
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fischer indolisation of N-(α-ketoacyl)anthranilic acids into 2-(indol-2-carboxamido)benzoic acids and 2-indolyl-3,1-benzoxazin-4-ones and their NMR study†‡
Karel
Proisl
a,
Stanislav
Kafka
*a,
Damijana
Urankar
b,
Martin
Gazvoda
b,
Roman
Kimmel
a and
Janez
Košmrlj
*b
aDepartment of Chemistry, Faculty of Technology, Tomas Bata University in Zlin, 762 72 Zlin, Czech Republic. E-mail: kafka@ft.utb.cz
bFaculty of Chemistry and Chemical Technology, University of Ljubljana, SI-1000 Ljubljana, Slovenia. E-mail: janez.kosmrlj@fkkt.uni-lj.si
First published on 1st October 2014
Abstract
N-(α-ketoacyl)anthranilic acids reacted with phenylhydrazinium chloride in boiling acetic acid to afford 2-(indol-2-carboxamido)benzoic acids in good to excellent yields and 2-indolyl-3,1-benzoxazin-4-ones as by-products. The formation of the latter products could easily be suppressed by a hydrolytic workup. Alternatively, by increasing the reaction temperature and/or time, 2-indolyl-3,1-benzoxazin-4-ones can be obtained exclusively. Optimisations of the reaction conditions as well as the scope and the course of the transformations were investigated. The products were characterized by 1H, 13C and 15N NMR spectroscopy. The corresponding resonances were assigned on the basis of the standard 1D and gradient selected 2D NMR experiments (1H–1H gs-COSY, 1H–13C gs-HSQC, 1H–13C gs-HMBC) with 1H–15N gs-HMBC as a practical tool to determine 15N NMR chemical shifts at the natural abundance level of 15N isotope.
Introduction
Molecules containing indole1 and anthranilic acid2 scaffolds are ubiquitous in many natural products with diverse biological activities. Thus, it is not surprising that the compounds featuring both the indole and the anthranilic acid fragments are encountered in nature. Alkaloids cephalandole C and cephalinone B, isolated from the native orchid plant Cephalanceropsis gracilis,3 and the alkaloids secofascaplysins, isolated from the sponge Fascaplysinopsis reticulate,4 consist of the 2-(indol-2-carboxamido)benzoic acid 1 substructures, for example (Fig. 1). Derivatives of 2-(indol-2-carboxamido)benzoic acid 1 are receiving attention because of their antibacterial activities.5 In materials sciences, the 2-(indol-2-carboxamido)benzoic acid derivatives are used as UV absorbers.6 Compounds with indol-2-carboxamide scaffold are potent inhibitors of HIV-1 replication (Delavirdine, Rescriptor®)7 and the androgen receptor binding function 3 (BF3),8 and were identified as hydrogen-bonding organocatalysts for the ring-opening polymerization of cyclic esters.9 | ||
| Fig. 1 Naturally occurring compounds containing 2-(indol-2-carboxamido)benzoic acid and 3,1-benzoxazin-4-one substructures (blue). | ||
3,1-Benzoxazin-4-ones serve as valuable precursors for the preparation of fused heterocycles including an important pharmacophore, quinazolin-4-one.10 It is also noteworthy that 3,1-benzoxazin-4-ones are potent inhibitors of the human leukocyte elastase,11 human chymase,12 chymotrypsin13 and proteases of herpes simplex type 1,14 human cathepsin G15 and serine,16 for example. Avenalumin I, phytoalexin of the 3,1-benzoxazin-4-one structure that is inhibitory to the growth of rust fungi, was isolated from oat leaves (Fig. 1).17
Surprisingly, despite the biological relevance and synthetic potential of 2-(indol-2-carboxamido)benzoic acids 1, to our knowledge, the only published approach to access these compounds makes use of N-acylation of anthranilic acids with 1H-indol-2-carboxylic acid derivatives.18 As an alternative to this, herein we report the protocol that is based on the Fischer indolisation of N-(α-ketoacyl)anthranilic acids 2 (Scheme 1). Optimization of the reaction conditions and the scope as well as multinuclear NMR spectral analysis of the products is reported.
 | ||
| Scheme 1 Two retrosynthetic approaches to 2-(indol-2-carboxamido)benzoic acids 1. | ||
Results and discussion
Synthesis
Out of the available methods to access indoles, the Fischer indole synthesis remains one of the most important and versatile methods.19 It is generally conducted by reacting an aryl hydrazine with a carbonyl compound.19,20 Thus, for the construction of 2-(indol-2-carboxamido)benzoic acid 1 through this pathway N-(α-ketoacyl)anthranilic acid 2 appears to be the most suitable carbonyl substrate. Based on our recent report,21 the latter is easily accessible through the consecutive oxidation protocol from 4-hydroxyquinolin-2-one as shown in Scheme 2. | ||
| Scheme 2 The preparation of the starting N-(α-ketoacyl)anthranilic acids 2 and the key of substituents R1–R6. | ||
The Fischer indolisation is known to proceed through an intermediately formed hydrazone, which then undergoes several consecutive transformations. It isomerises to an enamine, which after protonation rearranges to an imine and then a cyclic aminal. Acid catalysed elimination of ammonia from the latter finally gives rise to the indole ring. The above mentioned hydrazone can in principle be pre-assembled by condensation of aryl hydrazine with an appropriate carbonyl compound. Herein, we decided to compare the stepwise protocol that proceeds through the isolated phenylhydrazone 3 (Route A, Scheme 3) with the direct-one in which N-(α-ketoacyl)anthranilic acids 2 are treated with phenylhydrazine directly into the 2-(indol-2-carboxamido)benzoic acid 1 (Route B).
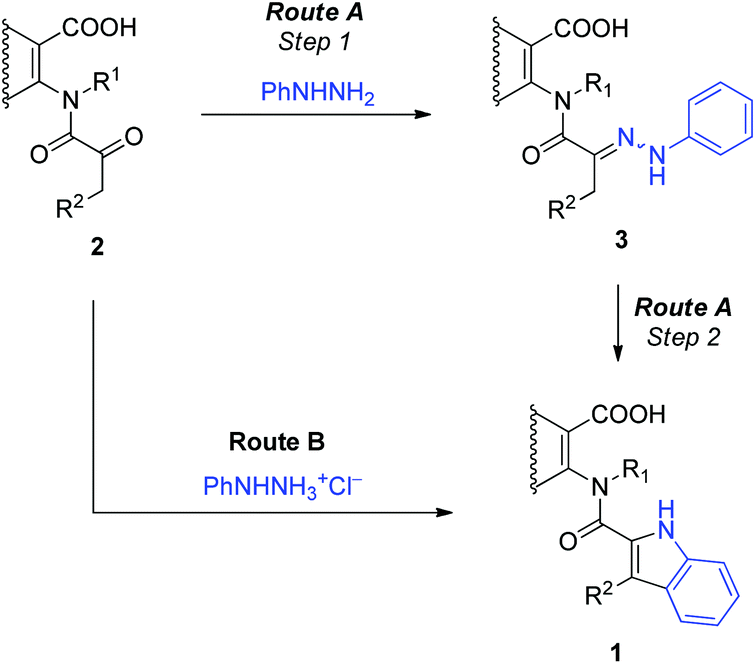 | ||
| Scheme 3 The stepwise (Route A) and direct (Route B) routes to the 2-(indol-2-carboxamido)benzoic acids 1 by the Fischer indolisation of N-(α-ketoacyl)anthranilic acids 2. | ||
To examine the stepwise protocol, Route A, phenylhydrazones 3a,c–e were initially prepared by the condensation of N-(α-ketoacyl)anthranilic acids 2a,c–e with phenylhydrazine. The isolated phenylhydrazones were in the subsequent step subjected to the thermally induced rearrangement into 2-(indol-2-carboxamido)benzoic acids 1a,c–e. The results are collected in Table 1. Although the preparation of the phenylhydrazones 3a,c–e proceeded in high 78–88% yields, the subsequent indolisation step into the target 2-(indol-2-carboxamido)benzoic acids 1a,c–e returned low 17–33% yields.
| Step 1 | Step 2 | ||
|---|---|---|---|
| Reaction conditions | 3, yielda | Reaction conditions | 1, yielda |
| a Percent yields of isolated pure products are given. | |||
| AcOH, 60 °C, 2 h | 3a, 88 | 235 °C, 20 min | 1a, 27 |
| AcOH, 60 °C, 3 h | 3c, 80 | 180 °C, 10 min | 1c, 17 |
| AcOH, 60 °C, 3 h | 3d, 79 | 245 °C, 15 min | 1d, 28 |
| AcOH, 60 °C, 1.25 h | 3e, 78 | 180 °C, 15 min | 1e, 33 |
The disappointing overall results of the stepwise procedure prompted us to test the direct approach, outlined as Route B in Scheme 3. In screening for the optimal reaction conditions, a mixture of selected N-(α-ketoacyl)anthranilic acid 2 and phenylhydrazinium chloride was heated under reflux in different solvents including acetonitrile, acetic acid, methanol and water. Since Lewis22 and Brønsted acids23 are well known to promote the Fischer indolisation, we selected to test hydrochloric acid, sulphuric acid, acetic acid, zinc(II) chloride and bismuth(III) nitrate pentahydrate (Bi(NO3)3·5H2O)24 as acid catalysts (Table 2). Good results in terms of the reaction time and the product yields were obtained for the reactions in boiling acetonitrile either in the presence of Bi(NO3)3·5H2O (entries 3, 4, 9–13) or acetic acid (entry 8). However, the highest yields of the products 1 were seen by conducting the reaction in boiling acetic acid in the absence of the additives (entry 1). As expected, the heating of N-(α-ketoacyl)anthranilic acid 2a in aqueous hydrochloric or sulphuric acid resulted in an amide bond hydrolysis to produce anthranilic acid (entries 6 and 7).
| Entry | 2 | Solventa | Additive, reaction time in h | 1 | Yieldb |
|---|---|---|---|---|---|
| a All reactions were conducted under reflux. b Percent yields of isolated pure products are given. c Anthranilic acid was isolated as the main product in 35% (entry 6) and 26% (entry 7) yield (Experimental). | |||||
| 1 | 2a | AcOH | None, 9 | 1a | 78 |
| 2 | 2a | MeOH | 20 mol% Bi(NO3)3·5H2O, 8 | 1a | 26 |
| 3 | 2a | MeCN | 20 mol% Bi(NO3)3·5H2O, 5 | 1a | 65 |
| 4 | 2a | MeCN | 10 mol% Bi(NO3)3·5H2O, 12 | 1a | 55 |
| 5 | 2a | MeCN | 20 mol% ZnCl2, 28 | 1a | 49 |
| 6 | 2a | H2O | 130 mol% HCl, 3 | 1a | 7c |
| 7 | 2a | H2O | 120 mol% H2SO4, 2 | 1a | 8c |
| 8 | 2a | MeCN | 2000 mol% AcOH, 22 | 1a | 70 |
| 9 | 2b | MeCN | 20 mol% Bi(NO3)3·5H2O, 12 | 1b | 53 |
| 10 | 2b | MeCN | 10 mol% Bi(NO3)3·5H2O, 19 | 1b | 52 |
| 11 | 2c | MeCN | 20 mol% Bi(NO3)3·5H2O, 16 | 1c | 71 |
| 12 | 2d | MeCN | 20 mol% Bi(NO3)3·5H2O, 11 | 1d | 75 |
| 13 | 2e | MeCN | 20 mol% Bi(NO3)3·5H2O, 11 | 1e | 69 |
Having identified the optimal reaction conditions we turned to examine the scope of the reaction. Several N-(α-ketoacyl)anthranilic acids (2a–l) were allowed to react with phenylhydrazinium chloride in boiling acetic acid, which afforded the target 2-(indol-2-carboxamido)benzoic acids (1a–l) in good to excellent yields of the isolated products (Table 3). In few instances, the reactions were accompanied by the formation of small amounts of by-products, which were isolated and identified as 3,1-benzoxazin-4-ones 4 and phenylhydrazides 5. The only exception to this was compound 2m, which afforded naphthoxazinone 4m as the sole product, with the anthranilic acid 1m being undetected in the reaction mixture (entry 13).
|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | 2 | Substituents | Reaction time (h) | Product, yielda | |||||||
| R1 | R2 | R3 | R4 | R5 | R6 | 1 | 4 | 5 | |||
| a Percent yields of isolated pure products are given. | |||||||||||
| 1 | a | H | Me | H | H | H | H | 9 | 78 | ||
| 2 | b | H | Me | H | OMe | H | H | 14 | 83 | 5 | 2 |
| 3 | c | H | Me | H | H | OMe | H | 15 | 89 | ||
| 4 | d | H | Me | H | H | H | OMe | 9 | 81 | 5 | |
| 5 | e | Me | Me | H | H | H | H | 8 | 91 | ||
| 6 | f | H | n-Pr | H | H | H | H | 10 | 91 | 2 | 1 |
| 7 | g | H | Me | H | H | H | Me | 14 | 55 | 14 | |
| 8 | h | H | Me | H | Me | H | H | 14 | 76 | ||
| 9 | i | H | Me | Cl | H | H | Me | 17 | 78 | 4 | |
| 10 | j | H | n-Pr | H | Me | H | Me | 12 | 51 | 12 | |
| 11 | k | H | n-Pr | H | OMe | H | OMe | 13 | 56 | 10 | |
| 12 | l | H | Ph | H | H | H | H | 19 | 62 | ||
| 13 | m | H | n-Pr | H | H | –(CH)4– | 9 | 0 | 87 | ||

In principle, the formation of 2-indolyl-3,1-benzoxazin-4-ones 4 could be realised by two different pathways as shown in Scheme 4. An initial formation of 2-acyl-3,1-benzoxazin-4-one 4′ through the Path a could be followed by the Fischer indolisation with phenylhydrazinium chloride. This pathway was, however, ruled out on the basis of the 3,1-benzoxazin-4-ones reactivity considerations. In the presence of nucleophiles these compounds are prone to undergo rapid ring opening into the anthranilic acid derivatives (vide infra) suggesting that somehow higher amounts of the phenylhydrazides 5 should have been formed in the reactions shown in Table 3. The Path a was also ruled out experimentally by heating compound 2a in neat acetic acid under the reflux conditions in the absence of phenylhydrazinium chloride, which resulted in no detectable formation of 2-acyl-3,1-benzoxazin-4-one 4′. This left the Path b, i.e. the initial Fischer indolisation of the N-(α-ketoacyl)anthranilic acid 2 with phenylhydrazinium chloride into 2-(indol-2-carboxamido)benzoic acid 1 and subsequent cyclodehydration into 2-indolyl-3,1-benzoxazin-4-one 4, as the most plausible.
 | ||
| Scheme 4 The proposed formation of 3,1-benzoxazin-4-ones 4. | ||
Generally, the 3,1-benzoxazin-4-one skeleton is formed by the reaction of anthranilic acids with an appropriate acid anhydride at the elevated temperatures.25 Mechanistically, the amino group of the anthranilic acid is N-acylated and the carboxylic group is transformed into a mixed anhydride intermediate. This intermediate then undergoes an intramolecular nucleophilic displacement of the carboxylate ion from the anhydride moiety by the amide in its iminol form.26 In turn, heating the 2-(indol-2-carboxamido)benzoic acid 1 in acetic acid is unlikely to produce mixed anhydride. Since an intramolecular nucleophilic attack of the carboxylic group to the amide is also unlikely because of the low electrophilicity of the amide, the formation of 3,1-benzoxazin-4-ones 4 could best be rationalized through the intramolecular nucleophilic attack of the iminol 1′ to the protonated carboxylic group as shown in Scheme 4. The formation of naphthoxazinone 4m as the sole product from 2m (Table 3, entry 13) could be accounted for by an enhanced resonance stabilisation of iminol 1m′, the result of an extended conjugation through the naphthalene ring.
If the mechanism shown in Scheme 4 is operating, it can be expected that a prolonged reaction time and/or an elevated reaction temperature would work beneficially to the formation of 3,1-benzoxazin-4-one 4. Indeed, by prolonging the heating of compound 1j with phenylhydrazinium chloride in acetic acid (bpAcOH = 118 °C) from 12 h to 40 h increased the yield of the expected product 4j from 15% to 65% (compare entries 1 and 2 in Table 4). The use of the higher boiling propanoic acid (bp = 141 °C) in place of acetic acid afforded the 3,1-benzoxazin-4-one 4j in 72% yield already within 17 h (entry 3). It is noteworthy that these reaction conditions can potentially be utilized as a convenient one-pot protocol for the preparation of 3,1-benzoxazin-4-ones 4 from N-(α-ketoacyl)anthranilic acids 2 and aryl hydrazines. The synthetic methodologies toward 3,1-benzoxazin-4-ones, other than those utilizing anthranilic acids, have been reviewed.27

Minute amounts of the hydrazides 5 also accompanied the formation of 2-(indol-2-carboxamido)benzoic acids 1 (Table 3). As it is less likely that under the applied reaction conditions phenylhydrazinium chloride reacts with the carboxyl group of either the starting N-(α-ketoacyl)anthranilic acids 2 or the product 2-(indol-2-carboxamido)benzoic acids 1, it is reasonable to assume that the formation of hydrazides 5 proceeds through the ring-opening at the 3,1-benzoxazin-4-ones 4. Smooth reactivity of the 3,1-benzoxazin-4-ones towards different nucleophiles is well documented.26,28 In our case the reactivity of compound 4f towards phenylhydrazine to form 5f was independently confirmed in boiling toluene as the reaction solvent (Table 5). Analogously, treatment with n-butylamine gave the appropriate amide 6f.
|
|
||||
|---|---|---|---|---|
| Entry | 4 | NuH | Reaction conditions, reaction time in h | Product, yielda |
| a Percent yields of isolated pure products are given. | ||||
| 1 | 4f | PhNHNH2 | Toluene, reflux, 2 | 5f, 86 |
| 2 | 4f | n-BuNH2 | Toluene, reflux, 2 | 6f, 84% |
| 3 | 4f | H2O | DMSO, H2O, NaOH, rt, 1 | 1f, 91% |
| 4 | 4f | H2O | dioxane, 0.1 M H2SO4, rt, 2 | 1f, 70% |
| 5 | 4j | H2O | DMSO, H2O, NaOH, rt, 3 | 1j, 98% |
| 6 | 4m | H2O | DMSO, H2O, NaOH, rt, 12 | 1m, 95% |
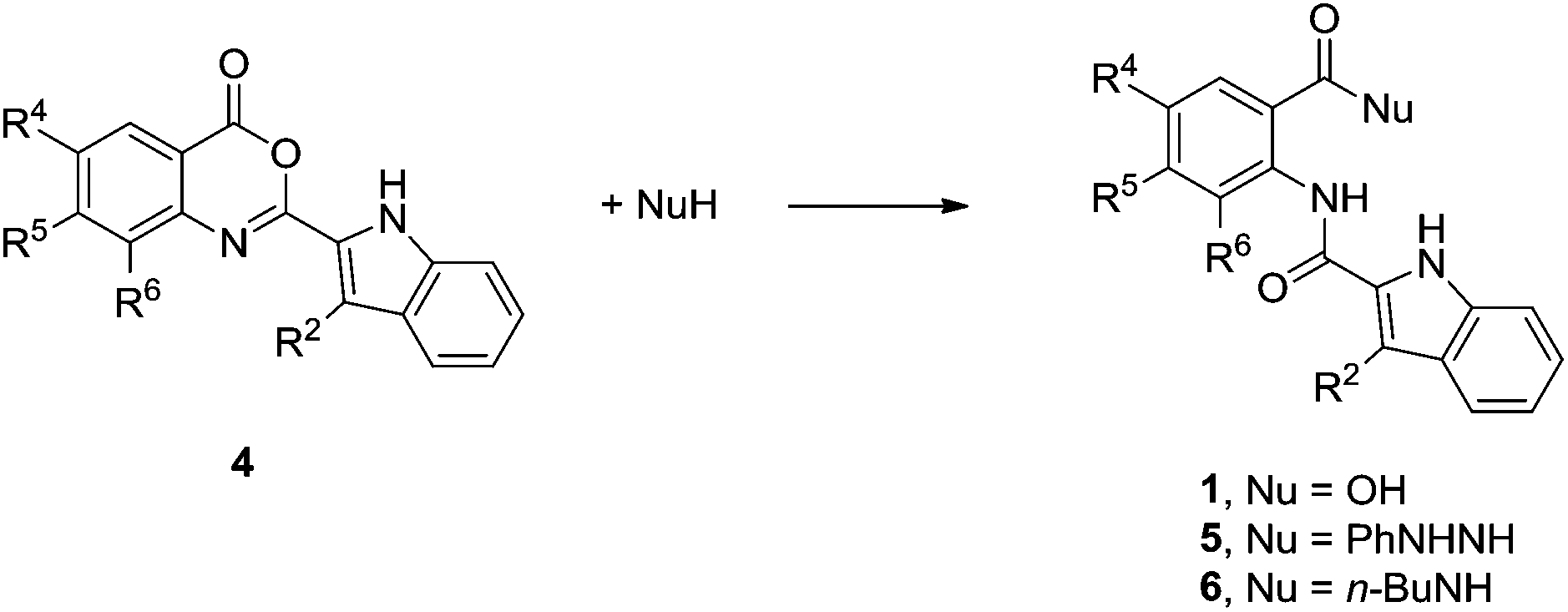
Aqueous sodium hydroxide in DMSO mediated complete hydrolysis of 3,1-benzoxazin-4-ones 4f,j,m to the corresponding 2-(indol-2-carboxamido)benzoic acids 1f,j,m (Table 5). High tendency of related 3,1-benzoxazin-4-ones for hydrolysis into the corresponding anthranilic acids has been documented.29,30 With these results in mind it can be assumed that higher quantities of the 3,1-benzoxazin-4-one products 4 are actually formed during the Fischer indolisation of N-(α-ketoacyl)anthranilic acids 2 shown in Table 3 as they were actually isolated. During the isolation workup the latter most probably partly hydrolyse back into the 2-(indol-2-carboxamido)benzoic acids 1.
The elemental composition of all the compounds under investigation was confirmed by combustion analysis and high-resolution mass spectrometry with electrospray ionization. In addition, low resolution mass spectra with electron impact ionization and the infrared spectra were provided. In the latter, the characteristic absorption bands belonging to the indole N–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CN bonds were identified, where appropriate.
O and CN bonds were identified, where appropriate.
NMR study
The compounds 1, 3–6 were fully characterized by 1H, 13C and 15N NMR spectroscopy. The corresponding resonances were assigned on the basis of gradient-selected 2D NMR experiments including 1H–1H gs-COSY, 1H–13C gs-HSQC, 1H–13C gs-HMBC and 1H–15N gs-HMBC. The spectra of compounds 1, 3, 5 and 6 were recorded in DMSO-d6. For 3,1-benzoxazin-4-ones 4, which proved to rapidly hydrolyse in DMSO-d6 into the 2-(indol-2-carboxamido)benzoic acids 1, less polar CDCl3 was identified as a suitable solvent. Acetone-d6 was used as an alternative for dissolving compound 4k due to its sparing solubility in CDCl3. Some characteristic spectral features are discussed below. For the atom numbering scheme, see Fig. 2.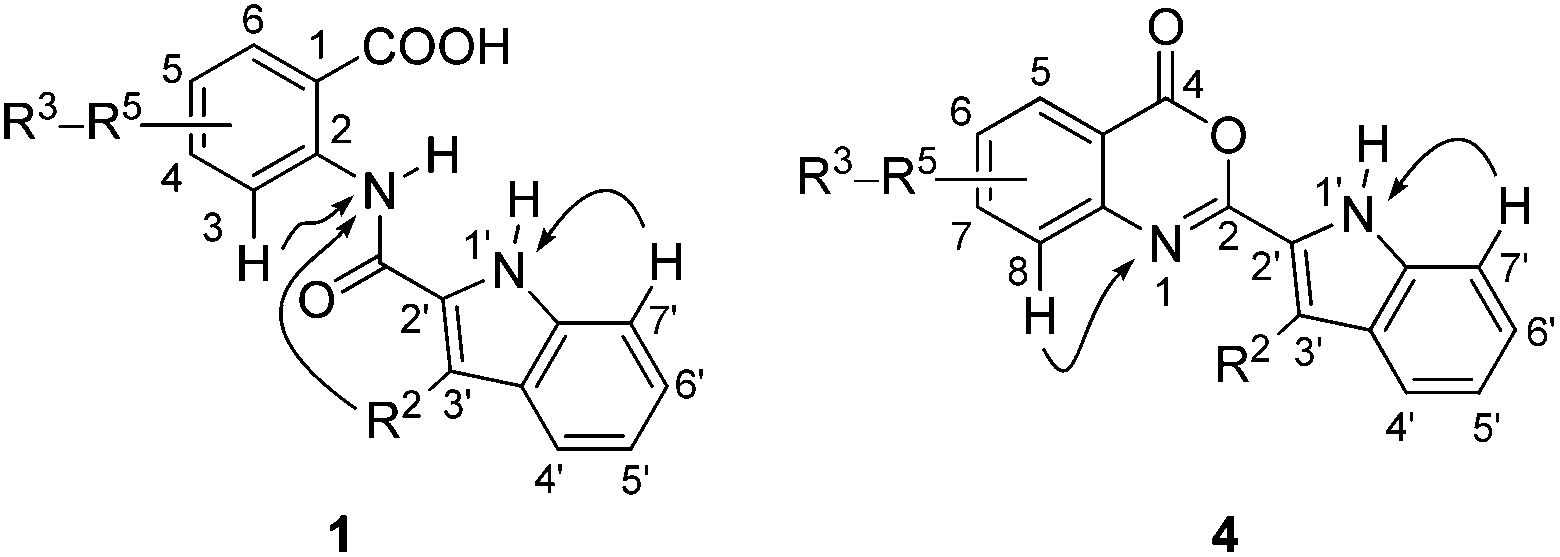 | ||
| Fig. 2 Atom numbering scheme for compounds 1 and 4. Curved arrows denote long-range 1H–15N gs-HMBC connectivities. | ||
There is a wealth of 1H and 13C data31 as well as 15N NMR data32 on indoles reported in the literature. The relatively unsubstituted indole ring in the compounds 1 and 4 enabled us to unequivocally assign all proton, carbon and nitrogen resonances via long-range 1H–13C and 1H–15N heteronuclear coupling pathways, which we found in agreement to those discussed in the literature.33 For the indole ring in 2-(indol-2-carboxamido)benzoic acids 1 the order of shielding in the 13C NMR spectra is C7′ > C3′ > C5′ > C4′ > C6′ > C2′ ≅ C3a′ > C7a′ (Table 6). By changing the indole C-2′ substituent from the carbamoyl group in compounds 1 into the 3,1-benzoxazin-4-one ring in 4, the most dramatic changes in the chemical shift are seen for the carbon atoms of the fused 5-membered ring. In comparison with compounds 1, the C2′ atoms of the indole ring in 4 are shielded by ca. 4 ppm, whereas the C3′ and C3a′ atoms are deshielded by ca. 6 ppm and ca. 1 ppm, respectively. By changing the R2 = Me to the R2 = n-Pr in either 1 or 4, the carbon atom C3′ becomes more shielded for ca. 5–6 ppm (Tables 6 and 7).
| Atomb | 1a | 1b | 1c | 1d | 1f | 1g | 1h | 1i | 1j | 1k | 1l | 1m |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a In DMSO-d6. b For atom numbering scheme, see Fig. 2. c Resonances for the C3′ phenyl ring: 132.9 ppm (C1′′), 130.2 ppm (C2′′, C6′′), 128.7 ppm (C3′′, C5′′), 127.2 ppm (C4′′). d For the complete set of resonances, see the Experimental section. e Could not be assigned unequivocally; for the complete list of resonances, see the Experimental section. f Not observed. | ||||||||||||
| C1 | 116.5 | 118.0 | 108.6 | —e | 116.4 | 126.8 | 116.4 | —e | —e | —e | 116.4 | |
| C2 | 141.0 | 134.3 | 143.0 | —e | 140.8 | 136.3 | 138.6 | —e | 133.7 | 118.8 | 140.4 | |
| C3 | 120.6 | 122.4 | 105.3 | 153.7 | 120.5 | 135.8 | 120.6 | —e | —e | 155.1 | 120.4 | |
| C4 | 134.2 | 120.3 | 163.6 | 115.0 | 134.1 | 134.4 | 134.7 | 131.6 | 134.8 | 102.4 | 134.0 | |
| C5 | 122.8 | 154.3 | 108.6 | 126.1 | 122.8 | 125.7 | 132.0 | —e | —e | 157.5 | 122.9 | |
| C6 | 131.3 | 114.8 | 133.2 | 121.5 | 131.2 | 128.1 | 131.3 | —e | 128.3 | 105.1 | 131.1 | |
| C3(CH3) | 18.7 | 17.5 | 18.4 | |||||||||
| C5(CH3) | 20.2 | 20.2 | ||||||||||
| C3(OCH3) | 56.1 | 56.2 | ||||||||||
| C4(OCH3) | 55.5 | |||||||||||
| C5(OCH3) | 55.4 | 55.6 | ||||||||||
| C2′ | 128.2 | 128.2 | 128.1 | 127.2 | 127.7 | 127.3 | 128.1 | 126.8 | 126.8 | —e | 128.0 | —e |
| C3′ | 113.3 | 112.9 | 113.4 | 115.2 | 119.3 | 114.6 | 113.1 | 114.6 | 120.2 | 120.6 | 118.5 | 121.2 |
| C3a′ | 128.0 | 128.1 | 128.1 | 128.0 | 127.5 | 128.1 | 128.1 | 127.8 | 127.6 | 127.7 | 126.7 | 127.7 |
| C4′ | 120.1 | 120.0 | 120.2 | 119.9 | 120.1 | 120.0 | 120.1 | 119.8 | 119.9 | 120.0 | 120.2 | 120.2 |
| C5′ | 119.4 | 119.3 | 119.4 | 119.2 | 119.4 | 119.3 | 119.4 | 119.1 | 119.2 | 119.2 | 120.6 | 119.4 |
| C6′ | 124.4 | 124.2 | 124.4 | 124.2 | 124.2 | 124.2 | 124.3 | 124.0 | 124.0 | 124.0 | 124.5 | 124.3 |
| C7′ | 112.2 | 112.2 | 112.2 | 112.0 | 112.2 | 112.1 | 112.2 | 111.9 | 112.0 | 112.0 | 112.6 | 112.2 |
| C7a′ | 136.0 | 135.9 | 136.1 | 135.6 | 136.0 | 135.7 | 136.0 | 135.4 | 135.5 | 135.5 | 136.0 | 135.7 |
| C3′CH2 | 25.7 | 26.0 | 26.1 | |||||||||
| C3′CH2CH2 | 24.0 | 23.9 | 23.9 | |||||||||
| C3′(CH2)2CH3 | 13.9 | 14.0 | 14.1 | |||||||||
| C3′CH3 | 9.7 | 9.7 | 9.7 | 9.8 | 9.8 | 9.7 | 9.6 | |||||
| CONH | 160.5 | 160.2 | 160.7 | 160.2 | 160.5 | 160.4 | 160.4 | 160.8 | 160.2 | 160.2 | 160.2 | 161.6 |
| COOH | 169.7 | 169.3 | 169.6 | 167.9 | 169.6 | 168.4 | 169.7 | 166.5 | 168.3 | 167.7 | 168.7 | 168.3 |
| N1′ | 132.7 | 132.7 | 132.7 | 130.3 | 132.2 | 131.3 | 132.7 | 130.9 | 130.8 | 130.2 | 136.9 | 130.5 |
| CONH | 126.9 | 125.1 | 127.7 | 116.1 | 126.6 | 122.8 | 126.3 | 120.7 | 121.4 | 113.9 | 129.5 | —f |
| Atomb | 4b | 4c | 4d | 4f | 4g | 4i | 4j | 4k |
|---|---|---|---|---|---|---|---|---|
| a In CDCl3, unless otherwise stated. b For atom numbering scheme, see Fig. 2. c In acetone-d6. d Could not be unequivocally assigned. e Not observed. | ||||||||
| C2 | 151.8 | 154.3 | 153.1 | 153.3 | 152.1 | 152.6 | 151.0 | 151.4 |
| C4 | 159.3 | 158.7 | 158.9 | 159.0 | 159.6 | 156.2 | 159.7 | 159.6 |
| C4a | 117.1 | —d | 117.4 | 116.6 | 116.4 | 113.8 | 116.1 | —d |
| C5 | 108.7 | —d | 120.0 | 128.7 | 126.3 | 133.1 | 125.9 | 100.6 |
| C6 | 158.9 | 130.4 | 127.8 | 127.6 | 127.1 | 129.3 | 137.4 | 160.6 |
| C7 | 126.1 | 166.3 | 117.1 | 136.6 | 137.4 | 136.8 | 138.7 | 108.0 |
| C8 | 128.1 | —d | 153.8 | 126.6 | 135.5 | 134.5 | 135.3 | 156.7 |
| C8a | 141.4 | —d | 137.4 | 147.3 | 145.6 | 147.8 | 143.6 | 133.1 |
| C6(CH3) | 17.2 | |||||||
| C8(CH3) | 17.3 | 17.7 | 21.2 | |||||
| C6(OCH3) | 56.0 | 56.4 | ||||||
| C7(OCH3) | 56.0 | |||||||
| C8(OCH3) | 56.4 | 57.0 | ||||||
| C2′ | 123.4 | 123.3 | 123.5 | 123.0 | 123.4 | 122.9 | 123.2 | —d |
| C3′ | 119.4 | 120.5 | 120.3 | 125.6 | 120.3 | 121.2 | 125.0 | 124.2 |
| C3a′ | 129.3 | 129.2 | 129.2 | 128.9 | 129.2 | 129.2 | 128.9 | 129.6 |
| C4′ | 120.5 | 120.6 | 120.6 | 120.8 | 120.5 | 120.6 | 120.6 | 121.0 |
| C5′ | 120.1 | 120.2 | 120.1 | 120.1 | 120.2 | 120.3 | 120.1 | 120.6 |
| C6′ | 125.6 | 125.9 | 125.9 | 125.8 | 125.8 | 126.1 | 125.5 | 125.7 |
| C7′ | 111.3 | 111.4 | 111.5 | 111.5 | 111.5 | 111.6 | 111.5 | 113.0 |
| C7a′ | 136.5 | 136.6 | 136.8 | 136.7 | 136.5 | 136.6 | 136.4 | 138.0 |
| C3′CH2 | 26.9 | 27.0 | 27.6 | |||||
| C3′CH2CH2 | 24.2 | 23.8 | 25.0 | |||||
| C3′(CH2)2CH3 | 14.2 | 14.3 | 14.7 | |||||
| C3′CH3 | 10.4 | 10.5 | 10.5 | 10.6 | 10.7 | |||
| N1 | 222.4 | 222.6 | 212.0 | 222.8 | —e | —e | 216.4 | |
| N1′ | 120.5 | 120.9 | 121.4 | 121.1 | 120.5 | 120.3 | 120.9 | —e |
The three-bond long-range couplings were observed in the 1H–15N gs-HMBC spectra from H7′ to the indole N1′ resonance in both 1 and 4 (Fig. 2). In addition, a one-bond direct NH doublet response corroborated the assignment of N1′ (Fig. 3). The chemical shifts of the N1′ atoms in indoles 1 appear at δ 130.2–136.9 ppm and are in good agreement with the literature values reported for Delavirdine.34 In compounds 4, the indole nitrogen atoms N1′ resonate in the narrow range of δ 120.3–121.4 ppm (Tables 6 and 7).
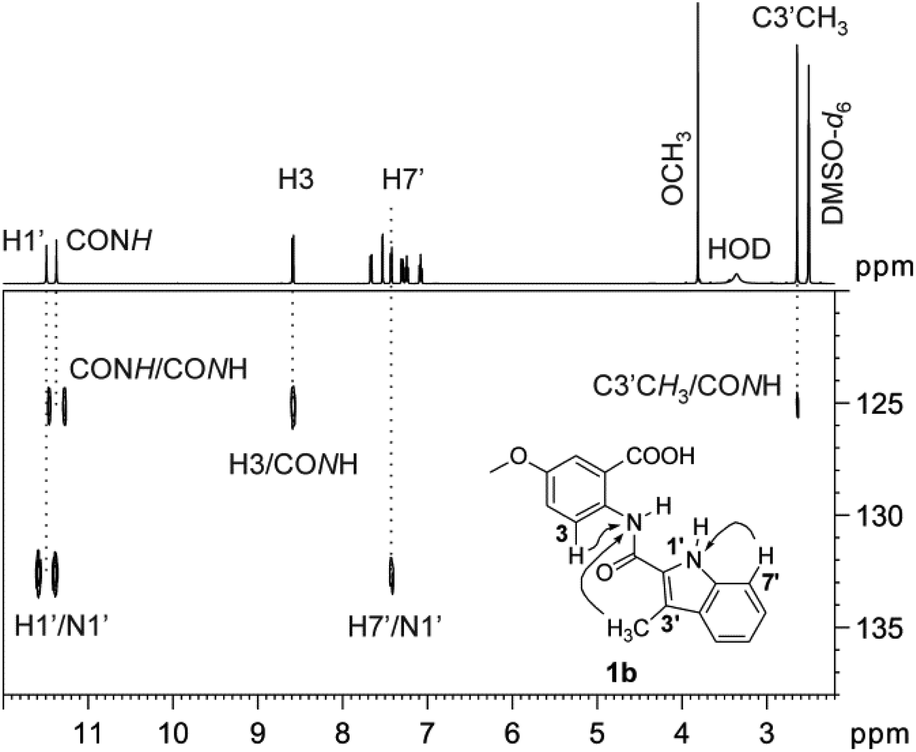 | ||
| Fig. 3 1H–15N gs-HMBC spectrum of 2-(indol-2-carboxamido)benzoic acid 1b recorded in DMSO-d6, optimized for 5 Hz long-range coupling. Direct responses for the N1′ and CONH resonances were observed as doublets at 132.7 ppm and 125.1 ppm, respectively. The spectrum also features three-bond long-range response to N1′ and CONH from H7′ and H3, respectively. Five-bond long-range correlation to CONH from C3′CH3 is also observed. | ||
Systematic NMR investigations of 3,1-benzoxazin-4-ones are more scarce.29,35 As pointed out by Osborne and Goolamali some early NMR data should be taken with care because these compounds often show high tendency for hydrolysis into the corresponding anthranilic acids, especially when measured in polar solvents such as DMSO-d6.29 High susceptibility towards hydrolysis has been illustrated by 2-methyl-3,1-benzoxazin-4-one that reacts with water into 2-acetylaminobenzoic acid already in the solid state.30 Osborne and Goolamali reported an unequivocal differentiation between anthranilic acids and 3,1-benzoxazin-4-ones that was achieved through determination of characteristic JCH coupling interactions in the carbonyl region of the proton coupled 13C NMR spectra.29
Herein, the proton, carbon and nitrogen resonances belonging to the anthranilic moiety in 1 and the 3,1-benzoxazin-4-one group in 4 were rapidly assigned using the gradient-selected 2D NMR experiments. The results are collected in Tables 6 and 7. Characteristic for the downfield regions of the 13C NMR spectra of the acylanthranilic acids 1 were two signals appearing at δ 160.2–161.6 ppm and δ 166.5–169.7 ppm for the amide carbonyl and for the carboxylic group, respectively. In 3,1-benzoxazin-4-one 4, the downfield region of the spectra were occupied with three signals for C8a (δ 133.1–147.8 ppm), C2 (δ 151.0–154.3 ppm) and C4 (δ 156.2–159.7 ppm). Our results are in agreement with the literature data.29
Chemical shifts for the 3,1-benzoxazin-4-one nitrogen atoms in compounds 4 were in the range of δ 212.0–222.8 ppm (Table 7). Due to the lack of the 15N NMR data no comparison with the literature could be done. In C8-unsubstituted 3,1-benzoxazin-4-ones 4b,c,f,h (R6 = H), three-bond long-range couplings were observed in the 1H–15N gs-HMBC spectra from H8 to the N1 resonance as illustrated in Fig. 2. The strongly electron donating C8-methoxy substituent in derivatives 4d,k (R6 = OMe) enabled a four-bond long-range couplings from H7 to the N1 whereas no such correlation could be observed in the C8-methyl substituent analogues 4g,i,j (R6 = Me).
The amide nitrogen atoms (CONH) in acylanthranilic acids 1 resonate in the range of δ 113.9–129.5 ppm (Table 6). In analogy with the above, the cross-peaks between H3 and CONH resonance were observed in 1H–15N gs-HMBC spectra of 1a–c,h,l. In the 4-methoxy substituted acylanthranilic acid 1c there was also a four-bond long-range coupling from H6 to the CONH. With the exception of 1l and 1m, the 1H–15N gs-HMBC spectra also featured the one-bond direct NH doublet response. Interestingly, in 3-methylindoles 1a,b,g,h, five-bond long-range couplings were observed in the 1H–15N gs-HMBC spectra from the C3′CH3 to the amide nitrogen (CONH) resonance. With the 2-(indol-2-carboxamido)benzoic acid 1b as a representative example, the above mentioned 1H–15N gs-HMBC spectral features are illustrated in Fig. 3.
Conclusions
We have demonstrated that Fischer indolisation of N-(α-ketoacyl)anthranilic acids can serve as a mild and highly efficient alternative for the preparation of 2-(indol-2-carboxamido)benzoic acids. By simple changes in the reaction conditions, this protocol offers a potential to access 2-indolyl-3,1-benzoxazin-4-ones. The anthranilic acid, indol-2-carboxamide and 3,1-benzoxazin-4-one structural motifs discussed herein were fully characterized by 1H, 13C and 15N NMR spectroscopy. The data presented will help in unequivocal identification of these classes of compounds.Experimental section
General
The reagents and solvents were used as obtained from commercial sources. Compounds 2a–k,m were prepared according to the literature procedure.21 Column chromatography was carried out on Fluka Silica gel 60 (particle size 0.063–0.2 mm, activity acc. Brockmann and Schodder 2–3). Melting points were determined on the microscope hot stage, Kofler, PolyTherm, manufacturer Helmut Hund GmbH, Wetzlar and are uncorrected. TLC was carried out on pre-coated TLC sheets ALUGRAM® SIL G/UV254 for TLC, MACHEREY-NAGEL. NMR spectra were recorded with a Bruker Avance III 500 MHz NMR instrument operating at 500 MHz (1H), 126 MHz (13C) and 51 MHz (15N). Proton spectra were referenced to TMS as an internal standard. Carbon chemical shifts were determined relative to the 13C signal of DMSO-d6 (39.5 ppm), CDCl3 (77.0 ppm) or acetone-d6 (high-field at 20.8 ppm). 15N chemical shifts were extracted from 1H–15N gs-HMBC spectra (with 20 Hz digital resolution in the indirect dimension and the parameters adjusted for a long-range 1H–15N coupling constant of 5 Hz) determined with respect to external nitromethane and are corrected to external ammonia by addition of 380.5 ppm. Nitrogen chemical shifts are reported to one decimal place as measured from the spectrum; however, the data should not be considered to be more accurate than ±0.5 ppm because of the digital resolution limits of the experiment. Chemical shifts are given on the δ scale (ppm). Coupling constants (J) are given in Hz. Multiplicities are indicated as follows: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet) or br (broadened). Infrared spectra were recorded on a Mattson 3000 FTIR Spectrometer or a Thermo Scientific Nicolet iS10 FT-IR Spectrometer using samples in potassium bromide disks and only the strongest/structurally most important peaks are listed; absorption band intensities are indicated as follows: s (strong), m (medium), w (weak) or b (broad). Electron impact mass spectra (EI) were recorded on a Shimadzu QP–2010 instrument at 70 eV. HRMS spectra were recorded with the Agilent 6224 Accurate Mass TOF LC/MS system with electrospray ionization (ESI). Elemental analyses (C, H, N) were performed with the FlashEA1112 Automatic Elemental Analyzer (Thermo Fisher Scientific Inc.).2-(2-Oxo-3-phenylpropanamido)benzoic acid (2l)
To a solution of 3-benzyl-3-hydroxyquinoline-2,4(1H,3H)-dione (669.4 mg, 2.50 mmol) in ethyl acetate (120 mL), a solution of sodium periodate (1.62 g, 7.55 mmol) in water (60 mL) was added. The two-phase reaction mixture was vigorously stirred for 7 hours at room temperature. The organic layer was separated, and extracted with 5% aqueous sodium thiosulphate (2 × 20 mL), 5% aqueous hydrochloric acid (2 × 20 mL) and finally with 5% aqueous potassium carbonate (7 × 10 mL). The potassium carbonate extract was washed with benzene (30 mL) and neutralised by addition of 10% aqueous hydrochloric acid. The resulting solution was extracted with ethyl acetate. The organic layer was separated, dried over sodium sulphate and concentrated in vacuo. The crude product was recrystallized from cyclohexane (85 mL) to give compound 2l (424.3 mg, 60%) as colourless microcrystals, mp 179–181 °C, Rf = 0.49 (10% ethanol in chloroform), 1H NMR (500 MHz, DMSO-d6) δ 4.32 (s, 2H), 7.23–7.37 (m, 6H), 7.67–7.72 (m, 1H), 8.07 (dd, 1H, J = 8.0, 1.6 Hz), 8.72 (d, 1H, J = 8.5 Hz), 12.39 (br s, 1H), 13.84 (br s, 1H); due to decomposition, no 13C NMR spectrum could be recorded. IR (cm−1): ν 3221 w, 3063 w, 3031 w, 1697 s, 1672 s, 1603 m, 1583 s, 1531 s, 1451 m, 1420 s, 1270 s, 1055 m, 755 m, 695 m; MS (EI) m/z (%): 284 (3), 283 (17, [M+]), 164 (20), 146 (100), 137 (25), 118 (14), 92 (15), 91 (54), 90 (25), 65 (16); HRMS (ESI+): m/z calcd for C16H14NO4+ [M + H]+ 284.0917, found 284.0917; HRMS (ESI−): m/z calcd for C16H12NO4− [M − H]− 282.0772, found 282.0773. Found: C, 67.9; H, 4.6; N, 4.9. Calc. for C16H13NO4: C, 67.8; H, 4.6; N, 4.9%.The synthesis of phenylhydrazones (3a,c–e) (Table 1)
To a solution of an appropriate N-(α-ketoacyl)anthranilic acid 2 (3.0 mmol) in acetic acid (25 mL), phenylhydrazine (0.49 g, 4.5 mmol) was slowly added and the resulting solution was stirred at 60 °C until TLC analysis revealed complete consumption of the starting compound (Table 1). The reaction mixture was cooled and poured into crushed ice (200 g). For phenylhydrazones 3a,d,e, the precipitated solid was collected by filtration and washed with water (3 × 20 mL) and cyclohexane (2 × 10 mL). For phenylhydrazone 3c the filter cake was dried at 50 °C and recrystallized from ethyl acetate to yield 3c·AcOH. The yields of compounds 3a,c–e are collected in Table 1.N), 141.2 (C2), 144.4 (C1 of phenyl), 163.2 (CONH), 169.4 (COOH); 15N NMR (51 MHz, DMSO-d6) δ 116.4 (CONH), 144.1 (NHPh), 327.6 (CN); IR (cm−1): ν 3287 m, 2970 w, 1665 s, 1650 s, 1604 m, 1586 m, 1577 m, 1518 s, 1496 s, 1449 m, 1241 s, 1225 s, 748 m, 694 m; MS (EI) m/z (%): 312 (8), 311 (40, [M+]), 147 (11), 146 (91), 145 (16), 93 (19), 92 (55), 91 (100), 90 (18), 65 (46). Found: C, 65.6; H, 5.5; N, 13.25. Calc. for C17H17N3O3: C, 65.6; H, 5.5; N, 13.5%.
General procedure for the Fischer indolisation of phenylhydrazones 3a,c–e into 2-(indol-2-carboxamido)benzoic acids 1a,c–e (Table 1)
Phenylhydrazones 3a,d,e or 3c· AcOH (2.0 mmol) were heated under a nitrogen atmosphere on a metal bath at the temperature and for the time given in Table 1. The course of reaction was monitored by a moistened pH test strip (alkaline reaction of released ammonia gas). After cooling the reaction mixture, the corresponding product was isolated as follows.Compound 1a was obtained from the solidified reaction mixture by two subsequent recrystallizations from ethanol and isopropyl alcohol.
Compound 1c was obtained from the oily reaction mixture by trituration with toluene (10 mL). The precipitate that formed was collected by filtration, recrystallized from isopropyl alcohol and additionally purified by column chromatography on silica gel using successive mixtures of ethyl acetate and ethanol as eluents.
Compound 1d was obtained by triturating the solidified reaction mixture with toluene for 1 hour at ambient temperature followed by filtration. Pure 1d was obtained after recrystallization of the filter cake from isopropyl alcohol.
Compound 1e was obtained by washing the solidified reaction mixture with toluene (10 mL) and cyclohexane (10 mL). The solid was subjected to column chromatography on silica gel using successive mixtures of ethyl acetate and ethanol as eluents. Recrystallisation from the benzene–ethyl acetate mixture afforded pure 1e.
Solvent and catalyst screening for the Fischer indolisation of 2 into 1 (Table 2)
A stirred mixture of N-(α-ketoacyl)anthranilic acid 2 (2.00 mmol), phenylhydrazinium chloride (310 mg, 2.15 mmol) and the catalyst in the solvent (entry 1: 7.5 mL; entries 2–13: 13 mL) was heated under reflux until TLC analysis indicated complete consumption of the starting material. The progress of the reaction was accompanied by the colour change of the reaction mixture from pale yellow to green and finally to dark red. The products were isolated as follows.Entries 1 and 5: the cooled reaction mixture was poured into ice water (35–60 mL). The solid was collected by filtration, washed with water (20–60 mL) and recrystallized from ethanol affording pure compound 1a.
Entries 2–4, 9–13: the cooled reaction mixture was poured into ice water (35 mL). If an oily organic phase was formed (entry 13), the resulting mixture was stirred overnight. The resulting solid was collected by filtration, washed with 10% HCl (16 mL) and water (25 mL), and recrystallized from ethanol affording the appropriate pure compound 1.
Entries 6 and 7: the cooled reaction mixture was filtered. The filter cake was washed with water (10 mL) and recrystallized from ethanol affording a pure compound 1a. The filtrate was evaporated to dryness and the residue was dissolved in a minimal amount of water (ca. 25 mL). The solution was made alkaline with 0.5 M NaOH, washed with benzene (3 × 5 mL) and carefully neutralised with 5% HCl. The precipitated anthranilic acid was filtered off as colourless crystals.
Entry 8: the reaction mixture was concentrated in vacuo and the oily residue was triturated with water (10 mL) to give white precipitate of pure 1a, which was collected by filtration and washed with water (2 × 3 mL).
General procedure for the Fischer indolisation of N-(α-ketoacyl)anthranilic acids 2 into 2-(indol-2-carboxamido)benzoic acids 1 (Table 3)
A mixture of N-(α-ketoacyl)anthranilic acid 2 (2.00 mmol) and phenylhydrazinium chloride (310 mg, 2.15 mmol) in acetic acid (7.5 mL) was heated under reflux until TLC indicated complete consumption of the starting material 2. In the case of 2m (entry 13), at the onset of the reaction the formation of solid prevented an efficient stirring of the reaction mixture. Thus an additional amount of acetic acid (5 mL) was added. The reaction mixture was cooled and diluted with ice water (60 mL). After stirring the resulting mixture for several hours, the Precipitate A was collected by filtration and washed with water (20 mL). The filtrates were combined into the Filtrate A. The Precipitate A and the Filtrate A were treated as follows.Entries 1–6, 8, 10–12: recrystallization of the Precipitate A from ethanol gave pure products 1a–f,h,j–l.
Entries 2, 4, 6, 11: the Filtrate A was concentrated in vacuo and the residue was subjected to column chromatography on silica gel using 30% ethyl acetate in chloroform as an eluent to isolate benzoxazinones 4d,f,k and phenylhydrazides 5b,f as indicated in Table 3.
Entry 7: recrystallisation of the Precipitate A from ethanol afforded the appropriate benzoxazinones 4g. The mother liquor was concentrated in vacuo. From the residue the second crop of 1g crystallized, which was collected by filtration and washed with a small amount of benzene and recrystallized from ethanol. The yield of the combined crops of 1g is given in Table 3, entry 7.
Entry 13: recrystallisation of the Precipitate A from ethanol afforded benzoxazinone 4m.
Entry 9: the Precipitate A was washed with boiling benzene (15 mL) and recrystallized from ethanol affording a pure compound 1i. The benzene extract was concentrated to the oily residue and subjected to column chromatography on silica gel using 10% ethyl acetate in chloroform as an eluent, to give benzoxazinone 4i.
Entry 10: the Filtrate A was evaporated to dryness and subjected to column chromatography on silica gel with chloroform as an eluent, affording benzoxazinone 4j.
Reaction of 4f with phenylhydrazine into hydrazide 5f (Table 5, entry 1)
A mixture of benzoxazinone 4f (332 mg, 1.09 mmol) and phenylhydrazine (143 mg, 1.32 mmol) in toluene (15 mL) was heated under reflux for 2 hours. After cooling, the precipitated solid was filtered off with suction and crystallized from benzene to give hydrazide 5f (382 mg, 86%). The spectral and analytical data were in agreement with those for the authentic sample, as prepared above.General procedure for the hydrolysis of benzoxazinones 4f,j,m into 2-(indol-2-carboxamido)benzoic acids 1f,j,m (Table 5, entries 3–6)
A mixture of benzoxazinone 4 (343 mg, 0.968 mmol), dimethyl sulfoxide (10 mL) and aqueous sodium hydroxide (0.5 M, 1 mL) was stirred at room temperature for the time indicated in Table 5 (for the reaction in entry 4, dioxane (3 mL) and aqueous sulphuric acid (0.1 M, 2 mL) were used). The reaction mixture was diluted with water (50 mL) and acidified with 10% hydrochloric acid to Congo red (for the reaction from entry 4, no additional acid was added). After stirring for 1 h, the precipitate was filtered off, washed with water and dried to afford 2-(indol-2-carboxamido)benzoic acid 1. Yields of the products are indicated in Table 5. The spectral and analytical data for 1f,j were in agreement with those for the authentic samples, prepared above.1m: Colourless solid, mp 227–230 °C (ethanol), Rf = 0.39 (10% ethanol in chloroform). 1H NMR (500 MHz, DMSO-d6) δ 0.94 (t, 3H, J = 7.1 Hz, C3′(CH2)2CH3), 1.69 (tq, 2H, J = 7.1, 7.1 Hz, C3′CH2CH2), 3.11 (t, 2H, J = 7.1 Hz, C3′CH2), 7.10 (dd, 1H, J = 7.5, 7.5 Hz, H5′), 7.28 (dd, 1H, J = 7.5, 7.5 Hz, H6′), 7.50 (d, 1H, J = 8.2 Hz, H7′), 7.61 (dd, 1H, J = 7.5, 7.5 Hz, H7), 7.67 (dd, 1H, J = 7.5, 7.5 Hz, H6), 7.70 (d, 1H, J = 8.0 Hz, H4′), 7.95 (d, 1H, J = 8.7 Hz, H4), 7.98 (d, 1H, J = 8.6 Hz, H3), 8.03 (d, 1H, J = 8.1 Hz, H5), 8.09 (d, 1H, J = 8.4 Hz, H8), 10.27 (br s, 1H, CONH), 11.54 (br s, 1H, H1′), 13.30 (br s, 1H, COOH); 13C NMR (126 MHz, DMSO-d6) δ 14.2 (C3′(CH2)2CH3), 24.1 (C3′CH2CH2), 26.2 (C3′CH2), 112.2 (C7′), 119.4 (C5′), 120.2 (C4′), 121.2 (C3′), 123.8 (C2), 124.3 (C6′), 125.5 (C8), 126.0 (C3), 126.2 (C4), 126.6 (C7), 126.7 (C2′), 127.7 (C3a′), 128.0 (C5), 128.1 (C6), 129.5 (C8a), 135.2 (C4a), 135.7 (C7a′), 135.8 (C1), 161.6 (CONH), 168.3 (COOH); IR (cm−1): ν 3311 s, 3051 w, 2957 m, 2927 m, 2865 w, 1675 s, 1641 s, 1604 w, 1571 m, 1530 w, 1482 m, 1465 w, 1451 w, 1416 m, 1335 m, 1285 m, 1259 m, 1238 s, 1224 w, 1204 w, 1178 w, 767 m, 749 s; MS (EI) m/z (%): 373 (2), 372 (7), 355 (23), 354 (87), 326 (28), 325 (100), 297 (23), 283 (19), 269 (19), 170 (46), 169 (26), 156 (27), 140 (48), 129 (20), 128 (58), 115 (25), 77 (16); HRMS (ESI+): m/z calcd for C23H21N2O3+ [M + H]+ 373.1547, found 373.1544. Found: C, 74.4; H, 5.5; N, 7.7. Calc. for C23H20N2O3: C, 74.2; H, 5.4; N, 7.5%.
Acknowledgements
This study was supported by the internal grant of the TBU in Zlin (no. IGA/FT/2014/010) funded from the resources of specific university research and The Ministry of Education, Science and Sport, Republic of Slovenia, the Slovenian Research Agency (P1-0230 and Post-doctoral Grant to M. G. (430-168/2013/114). This work was also partly supported through the infrastructure of the EN-FIST Centre of Excellence, Ljubljana.Notes and references
- (a) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS PubMed; (b) R. J. Sundberg, in Indoles, Academic Press, San Diego, 1996 Search PubMed; (c) G. W. Gribble, Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Ress, E. F. V. Scriven and C. W. Bird, Pergamon Press, Oxford, 1996, vol. 2, p. 207 Search PubMed.
- P. Wiklund and J. Bergman, Curr. Org. Synth., 2006, 3, 379–402 CrossRef CAS.
- P.-L. Wu, Y.-L. Hsu and C.-W. Jao, J. Nat. Prod., 2006, 69, 1467–1470 CrossRef CAS PubMed.
- C. Jimènez, E. Quiñoa, M. Adamczeski, L. M. Hunter and P. Crews, J. Org. Chem., 1991, 56, 3403–3410 CrossRef.
- (a) M. S. Shengelia, V. G. Avramenko, L. N. Kuleshova, N. N. Suvorov, V. A. Silin and E. N. Padeiskaya, Khim.-Farm. Zh., 1985, 19, 1075–1078 Search PubMed; (b) A. Thorarensen, C. J. Ruble, J. F. Fisher, D. L. Romero, T. J. Beauchamp and J. M. Northuis, PCT Int. Appl, WO 2004018428 A1 Search PubMed; Chem. Abstr. 2004 140 235498 Search PubMed; (c) J. Li, B. D. Wakefield, J. C. Ruble, C. M. Stiff, D. L. Romero, K. R. Marotii, M. T. Sweeney, G. E. Zurenko, D. C. Rohrer and A. Thorarensen, Bioorg. Med. Chem. Lett., 2007, 17, 2347–2350 CrossRef CAS PubMed; (d) C. Stiff, D. R. Graber, A. Thorarensen, B. D. Wakefield, K. R. Marotti, E. P. Melchior, M. T. Sweeney, F. Han, D. C. Rohrer, G. E. Zurenko and D. L. Romero, Bioorg. Med. Chem. Lett., 2008, 18, 6293–6297 CrossRef CAS PubMed.
- I. Amasaki and N. Hanaki, U.S. Pat. Appl. Publ, US 2009/0239982 A1 Search PubMed; Chem. Abstr. 2008 149 567210 Search PubMed.
- (a) T. J. Dueweke, S. M. Poppe, D. L. Romero, S. M. Swaney, A. G. So, K. M. Downey, I. W. Althaus, F. Reusser, M. Busso, L. Resnick, D. L. Mayers, J. Lane, P. A. Aristoff, R. C. Thomas and W. G. Tarpley, Antimicrob. Agents Chemother., 1993, 37, 1127–1131 CrossRef CAS; (b) M.-P. de Béthune, Antiviral Res., 2010, 85, 75–90 CrossRef PubMed.
- F. Ban, E. Leblanc, H. Li, R. S. N. Munuganti, K. Frewin, P. S. Rennie and A. Cherkasov, J. Med. Chem., 2014, 57, 6867–6872 CrossRef CAS PubMed.
- S. Koeller, J. Kadota, F. Peruch, A. Deffieux, N. Pinaud, I. Pianet, S. Massip, J. M. Leger, J. P. Desvergne and B. Bibal, Chem. – Eur. J., 2010, 16, 4196–4205 CrossRef CAS PubMed.
- For recent examples, see: (a) A. M. Alanazi, A. A. M. Abdel-Aziz, I. A. Al-Suwaidan, S. G. Abdel-Hamide, T. Z. Shawer and A. S. El-Azab, Eur. J. Med. Chem., 2014, 79, 446–454 CrossRef CAS PubMed; (b) M. A. El-Hashash and Y. A. El-Badry, Helv. Chim. Acta, 2011, 94, 389–396 CrossRef CAS.
- (a) A. Krantz, R. W. Spencer, T. F. Tam, T. J. Liak, L. J. Copp, E. M. Thomas and S. P. Rafferty, J. Med. Chem., 1990, 33, 464–479 CrossRef CAS; (b) A. Arcadi, C. Asti, L. Brandolini, G. Caselli, F. Marinelli and V. Ruggieri, Bioorg. Med. Chem. Lett., 1999, 9, 1291–1294 CrossRef CAS.
- U. Neumann, N. M. Schechter and M. Gütschow, Bioorg. Med. Chem., 2001, 9, 947–954 CrossRef CAS.
- L. Hedstrom, A. R. Moorman, J. Dobbs and R. H. Abeles, Biochemistry, 1984, 23, 1753–1759 CrossRef CAS.
- R. L. Jarvest, M. J. Parratt, C. M. Debouck, J. G. Gorniak, J. L. Jennings, H. T. Serafinowska and J. E. Strickler, Bioorg. Med. Chem. Lett., 1996, 6, 2463–2466 CrossRef CAS.
- M. Gütschow and U. Neumann, Bioorg. Med. Chem., 1997, 5, 1935–1942 CrossRef.
- (a) R. Krantz, E. W. Spencer, T. F. Tam, E. Thomas and L. J. Copp, J. Med. Chem., 1987, 30, 589–591 CrossRef; (b) J. L. Gilmore, S. J. Hays, B. W. Caprathe, C. Lee, M. R. Emmerling, W. Michael and J. C. Jaen, Bioorg. Med. Chem. Lett., 1996, 6, 679–682 CrossRef CAS; (c) S. J. Hays, B. W. Caprathe, J. L. Gilmore, N. Amin, M. R. Emmerling, W. Michael, R. Nadimpali, R. Nath, K. J. Raser, D. Stafford, D. Watson, K. Wang and J. C. Jaen, J. Med. Chem., 1998, 41, 1060–1067 CrossRef CAS PubMed.
- S. Mayama, T. Tani, T. Ueno, K. Hirabayashi, T. Nakashima, H. Fukami, Y. Mizuno and H. Irie, Tetrahedron Lett., 1981, 22, 2103–2106 CrossRef CAS.
- For example, see: (a) M. S. Shengelia, V. G. Avramenko, L. N. Kuleshova, N. N. Suvorov, V. A. Silin and E. N. Padeiskaya, Khim.-Farm. Zh., 1985, 19, 1075–1078 Search PubMed; (b) S. P. Hiremath, A. C. Bajji and J. S. Biradar, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1988, 27, 756–757 Search PubMed; (c) S. A. Samsoniya, M. V. Trapaidze, N. A. Kuprashvili, D. S. Zurabishvili and N. N. Suvorov, Chem. Heterocycl. Compd., 1998, 34, 816–821 CrossRef CAS; (d) E. S. Lee, J. K. Son, Y. H. Na and Y. Jahng, Heterocycl. Commun., 2004, 10, 325–330 CrossRef CAS PubMed.
- For reviews of the Fischer indole synthesis, see: (a) B. Robinson, Chem. Rev., 1963, 63, 373–401 CrossRef; (b) B. Robinson, Chem. Rev., 1969, 69, 227–250 CrossRef CAS; (c) G. W. Gribble, J. Chem. Soc., Perkin Trans. 1, 2000, 1045–1075 RSC.
- For seminal studies of the Fischer indole synthesis, see: (a) E. Fischer and F. Jourdan, Ber. Dtsch. Chem. Ges., 1883, 16, 2241–2245 CrossRef; (b) E. Fischer and O. Hess, Ber. Dtsch. Chem. Ges., 1884, 17, 559–568 CrossRef.
- S. Kafka, K. Proisl, V. Kašpárková, D. Urankar, R. Kimmel and J. Košmrlj, Tetrahedron, 2013, 69, 10826–10835 CrossRef CAS PubMed.
- (a) M. Nakazaki and K. Yamamoto, J. Org. Chem., 1976, 41, 1877 CrossRef CAS; (b) G. Baccolini and P. E. Todesco, J. Chem. Soc., Chem. Commun., 1981, 563 RSC.
- (a) K. G. Liu, A. J. Robichaud, J. R. Lo, J. F. Mattes and Y. Cai, Org. Lett., 2006, 8, 5769–5771 CrossRef CAS PubMed; (b) S. Gore, S. Baskaran and B. König, Org. Lett., 2012, 14, 4568–4571 CrossRef CAS PubMed.
- A. Sudhakara, H. Jayadevappa, H. N. H. Kumar and K. M. Mahadevan, Lett. Org. Chem., 2009, 6, 159–164 CrossRef CAS.
- M. Shariat, M. W. Samsudin and Z. Zakaria, Chem. Cent. J., 2013, 7(58), 1–6 Search PubMed.
- G. M. Coppola, J. Heterocycl.. Chem., 1999, 36, 563–588 CrossRef CAS.
- (a) X. L. Lian, H. Lei, X. J. Quan, Z. H. Ren, Y. Y. Wang and Z. H. Guan, Chem. Commun., 2013, 49, 8196–8198 RSC; (b) Z. Y. Ge, Q. M. Xu, X. D. Fei, T. Tang, Y. M. Zhu and S. J. Ji, J. Org. Chem., 2013, 78, 4524–4529 CrossRef CAS PubMed.
- H. M. F. Madkour, ARKIVOC, 2004, i, 36–54 CrossRef.
- A. G. Osborne and Z. Goolamali, Spectrochim. Acta, Part A, 2000, 56, 1079–1100 CrossRef CAS.
- J. Vicens, M. C. Etter and L. A. Errede, Tetrahedron Lett., 1983, 24, 723–724 CrossRef CAS.
- (a) E. Wenkert, E. W. Hagaman, N. Kunesch, N. Y. Wang and B. Zsadon, Helv. Chim. Acta, 1976, 59, 2711–2723 CrossRef CAS; (b) M. S. Morales-Rios, J. Espiñeira and P. Joseph-Nathan, Magn. Reson. Chem., 1987, 25, 377–395 CrossRef CAS; (c) M. S. Morales-Rios and P. Joseph-Nathan, Magn. Reson. Chem., 1987, 25, 911–918 CrossRef CAS; (d) P. Joseph-Nathan, R. E. del Río and M. S. Morales-Ríos, Heterocycles, 1988, 27, 377–383 CrossRef CAS; (e) A. A. Panasenko, A. F. Caprosh, O. M. Radul and M. A. Rekhter, Russ. Chem. Bull., 1994, 43, 60–63 CrossRef.
- (a) E. Rosenberg, K. L. Williamson and J. D. Roberts, Org. Magn. Reson., 1976, 8, 117–119 CrossRef CAS; (b) R. M. Claramunt, D. Sanz, C. Lopez, J. Jimenez, M. L. Jimeno, J. Elguero and A. Fruchier, Magn. Reson. Chem., 1997, 35, 35–75 CrossRef CAS; (c) M. d'Ischia, A. Napolitano and A. Pezzella, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, vol. 3 Search PubMed.
- D. C. G. A. Pinto, C. M. M. Santos and A. M. S. Silva, in Recent Research Developments in Heterocyclic Chemistry 2007, ed. T. M. V. D. Pinho e Melo, 2007, pp. 397–475 Search PubMed.
- K. A. Farley, G. S. Walker and G. E. Martin, Magn. Reson. Chem., 1997, 35, 671–679 CrossRef CAS.
- L. Lázár and F. Fülöp, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, vol. 8 Search PubMed.
Footnotes |
| † Dedicated to Professor Marijan Kočevar on his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra. See DOI: 10.1039/c4ob01714e |
| This journal is © The Royal Society of Chemistry 2014 |