
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conformational effects due to stereochemistry and C3-substituents in xylopyranoside derivatives as studied by NMR spectroscopy
Jerk
Rönnols
a,
Sophie
Manner
b,
Ulf
Ellervik
b and
Göran
Widmalm
*a
aDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, SE-106 91 Stockholm, Sweden. E-mail: gw@organ.su.se
bCentre for Analysis and Synthesis, Chemical Center, Lund University, PO Box 124, SE-221 00 Lund, Sweden
First published on 26th August 2014
Abstract
Glycosaminoglycans contain a β-D-xylopyranose residue at its reducing end, which links the polysaccharide to the protein in proteoglycans. 2-Naphthyl β-D-xylopyranosides have shown inhibition of tumor growth and we herein investigate conformation and dynamics of compounds structurally and stereochemically modified at the C3 position as well as the influence of solvent. The 3-deoxygenated compound, the 3-C-methyl-substituted β-D-xylopyranoside, β-D-ribopyranoside, the 3-C-methyl-substituted β-D-ribopyranoside as well as 2-naphthyl β-D-xylopyranoside were analyzed by NMR spectroscopy. Conformational equilibria were dependent on the solvent of choice, either methanol-d4 or chloroform-d, with mainly 4C1 and 1C4 conformations present but also skew conformations to some extent. Intramolecular hydrogen bonding was concluded to be important for the 3-C-methyl-substituted β-D-xylopyranosides in the non-polar solvent. Dynamic NMR (DNMR) spectroscopy was carried out for the 3-deoxygenated compound, which at 25 °C in methanol-d4 exists with equally populated states of the 4C1 and the 1C4 conformations, but at −100 °C only a few percent is present of the latter. Using 13C NMR detection for DNMR, resonance lines were shown to broaden at −40 °C and to sharpen again below −90 °C, without the emergence of a second set of NMR resonances, a typical behavior for an unequally populated equilibrium. The enthalpy and entropy activation barriers were calculated and resulted in ΔH‡ = 47.3 kJ mol−1 and ΔS‡ = 54 J mol−1 K−1.
Introduction
The three-dimensional structure of carbohydrates and their interactions with proteins1 play important roles in regulation of biochemical processes. One class of highly complex carbohydrates is glycosaminoglycans (GAGs) being constituents of proteoglycans.2 Xylopyranosides, bearing a hydrophobic aglycone, can act as an acceptor in the biosynthesis of GAGs in cells, with different results depending on the aglycone, cell-type, and the substitution pattern of the xyloside.3 We have previously reported on the conformational properties of XylNap (1) (Fig. 1), with a 2-naphthyl aglycone, in methanol-d4 (ref. 3) and a wide range of other deuterated solvents,4 where it displayed a conformational dependence on the polarity and hydrogen bond accepting ability of the solvents. Solvents of low polarity and low hydrogen bond accepting ability induced conformational transformations from the otherwise preferred 4C1 conformation to the 2SO and 1C4 conformations, with a maximum of ∼20% of the 1C4 conformation in chloroform, benzene, and toluene. An intramolecular hydrogen bond was detected with the HO2 hydroxyl proton as the donor and an electronegative substituent (F or OH) on C4 as an acceptor; this interaction was suggested to stabilize the 1C4 conformation. | ||
| Fig. 1 Schematic of compounds 1–5. | ||
It was observed that the 3-deoxy-compound (2) resides in equal amounts of the 4C1 and 1C4 conformations in methanol solution at 37 °C, in stark contrast to the other substances in the study.3 In a subsequent study of the 3-C-methylated compounds 3 and 4 in methanol-d4 solution, an increased conformational flexibility was observed, compared to 1.5 Compound 3 was the most flexible of the two, due to the larger 1,3-diaxial interactions arising from the methyl group in the 4C1 conformation of 3 compared to that of the hydroxyl group in 4. Herein, we elaborate further on the effects of the stereochemistry and substituents at C3 on the conformation of xylopyranoside derivatives (Fig. 1).
Results and discussion
1H NMR spectra of compounds 1–5 in chloroform-d solution at 37 °C were recorded and resonances assigned; NMR assignments were also carried out for compound 5 in methanol-d4 solution. Scalar couplings were extracted through NMR spin simulation6 and 13C NMR data were also acquired for some of the compounds (Table 1). The resulting coupling constants were fitted to coupling constants calculated from the Haasnoot–Altona equation7 and molecular models of the canonical ring conformations 4C1, 1C4, 1S3, 1S5, OS2, 3S1, 5S1 and 2SO in accordance with the previously devised methodology.3,4 The resulting equilibrium populations and those previously reported for compounds 1–4 in methanol-d4 (ref. 2 and 4) and of 1 in chloroform-d solution2 are listed in Table 2. In chloroform-d solution, compounds 2 and 3 were observed to occupy the 1C4 conformation to a large extent, ∼90%, supported by the long-range 4JH2,H4 coupling in the latter, whereas the 4C1 and 1C4 conformations are close to equally populated for compound 4. The intermediate 3JH1,H2 coupling for compound 5 in methanol-d4 solution suggests a large population of the 1C4 conformation, >50%, and in the non-polar chloroform-d solution, 3JH1,H2 = 2.2 Hz is consistent with a large population of the 1C4 conformation, ∼90%.| Compound | 1 | 2 | 3 | 4 | 5 | Me | HO2 | HO3 | HO4 |
|---|---|---|---|---|---|---|---|---|---|
| a In methanol-d4. b 4 J H2,H4. | |||||||||
| 1 | 5.109 | 3.709 | 3.675 | 3.809 | 4.112, 3.468 | 2.704 | 2.768 | 2.266 | |
| (6.224) | (7.786) | (7.634) | (4.501, 8.268) | (−11.880) | (4.198) | (3.736) | (3.447) | ||
| [100.91] | [74.64] | [74.56] | [69.42] | [64.54] | |||||
| 2 | 5.513 | 3.888 | 2.050, 2.267 | 3.940 | 3.988, 3.635 | 3.461 | 2.452 | ||
| (2.050) | (3.092, 3.556) | (3.570, 3.156, −14.733) | (1.575, 2.125) | (−12.427) | (8.279) | (4.963) | |||
| [98.07] | [66.92] | [30.82] | [65.77] | [65.38] | |||||
| 3 | 5.590 | 3.625 | 3.608 | 4.265, 3.641 | 1.438 | 3.172 | 3.631 | 2.282 | |
| (2.559) | (1.441)b | (1.937, 3.117) | (−12.753) | (8.678) | (5.091) | ||||
| 4 | 5.555 | 3.666 | 3.632 | 4.037, 3.814 | 1.531 | n.d. | n.d. | n.d. | |
| (4.443) | (3.268, 6.124) | (−11.956) | |||||||
| 5 | 5.759 | 4.067 | 4.134 | 4.012 | 4.012, 3.905 | 3.433 | 3.159 | 3.095 | |
| (2.185) | (1.295) | (3.009) | (1.655, 2.624) | (−12.527) | (4.544) | (3.826) | (3.815) | ||
| 5 | 5.592 | 3.860 | 4.074 | 3.874 | 3.900, 3.815 | ||||
| (3.881) | (3.197) | (3.184) | (2.662, 4.825) | (−11.852) | |||||
| [100.65] | [72.37] | [68.26] | [70.46] | [65.57] | |||||
Compounds 2–4 were also investigated by 1D 1H, 1H-NOESY experiments.5,8 In methanol-d4 solution, NOE correlations were observed for compound 2 from H1 to H3pro-R and H5pro-S (Fig. 2a), which are significant in the 4C1 conformation (α-face of the ring), and between H3pro-S and H5pro-R (Fig. 2b), which is significant only in the 1C4 conformation (β-face of the ring), consistent with approximately equally populated 4C1 and 1C4 conformations. In chloroform-d solution, correlations between H1 and H3pro-R and H5pro-S were not observed, while a strong NOE correlation between H3pro-S and H5pro-R was present, thus verifying the strong prevalence for the 1C4 conformation in the non-polar solvent. NOE experiments were pursued for compounds 3 and 4 in chloroform-d solution, with selective excitation of the respective methyl groups. Strong NOEs were detected for compound 3 from the methyl group to the H2 and H4 atoms (Fig. 2c), indicating a preference for the 1C4 conformation. In methanol-d4 solution, where the 4C1 conformation is highly favored, strong inter-nuclear correlations were observed from the methyl group to the H1 and H5pro-S atoms5 (Fig. 2d), none of which were observed in chloroform-d solution, further underlining the conformational dependence on solvent properties. In compound 4, NOE correlations from the methyl group to the H2 and H4 atoms (Fig. 2e) were observed both in methanol-d4 solution5 and chloroform-d solution, consistent with a 4C1 conformation. In chloroform-d, an NOE correlation to the H5pro-R resonance was also observed, which indicates a large population of the 1C4 conformation (Fig. 2f).
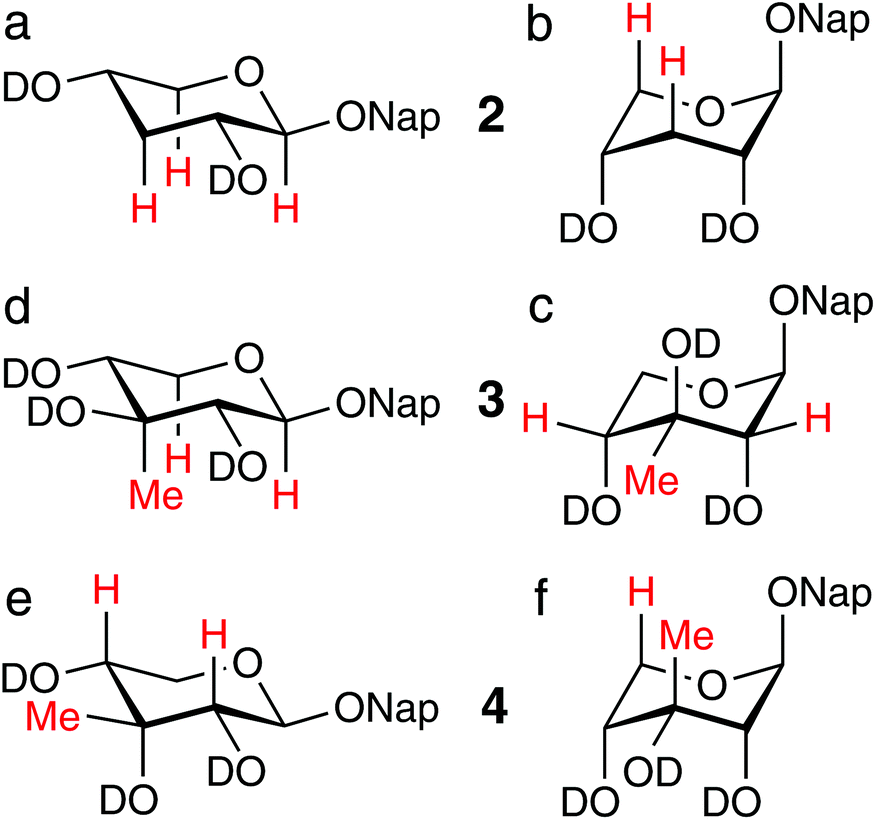 | ||
| Fig. 2 1H,1H-NOE correlations between atoms (colored red), observed from selective excitations of, inter alia, H1 or CH3. Compound 2 in 4C1 conformation (a) and in 1C4 conformation (b); 3 in 1C4 (c) and in 4C1 (d); 4 in 4C1 (e) and in 1C4 (f). | ||
Complementary to the NOE-based information for compound 3 in chloroform-d solution, chemical shift information and scalar coupling data are also illustrative. The 1H NMR chemical shift for the hydroxyl proton HO3 is unusually high, δH 3.63, which may be compared to that of the 4-fluoro-4-deoxy-β-D-Xylp-Nap derivative, having δH 2.92 being present to 26% in the 1C4 conformation.5 In this compound, an F4⋯HO2 hydrogen bond was proven by JF4,HO2 = 2 Hz; additionally, the 3JH2,HO2 = 6.1 Hz. In compound 3, the latter coupling constant was also large being 3JH2,HO2 = 8.7 Hz, indicating hindered rotation. In order to obtain information on the conformational preference (if any) of the hydroxyl proton HO3, we rely on the heteronuclear 1H,13C spin–spin coupling constant to the methyl group on C3, since the compound is devoid of a hydrogen atom in this position. Employing a J-HMBC experiment9 and detecting the interaction between the methyl 13C group and the HO3 group, we obtained 3JC(Me),HO3 = 6.5 Hz, consistent with an antiperiplanar arrangement (cf. eqn (10) in ref. 10). Taken together, these results support that the 1C4 conformation for compound 3 in chloroform-d solution is stabilized by the intramolecular hydrogen bonds O4⋯HO2 and O1⋯HO3.
To elaborate on the dynamics of the 3-deoxygenated compound (2), 1H NMR spectra were recorded at a 1H resonance frequency of 500 MHz in a temperature range from 50 °C to −68 °C in methanol-d4 solution. The equilibrium populations were altered significantly with temperature, observed through changes in e.g. the 3JH1,H2 and the 3JH4,H5pro-S coupling constants (Fig. 3). At temperatures below ∼25 °C, the 4C1 conformation is predominantly occupied, reflected by increased values of the 3JH1,H2 coupling constant (Table 3). At temperatures below −68 °C, where the population of the 4C1 conformation was >80%, some of the signals, e.g. H1 and H3pro-R, were significantly broadened, thus disabling coupling constant analysis. The δH1 and JH1,H2 for the separate conformers could, however, be extrapolated to give for the 4C1 conformation δH1 5.02 and JH1,H2 = 7.64 Hz and for the 1C4 conformation δH1 5.50 and JH1,H2 = 1.56 Hz.
 | ||
| Fig. 3 The 1H NMR resonance at 500 MHz of H5pro-S of compound 2 in methanol-d4 solution as a function of temperature. | ||
| Temp/°C | 4 C 1/% | 1 C 4 | δ H1/ppm | J H1,H2/Hz | k ex/s−1 |
|---|---|---|---|---|---|
| 50 | 46 | 54 | 5.279 | 4.416 |
100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) 000000 000000
|
| 39 | 48 | 52 | 5.271 | 4.513 |
52000000
|
| 23 | 51 | 49 | 5.255 | 4.717 |
19000000
|
| −4 | 59 | 41 | 5.225 | 5.015 |
2500000
|
| −26 | 65 | 35 | 5.193 | 5.452 |
340000
|
| −47 | 73 | 27 | 5.158 | 5.850 |
37000
|
| −66 | 80 | 20 | 5.13 | 6.42 | 2800 |
| −68 | 80 | 20 | 5.127 | 6.580 | 2500 |
| −76 | 85 | 15 | 5.10 | 6.73 | 840 |
| −86 | 90 | 10 | 5.04 | 7.03 | 180 |
| −95 | 92 | 8 | 5.02 | 7.15 | 44 |
| −101 | 93 | 7 | 5.02 | 7.21 | 9.7 |
At this point, we turned to a higher magnetic field strength of 18.8 T, which enabled extension of the above dynamic NMR (DNMR) measurements.11 Using 13C NMR spectra at 200 MHz spectrometer frequency, we observed a broadening of the resonances corresponding to C1, C3, and C5 at −40 °C that were observed to coalesce at −60 °C (Fig. 4). Well defined sharp peaks were observed below −80 °C, corresponding to the 4C1 conformer, while resonances from the less populated conformer were absent even after prolonged experimental time; a typical behavior for an unequally populated equilibrium.12,13 The two H3 resonances of 2 were unequally broadened in 1H NMR spectra below −60 °C at 800 MHz spectrometer frequency. The H3pro-R resonance (axially oriented in the 4C1 conformation) was significantly broadened and eventually split up at −95 °C, yielding a major and a minor signal (∼5%) at −101 °C (Fig. 5). The conformational exchange rates kex = k1 + k−1, where k1 and k−1 are the forward and reverse rate constants for the 4C1 ⇌ 1C4 conformational equilibrium, respectively, were subsequently investigated by NMR line-shape analysis of the H3 resonances, with emphasis on exchange rates and population distribution, and yielded the data reported in Table 3. The enthalpy and entropy activation barriers were calculated from an Eyring plot14 resulting in ΔH‡ = 47.3 kJ mol−1 and ΔS‡ = 54 J mol−1 K−1. The free energy of activation barrier ΔG‡ can subsequently be calculated at a given temperature, under the assumption that ΔH‡ and ΔS‡ were not temperature dependent. For example, at −70 °C one obtains ΔG‡ = 36.3 kJ mol−1, which can be compared to protected O-methyl glycosides of α-D-mannuronic acid esters in CD2Cl2 at the same temperature showing ΔG‡ ≈ 46 kJ mol−1 for the 4C1 ⇌ 1C4 ring interconversion.15 Thus, the free energy of activation barrier ΔG‡ for the 3-deoxygenated compound (2), is indeed low, but can be determined by DNMR spectroscopy.
 | ||
| Fig. 4 The 13C NMR resonances at 200 MHz of compound 2 in methanol-d4 solution as a function of temperature. | ||
 | ||
| Fig. 5 The 1H NMR resonances at 800 MHz of H3pro-R (∼1.6 ppm) and H3pro-S (∼2.4 ppm) of compound 2 in methanol-d4 solution as a function of temperature and the corresponding simulated spectra. Note the small peak at ∼2.0 ppm in the spectrum obtained at the lowest temperature. | ||
Complementary spectroscopic techniques include optical rotation, which facilitates additional information about conformational preferences. The technique can be used to differentiate anomeric configuration between methyl glycosides and has been applied in conformational studies of oligosaccharides.16,17 Additionally, it has been suggested that the extent of a particular chair conformation can be deduced in this way.18 For compound 1 in methanol the specific rotation [α]20D = −30.04° corresponding to a molecular rotation [M]20D = −83.0°, and is similar to that of the corresponding methyl xyloside in water, [M]20D = −108°, but numerically quite different to those of other methyl pentopyranosides.18 For β-D-Xylp-OMe in water the calculated molecular rotation for the 1C4 chair conformation has a value that is more negative than that corresponding to the 4C1 conformation. In methanol solution compound 3 has [α]20D = −44° and compound 4 has [α]20D = −18°,5 corresponding to [M]20D = −128°, and [M]20D = −52°, respectively. A direct comparison to compound 1, with conformational preferences determined by NMR spectroscopy (Table 2), is difficult due to the additional 3-C-methyl group in 3 and 4. However, the significantly more negative value of 3 is consistent with the presence of a larger proportion of a 1C4 conformation, harmonizing with the NMR-based results (Table 2), where the extent of the 4C1 conformation is 70% in 3 compared to 90% in 4. It is anticipated that these types of changes in molecular rotation will be able to report on alterations of conformational equilibria and we foresee that NMR studies in the future may be complemented by determination of the molecular rotation.
We conclude by noting that, in comparison to the 2-naphthyl β-D-xylopyranoside, stereochemical and substituent modifications at C3 lead to large changes in flexibility and/or conformational equilibria being dependent on the solvent. Notably, the flexibility is increased if the hydroxyl group at C3 is removed, leading to lower steric interactions, or if a C-methyl substituent is added, leading to higher steric interactions, or if the stereochemistry is inverted at the C3 position. The conformational equilibrium of the 3-deoxygenated compound was found to be highly temperature dependent with an almost exclusive 4C1 conformation present at −100 °C and only a few percent of the 1C4 conformation compared to equally populated states at 25 °C. Thus, the enthalpy-favored 4C1 conformer of compound 2 becomes evident at low temperature and only at high temperature is the 1C4 conformer favored in methanol solution as a result of significant entropic contributions to the conformational state. The importance of sugar puckering19 and the implications of the herein obtained results for glycosyl transferase activity and antiproliferative properties of these compounds is a challenging area for future research where progress recently has been made.5
Experimental section
Experimental procedures and analysis protocols were the same as previously described.3–5,15 NMR experiments were performed on a Bruker AVANCE II 500 MHz and AVANCE III 600 MHz spectrometers and a Varian Inova 800 MHz spectrometer. 1H and 13C chemical shifts are reported in ppm using residual solvent signals as references, viz., in methanol-d4: δH 3.31 and δC 49.0; in chloroform-d: δH 7.26 and δC 77.16. Two J-HMBC experiments were performed as described9,20 using a scaling factor κ of 18.1 and 21.4. For compound 1 the specific rotation was measured on five separate samples with a PerkinElmer 341 polarimeter at 589 nm and 20 °C in a quartz cuvette of 10 cm length; SD 0.46, c 1.01, MeOH.Acknowledgements
This work was supported by grants from the Swedish Research Council, The Knut and Alice Wallenberg Foundation, and the Royal Physiographic Society in Lund. The Swedish NMR Centre at Göteborg University is thanked for access to a high field spectrometer.References
- G. Widmalm, Carbohydr. Res., 2013, 378, 123–132 CrossRef CAS PubMed.
- N. S. Gandhi and R. L. Mancera, Chem. Biol. Drug Des., 2008, 72, 455–482 CAS.
- A. Siegbahn, U. Aili, A. Ochocinska, M. Olofsson, J. Rönnols, K. Mani, G. Widmalm and U. Ellervik, Bioorg. Med. Chem., 2011, 19, 4114–4126 CrossRef CAS PubMed , and references therein.
- J. Rönnols, S. Manner, A. Siegbahn, U. Ellervik and G. Widmalm, Org. Biomol. Chem., 2013, 11, 5465–5472 Search PubMed.
- A. Siegbahn, S. Manner, A. Persson, E. Tykesson, K. Holmqvist, A. Ochocinska, J. Rönnols, A. Sundin, K. Mani, G. Westergren-Thorsson, G. Widmalm and U. Ellervik, Chem. Sci., 2014, 5, 3501–3508 RSC.
- R. Laatikainen, M. Niemitz, U. Weber, J. Sundelin, T. Hassinen and J. Vepsäläinen, J. Magn. Reson., Ser. A, 1996, 120, 1–10 CrossRef CAS.
- C. A. G. Haasnoot, F. A. A. M. De Leeuw and C. Altona, Tetrahedron, 1980, 36, 2783–2792 CrossRef CAS.
- K. Stott, J. Keeler, Q. N. Van and A. J. Shaka, J. Magn. Reson., 1997, 125, 302–324 CrossRef CAS.
- A. Meissner and O. W. Sørensen, Magn. Reson. Chem., 2001, 39, 49–52 CrossRef CAS.
- H. Zhao, Q. Pan, W. Zhang, I. Carmichael and A. S. Serianni, J. Org. Chem., 2007, 72, 7071–7082 CrossRef CAS PubMed.
- J. Sandström, Dynamic NMR Spectroscopy, Academic Press, New York, 1982 Search PubMed.
- F. A. L. Anet, I. Yavari, I. J. Ferguson, A. R. Katritzky, M. Moreno-Mañas and M. J. T. Robinson, J. Chem. Soc., Chem. Commun., 1976, 399–400 RSC.
- N. Okazawa and T. S. Sorensen, Can. J. Chem., 1978, 56, 2737–2742 CrossRef CAS.
- K. D. Zimmer, R. Shoemaker and R. R. Ruminski, Inorg. Chim. Acta, 2006, 359, 1478–1484 CrossRef CAS PubMed.
- J. Rönnols, M. T. C. Walvoort, G. A. van der Marel, J. D. C. Codée and G. Widmalm, Org. Biomol. Chem., 2013, 11, 8127–8134 Search PubMed.
- C. A. Duda and E. S. Stevens, J. Am. Chem. Soc., 1990, 112, 7406–7407 CrossRef CAS.
- E. Säwén, T. Massad, C. Landersjö, P. Damberg and G. Widmalm, Org. Biomol. Chem., 2010, 8, 3684–3695 Search PubMed.
- J. F. Stoddart, Stereochemistry of Carbohydrates, John Wiley & Sons, Inc., New York, 1971, pp. 147–155 Search PubMed.
- H. B. Mayes, L. J. Broadbelt and G. T. Beckham, J. Am. Chem. Soc., 2014, 136, 1008–1022 CrossRef CAS PubMed.
- K. H. M. Jonsson, R. Pendrill and G. Widmalm, Magn. Reson. Chem., 2011, 49, 117–124 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |