
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile diverted synthesis of pyrrolidinyl triazoles using organotrifluoroborate: discovery of potential mPTP blockers†
Sun hwa
Jung
ab,
Kihang
Choi
b,
Ae Nim
Pae
ac,
Jae Kyun
Lee
a,
Hyunah
Choo
ac,
Gyochang
Keum
*ac,
Yong Seo
Cho
ac and
Sun-Joon
Min
*ac
aCenter for Neuro-Medicine, Brain Science Institute, Korea Institute of Science and Technology (KIST), Hwarangno 14-gil 5, Seongbuk-gu, Seoul 136-791, South Korea
bDepartment of Chemistry, Korea University, Seoul 136-701, South Korea
cDepartment of Biological Chemistry, Korea University of Science and Technology (UST), 176 Gajung-dong, 217 Gajungro, Yuseong-gu, Daejeon, 305-333, South Korea. E-mail: gkeum@kist.re.kr; sjmin@kist.re.kr
First published on 7th October 2014
Abstract
This article describes the rapid and diversified synthesis of pyrrolidinyl triazoles for the discovery of mitochondrial permeability transition pore (mPTP) blockers. The 1,3-dipolar cycloaddition of ethynyl trifluoroborate with azidopyrrolidine produced a key intermediate, triazolyl trifluoroborate 4, which subsequently underwent a Suzuki–Miyaura coupling reaction to afford a series of 1,4-disubstituted triazoles 2. Subsequent biological evaluation of these derivatives indicated 2ag and 2aj as the most potent mPTP blockers exhibiting excellent cytochrome P450 (CYP) stability when compared to the previously reported oxime analogue 1. The present work clearly demonstrates that a 1,2,3-triazole can be used as a stable oxime surrogate. Furthermore, it suggests that late-stage diversification through coupling reactions of organotrifluoroborates is suitable for the rapid discovery of biologically active molecules.
1. Introduction
1,2,3-Triazoles are an important class of heterocycles in pharmaceutical science, chemical biology, supramolecular chemistry and materials science.1–5 In particular, various medicinal agents contain triazoles as important pharmacophores, which can be favorable in the binding to biomolecular targets.6 Triazoles are often used as peptide bond isosteres because of their planarity and enhanced physiochemical properties such as increased metabolic stability and solubility.7–9 The nitrogen atoms in the triazole rings have hydrogen bonding capabilities and are not protonated at physiological pH because of their low basicity.The Huisgen 1,3-dipolar cycloaddition of azides and alkynes is widely used for the synthesis of 1,2,3-triazoles.9–13 The copper(I) catalyzed version of this reaction, using azides and terminal alkynes, provides 1,4-disubstituted 1,2,3-triazoles with high regioselectivity,14–18 making this reaction a powerful tool in medicinal chemistry for the structural modification of lead compounds.
Recently, we have identified pyrrolidinyl oxime, 1, as an mPTP (mitochondrial permeability transition pore) blocker, which is a viable therapeutic target for the treatment of Alzheimer's disease (AD).19 It effectively recovered amyloid beta-induced mitochondrial dysfunction; however, the cytochrome P450 (CYP) enzyme inhibition study revealed that it significantly reduced CYP enzymatic activities, which can cause side effects such as a drug–drug interaction. In this regard, we proposed that the stable 1,2,3-triazole could serve as an effective isostere of oxime to improve the CYP stability without reducing the mPTP inhibitory activity.20 In addition, incorporation of substituents on the aromatic ring can alter the effect of CYP inhibition. Considering these structural modifications, we propose triazole derivatives 2 as new candidates for mPTP blockers (Fig. 1).
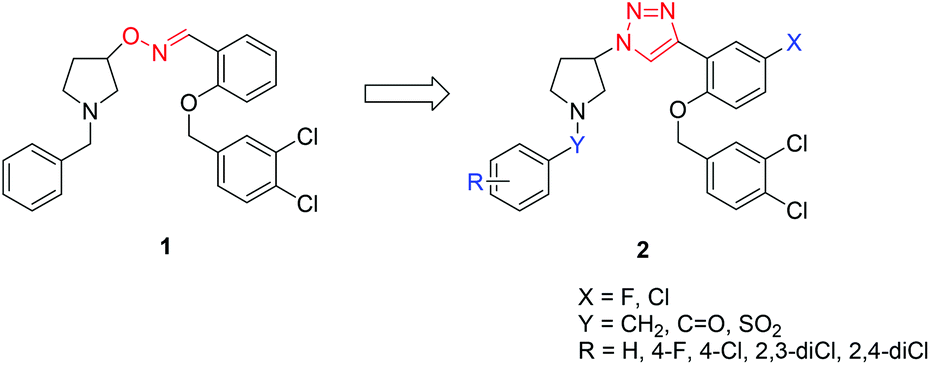 | ||
| Fig. 1 Structures of pyrrolidinyl oxime 1 and triazole derivatives 2. | ||
As described in Scheme 1, we designed an efficient, step-economical synthetic approach toward a chemical library of diverse 1,2,3-triazole derivatives. The synthesis of a triazole via a typical 1,3-cycloaddition requires the preparation of a terminal alkyne as a precursor; hence, this strategy is not effective for achieving molecular diversity. Alternatively, we envisioned that triazolyl trifluoroborate 4, derived from the annulation of ethynyl trifluoroborate 621,22 and azide 7, can undergo a rapid Suzuki–Miyaura coupling reaction to afford various triazole analogues 3.23–25 A recent report showed the transformation of 6 to the corresponding triazolyl trifluoroborates, and subsequent cross-coupling reactions, to afford 1,4-disubstituted 1,2,3-triazoles.26 However, this protocol was only applied to a limited number of substrates, such as primary or aromatic azides, and no further applications of the synthesized compounds were pursued.
 | ||
| Scheme 1 Proposed synthesis of 1-pyrrolidinyl-1,2,3-triazoles 2. | ||
In this paper, we report an efficient and divergent synthesis of various highly functionalized 1-pyrrolidinyl 4-aryl 1,2,3-triazoles 2via triazolyl trifluoroborates for the discovery of mPTP blockers. Furthermore, we investigate the effect of structural modification of oximes into triazoles on the target efficacy and CYP enzyme stability.
2. Results and discussion
As depicted in Table 1, we performed a model study through the cycloaddition of 6, using benzyl azide 8 as a substrate. Based on the reaction conditions reported in the literature,27 we employed copper(I) halides as catalysts as well as various bases and additives. Initially, the desired triazole trifluoroborate 9 was obtained in low yields when copper(I) iodide was used (entries 1–3). We explored several bases in order to improve the reaction yield, but the cycloaddition reactions did not afford 9 in any of the cases (entries 4–8). However, 9 was formed in high yield when the reaction with copper(I) bromide and cesium carbonate in the presence of N,N-dimethylethylenediamine (DMEDA) was performed at 90 °C for 30 min (entry 11).28,29 The choice of solvent system is crucial to prevent the generation of side product 10, which is most likely formed by protodeboronation via a solvolysis mechanism.30|
|
||||
|---|---|---|---|---|
| Entry | Conditions | Temp. (°C) | Time (h) | Yield (%) |
| a Reaction was performed in acetone-d6. b The deboronated product 10 was formed exclusively. | ||||
| 1 | CuI | 60 | 17 | 24 |
| 2a | CuI | 60 | 20 | 40 |
| 3 | CuI | 90 | 24 | 28 |
| 4 | CuI, TEA | 50 | 24 | — |
| 5 | CuI, K2CO3 | 50 | 24 | — |
| 6 | CuI, Cs2CO3 | 50 | 24 | — |
| 7 | CuI, iPr2NEt | 50 | 24 | — |
| 8 | CuI, iPr2NH | 50 | 24 | — |
| 9a | CuBr, Cs2CO3 | 60 | 15 | —b |
| 10 | CuBr, Cs2CO3, DMEDA | 60 | 3 | 40 |
| 11 | CuBr, Cs2CO3, DMEDA | 90 | 0.5 | 85 |
| 12a | CuBr, Cs2CO3, DMEDA | 60 | 15 | Trace |
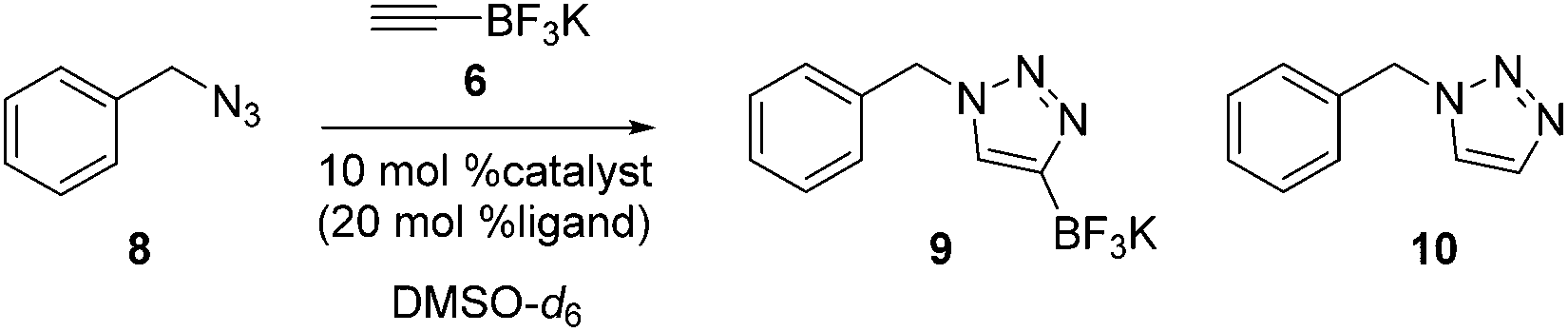
After the optimal reaction conditions were selected, the scope of the cycloaddition reaction of 6 with various substrates was briefly investigated (Table 2). In general, the reactions of aromatic or aliphatic azides 11 afforded the corresponding triazoles 12 in good to excellent yields. It was found that various functional groups, such as alcohol, ester, allyl, and carbamate, were tolerated under these reaction conditions (entries 1–3, and 5). In particular, secondary pyrrolidine/piperizine azides 11d and 11e gave the desired products 12d and 12e in good yields (entries 4, 5), which could then be applied to the divergent synthesis of the proposed 1,4-disubstituted triazole library. While the sterically demanding 2,6-dimethylphenyl azide 11i was readily converted to triazole 12i within 3 h (entry 9), the use of adamantyl azide 11f did not provide the desired product 12f because of the high steric hindrance (entry 6).
|
|
||||
|---|---|---|---|---|
| Entry | R1 | Time (h) | Product | Yield (%) |
| 1 | HOCH2CH2CH2 (11a) | 0.5 | 12a | 72 |
| 2 | EtO2CCH2 (11b) | 1 | 12b | 62 |
| 3 | PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2 (11c) CHCH2 (11c) |
0.5 | 12c | 87 |
| 4 |

|
1.5 | 12d | 71 |
| 5 |

|
1.5 | 12e | 45 |
| 6 |

|
24 | 12f | No reaction |
| 7 |

|
0.5 | 12g | 90 |
| 8 |

|
1 | 12h | 72 |
| 9 |
|
3 | 12i | 69 |

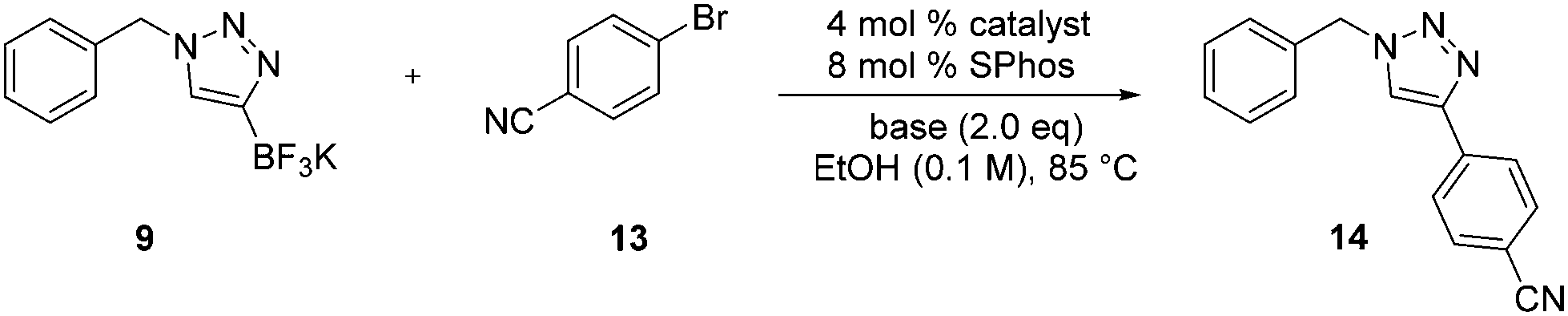
The Suzuki–Miyaura coupling reaction was then explored in order to produce a variety of derivatives in the late-stage functionalization, as suggested in Scheme 1. The reaction of 9 with 4-bromobenzonitrile 13 was tested, and the results are summarized in Table 3. Following a literature procedure,31 the initial reactions were attempted in the presence of Pd(OAc)2 and SPhos using various bases. When K2CO3, Cs2CO3, and K3PO4 were used as bases, 14 was not formed at all, or was obtained in very low yield. However, the use of Na2CO3 as a base dramatically increased the yield to up to 99%. A variety of catalysts were also screened in order to optimize the reaction conditions. Among the catalysts tested, Pd2(dba)3 provided the best results, affording triazole 14 in quantitative yield. Many solvent systems were used, such as DMF/H2O, DME, THF, and dioxane; however, all these afforded lower yields of the coupled product 14. Alternatively, ethanol gave higher yields, suggesting that it is a critical component in the reaction efficiency.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Base | Time (h) | Yield (%) |
| a No ligand was used. b XPhos was used as a ligand. c Catalyst (2 mol%), ligand (4 mol%). | ||||
| 1 | Pd(OAc)2 | K2CO3 | 3 | — |
| 2 | Pd(OAc)2 | Cs2CO3 | 3 | — |
| 3 | Pd(OAc)2 | K3PO4 | 3 | 8 |
| 4 | Pd(OAc)2 | Na2CO3 | 18 | 99 |
| 5 | PdCl2(dppf)·CH2Cl2 | Na2CO3 | 12 | 78 |
| 6 | Pd(PPh3)4 | Na2CO3 | 18 | 76 |
| 7 | Pd(dba)2 | Na2CO3 | 7 | 96 |
| 8 | Pd2(dba)3 | Na2CO3 | 18 | 100 |
| 9a | Pd2(dba)3 | Na2CO3 | 24 | 9 |
| 10b | Pd2(dba)3 | Na2CO3 | 18 | 94 |
| 11c | Pd2(dba)3 | Na2CO3 | 18 | 88 |
Given the positive results from the optimized cycloaddition reaction and the Suzuki–Miyaura coupling reaction in the model study, these reactions were applied to the synthesis of triazole derivatives 2 as mPTP blockers (Scheme 2). Starting from commercially available 3-hydroxy N-Boc-pyrrolidine 15, 3-azidopyrrolidine 7 was easily prepared by mesylation and consecutive SN2 displacement of the azide. According to the experimental procedure previously established in Table 1, we attempted the 1,3-dipolar cycloaddition of potassium ethynyl trifluoroborate 6 with azide 7. As anticipated, the desired triazolyl trifluoroborate 4 was obtained in 70% yield. In the meantime, 2-bromo-1-benzyloxybenzenes 17, containing either a fluoride or chloride substituent at the 4-position, were easily prepared as coupling partners by benzylation of bromophenols 16. A coupling reaction of 4 and 17 could then be completed under the optimized conditions. This reaction proceeded successfully, giving 1,4-disubstituted triazoles 3 in moderate yields. Removal of the Boc protecting group on 3 using trifluoroacetic acid afforded the secondary amine 18 as a salt. This was then subjected to reductive amination, acylation, and sulfonylation to provide N-functionalized pyrrolidines 2 in good yields. Thus, we obtained a series of twenty new triazole derivatives in this concise and divergent manner using 4 as a key intermediate.
 | ||
| Scheme 2 Divergent synthesis of 1-pyrrolidynyl triazoles 2: (a) (i) MsCl, TEA, CH2Cl2, rt, 18 h, (ii) NaN3, DMF, 80 °C, 3 h, 85%; (b) 6, CuBr, Cs2CO3, DMEDA, DMSO, 90 °C, 1.5 h, 70%; (c) 3,4-dichlorobenzyl chloride, K2CO3, KI, acetone, 60 °C, 75% (17a), and 95% (17b); (d) Pd2(dba)3, SPhos, Na2CO3, 85 °C, 18 h, 54% (3a), and 53% (3b); (e) TFA, CH2Cl2, rt, 0.5 h, 99% (18a), and 99% (18b); (f) ArCHO, NaBH(OAc)3, AcOH, THF, rt, 24 h (for 2aa–e and 2ba–e), or ArCOCl, Et3N, THF, rt, 24 h (for 2af–h and 2bf–h), or ArSO2Cl, Et3N, THF, rt, 24 h (for 2ai–j and 2bi–j). | ||
The inhibitory activity of pyrrolidinyl triazoles 2 against amyloid beta-induced mPTP opening was evaluated by a cell-based JC-1 assay (Fig. 2).32,33 In this assay, the color of the JC-1 dye changed from green to red as the mitochondrial membrane potential increased. Thus, the lower the percent increase of the green/red (g/r) ratio of the compound, the higher is the recovery of mitochondrial function in the cell. Twelve of the triazole derivatives 2 increased the inhibition of mPTP opening with less than 40% increase in the g/r ratio, indicating that the triazole is an effective structural surrogate of oxime. Further consideration of the structure–activity relationship (SAR) demonstrated the 2a series containing fluorine on the internal phenyl ring (X = F) to be more active than the chlorine-substituted 2b compounds. Smaller substituents at the R position were found to increase the inhibitory activity against mPTP opening. In particular, electron-withdrawing groups, such as carbonyl or sulfonyl, between pyrrolidine and the terminal phenyl ring significantly enhanced the inhibitory activity when compared to the initial lead compound 1.
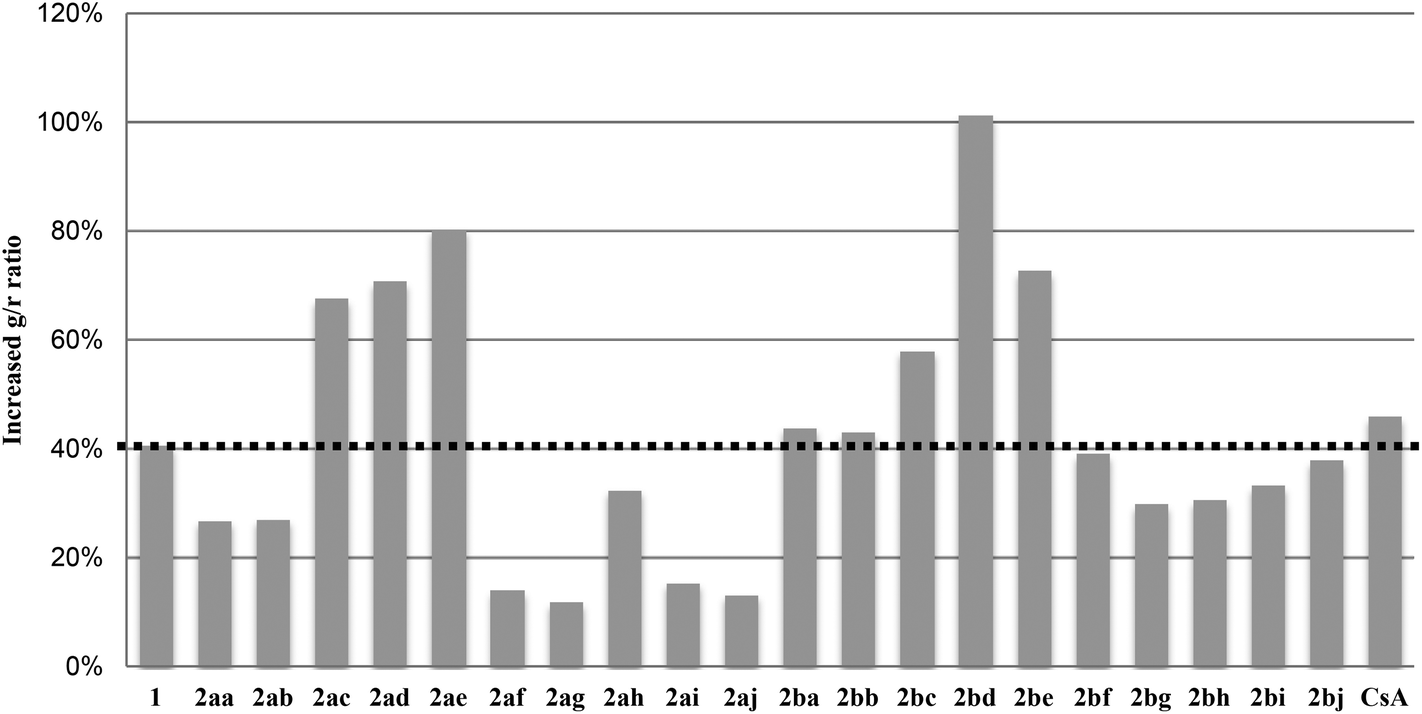 | ||
| Fig. 2 Inhibitory activity of 2 against amyloid beta-induced mPTP opening. Percent increase in the fluorescence ratio (green/red) after treatment of each compound with amyloid beta with respect to that of amyloid beta alone (100%). Cyclosporin A (CsA) was used as a control. | ||
In order to determine the effect of triazoles on the CYP stability, the inhibitory activities of selected compounds 2 against five CYP450 isozymes in the human liver were tested. Using the CYP450 screening kits, the percent remaining activity of the CYP enzymes after treatment with 2 was obtained, as shown in Table 4. The selected compounds 2 significantly increased the percent CYP remaining activities in comparison with oxime 1. In particular, the most active triazole derivatives 2ag and 2aj, containing 4-fluorobenzoyl and 4-fluorobenzenesulfonyl groups, exhibited relatively low inhibitory activities against the tested CYP isozymes. This suggested that this triazole-based structural modification played an important role in the stability of the CYP enzymes as well as in blocking the mPTP opening.
| Compds | % CYP isozyme remaining activityc | ||||
|---|---|---|---|---|---|
| 1A2 | 2D6 | 2C9 | 3A4 | 2C19 | |
| a Each value represents the mean of triplicate experiments. b Each value represents the mean of duplicated experiments. c The remaining activity of each isozyme was obtained at a concentration of 10 μM using a fluorogenic Vivid® CYP450 screening kit. d The reference compounds for each CYP isozyme assay are as follows: α-naphthoflavone (1A2), quinidine (2D6), sulfaphenazole (2C9), ketoconazole (3A4), and miconazole (2C19). | |||||
| 2ab | 67.97 | 47.00 | 32.74 | 12.34 | 11.21 |
| 2ag | 83.44 | 102.41 | 20.66 | 36.74 | 20.47 |
| 2aj | 86.97 | 137.73 | 33.77 | 38.72 | 13.64 |
| 2bb | 62.32 | 44.25 | 80.28 | 8.32 | 14.06 |
| 2bg | 65.10 | 78.86 | 23.30 | 26.72 | 20.67 |
| 2bj | 68.08 | 37.66 | 41.60 | 16.83 | 5.37 |
| 1 | 3.24 | 6.25 | 34.67 | −0.06 | — |
| Controld | 4.27 | 6.66 | 10.44 | 9.85 | 5.77 |
3. Conclusion
In this study, we have demonstrated the synthesis of twenty pyrrolidinyl triazoles 2via cycloaddition of ethynyl trifluoroborate 6, followed by the Suzuki–Miyaura coupling reaction of the corresponding cycloadducts. These were then evaluated for their inhibitory activity against amyloid beta-induced mPTP opening. The reaction conditions that were optimized in the model system were successfully applied to the synthesis of key intermediates 4, which were transformed to a library of N-functionalized pyrrolidinyl triazoles 2 in a divergent manner with high overall yields. Thus, we believe that this methodology is useful for the rapid preparation of a large number of 1,4-disubstituted 1,2,3-triazole analogs.Screening of the triazole analogues 2 in the JC-1 assay identified 2ag and 2aj as potent mPTP blockers. These also showed excellent CYP stability in comparison with oxime 1. These findings indicate that the 1,2,3-triazole functional group can be used as a stable isostere of oxime to improve the CYP stability while maintaining the inhibitory activity against a molecular target.
Experimental section
General
All reactions were carried out under dry nitrogen unless otherwise indicated. Commercially available reagents were used without further purification. Solvents and gases were dried according to standard procedures. Organic solvents were evaporated with reduced pressure using a rotary evaporator. Analytical thin layer chromatography (TLC) was performed using glass plates precoated with silica gel (0.25 mm). TLC plates were visualized by exposure to UV light (UV), and then were visualized with a p-anisaldehyde stain followed by brief heating on a hot plate. Flash column chromatography was performed using silica gel 60 (230–400 mesh, Merck) with the indicated solvents. 1H, 11B, 13C and 19F spectra were recorded on Bruker 300, Bruker 400 (376 MHz) or Varian 300 NMR spectrometers. 1H NMR spectra are represented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), integration, and coupling constant (J) in hertz (Hz). 1H NMR chemical shifts are reported relative to CDCl3 (7.26 ppm). 13C NMR spectra were recorded relative to the central line of CDCl3 (77.0 ppm). 19F NMR chemical shifts were referenced to external CFCl3 (0.0 ppm). 11B NMR spectra at 128 MHz were obtained on a spectrometer equipped with the appropriate decoupling accessories. All 11B NMR chemical shifts were referenced to external BF3·OEt2 (0.0 ppm) with a negative sign indicating an upfield shift.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) to give the title compound 14 (25.7 mg, 99.9%) as a white solid; mp 140.4–142.0 °C; 1H NMR (CDCl3, 400 MHz) δ 7.90 (d, J = 8.5 Hz, 2H), 7.77 (s, 1H), 7.66 (d, J = 8.5 Hz, 2H), 7.38 (m, 3H), 7.31 (m, 2H), 5.58 (s, 2H); 13C NMR (CDCl3, 100 MHz) δ 146.3, 134.9, 134.2, 132.6, 129.2, 129.0, 128.1, 126.0, 120.7, 118.7, 111.4, 54.4.
:1) to give the title compound 7 (0.49 mg, 85%) as a colorless oil; 1H NMR (CDCl3, 300 MHz) δ 4.11 (m, 1H), 3.52–3.42 (m, 4H), 2.12–1.95 (m, 2H), 1.45 (s, 9H); 13C NMR (CDCl3, 100 MHz) δ 153.7, 78.8, 60.1, 59.4, 50.6, 50.3, 43.5, 43.2, 30.9, 30.1, 27.9.
:15) to give the title compound 17a (1.38 g, 75%) as a yellow oil; 1H NMR (CDCl3, 300 MHz) δ 7.56 (d, J = 1.9 Hz, 1H), 7.45 (d, J = 8.2 Hz, 1H), 7.31 (td, J = 2.6, 7.8 Hz, 2H), 7.99–6.93 (m, 1H), 6.84 (q, J = 4.6 Hz, 1H), 4.99 (s, 2H).
:1) to give the title compound 2aa (14.8 mg, 62%) as a yellow solid; mp 148.6–149.9 °C; 1H NMR (CDCl3, 300 MHz) δ 8.16 (s, 1H), 8.07 (dd, J = 3.0, 9.5 Hz, 1H), 7.54 (s, 1H), 7.37 (d, J = 8.2 Hz, 1H), 7.25–7.23 (m, 6H), 7.00–6.91 (m, 2H), 5.25 (m, 1H), 5.05 (s, 2H), 3.66 (q, J = 13.1 Hz, 2H), 2.91 (m, 3H), 2.58 (m, 2H), 2.06 (m, 1H); 13C NMR (CDCl3, 75 MHz) δ 157.7 (d, 1J = 237.8 Hz), 150.6 (d, 4J = 2.0 Hz), 142.5 (d, 4J = 2.8 Hz), 136.7, 132.9, 132.5, 130.8, 129.5, 128.9, 128.6, 127.9, 126.8, 122.1, 121.5 (3d, J = 8.7 Hz), 115.0 (d, 2J = 23.3 Hz), 114.5 (2d, J = 25.0 Hz), 113.4 (d, 3J = 8.4 Hz), 77.2, 69.9, 59.2, 59.0, 52.6, 32.7; HRMS-ESI (m/z): [M + H]+ calcd for C26H24Cl2FN4O 497.13057, found 497.13019.
:1) to give the title compound 2af (18.7 mg, 67%) as a white solid; mp 55.4–57.5 °C; 1H NMR (CDCl3, 300 MHz) δ 8.04 (d, J = 7.3 Hz, 1H), 7.90 (s, 1H), 7.52–7.41 (m, 7H), 7.23 (s, 1H), 6.98 (td, J = 3.0, 8.2 Hz, 1H), 6.89 (q, J = 4.4 Hz, 1H), 5.21 (bs, 1H), 5.10 (s, 2H), 4.19–3.65 (m, 4H), 2.51 (bs, 2H); 13C NMR (CDCl3, 75 MHz) δ 170.1, 157.7 (d, 1J = 238.0 Hz), 150.5, 142.4, 136.7, 135.9, 133.0, 132.6, 130.9, 130.4, 129.4, 128.4, 127.2, 126.7, 122.3, 120.9 (d, 3J = 9.2 Hz), 115.3 (d, 2J = 23.4 Hz), 114.5 (d, 2J = 25.3 Hz), 113.4 (d, 3J = 8.3 Hz), 77.2, 69.8, 65.8, 63.7, 59.3, 58.4, 54.2, 51.8, 47.7, 44.3, 32.7, 30.5; HRMS-ESI (m/z): [M + H]+ calcd for C26H22Cl2FN4O2 511.10984, found 511.10888.
:3) to give the title compound 14 (25.7 mg, 99.9%) as a white solid; mp 140.4–142.0 °C; 1H NMR (CDCl3, 400 MHz) δ 7.90 (d, J = 8.5 Hz, 2H), 7.77 (s, 1H), 7.66 (d, J = 8.5 Hz, 2H), 7.38 (m, 3H), 7.31 (m, 2H), 5.58 (s, 2H); 13C NMR (CDCl3, 100 MHz) δ 146.3, 134.9, 134.2, 132.6, 129.2, 129.0, 128.1, 126.0, 120.7, 118.7, 111.4, 54.4.
:1) to give the title compound 7 (0.49 mg, 85%) as a colorless oil; 1H NMR (CDCl3, 300 MHz) δ 4.11 (m, 1H), 3.52–3.42 (m, 4H), 2.12–1.95 (m, 2H), 1.45 (s, 9H); 13C NMR (CDCl3, 100 MHz) δ 153.7, 78.8, 60.1, 59.4, 50.6, 50.3, 43.5, 43.2, 30.9, 30.1, 27.9.
:15) to give the title compound 17a (1.38 g, 75%) as a yellow oil; 1H NMR (CDCl3, 300 MHz) δ 7.56 (d, J = 1.9 Hz, 1H), 7.45 (d, J = 8.2 Hz, 1H), 7.31 (td, J = 2.6, 7.8 Hz, 2H), 7.99–6.93 (m, 1H), 6.84 (q, J = 4.6 Hz, 1H), 4.99 (s, 2H).
:1) to give the title compound 2aa (14.8 mg, 62%) as a yellow solid; mp 148.6–149.9 °C; 1H NMR (CDCl3, 300 MHz) δ 8.16 (s, 1H), 8.07 (dd, J = 3.0, 9.5 Hz, 1H), 7.54 (s, 1H), 7.37 (d, J = 8.2 Hz, 1H), 7.25–7.23 (m, 6H), 7.00–6.91 (m, 2H), 5.25 (m, 1H), 5.05 (s, 2H), 3.66 (q, J = 13.1 Hz, 2H), 2.91 (m, 3H), 2.58 (m, 2H), 2.06 (m, 1H); 13C NMR (CDCl3, 75 MHz) δ 157.7 (d, 1J = 237.8 Hz), 150.6 (d, 4J = 2.0 Hz), 142.5 (d, 4J = 2.8 Hz), 136.7, 132.9, 132.5, 130.8, 129.5, 128.9, 128.6, 127.9, 126.8, 122.1, 121.5 (3d, J = 8.7 Hz), 115.0 (d, 2J = 23.3 Hz), 114.5 (2d, J = 25.0 Hz), 113.4 (d, 3J = 8.4 Hz), 77.2, 69.9, 59.2, 59.0, 52.6, 32.7; HRMS-ESI (m/z): [M + H]+ calcd for C26H24Cl2FN4O 497.13057, found 497.13019.
:1) to give the title compound 2af (18.7 mg, 67%) as a white solid; mp 55.4–57.5 °C; 1H NMR (CDCl3, 300 MHz) δ 8.04 (d, J = 7.3 Hz, 1H), 7.90 (s, 1H), 7.52–7.41 (m, 7H), 7.23 (s, 1H), 6.98 (td, J = 3.0, 8.2 Hz, 1H), 6.89 (q, J = 4.4 Hz, 1H), 5.21 (bs, 1H), 5.10 (s, 2H), 4.19–3.65 (m, 4H), 2.51 (bs, 2H); 13C NMR (CDCl3, 75 MHz) δ 170.1, 157.7 (d, 1J = 238.0 Hz), 150.5, 142.4, 136.7, 135.9, 133.0, 132.6, 130.9, 130.4, 129.4, 128.4, 127.2, 126.7, 122.3, 120.9 (d, 3J = 9.2 Hz), 115.3 (d, 2J = 23.4 Hz), 114.5 (d, 2J = 25.3 Hz), 113.4 (d, 3J = 8.3 Hz), 77.2, 69.8, 65.8, 63.7, 59.3, 58.4, 54.2, 51.8, 47.7, 44.3, 32.7, 30.5; HRMS-ESI (m/z): [M + H]+ calcd for C26H22Cl2FN4O2 511.10984, found 511.10888.
Biological evaluation
JC-1 mitochondrial membrane potential assay
HT-22 cells (30000 per well) were seeded into a clear 96-well plate (FALCON) at 200 μL per well one day prior to assay. 750 μM of JC-1 (Stratagene) in DMSO stock solution was dissolved in phenol red-free Opti-MEM (GIBCO) medium to make a final concentration of 7.5 μM JC-1 per well. The medium was removed from the plate, and 100 μL per well of JC-1 was added. Plates were incubated for 1 h and 15 min at 37 °C and washed twice with 100 μL per well PBS. Subsequently, cells were treated with 25 μL solution of each compound at 10 μM in Opti-MEM and incubated at 37 °C for 10 min followed by addition of 25 μL of amyloid beta (American peptide, 1–42) solution at 10 μM. Fluorescence was measured at every one hour for three hours at ex/em 530 nm/580 nm (‘red’) and ex/em 485 nm/530 nm (‘green’). The ratio of green to red fluorescence was recorded and the percent changes in the ratio for each compound were calculated and normalized using the vehicle control as 100%.
CYP inhibition assay
The test compound (40 μL of a 25 μM solution of the compound in distilled water) into a 96-well plate and then 50 μL of the Master Pre-Mix (CYP450 BACULOSOMES® Reagent and Regeneration System) were added. The plate was incubated for 20 min at 37 °C to allow the compound to interact with the CYP450. The reaction was initiated by adding Vivid® CYP450 substrate/NADP+ buffer (10 μL). The remaining enzyme activity was measured by reading the amount of fluorescent product using a fluorescence plate reader. The reference inhibitor for each CYP isozyme is indicated in Table 4.Acknowledgements
This research was supported by the Korea Institute of Science and Technology (KIST-2E24670, 2E25023, 2E24510, 2V03370) and the National Research Foundation of Korea (NRF-2013R1A1A2005550 and NRF-2014M3C1A3054141) funded by the Ministry of Science, ICT and Future Planning. We thank Mr Beoung-Geon Park and Ms Min Kyung Ko for the JC-1 assay and the CYP stability experiments. We thank Dr Jungyeob Ham for helpful discussion on organotrifluoroborates.Notes and references
- K. B. Sharpless and R. Manetsch, Expert Opin. Drug Discovery, 2006, 1, 525–538 CrossRef CAS PubMed.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC.
- P. Wu and V. V. Fokin, Aldrichimica Acta, 2007, 40, 7–17 CAS.
- A. B. Braunschweig, W. R. Dichtel, O. S. Miljanic, M. A. Olson, J. M. Spruell, S. I. Khan, J. R. Heath and J. F. Stoddart, Chem. – Asian J., 2007, 2, 634–647 CrossRef CAS PubMed.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS.
- S. G. Agalave, S. R. Maujan and V. S. Pore, Chem. – Asian J., 2011, 6, 2696–2718 CrossRef CAS PubMed.
- D. K. Dalvie, A. S. Kalgutkar, S. C. Khojasteh-Bakht, R. S. Obach and J. P. O'Donnell, Chem. Res. Toxicol., 2002, 15, 269–299 CrossRef CAS.
- W. S. Horne, M. K. Yadav, C. D. Stout and M. R. Ghadiri, J. Am. Chem. Soc., 2004, 126, 15366–15367 CrossRef CAS PubMed.
- Y. L. Angell and K. Burgess, Chem. Soc. Rev., 2007, 36, 1674–1689 RSC.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 633–645 CrossRef.
- R. Huisgen, Pure Appl. Chem., 1989, 61, 613–628 CrossRef CAS.
- K. V. Gothelf and K. A. Jorgenson, Chem. Rev., 1998, 98, 863–909 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3062 CrossRef PubMed.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS PubMed.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- Y. S. Kim, S. H. Jung, B.-G. Park, M. K. Ko, H.-S. Jang, K. Choi, J.-H. Baik, J. Lee, J. K. Lee, A. N. Pae, Y. S. Cho and S.-J. Min, Eur. J. Med. Chem., 2013, 62, 71–83 CrossRef CAS PubMed.
- R. Kharb, P. C. Sharma and M. S. Yar, J. Enzyme Inhib. Med. Chem., 2011, 26, 1–21 CrossRef CAS PubMed.
- R. A. Oliveira, R. O. Silva, G. A. Molander and P. H. Menezes, Magn. Reson. Chem., 2009, 47, 873–878 CrossRef CAS PubMed.
- Y. Yamamoto, K. Hattori, J.-I. Ishii and H. Nishiyama, Tetrahedron, 2006, 62, 4294–4305 CrossRef CAS PubMed.
- G. A. Molander and N. Ellis, Acc. Chem. Res., 2007, 40, 275–286 CrossRef CAS PubMed.
- S. Darses and J.-P. Genet, Chem. Rev., 2008, 108, 288–325 CrossRef CAS PubMed.
- S. Darses and J.-P. Genet, Eur. J. Org. Chem., 2003, 4313–4327 CrossRef CAS.
- T. Kim, J. H. Song, K. H. Jeong, S. Lee and J. Ham, Eur. J. Org. Chem., 2013, 3992–3996 CrossRef CAS.
- K. Bolla, T. Kim, J. H. Song, S. Lee and J. Ham, Tetrahedron, 2011, 67, 5556–5563 CrossRef CAS PubMed.
- L. Ackermann, H. K. Potukuchi, D. Landsberg and R. Vicente, Org. Lett., 2008, 10, 3081–3084 CrossRef CAS PubMed.
- L. Ackermann and H. K. Potukuchi, Org. Biomol. Chem., 2010, 8, 4503–4513 CAS.
- A. J. J. Lennox and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2012, 134, 743–7441 CrossRef PubMed.
- W. Ren, J. Li, D. Zou, Y. Wu and Y. Wu, Tetrahedron, 2012, 68, 1351–1358 CrossRef CAS PubMed.
- B. K. Wagner, T. Kitami, T. J. Gilbert, D. Peck, A. Ramanathan, S. L. Schreiber, T. R. Golub and V. K. Mootha, Nat. Biotechnol., 2008, 26, 343–351 CrossRef CAS PubMed.
- S. T. Smiley, M. Reers, C. Mottola-Hartshorn, M. Lin, A. Chen, T. W. Smith, G. D. Steele and L. B. Chen Jr., Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 3671–3675 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob01967a |
| This journal is © The Royal Society of Chemistry 2014 |