
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion binding of a neutral bis(cyclopeptide) in water–methanol mixtures containing up to 95% water†
Fabian
Sommer
and
Stefan
Kubik
*
Technische Universität Kaiserslautern, Fachbereich Chemie – Organische Chemie, Erwin-Schrödinger-Straße, D-67663 Kaiserslautern, Germany. E-mail: kubik@chemie.uni-kl.de
First published on 8th September 2014
Abstract
Anion receptor 2b was designed and synthesized, which was structurally derived from a previously described bis(cyclopeptide) 2a comprising two covalently linked cyclic hexapeptide rings with alternating L-proline and 6-aminopicolinic acid subunits. Solubilizing groups attached to the aromatic cyclopeptide subunits of 2b cause a substantial improvement of water solubility with respect to 2a, but have negligible effects on anion binding properties. Thus, anion affinity of 2b could be evaluated in aqueous solvent mixtures in which 2a is not sufficiently soluble, namely in water–methanol with a water content of up to 95 vol%. The solvent-dependent characterization of anion binding showed that the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ka values of the iodide and sulfate complexes of 2b decrease linearly with increasing water content while the individual contributions of complexation enthalpy and entropy correlate with the solvent composition in a more complex manner. The obtained results provide insight into the factors that control anion affinity and selectivity of these neutral receptors in aqueous media. In addition, they show that substantial anion affinity can be expected even in 100% water.
Ka values of the iodide and sulfate complexes of 2b decrease linearly with increasing water content while the individual contributions of complexation enthalpy and entropy correlate with the solvent composition in a more complex manner. The obtained results provide insight into the factors that control anion affinity and selectivity of these neutral receptors in aqueous media. In addition, they show that substantial anion affinity can be expected even in 100% water.
Introduction
Molecular recognition phenomena such as the interaction of a synthetic receptor with its substrate are very sensitive to the medium in which they take place.1 While polar binding partners generally like to associate in non-polar solvents where interactions are strong and solvation is weak, the strength of interactions generally significantly drops upon increasing solvent polarity. Polar solvents not only reduce the strength of direct non-covalent interactions, they usually also solvate the binding partners more efficiently than non-polar ones, causing complex stability to suffer from an increasing enthalpic penalty associated with the desolvating receptor and/or substrate. Desolvating binding partners in polar solvents, particularly in water, can also become the driving force of complex formation, however, if it involves the release of high-energy water,2 which results in an enthalpic advantage of binding, or leads to overall higher disorder, which is entropically favourable.3Solvent-dependent evaluation of the binding properties of a synthetic receptor can shed light on the subtle interplay of the factors that control receptor affinity and selectivity. Such investigations were performed, for example, to characterize glucose affinity of a synthetic carbohydrate receptor in chloroform–methanol and water–methanol mixtures.4 A more recent investigation has addressed anion binding to diols in acetonitrile–chloroform mixtures revealing a non-linear correlation between complex stability and solvent composition.5 In the area of anion coordination chemistry6 solvent-dependent binding studies are routinely performed if a receptor turns out to possess an anion affinity in a specific solvent that is too high to be evaluated. If binding is too strong in DMSO, for example, more competitive water–DMSO mixtures are often used in which complex stability is lower and can thus be quantified.7 The maximum amount of water in the solvent mixture is determined by receptor solubility and is usually chosen so as to not lower complex stability below the detection limit. The increase of the water content of the solvent mixture sometimes also simplified the receptor's mode of interaction, for example, from the formation of higher to well defined 1:1 complexes,7e,f,h but the consequences of changing the solvent composition on the thermodynamics of binding have not often been considered.
Here, we describe a bis(cyclopeptide) whose solubility allowed a detailed thermodynamic characterization of anion binding properties in a wide range of water–methanol mixtures. Such bis(cyclopeptides) were previously shown by us to interact with halides or sulfate anions in aqueous solvent mixtures.8 They derive from cyclopeptide 1 whose mode of binding involves sandwiching an inorganic anion between two cyclopeptide rings.9 These 2:1 complexes could be converted into 1:1 complexes by covalently connecting two cyclopeptides via appropriate linking units. While anion binding of 1 could be evaluated in water–methanol mixtures containing up to 80% of water, the respective bis(cyclopeptides) are, unfortunately, considerably less soluble so that the water content of the mixture used to study the properties of, for example, 2a could not exceed 50%.8c Other bis(cyclopeptides) allowed binding studies in water–acetonitrile mixtures containing up to 75% of water.8e The respective investigations indicated that hydrophobic interactions between the two cyclopeptide rings not involving direct receptor/substrate interactions contribute to the overall stability of the complexes formed. To what extent anion affinity of these receptors is retained in 100% water remained an open question, however.
Results and discussion
Structural design and synthesis
Our approach to improving the water solubility of 2a without compromising binding properties relied on the introduction of solubilizing groups into positions of the receptor in which their impact on binding properties was expected to be minimal. The corresponding structural considerations were based on the previously reported calculated structure of the iodide complex of 2a as shown in Fig. 1.8c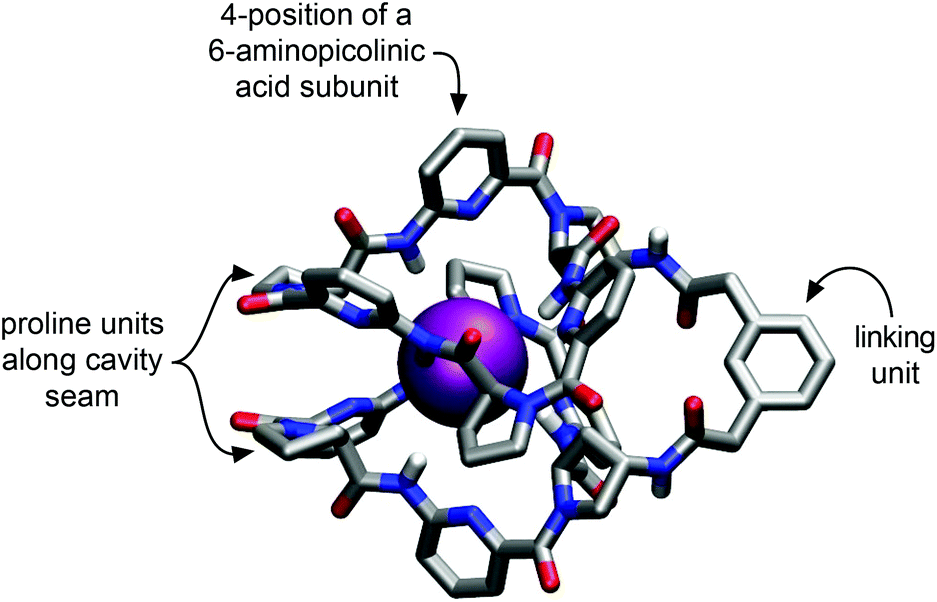 | ||
| Fig. 1 Calculated geometry of the iodide complex of 2a obtained with the MMFF94 force field as implemented in PCModel, Serena Software, Inc. Hydrogen atoms at the carbon atoms of the receptor have been omitted for clarity.8c | ||
This structure indicates that additional substituents on the bis(cyclopeptide) proline residues would be oriented close to the seam of the cavity where the two cyclopeptide moieties approach each other. Substituents in these positions were therefore expected to affect complex formation by steric effects. The phenylene moiety of the linker is oriented away from the cavity and would therefore make a better modification site. The number of groups that could be introduced here is limited, however, and it was therefore decided to incorporate the solubilizing groups into the 4-positions of the aminopicolinic acid subunits. This strategy allowed introduction of overall six additional groups expected to exhibit no strong effect on anion affinity because they all diverge from the receptor cavity. Triethylene glycol residues were chosen as solubilizing groups, which should increase water solubility without introducing charges so that competing effects on anion binding of other negatively charged species in solution could be avoided.
The 4-substituted 6-aminopicolinic acid derivative 3 required for the synthesis of 2b was obtained from the commercially available chelidamic acid according to a reported procedure.11 This amine was coupled to (2S)-1-(tert-butyloxycarbonyl) proline and (2S,4S)-4-(benzyloxycarbonylamino)-1-(tert-butyloxycarbonyl)proline12 using PyCloP as a coupling reagent to yield, respectively, the dipeptides 4 and 5 required for the synthesis of the cyclopeptide moieties of 2b (Scheme 1). Appropriate chain elongation of these dipeptides afforded the linear cyclopeptide precursor, which was deprotected at both ends and cyclized by following established protocols.8a,13 The resulting cyclopeptide 6, containing one Z-protected 4-aminoproline unit, was subjected to hydrogenation and then reacted with 2,2′-(1,3-phenylene)diacetic acid in the presence of TBTU.8c The coupling product 2b was obtained after chromatographic purification in analytically pure form and in sufficient amounts for the following binding studies.
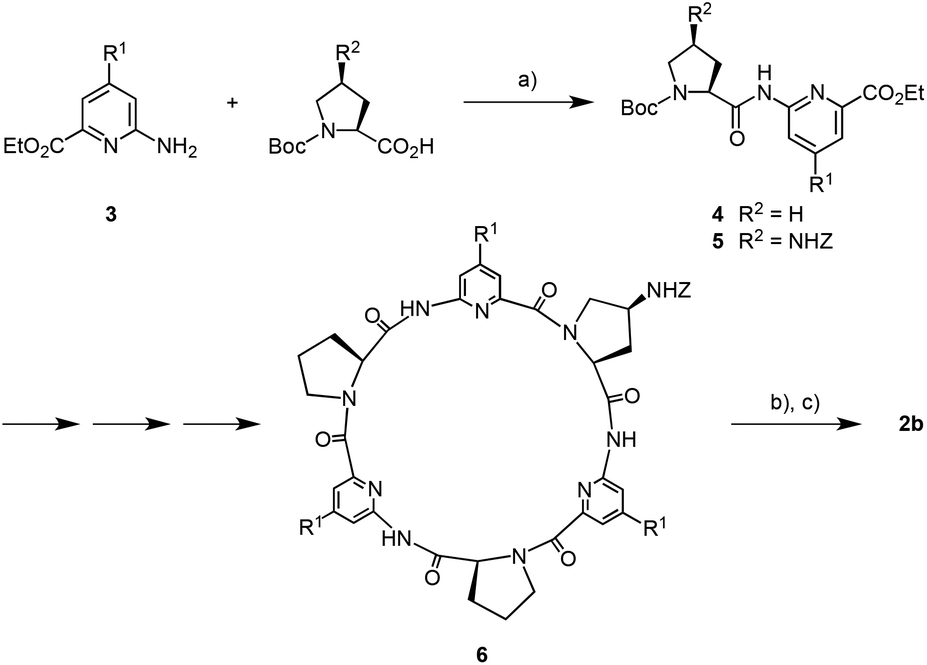 | ||
| Scheme 1 Synthesis of bis(cyclopeptide) 2b (R1 = (OCH2CH2)3OCH3). (a) Chlorotripyrrolidinophosphonium hexafluorophosphate (PyCloP), N,N-diisopropylethylamine, CH2Cl2, 25 °C, 10 d, 4 82%, 5 96%; (b) H2, 1.2 equiv. HCl, 10% Pd/C, CH2Cl2–CH3OH, 1:1 (v/v), 25 °C, 18 h; (c) 2,2′-(1,3-phenylene)diacetic acid, 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium (TBTU), N,N-diisopropylethylamine, dimethylformamide, 25 °C, 2 h, 47% over 2 steps. Z = benzyloxy carbonyl, Boc = tert-butoxy carbonyl. | ||
Qualitative binding studies
Qualitative information about the binding properties of 2b was derived from ESI mass spectrometric and NMR spectroscopic investigations. Because the water solubility of 2b, although considerably improved by the presence of the six triethylene glycol residues, unfortunately turned out to be still insufficient to allow binding studies in 100% water, these experiments were performed in 90% water–methanol. In addition, measurements in 50% water–methanol mixtures were carried out to compare the properties of 2b with those of 2a. These binding studies concentrated on sulfate and iodide as substrates because both anions generally exhibit strongest interactions with our bis(cyclopeptides) and represent prototypes of strongly and weakly coordinating anions, respectively.8,13
Fig. 2 shows the ESI-TOF MS spectra of 2b in the presence of sodium iodide or sulfate. In all spectra, the most prominent peak is that of the 1:1 complex between the bis(cyclopeptide) and the respective anion ([M + I]− = 2590.0, [M + SO4]2– = 1279.5). Minor additional peaks could be assigned to the chloride complex ([M + Cl]− = 2498.1). Mass spectrometry thus indicated that 2b binds to iodide and sulfate anions independent of the composition of the solvent mixture. Moreover, the major complex species are 1:1 complexes as in the case of other structurally related bis(cyclopeptides).8
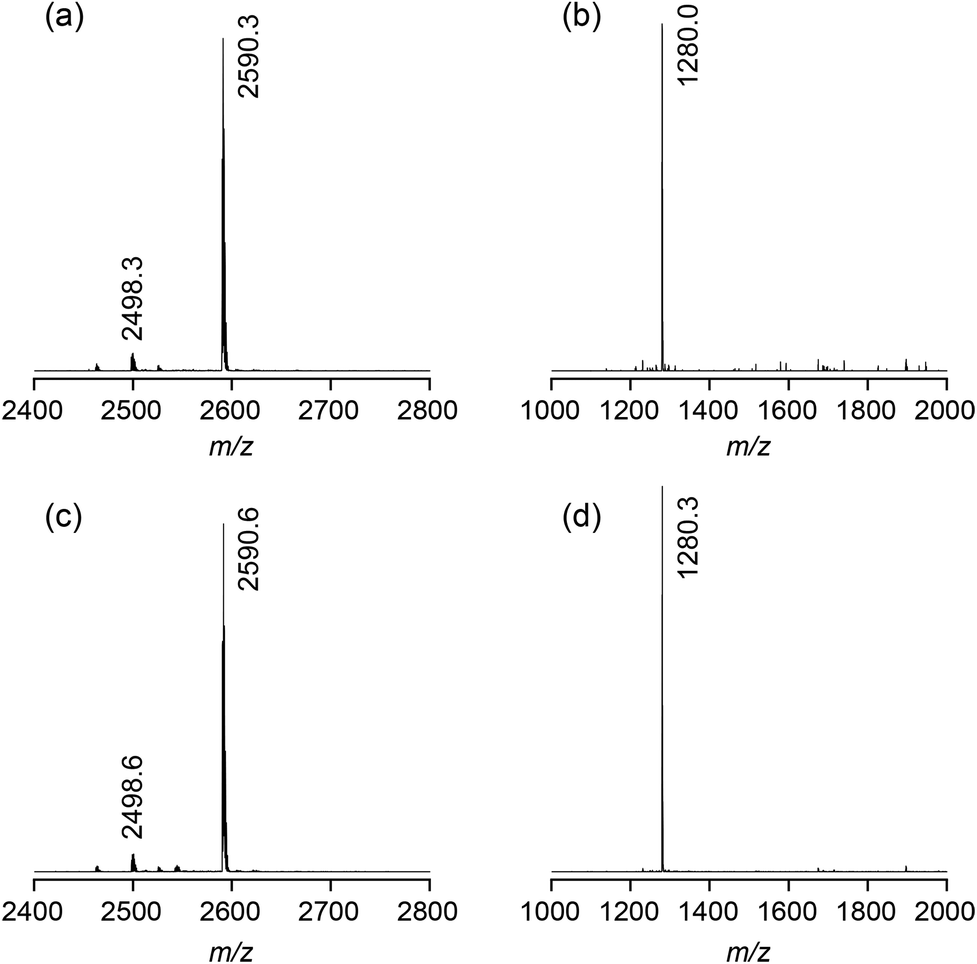 | ||
| Fig. 2 ESI-TOF MS spectra (negative mode) of 2b (1 mM) in the presence of 1 equiv. of NaI (a) or Na2SO4 (b) in 50% water–methanol and 1 equiv. of NaI (c) or Na2SO4 (d) in 90% water–methanol. | ||
Additional evidence for the ability of 2b to interact with anions in 90% water–methanol came from NMR spectroscopy. Fig. 3 shows the effect of sulfate anions on the 1H NMR spectra of 2b in 50% D2O–CD3OD and 90% D2O–CD3OD. In the absence of sulfate anions the H(α) protons on the proline residues of 2b absorb between 5.60 and 5.76 ppm in 50% D2O–CD3OD. In 90% D2O–CD3OD these signals are slightly shifted upfield and those of the unsubstituted proline rings collapse into one. Addition of sulfate causes a substantial downfield shift of the proline H(α) signals in both solvents by ca. 1 ppm. In addition, spreading out of the signals in the aromatic region of the spectra is observed. Both effects are characteristic features of all anion binding bis(cyclopeptides) studied so far.8,13 The shifts of the proline H(α) signals, for example, are due to the spatial proximity of the corresponding protons to the anion once it is included into the bis(cyclopeptide) cavity. NMR spectroscopy thus indicates that the mode of anion binding of 2b does not differ from that of structurally related receptors and that it is independent of solvent composition. Analogous albeit slightly smaller shifts of the proline H(α) signals were observed in the 1H NMR spectra of 2b after addition of iodide anions in both solvent mixtures, consistent with the more weakly coordinating nature of the halide in comparison with sulfate anions (see ESI†).
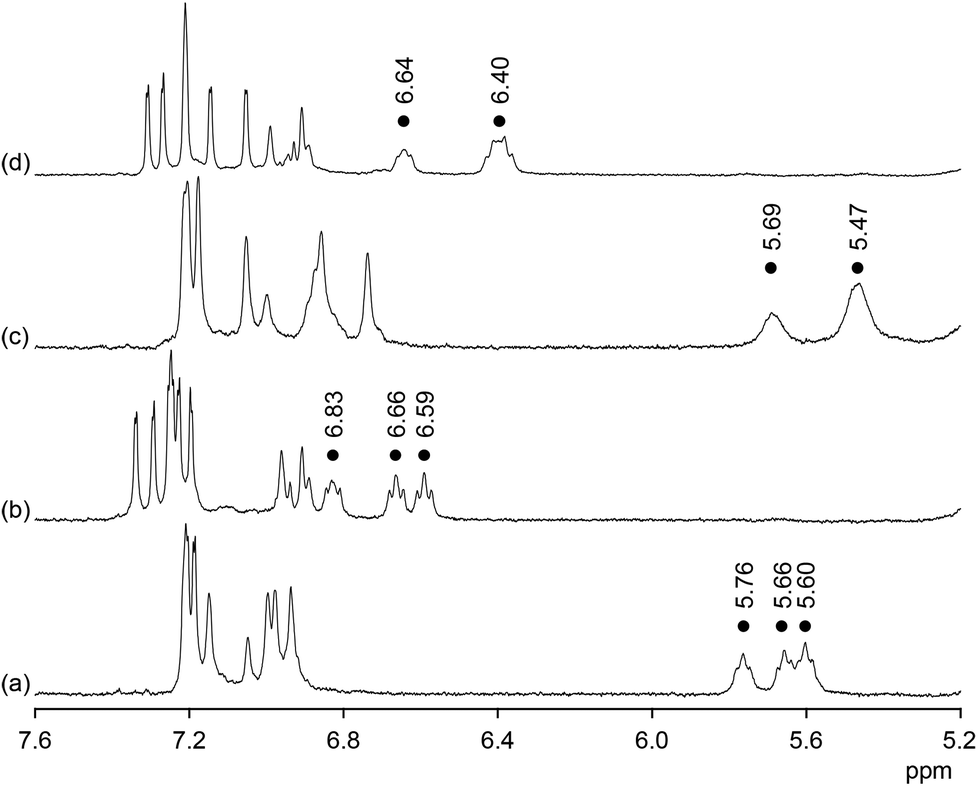 | ||
| Fig. 3 1H NMR spectra of 2b (1.8 mM) prior (a) and after (b) the addition of 2 equiv. of Na2SO4 in 50% D2O–CD3OD and of 2b (1.0 mM) prior (c) and after (d) the addition of 2 equiv. of Na2SO4 in 90% D2O–CD3OD. The dots indicate the signals of the H(α) protons on the proline residues of 2b. | ||
Quantitative binding studies
Anion binding of 2b was quantified by isothermal titration calorimetry (ITC).14 Initial investigations addressed the question if and to what extent anion affinity of 2b is affected by the presence of the triethylene glycol units. To this end, binding of 2b to iodide and sulfate was evaluated in a solvent mixture, in which quantitative information about the properties of 2a is available, namely in 50% water–methanol. The obtained results together with the ones previously reported for bis(cyclopeptide) 2a are summarized in Table 1.| Sulfate | Iodide | |||||
|---|---|---|---|---|---|---|
| logKa |
ΔH | TΔS | logKa |
ΔH | TΔS | |
| a Recorded in 50% water–methanol at 298 K; energies in kJ mol−1; the values are means of at least three independent measurements with the standard deviations specified. | ||||||
|
2a8c |
5.97 ± 0.02 | −13.2 ± 1.1 | 20.9 ± 1.2 | 4.43 ± 0.11 | −15.4 ± 0.7 | 9.8 ± 1.2 |
| 2b | 5.87 ± 0.02 | −14.7 ± 0.8 | 18.8 ± 0.8 | 4.59 ± 0.01 | −16.2 ± 0.8 | 10.0 ± 0.7 |
Table 1 shows that iodide and sulfate affinity of 2b in 50% water–methanol is practically indistinguishable from that of 2a. Moreover, no major differences are apparent within the error limits in the thermodynamics of complex formation. The conclusion that can be drawn from this result is that, in accordance with the design strategy, the impact of the triethylene glycol residues in 2b on anion affinity is negligible. Compound 2b can thus be expected to behave like previously described bis(cyclopeptides) with the important advantage that it allows anion recognition to be followed in much more competitive media.
Anion binding of 2b was subsequently studied in a range of different water–methanol mixtures. While iodide recognition could be evaluated in mixtures containing between 20% and 95% of water, solubility problems prevented titrations with sodium sulfate in mixtures containing less than 30% of water. In addition, no reliable results could be derived from the titration with sodium sulfate in 90% water–methanol because the enthalpy of sulfate complexation changes sign close to this solvent composition. The results of all measurements are summarized in Table 2 and depicted graphically in Fig. 4. The stoichiometric factors n deriving from these titrations support the 1:1 stoichiometry of both anion complexes of 2b.
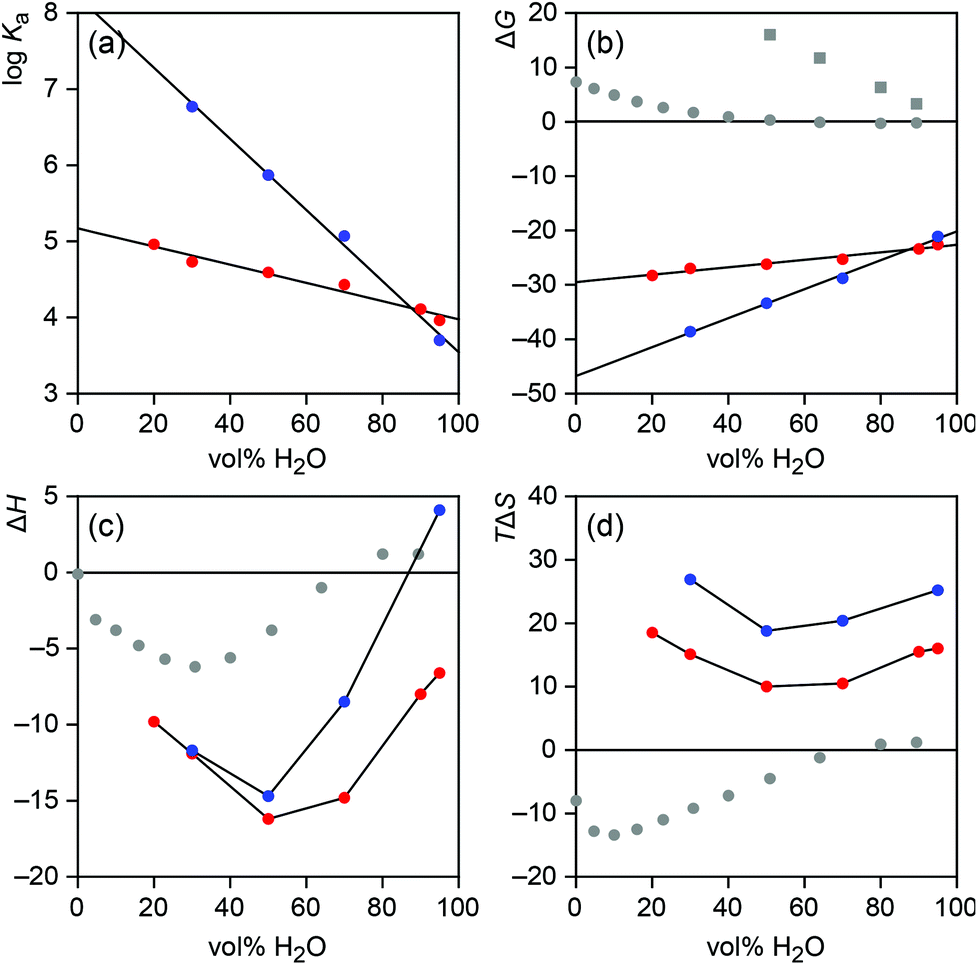 | ||
| Fig. 4 Solvent dependence of the stability (a), Gibbs free energy of formation (b), formation enthalpy (c), and formation entropy (d) of the iodide (red) and sulfate (blue) complexes of 2b. In addition, b–d contain data about the Gibbs energy (b), enthalpy (c), and entropy (d) of the transfer of iodide (grey dots) and sulfate (grey squares) anions from water to water–methanol mixtures.16 All energies in kJ mol−1. | ||
| Vol%b | Sulfate | Iodide | ||||||
|---|---|---|---|---|---|---|---|---|
| logKa |
ΔH | TΔS | n | logKa |
ΔH | TΔS | n | |
| a Recorded at 298 K; energies in kJ mol−1; the values are means of at least three independent measurements with the standard deviations specified. b Vol% of water in the water–methanol mixture. c Not determined. | ||||||||
| 20 | n.d.c | n.d. | n.d. | n.d. | 4.96 ± 0.02 | −9.8 ± 0.3 | 18.5 ± 0.3 | 1.01 |
| 30 | 6.77 ± 0.10 | −11.7 ± 0.2 | 26.9 ± 0.7 | 0.93 | 4.73 ± 0.01 | −11.9 ± 0.2 | 15.1 ± 0.2 | 0.99 |
| 50 | 5.87 ± 0.02 | −14.7 ± 0.8 | 18.8 ± 0.8 | 0.93 | 4.59 ± 0.01 | −16.2 ± 0.8 | 10.0 ± 0.7 | 1.03 |
| 70 | 5.07 ± 0.02 | −8.5 ± 0.3 | 20.4 ± 0.4 | 0.93 | 4.43 ± 0.02 | −14.8 ± 0.2 | 10.5 ± 0.3 | 1.03 |
| 90 | n.d. | n.d. | n.d. | n.d. | 4.12 ± 0.02 | −8.0 ± 0.4 | 15.5 ± 0.5 | 1.08 |
| 95 | 3.70 ± 0.05 | 4.1 ± 0.8 | 25.2 ± 0.5 | 0.77 | 3.96 ± 0.03 | −6.6 ± 0.3 | 16.0 ± 0.5 | 1.18 |
Fig. 4a and 4b show that, as expected, iodide and sulfate affinity of 2b decrease upon increasing the water content of the solvent. For both anions the correlation is linear (r2 > 0.97), with a more pronounced drop observed for sulfate binding. The different slopes of the lines cause an inversion of anion affinity from sulfate as the better substrate across the major range of solvent compositions to iodide in mixtures containing more than 90% of water. Thus, the intrinsically less strongly coordinating anion forms the more stable complex in the most competitive solvent mixtures. Assuming that the observed linear correlation between complex stability and solvent composition extends to 100%, an iodide affinity logKa of 4.0 can be expected for 2b in water and a sulfate affinity of 3.5.
While the reduction of complex stability in the more competitive solvent mixtures has several reasons, one being the weakened receptor/anion interactions, the different extents to which sulfate and iodide affinity of 2b decrease with increasing water content of the solvent are likely partially due to the different nature of both anions, reflected in the ease with which they are desolvated. With a free energy of hydration ΔGhydr of −1090 kJ mol−1, sulfate anions are significantly more strongly solvated in water than iodide anions whose free energy of hydration amounts to −283 kJ mol−1.15 Information about how these values change in water–methanol mixtures can be derived from the literature known Gibbs energies required for transferring the respective anion from water into a water–methanol mixture.16 The corresponding data, included in Fig. 4b, show that transferring iodide or sulfate anions from water to water–methanol mixtures becomes increasingly endergonic as the methanol content of the solution rises. Thus, desolvating both anions becomes easier in terms of ΔG in less polar media, where interactions can also be assumed to be stronger. Not surprisingly, this effect is more pronounced for the strongly coordinating, doubly charged sulfate anion, which is consistent with the stronger dependence of sulfate affinity on solvent composition. Thus, sulfate affinity of 2b presumably benefits stronger than iodide affinity from the increasingly more facile anion desolvation as the methanol content of the solvent rises. Conversely, in the high water content solvents, anion desolvation has a stronger detrimental effect on complex stability in the case of sulfate. In these solvents, the free energy required for desolvating the sulfate anion seems to compensate sulfate/receptor interactions to such an extent that the overall logKa of the sulfate complex approaches or even falls behind that of the complex with the intrinsically weaker bound but less efficiently hydrated iodide anion.
In this context it is worth noting that the stability of the sulfate complex of 2b drops less steeply with increasing water content of the solvent mixture than expected on the basis of the solvent dependence of the Gibbs energy of sulfate desolvation. Specifically, one would expect sulfate affinity of 2b to drop by 3.3 kJ mol−1 with every 10% increase of water based on the extent to which the Gibbs energy of the transfer of sulfate anions changes with solvent composition, yet the observed decrease of the complex stability only amounts to 2.7 kJ mol−1. This result is consistent with previous results obtained for iodide complexation of another bis(cyclopeptide) in water–acetonitrile mixtures, and supports the notion that hydrophobic interactions between the apolar subunits of the cyclopeptide moieties of these bis(cyclopeptides) contribute to complex stability in solvents with a high water content.8e These hydrophobic interactions reinforce complex stability beyond the strength of the direct receptor/substrate interactions.17a Unfortunately, analogous considerations for iodide complexation are less clear because the Gibbs energy of the transfer of iodide anions from water to water–methanol mixtures does not change linearly with solvent composition (Fig. 4b).
Further insights into the solvent-dependent thermodynamics of anion binding to 2b were obtained by considering the enthalpic and entropic contributions independently. Interestingly, it turned out that the complexation enthalpy and entropy do not correlate with solvent composition in a linear fashion (Fig. 4c and 4d). Both thermodynamic parameters exhibit U-shaped curves with minima at 50% water–methanol independent of the anion. In mixtures containing more or less water, complex formation is enthalpically less favourable and entropically more favourable. These opposing enthalpic and entropic terms produce an overall monotonous decrease of the absolute values of the Gibbs energies of complex formation across the whole investigated solvent range.17b
Comparing the curves in Fig. 4b and 4c shows that the more pronounced drop of the sulfate affinity of 2b upon increasing the water content of the solvent is mainly enthalpic in origin. While the difference in the complexation entropy of sulfate and iodide binding remains relatively constant over the range of solvent mixtures studied, sulfate binding becomes enthalpically significantly less favourable in more competitive media than iodide binding, eventually causing sulfate complexation to become endothermic in 95% water–methanol. To relate this effect to the desolvation enthalpies of iodide and sulfate anions is not straightforward, however.
In water, desolvating the more strongly coordinating doubly charged sulfate anion requires more energy than dehydrating iodide (ΔHhydr (SO42–) = −1035 kJ mol−1, ΔHhydr (I−) = −291 kJ mol−1). Conversely, dehydration of sulfate is entropically more favourable (ΔShydr (SO42−) = −200 J K−1 mol−1, ΔShydr (I−) = −36 J K−1 mol−1).15 Assuming that these trends hold over the whole range of solvent mixtures studied, they are consistent with the generally less favourable enthalpy and more favourable entropy of sulfate over the iodide binding observed for 2b. Exact information about the dependence of the thermodynamics of anion desolvation on solvent composition can again be derived from the enthalpies and entropies associated with transferring an anion from water to water–methanol mixtures. These values are reported for iodide and have been included in Fig. 4c and 4d.16a Respective thermodynamic data for sulfate are not available.
Interestingly, Fig. 4c and 4d show that the enthalpy and entropy of transferring iodide from water into water–methanol mixture also do not correlate linearly with solvent composition. Transferring iodide into a mixture with higher water content is slightly endothermic. At ca. 70% water content desolvation enthalpy changes sign and the exothermicity of desolvation reaches a maximum in ca. 30% water–methanol. Thus, desolvating iodide is more difficult in water–methanol mixtures than in 100% water below a water content of 70%. This trend is clearly not reflected in the observed enthalpic contribution to iodide complexation of 2b, which is most favourable in 50% water–methanol. A correlation of the enthalpy of iodide binding and desolvation is therefore not directly evident. In addition, due to the lack of respective data for sulfate anions a comparison of how desolvation enthalpies of the two anions differ when varying the solvent composition cannot be made.
One also has to consider that ITC titrations yield a global picture of complex formation involving several contributions, which makes it problematic to attribute an observed effect to a single cause. Besides anion desolvation, the thermodynamics of forming a complex such as the one shown in Fig. 1 are also affected by desolvation of parts of the receptor and by direct receptor/substrate interactions. In addition, binding of the anion could require separating an ion pair, particularly in less polar media, which could have an additional impact on the overall thermodynamic signature of complex formation.
It can be expected that direct interactions between an anion and 2b are favoured by enthalpy. The increase of the water content of the solvent should diminish this favourable term while desolvation of the binding partners should produce a favourable entropic term for complex formation. These qualitative trends are indeed visible in solvents containing more than 50% of water (Fig. 4c and 4d), but to explain the increasingly unfavourable enthalpy of binding and the increasingly favourable entropy of binding in solvent mixtures with a high methanol content other factors have to be considered. One could relate to ion pairing, which could make it more difficult to bind the anion in less polar media. To test this assumption, iodide complexation was also characterized in the least polar solvent mixture by using sodium iodide in the presence of an excess of 15-crown-5 and tetramethylammonium iodide. Should ion pairing have an effect of iodide binding, changing the nature of the cation was expected to result in characteristic changes in the thermodynamic signature of iodide complexation. Interestingly, it turned out that the interactions between 2b and iodide anions are practically independent of the counterion (Table 3), which strongly suggests that ion pairing is not responsible for the reduction of the enthalpic contribution to complex formation observed in solvent mixtures containing more than 50% of methanol.
| Counterion | logKa |
ΔH | TΔS |
|---|---|---|---|
| a Recorded at 298 K; energies in kJ mol−1. | |||
| Sodium | 4.96 | −9.8 | 18.5 |
| Sodium + 15-crown-5 | 4.92 | −10.1 | 18.0 |
| Tetramethylammonium | 5.02 | −10.7 | 17.9 |
An indirect indication that the solvent-dependent changes of complexation enthalpy and entropy are not only related to the properties of the anions or the salts is the fact that these trends are relatively anion independent. Complexation enthalpy is the best and entropy the worst in 50% water–methanol for both anions, in spite of their significantly different properties, suggesting that the reasons for the U-shapes of the curves in Fig. 4c and 4d lie also with the receptor.
Based on these arguments, we attribute the observed reduction of the favourable binding enthalpies at high methanol content to receptor desolvation, specifically the release of solvent molecules that surround the NH groups inside the binding site of 2b. Assuming that the binding site of 2b is preferentially solvated by water molecules, releasing these molecules upon anion binding should become enthalpically more difficult (and entropically more favourable) as the methanol content of the solvent mixture rises. That receptor desolvation has indeed a pronounced effect on the anion binding of our bis(cyclopeptides) has been shown previously. The endothermicity of sulfate complexation of a triply-linked bis(cyclopeptide) has, for example, been partly ascribed to the enthalpically difficult desolvation of the efficiently hydrated binding site of this receptor13 and the highly favourable binding entropy of this and related doubly-linked bis(cyclopeptides)8f to the associated entropic advantage.
The solvent-dependence of binding enthalpy observed for 2b can thus best be rationalized by strong binding site hydration at high methanol content and weakened receptor/substrate interactions at high water content combined with the effects of anion desolvation in the more competitive solvent mixtures.‡ This interpretation is consistent with the observed trends in complexation entropy. Binding becomes entropically more favourable in solvents with a high water content because anion desolvation and hydrophobic interactions within the anion complexes of 2b contribute favourably to complex formation while the release of water molecules from the binding site of the receptor explains the favourable entropic term in solvents containing an excess of methanol.
One additionally has to consider that enthalpy and entropy generally correlate with enthalpic improvements of complex stability being associated with negative effects on complexation entropy and vice versa.17b This correlation, which can be rationalized in terms of entropic disadvantages resulting from the tightening of receptor/substrate interactions, is also evident when comparing the influence of solvent composition on the enthalpy and the entropy of anion binding of 2b. Changes in solvent composition that lead to a more favourable binding enthalpy evidently have a negative influence on the entropy of binding.
Conclusions
In conclusion, by introducing solubilizing groups into the 6-aminopicolinic acid subunits of 2a considerable progress has been made in improving the water solubility of these bis(cyclopeptides). While solubility in 100% water could not be achieved, the corresponding bis(cyclopeptide) 2b allowed anion binding to be quantified in water–methanol mixtures containing up to 95% water. These investigations revealed subtle effects of the composition of the solvent mixture on the thermodynamics of binding. The obtained results provide insight into the factors that influence the interactions of these neutral receptors with anions in aqueous media, demonstrating, for example, the importance of solvation effects for complex formation, which could cause a weakly coordinating anion to bind more strongly to a receptor in competitive aqueous media than a strongly coordinating one.Moreover, extrapolation of the binding properties of 2b to 100% water suggested substantial iodide and sulfate affinity even in water. With a logKa of ca. 4, the expected iodide affinity of these bis(cyclopeptides) in water is of the same order, potentially even higher than that of other previously described neutral anion receptors such as the bambusurils or related analogues.§ The next generation of water-soluble bis(cyclopeptides) is now required to confirm these results. These receptors will likely feature other solubilizing groups in the aminopicolinic acid subunits or additional groups in the linker. Work in both directions is currently in progress.
Experimental
General details
Analyses were carried out as follows: melting points, Müller SPM-X 300; NMR, Bruker DPX 600, Bruker DPX 400 (peak assignments were confirmed by using H,H-COSY and HMQC spectra, spectra of Boc-protected linear peptides were measured at 100 °C in d6-DMSO because the hindered rotation around the bond between the Boc group and the proline nitrogen atom complicates the spectra at 25 °C); MALDI-TOF-MS, Bruker Ultraflex TOF/TOF; ESI-MS, Bruker Esquire 6000; elemental analysis, Elementar vario Micro cube; optical rotation, Perkin Elmer 241 MC digital polarimeter (d = 10 cm); RP chromatography, MERCK LiChroprep RP-8 (40–63 μm) prepacked column size B (310-25); preparative HPLC, Dionex Ultimate 3000 (column, Supelco, Ascentis C18, 250 × 21.2 mm, 5 μm particle size; flow, 10 mL min−1; eluent, aqueous: water, organic: acetonitrile; the following gradients were used for the isolation of 6 and 2b: 6: 0 min, 25% organic; 0–38 min, linear increase to 45% organic; 38–40 min, linear increase to 50% organic; 40–42 min, 50% organic; 42–45 min, linear decrease to 25% organic; 45–50 min, 25% organic; 2b: 0 min, 25% organic; 0–35 min, linear increase to 40% organic; 35–48 min, 40% organic; 48–50 min, linear decrease to 25% organic; 50–55 min, 25% organic); ITC, Microcal VP-ITC.The following abbreviations are used: Boc, tert-butoxy carbonyl; Z, benzyloxy carbonyl; DIEA, N-ethyldiisopropylamine; PyCloP, chlorotripyrrolidinophosphonium hexafluorophosphate; TBTU, O-(1H-benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate; Pro, L-proline; Apro, (4S)-4-amino-L-proline; Pda, 2,2′-(1,3-phenylene)diacetic acid, Mapa, 4-[2-[2-(2-methoxyethoxy)ethoxy]ethoxy]-2-aminopyridine-6-carboxylic acid; Teg, triethylene glycol.
Materials
All solvents were dried according to standard procedures prior to use. DMF p.a. was purchased and used without further purification. PyCloP was prepared according to the literature procedures while TBTU was purchased.11 (0.82 g, 2.5 mmol) and (2S)-1-(tert-butyloxycarbonyl)proline (0.65 g, 3.0 mmol) were dissolved in dry dichloromethane (50 mL). To this solution PyCloP (1.26 g, 3.0 mmol) and DIEA (1.04 mL, 6.0 mmol) were added. The mixture was stirred at 25 °C for 10 d. Afterwards the solvent was removed in vacuo and the residue purified by column chromatography (SiO2, ethyl acetate). Yield 1.08 g (82%), colourless solid; m.p. 44–48 °C; [α]22D = −33.3 (c = 1, chloroform); 1H NMR (600 MHz, 100 °C, [D6]DMSO): δ = 10.10 (s, 1H, NH), 7.86 (d, 4J = 2.1 Hz, 1H, MapaH(3)), 7.29 (d, 4J = 2.1 Hz, 1H, MapaH(5)), 4.47–4.49 (m, 1H, ProH(α)), 4.36 (q, 3J = 7.0 Hz, 2H, ethyl-CH2), 4.27 (t, 3J = 4.8 Hz, 2H, TegCH2), 3.81 (t, 3J = 4.8 Hz, 2H, TegCH2), 3.54–3.63 (m, 6H, TegCH2), 3.36–3.46 (m, 4H, ProH(δ)/TegCH2), 3.27 (s, 3H, TegCH3), 2.19–2.25 (m, 1H, ProH(β)), 1.89–1.98 (m, 2H, ProH(γ)/ProH(β)), 1.81–1.85 (m, 1H, ProH(γ)), 1.38 + 1.41 (2s, 9H, Boc-H), 1.35 (t, 3J = 7.1 Hz, 3H, ethyl-CH3) ppm; 13C NMR (151 MHz, 100 °C, [D6]DMSO): δ = 171.8 (Pro-CO), 166.4 (MapaC(4)), 163.8 (COOEt), 152.8 + 153.0 (MapaC(2)/Boc-CO), 147.5 (MapaC(6)), 107.4 (MapaC(5)), 101.9 (MapaC(3)), 78.3 (Boc-C), 67.6 + 68.1 + 69.2 + 69.3 + 69.6 + 70.8 (TegC), 60.5 (ethyl-CH2), 59.7 (ProC(α)), 57.3 (TegCH3), 46.1 (ProC(δ)), 29.7 (ProC(β)), 27.5 (Boc-CH3), 22.8 (ProC(γ)), 13.3 (ethyl-CH3) ppm; MS (MALDI-TOF) m/z (%) 526.3 (100) [M + H]+, 548.3 (75) [M + Na]+, 564.3 (13) [M + K]+; elemental analysis calcd (%) for C25H39N3O9·H2O: C 55.24, H 7.60, N 7.73; found C 55.42, H 7.38, N 7.36.
11 (0.56 g, 1.7 mmol) and (2S,4S)-4-(benzyloxycarbonylamino)-1-(tert-butyloxycarbonyl)proline (0.66 g, 1.8 mmol) were dissolved in dry dichloromethane (40 mL). To this solution PyCloP (0.76 g, 1.8 mmol) and DIEA (0.76 mL, 4.4 mmol) were added. The mixture was stirred at 25 °C for 10 d. Afterwards the solvent was removed in vacuo and the residue purified by column chromatography (SiO2, ethyl acetate). Yield 1.09 g (96%), colourless solid; m.p. 74–77 °C; [α]22D = −16.7 (c = 1, chloroform); 1H NMR (600 MHz, 100 °C, [D6]DMSO): δ = 10.28 (s, 1H, NH), 7.85 (d, 4J = 2.0 Hz, 1H, MapaH(3)), 7.29–7.35 (m, 6H, MapaH(5)/PhH), 6.91 (d, 3J = 6.1, 1H, Z-NH), 5.07 (d, 2J = 12.7 Hz, 1H, Z-CH2), 5.04 (d, 2J = 12.7 Hz, 1H, Z-CH2), 4.49–4.51 (m, 1H, AproH(α)), 4.36 (q, 3J = 7.1 Hz, 2H, ethyl-CH2), 4.26 (t, 3J = 4.8 Hz, TegCH2), 4.08–4.12 (m, 1H, Apro(γ)), 3.81 (t, 3J = 4.8 Hz, 2H, TegCH2), 3.72–3.75 (m, 1H, AproH(δ)), 3.61–3.63 (m, 2H, TegCH2), 3.54–3.58 (m, 4H, TegCH2), 3.44–3.46 (m, 2H, TegCH2), 3.25–3.28 (m, 4H, AproH(δ)/TegCH3), 2.53–2.58 (m, 1H, AproH(β)), 1.91–1.96 (m, 1H, AproH(β)), 1.37 + 1.40 (2s, 9H, Boc-H), 1.35 (t, 3J = 7.1 Hz, 3H, ethyl-CH3) ppm; 13C NMR (151 MHz, 100 °C, [D6]DMSO): δ = 171.7 (Apro-CO), 166.4 (MapaC(4)), 163.7 (COOEt), 154.9 (Z-CO), 152.6 + 152.8 (MapaC(2)/Boc-CO), 147.5 (MapaC(6)), 136.5 (PhC(1)), 127.6 (PhC(3)), 127.0 (PhC(4)), 126.8 (PhC(2)), 107.5 (MapaC(5)), 102.1 (MapaC(3)), 78.7 (Boc-C), 67.6 + 68.1 + 69.2 + 69.3 + 69.6 + 70.8 (TegC), 65.0 (Z-CH2), 60.5 (ethyl-CH2), 58.6 (AproC(α)), 57.3 (TegCH3), 51.4 (AproC(δ)), 48.9 (AproC(γ)), 35.2 (AproC(β)), 27.5 (Boc-CH3), 13.3 (ethyl-CH3) ppm; MS (MALDI-TOF) m/z (%) 675.5 (100%) [M + H]+, 697.5 (60) [M + Na]+; elemental analysis calcd (%) for C33H46N4O11: C 58.74, H 6.87, N 8.30; found C 58.65, H 6.93, N 8.04.
:10. The eluent composition was gradually changed until the pure product eluted (1,4-dioxane–H2O, 1:1). Analytically pure product was obtained by preparative HPLC. Yield 0.41 g (32%), colourless solid. m.p. 63–66 °C; [α]22D = −228.9 (c = 3, methanol); 1H NMR (600 MHz, 25 °C, [D4]MeOD–D2O, 1:1 (v/v): δ = 7.00–7.26 (m, 11H, MapaH(3)/MapaH(5)/PhH), 5.79–5.87 (m, 1H, AproH(α)), 5.63–5.72 (m, 2H, ProH(α)), 4.94 (d, 2J = 12.5 Hz, 1H, Z-CH2), 4.90 (d, 2J = 12.4 Hz, 1H, Z-CH2), 4.24–4.29 (m, 1H, AproH(γ)), 3.95–4.12 (m, 7H, TegCH2/AproH(δ)), 3.69–3.81 (m, 11H, TegCH2/ProH(δ)/AproH(δ)), 3.57–3.67 (m, 18H, TegCH2), 3.48–3.53 (m, 6H, TegCH2), 3.29 (s, 6H, TegCH3), 3.28 (s, 3H, TegCH3), 2.85–2.94 (m, 1H, AproH(β)), 2.58–2.67 (m, 2H, ProH(β)), 2.16–2.23 (m, 1H, AproH(β)), 2.02–2.12 (m, 2H, ProH(β)), 1.83–1.99 (m, 4H, ProH(γ)) ppm; 13C NMR (151 MHz, 25 °C, [D4]MeOD–D2O, 1:1 (v/v)): δ = 172.5 + 173.1 + 173.2 (Pro-CO/Apro-CO), 168.1 + 168.2 + 168.3 + 168.4 (MapaC(4)/Mapa-CO), 158.2 (Z-CO), 153.4 + 154.0 + 154.1 (MapaC(2)), 151.6 (MapaC(6)), 137.3 (PhC(1)), 129.4 (PhC(3)), 129.0 (PhC(4)), 128.4 (PhC(2)), 109.0 (MapaC(5)), 103.1 + 103.4 (MapaC(3)), 68.8 + 69.7 + 70.7 + 70.8 + 71.2 + 72.3 (TegC), 67.6 (Z-CH2), 63.4 + 63.5 (ProC(α)), 62.1 (AproC(α)), 59.0 (TegCH3), 53.9 (AproC(δ)), 50.0 (ProC(δ)), 49.1 (AproC(γ)), 38.6 (AproC(β)), 33.7 (ProC(β)), 23.4 (ProC(γ)) ppm; MS (ESI/TOF positive mode) m/z (%) 1287.4 (16%) [M + H]+, 1309.5 (100%) [M + Na]+, 1325.4 (50%) [M + K]+; MS (ESI/TOF negative mode) m/z (%) 1285.8 (50%) [M − H]−, 1321.9 (100%) [M + Cl]−; elemental analysis calcd (%) for C62H82N10O20·2H2O: C 56.27, H 6.55, N 10.58; found C 56.52, H 6.47, N 10.63.
:1 (v/v) (10 mL). After the addition of 1 N aqueous HCl (1.2 equiv., 0.31 mL) and 10% palladium–charcoal (25 mg) the mixture was stirred under a hydrogen atmosphere at atmospheric pressure until full conversion, typically for 18 h. Afterwards, the catalyst was filtered off through a pad of celite and washed thoroughly with methanol. The filtrate was evaporated to dryness and the residue was used without further purification.
The deprotected cyclopeptide was dissolved in degassed DMF (20 mL). 2,2′-(1,3-Phenylene)diacetic acid (25 mg, 0.13 mmol), TBTU (99 mg, 0.31 mmol), and DIEA (0.14 mL, 0.80 mmol) were added and the mixture was stirred at 25 °C for 2 h. After evaporation of the solvent in vacuo the residue was purified by preparative HPLC. Yield 0.15 g (47%), colourless solid; m.p. 100–105 °C; [α]22D = −211.1 (c = 1.5, methanol); 1H NMR (600 MHz, 25 °C, [D4]MeOD–D2O, 1:1 (v/v)): δ = 7.09–7.19 (m, 9H, MapaH(5)/PdaH(4)/PdaH(5)/PdaH(6)), 6.89–7.01 (m, 7H, MapaH(3)/PdaH(2)), 5.65–5.74 (m, 4H, ProH(α)), 5.56–5.62 (m, 2H, AproH(α)), 4.36–4.44 (m, 2H, AproH(γ)), 4.00–4.14 (m, 12H, TegCH2), 3.88–3.95 (m, 2H, AproH(δ)), 3.71–3.85 (m, 20H, TegCH2/ProH(δ)), 3.55–3.68 (m, 38H, TegCH2/AproH(δ)), 3.39–3.53 (m, 16H, TegCH2/Pda-CH2), 3.22–3.30 (m, 18H, TegCH3), 2.70–2.78 (m, 2H, AproH(β), 2.61–2.68 (m, 2H, ProH(β)), 2.51–2.59 (m, 2H, ProH(β)), 2.10–2.18 (m, 2H, AproH(β)), 2.00–2.09 (m, 4H, ProH(β)), 1.87–1.98 (m, 8H, ProH(γ)) ppm; 13C NMR (151 MHz, 25 °C, [D4]MeOD–D2O, 1:1 (v/v): δ = 172.4 + 173.0 + 174.7 (Pro-CO/Apro-CO), 168.1 + 168.2 + 168.5 + 168.6 (MapaC(4)/Mapa-CO), 153.0 + 154.0 (MapaC(2)), 151.5 + 151.7 (MapaC(6)), 136.5 (PdaC(1)/PdaC(3)), 129.9 (PdaC(2)), 128.6 (PdaC(5)), 128.5 (PdaC(4)/PdaC(6)), 108.8 + 108.9 + 109.0 (MapaC(5)), 102.9 + 103.2 + 103.3 + 103.4 + 103.5 (MapaC(3)), 68.8 + 69.7 + 70.7 + 70.8 + 71.2 + 72.3 (TegC), 63.4 + 63.5 (ProC(α)), 62.0 (AproC(α)), 59.0 (TegCH3), 53.7 (AproC(δ)), 50.0 + 50.1 (ProC(δ)), 47.7 + 47.8 (AproC(γ)), 43.2 (Pda-CH2), 37.9 (AproC(β)), 33.6 (ProC(β)), 23.5 (ProC(γ)) ppm; MS (ESI/TOF positive mode) m/z (%) 1244.0 (42%) [M + H + Na]2+, 1255.0 (100%) [M + 2Na]2+, 2464.9 (9%) [M + H]+, 2486.9 (72%) [M + Na]+, 2502.8 (7%) [M + K]+; elemental analysis calcd (%) for C118H158N20O38·5H2O: C 55.47, H 6.63, N 10.96; found C 55.47, H 6.55, N 10.91.
Qualitative NMR spectroscopic binding studies
For the measurements in 50% D2O–CD3OD, stock solutions of 2b in CD3OD (3.6 mM) and of NaI or Na2SO4 in D2O (7.2 mM) were prepared. After adding 250 μL of a salt solution to 250 μL of the receptor solution in an NMR tube, the tube was thoroughly shaken, and the 1H NMR spectrum was recorded (25 °C, 16 scans). For the measurements in 90% D2O–CD3OD, stock solutions of 2b in 20% CD3OD–D2O (2.0 mM) and of the salts in D2O (4.0 mM) were used. Equal volumes (500 μL) of the receptor solution and a salt solution were mixed in an NMR tube and the 1H NMR spectrum was recorded (25 °C, 128 scans). The spectra of pure receptor were obtained by mixing the respective stock solution of 2b with the appropriate volume of D2O.ITC titrations
The ITC titrations were carried out in water–methanol mixtures of varying compositions. Solvent mixtures were not degassed. The anionic substrates as their sodium salts (Na2SO4, NaI) and 2b were weighed using an analytical precision balance, dissolved in known volumes of the respective solvent mixture, and loaded into the system for immediate analysis. Solutions involved in the same titration experiment were made up from the same batch of solvent mixture. For the concentrations of 2b and the different salts used in the measurements, see ESI.†A standard ITC experiment involved the titration of a solution of the salt into a solution of the receptor at 25 °C using 30 injections of 8 μL, separated by an interval of 180 s, with the exception of the first injection, which was 2 μL. Binding constants and enthalpies of binding were obtained by curve fitting of the titration data using the one-site binding model. Data processing involved initial optimisation of the raw thermogram with NITPIC18a and subsequent non-linear regression of the binding isotherm with Sedphat.18b,c The peak produced by the first injection was discarded prior to data processing.
Acknowledgements
The support of this work by COST action CM1005 is kindly acknowledged. We also thank Prof. Dr S. Keller, Molecular Biophysics, Technische Universität Kaiserslautern for helpful discussions.Notes and references
- M. Rekharsky and Y. Inoue, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, Wiley-VCH, Chichester, 2012, vol. 1, pp. 117–134 Search PubMed.
- (a) D. B. Smithrud, E. M. Sanford, I. Chao, S. B. Ferguson, D. R. Carcanague, J. D. Evanseck, K. N. Houk and F. Diederich, Pure Appl. Chem., 1990, 62, 2227–2236 CrossRef CAS; (b) E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed; (c) F. Biedermann, M. Vendruscolo, O. A. Scherman, A. De Simone and W. M. Nau, J. Am. Chem. Soc., 2013, 135, 14879–14888 CrossRef CAS PubMed; (d) F. Biedermann, W. M. Nau and H.-J. Schneider, Angew. Chem., Int. Ed., 2014, 53 DOI:10.1002/anie.201310958.
- (a) W. Blokzijl and J. B. F. N. Engberts, Angew. Chem., Int. Ed. Engl., 1993, 32, 1545–1579 CrossRef; (b) D. Chandler, Nature, 2005, 437, 640–647 CrossRef CAS PubMed.
- E. Klein, Y. Ferrand, N. P. Barwell and A. P. Davis, Angew. Chem., Int. Ed., 2008, 47, 2693–2696 CrossRef CAS PubMed.
- A. Shokri and S. R. Kass, Chem. Commun., 2013, 49, 11674–11676 RSC.
- (a) A. Bianchi, K. Bowman-James and E. García-España, Supramolecular Chemistry of Anions, Wiley-VCH, New York, 1997 Search PubMed; (b) J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry, RSC, Cambridge, 2006 Search PubMed; (c) A. Bianchi, K. Bowman-James and E. García-España, Anion Coordination Chemistry, Wiley-VCH, Weinheim, 2012 Search PubMed.
- (a) B. R. Linton, M. S. Goodman, E. Fan, S. A. van Arman and A. D. Hamilton, J. Org. Chem., 2001, 66, 7313–7319 CrossRef CAS PubMed; (b) I. E. D. Vega, S. Camiolo, P. A. Gale, M. B. Hursthouse and M. E. Light, Chem. Commun., 2003, 1686–1687 RSC; (c) R. J. Fitzmaurice, F. Gaggini, N. Srinivasan and J. D. Kilburn, Org. Biomol. Chem., 2007, 5, 1706–1714 RSC; (d) C. Caltagirone, J. R. Hiscock, M. B. Hursthouse, M. E. Light and P. A. Gale, Chem. – Eur. J., 2008, 14, 10236–10243 CrossRef CAS PubMed; (e) P. A. Gale, J. R. Hiscock, C. Z. Jie, M. B. Hursthouse and M. E. Light, Chem. Sci., 2010, 1, 215–220 RSCV. J. Dungan, H. T. Ngo, P. G. Young and K. A. Jolliffe, Chem. Commun., 2013, 49, 264–266 RSC; (f) D. Curiel, G. Sánchez, C. Ramirez de Arellano, A. Tárraga and P. Molina, Org. Biomol. Chem., 2012, 10, 1896–1904 RSC; (g) G. Sanchez, A. Espinosa, D. Curiel, A. Tárraga and P. Molina, J. Org. Chem., 2013, 78, 9725–9737 CrossRef CAS PubMed; (h) R. B. P. Elmes, K. K. Y. Yuen and K. A. Jolliffe, Chem. – Eur. J., 2014, 20, 7373–7380 CrossRef CAS PubMed.
- (a) S. Kubik, R. Kirchner, D. Nolting and J. Seidel, J. Am. Chem. Soc., 2002, 124, 12752–12760 CrossRef CAS PubMed; (b) S. Otto and S. Kubik, J. Am. Chem. Soc., 2003, 125, 7804–7805 CrossRef CAS PubMed; (c) C. Reyheller, B. P. Hay and S. Kubik, New J. Chem., 2007, 31, 2095–2102 RSC; (d) C. Reyheller and S. Kubik, Org. Lett., 2007, 9, 5271–5274 CrossRef CAS PubMed; (e) Z. Rodriguez-Docampo, S. I. Pascu, S. Kubik and S. Otto, J. Am. Chem. Soc., 2006, 128, 11206–11210 CrossRef CAS PubMed; (f) Z. Rodriguez-Docampo, E. Eugenieva-Ilieva, C. Reyheller, A. Belenguer, S. Kubik and S. Otto, Chem. Commun., 2011, 47, 9798–9800 RSC.
- (a) S. Kubik, R. Goddard, R. Kirchner, D. Nolting and J. Seidel, Angew. Chem., Int. Ed., 2001, 40, 2648–2651 CrossRef CAS; (b) S. Kubik and R. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5127–5132 CrossRef CAS PubMed.
- (a) G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366–2393 CrossRef CAS PubMed; (b) M. T. Albelda, J. C. Frías, E. García-España and H.-J. Schneider, Chem. Soc. Rev., 2012, 41, 3859–3877 RSC; (c) C. Warwick, A. Guerreiro and A. Soares, Biosens. Bioelectron., 2013, 41, 1–11 CrossRef CAS PubMed.
- (a) A.-S. Chauvin, S. Comby, B. Song, C. D. B. Vandevyver and J.-C. Bünzli, Chem.– Eur. J., 2008, 14, 1726–1739 CrossRef CAS PubMed; (b) W. Q. Ong, H. Zhao, Z. Du, J. Z. Y. Yeh, C. Ren, L. Z. W. Tan, K. Zhang and H. Zeng, Chem. Commun., 2011, 47, 6416–6418 RSC.
- A. Fisher, A. Mann, V. Verma, N. Thomas, R. K. Mishra and R. L. Johnson, J. Med. Chem., 2006, 49, 307–317 CrossRef CAS PubMed.
- T. Fiehn, R. Goddard, R. W. Seidel and S. Kubik, Chem. – Eur. J., 2010, 16, 7241–7255 CrossRef CAS PubMed.
- F. P. Schmidtchen, in Analytical Methods in Supramolecular Chemistry, ed. C. A. Schalley, Wiley-VCH, Weinheim, 2007, pp. 55–78 Search PubMed.
- Y. Marcus, Ion Properties, Marcel Dekker, New York, 1997 Search PubMed.
- (a) G. Hefter, Y. Marcus and W. E. Waghorne, Chem. Rev., 2002, 102, 2773–2836 CrossRef CAS PubMed; (b) Y. Marcus, Chem. Rev., 2007, 107, 3880–3897 CrossRef CAS PubMed.
- (a) D. H. Williams, E. Stephens and M. Zhou, Chem. Commun., 2003, 1973–1976 RSC; (b) D. H. Williams and M. S. Westwell, Chem. Soc. Rev., 1998, 27, 57–63 RSC.
- (a) S. Keller, C. Vargas, H. Zhao, G. Piszczek, C. A. Brautigam and P. Schuck, Anal. Chem., 2012, 84, 5066–5073 CrossRef CAS PubMed; (b) J. C. D. Houtman, P. H. Brown, B. Bowden, H. Yamaguchi, E. Appella, L. E. Samelson and P. Schuck, Protein Sci., 2007, 16, 30–42 CrossRef CAS PubMed; (c) http://www.analyticalultracentrifugation.com/sedphat/download.htm .
- (a) J. Svec, M. Dusek, K. Fejfarova, P. Stacko, P. Klán, A. E. Kaifer, W. Li, E. Hudeckova and V. Sindelar, Chem. – Eur. J., 2011, 17, 5605–5612 CrossRef CAS PubMed; (b) M. Lisbjerg, B. M. Jessen, B. Rasmussen, B. E. Nielsen, A. Ø. Madsen and M. Pittelkow, Chem. Sci., 2014, 5, 2647–2650 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details for the chain elongation of dipeptides 4 and 5, NMR and mass spectra of compounds 4, 5, 6, and 2b, and thermograms of selected ITC titrations. See DOI: 10.1039/c4ob01497a |
| ‡ Additional contributions could come from effects of solvent composition on receptor preorganization, which are, however, difficult to assess, although the effects of solvent composition on the NMR spectra of 2b (Fig. 2) indicate that receptor conformation is affected by the solvent. |
| § The logKa of the iodide complex of bambus[6]uril in acetonitrile–water 1:1 amounts to 5.919a and that of biotin[6]uril in carbonate buffer at pH 10.8 to 3.3.19b |
| This journal is © The Royal Society of Chemistry 2014 |