus obtained was 5.70 (±0.30) ng ml−1, which equate to that of endogenous ESH. This concentration was above the limit of detection (0.78 ng ml−1), thus any increase in the concentration of ESH in the experiment above 1 ng ml−1 is considered significant enough to be ascribed to basal levels or biotransformation of the respective substrates by the crude endogenous enzymes of the ESH pathway.
When hercynine-d3 (7) was used as the substrate, as expected, no change in baseline ESH was observed. However, as expected, the LCMS analysis of the mixture revealed the production of ESH-d3 (10). The HRMS (ESI+) displayed a peak at m/z 233.1161 corresponding to [M + d3]+ (S4.2.1†). Peak values at m/z 230.0958 [M]+ or at m/z 231.0980 [M + H]+ belonging to the natural ESH were not present in the chromatogram. This clearly shows that the crude M. smegmatis enzymes (dialyzed extract) transformed the hercynine-d3 (7) into ESH-d3 (10). This proved that the isolated crude enzyme extract was fully functional in the reconstitution of ESH synthesis.
(β-Amino-β-carboxyethyl)ergothioneine sulfoxide (II) biosynthetically produced the highest concentration of ESH (22.6 ng ml−1) (Fig. 1). The (β-amino-β-carboxyethyl)ergothioneine sulfide (15) appeared to be almost as good a substrate as the (β-amino-β-carboxyethyl)ergothioneine sulfoxide (II) (19.2 ng ml−1 ESH). It is well known that PLP-dependent transformations can also undergo enzyme free conversion albeit with a much slower rate and specificity.22 Thus, the non-enzymatic treatment of the sulfide (15) with 50 mM PLP at 37 °C resulted in an efficient formation of ESH (96.3 ng ml−1) (Fig. S4.5.1†). However, under the same conditions, the sulfoxide (II) produced no ESH at all (Fig. S4.5.2†).
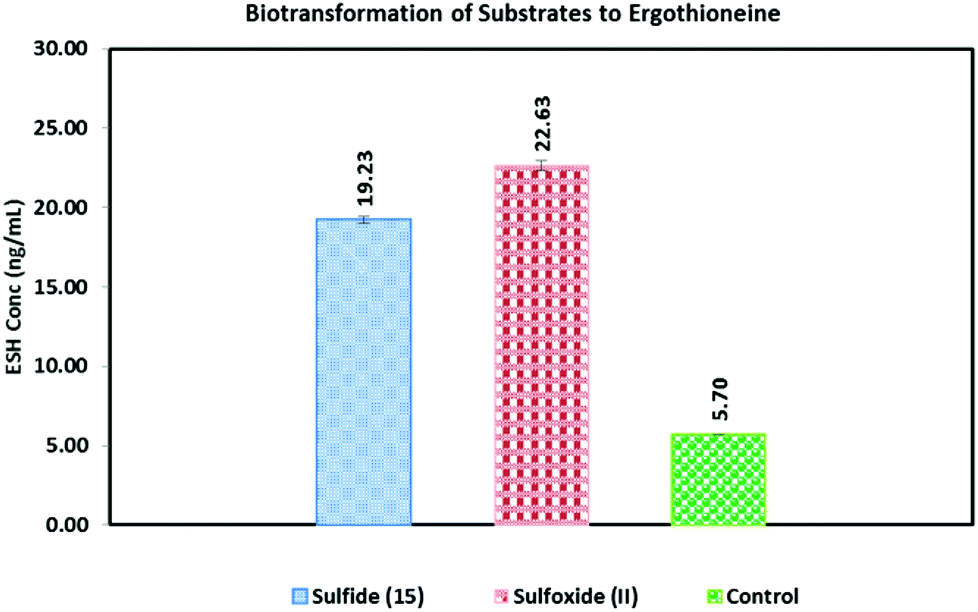 | ||
| Fig. 1 In vitro reconstitution of ESH; 100 μl reactions containing 20 mM Tris HCl pH = 7.4, 20 mM NaCl, 0.2 mM FeSO4·7H2O, 0.5 mM mercaptoethanol, 83 μl of crude M. smeg enzymes and 50 mM of either (a) S-(β-amino-β-carboxyethyl)ergothioneine sulfide (15); (b) S-(β-amino-β-carboxyethyl)ergothioneine sulfoxide (II) and (c) control. The crude enzyme reactions were incubated for 1 day at 37 °C followed by analysis by LC/MS. | ||
The pKa value of the amino acid α-hydrogen usually is in the range 20–30 and many enzymes effectively increase the α-hydrogen acidity with the aid of a coenzyme, PLP. PLP-dependent enzymes exist in their native state as an internal aldimine (Schiff base) (S-5.1–5.3†) with the ε-amino group of a lysine residue present in the catalytic site. The amino acid substrate displaces the lysine from the internal aldimine to form a new aldimine, termed external aldimine. The formation of such an external aldimine can reduce the pKa value of the α-proton of the substrate from 30 to as low as 6.22
PLP-dependent enzymes have been differentiated on the basis of their tertiary structures, more specifically according to fold types I–V. EgtE is classified as a fold type V which is found mainly in enzymes that catalyze β-replacement and β-elimination reactions. In the absence of a crystal structure of EgtE, structure homology analysis amongst mycobacteria is not possible. However, a putative β-lyase from E. tasmaniensis appeared to catalyze this step but has a sequence similarity to EgtE of M.Smeg and M.tb of only 14%.12 Thus, the proposed mechanism of this C–S β-lyase utilizing (β-amino-β-carboxyethyl)ergothioneine sulfide (15) as a substrate was based on that of the well characterized cystalysin from Treponema denticola which shares 31% sequence identity to the β-lyase from E. tasmaniensis.23 It may well be that actinomycetes have similar β-lyases at work.24
We also observed a rather facile non-enzymatic (PLP only) β-elimination with (β-amino-β-carboxyethyl)ergothioneine sulfide (15). However, the conversion of the natural substrate, (β-amino-β-carboxyethyl)ergothioneine sulfoxide (II), to ESH is not that straight forward. Elimination of an unstable (β-amino-β-carboxyethyl)ergothioneine sulfinate (IV) is envisaged, whereby self-condensation leads to the thiosulfinate, which in turn decomposes to ESH and an equivalent amount of relatively stable (β-amino-β-carboxyethyl)ergothioneine sulfinic acid (V). The latter sulfinic acid can subsequently be reduced to the thiol, ESH, with the aid of excess mercaptoethanol, but this may be difficult under the current experimental conditions.25 Note that the (β-amino-β-carboxyethyl)ergothioneine sulfoxide (II) did not produce ESH (in the absence of mercaptoethanol).
Efforts are underway to purify and crystallize EgtE, which would allow a better understanding of enzyme–substrate binding, specificity and the potential for inhibitor design. An EgtD deletion mutant of M. smegmatis and a mycothiol-deficient mutant did not affect their susceptibility to antibiotics.4 However, the ESH/mycothiol-deficient double mutant was significantly more sensitive to peroxide than either of the single mutants lacking either ESH or mycothiol, suggesting that both thiols play a role in protecting M. smegmatis against oxidative stress. Thus, an inhibitor of ESH synthesis will be valuable in drug susceptibility studies of mycobacteria.
Competing interest
The authors declare no competing financial interest.Acknowledgements
We would like to thank Dr Z. MacDonald for mass spectroscopy and the University of Cape Town Research and postgraduate committees for funding as well as Carine Sao and Dr B Baker for mycobacterium cultures.References
- C. F. Robert, Biochim. Biophys. Acta, 2013, 1830, 3182–3198 CrossRef PubMed.
- K. van Laer, C. J. Hamilton and J. Messens, Antioxid. Redox Signaling, 2013, 18, 1642–1653 CrossRef CAS PubMed.
- P. Ta, N. Buchmeier, G. L. Newton, M. Rawat and R. C. Fahey, J. Bacteriol., 2011, 8, 1981–1990 CrossRef PubMed.
- C. S. Emani, M. J. Williams, I. J. Wiid, N. F. Hiten, A. J. Viljoen, R.-D. Pietersen, P. D. van Helden and B. Baker, Antimicrob. Agents Chemother., 2013, 57, 3202–3207 CrossRef CAS PubMed.
- M. R. Ariyanayagam and A. H. Fairlamb, Mol. Biochem. Parasitol., 2001, 115, 189–198 CrossRef CAS.
- D. Grundemann, S. Harlfinger, S. Golz, A. Geerts, A. Lazar, R. Berkels, N. Jung, A. Rubbert and E. Schomig, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5256–5261 CrossRef PubMed.
- I. K. Cheah and B. Halliwell, Biochim. Biophys. Acta, 2012, 1822, 784–793 CrossRef CAS PubMed.
- H. Heath and J. Wildy, Biochem. J., 1957, 65, 220–222 Search PubMed.
- D. B. Melville, S. Eich and M. L. Ludwig, J. Biol. Chem., 1957, 224, 871–877 CAS.
- D. S. Genghof and O. van Damme, J. Bacteriol., 1968, 95, 340–344 CAS.
- Y. Ishikawa, S. E. Israel and D. B. Melville, J. Biol. Chem., 1974, 249, 4420–4427 CAS.
- F. P. Seebeck, J. Am. Chem. Soc., 2010, 132, 6632–6633 CrossRef CAS PubMed . Also Blast sequence alignment analysis of M. smeg EgtE against the β-lyases of T. denticola and E. tasmaniensis (not shown).
- G. T. M. Mashabela and F. P. Seebeck, Chem. Commun., 2013, 49, 7714–7716 RSC.
- H. Song, M. Leninger, N. Lee and P. Liu, Org. Lett., 2013, 15, 4854–4857 CrossRef CAS PubMed.
- Y. Ishikawa, S. E. Israel and D. B. Melville, J. Biol. Chem., 1974, 249, 4420–4427 CAS.
- S. Schwimmer, C. A. Ryan and F. Wong, J. Biol. Chem., 1964, 239, 777–782 CAS.
- J. J. Bryan, R. S. Hinks and P. G. Hultin, Can. J. Chem., 1985, 63, 452–456 CrossRef.
- H. Wild, J. Org. Chem., 1994, 59, 2748–2761 CrossRef CAS.
- K. Sato, M. Omote, A. Ando and I. Kumadaki, Org. Lett., 2004, 6, 4359–4361 CrossRef CAS PubMed.
- (a) I. Erdelmeier, S. Daunay, R. Lebel, L. Farescour and J.-C. Yadan, Green Chem., 2012, 14, 2256–2265 RSC; (b) I. Erdelmeier, The method of synthesizing ergothioneine and analogs, US 20120136159 A1, 2012 Search PubMed.
- M. Trampota, United States Patent, US 7,767,826, B2, 2010 Search PubMed.
- K. Toth and J. P. Richard, J. Am. Chem. Soc., 2007, 129, 3013–3021 CrossRef CAS PubMed.
- H. I. Krupka, R. Huber, S. C. Holt and T. Clausen, EMBO J., 2000, 19, 3168–3178 CrossRef CAS PubMed.
- M. Flavin and A. Segal, J. Biol. Chem., 1964, 239, 2220–2227 CAS.
- S. Sivaramakrishnan, A. H. Cummings and K. S. Gates, Bioorg. Med. Chem. Lett., 2010, 20, 444–447 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental materials, methods, and figures. See DOI: 10.1039/c4ob02023e |
| This journal is © The Royal Society of Chemistry 2015 |