
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrustrated Lewis pair catalyzed hydrosilylation and hydrosilane mediated hydrogenation of fulvenes†
Sergej
Tamke
a,
Constantin-G.
Daniliuc
b and
Jan
Paradies
*a
aKarlsruhe Institute of Technology, Institute of Organic Chemistry, Fritz-Haber-Weg 6, 76131, Karlsruhe, Germany. E-mail: jan.paradies@kit.edu; Fax: +49 721 608 45637; Tel: +49 721 608 45344
bWestfälische Wilhelms-Universität Münster, Organisch-Chemisches Institut, Corrensstr. 40, 48149, Münster, Germany. E-mail: constantin.daniliuc@uni-muenster.de; Fax: +49 251 83-39772; Tel: +49 251 83-33293
First published on 12th September 2014
Abstract
The frustrated Lewis pair (FLP) mediated hydrosilylation of pentafulvenes is described yielding allyl silanes with high regioselectivity in excellent yields. While phenyl substituted allyl silanes undergo B(C6F5)3-mediated rearrangement to vinyl silanes, dimethyl derivatives experience FLP-catalyzed hydrogenation followed by an unprecedented protodesilylation. This observation allowed the metal-free hydrogenation of 6,6-dimethylfulvene to iso-propyl cyclopentene according to a FLP-catalyzed triple domino reaction consisting of hydrosilylation, hydrogenation and protodesilylation. The mechanisms were investigated by deuteration experiments.
Introduction
Hydrogenations and hydrosilylations are two of the most applied processes in industrial chemistry.1 Although both transformations are efficiently catalyzed by transition metal complexes,2 research in the field of metal-free alternatives has flourished in the past few decades. Piers and co-workers demonstrated that the strong Lewis acid B(C6F5)3 (1) is a powerful hydrosilylation catalyst of a number of substrates including aldehydes, ketones, imines and enones.3 Also, less polarized substrates such as olefins were susceptible to borane and phosphonium-mediated hydrosilylation.4 We have reported earlier5 that the B(C6F5)3-catalyzed 1,4-hydrosilylation of enones3c can be combined with frustrated Lewis pair (FLP) catalyzed hydrogenations6,7 of the in situ generated silylenol ethers.8 Also, allyl silanes were susceptible to FLP-catalyzed hydrogenation,7d,9 providing access to saturated silanes. We sought to expand this methodology to compounds bearing two or more non-polar double bonds. Particularly pentafulvenes are highly reactive, electron-rich compounds which readily undergo cycloaddition reactions10 to construct complex molecules.10a,d In particular, this reactivity obstructs the use of pentafulvenes as cyclopentyl building blocks for natural products or fragrant syntheses.11 Erker has shown that FLPs can influence the fulvene-reactivity to yield unusual [6 + 4] cycloaddition products.12Herein we report the FLP-catalyzed hydrosilylation and subsequent hydrogenation of pentafulvenes to access silylated cyclopentene-derivatives according to a domino reaction sequence.13
Results and discussion
Hydrosilylation of fulvenes
We initiated our investigations by reacting 6,6-dimethylfulvene (2) with 1 equiv. of methyl(diphenyl) silane (3) in the presence of 10 mol% of the Lewis-acid B(C6F5)3 (1) (Table 1, entry 1).|
|
|||
|---|---|---|---|
| Entry | Lewis base | Time (h) | Yield (%) |
| a Reactions performed on a 0.1 mmol scale in CD2Cl2 (0.5 mL, 0.2 M) at r.t. using 1 equiv. of hydrosilane 3 and 10 mol% of B(C6F5)3 (1) and the corresponding Lewis base. Yields were determined by 1H NMR spectroscopy using the residual solvent signal as an internal standard. b Significant amounts of the oligomerization product were observed. c No hydrosilylation was observed even at 70 °C. d Isolated yield. | |||
| 1b | — | — | — |
| 2 | P(1-naphth)3 (4) | 1 | >95 |
| 3c | P(t-Bu)3 (6) | 78 | 0 |
| 4 | P(Mes)3 (7) | 24 | >95 (79%)d |
| 5 | P(C6F5)Ph2 (8) | 0.5 | >95 |
| 6 | PhNMe2 (9) | 24 | >95 |
| 7 | (1-naphth)NMe2 (10) | 1 | >95 |
| 8b | (p-tol)2NMe (11) | 1 | <5 |
Instantaneous conversion of the starting material was observed, yielding a mixture of hydrosilylation and oligomerization products. Remarkably, employing 10 mol% of the FLP consisting of P(1-naphth)3 (4) and B(C6F5)3 (1) (Table 1, entry 2) as the catalyst provided exclusively the hydrosilylation product 5 as a single regioisomer after 1 h in quantitative yield. The hydrosilylation product 5 was unambiguously characterized by NMR spectroscopy and X-ray crystallography14 as the addition product to the C2–C3 position (Fig. 1). Alternative products arising from FLP mediated cycloaddition12 were not detected throughout our study. Evidently the choice of the Lewis base has a dramatic impact on the outcome of the reaction, and we expanded our investigation to other phosphines and amines (Table 1, entries 3–8). Indeed, the nature of the employed Lewis base has a significant influence on the reaction rate. In the presence of P(t-Bu)3 (6)/1 the fulvene 2 was not consumed even after prolonged reaction time (78 h, entry 3), most likely due to the formation of the stable silylium salt [(t-Bu)3P-SiPh2Me][HB(C6F5)3].15 Phosphines or amines comprising less basic heteroatomic sites are viable Lewis bases for the FLP-catalyzed hydrosilylation of 2 providing the product 5 in quantitative yield. The FLP consisting of PMes3 (7)/(1) required 24 h for the quantitative hydrosilylation of 2 whereas the corresponding P(C6F5)Ph2 (8)/(1) needed only 0.5 h for completion. The electronic modification of the aniline derivatives exhibited even more drastic effects on the reaction rates (Table 1, entries 6–8). While N,N-dimethylaniline (9) as a Lewis base component gave 5 in 24 h (entry 6, >95%), the slightly less basic N,N-dimethyl-1-naphthylamine (10) provided the product in quantitative yield in 1 hour (Table 1, entry 7). In contrast, the FLP derived from N,N-di(p-toloyl)methylamine (11) and B(C6F5)3 (1) furnished only traces of the hydrosilylation product 5 accompanied by significant amounts of oligomerization products (Table 1, entry 8). From these observations some tentative conclusions can be drawn.
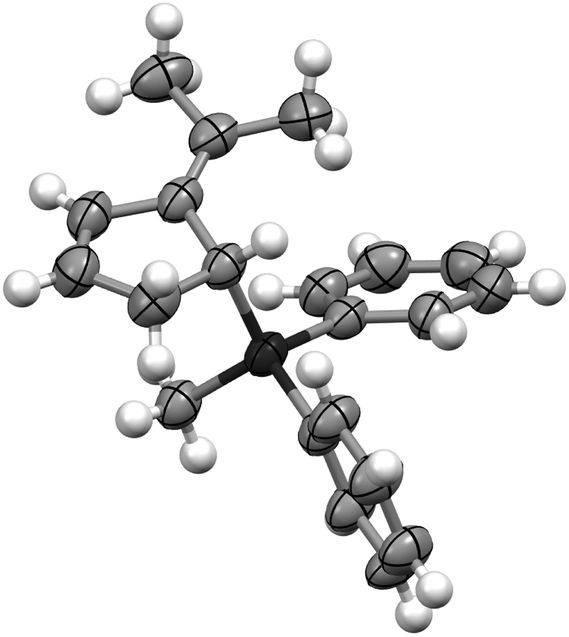 | ||
| Fig. 1 Crystal structure of allyl silane 5. | ||
The dependence of the reaction rates may be attributed to the formation of the encounter complex.16 Precoordination of borane 1 by the corresponding Lewis base significantly decelerates the undesired oligomerization, thus favoring the hydrosilane activation.
In order to investigate the substrate scope of the reaction, different pentafulvenes were reacted with hydrosilane 3 in the presence of 10 mol% 4/1 at room temperature. The aryl substituted pentafulvenes, 6,6-diphenylfulvene (12) and 6-methyl-6-phenylfulvene (13), readily underwent regioselective 1,2-hydrosilylation in 98% and 93% yield for 14 and 15 respectively with remarkable C2 regioselectivity (Scheme 1). The hydrosilylation of the unsymmetric fulvene 13 provided a mixture of (E)-15 and (Z)-15 in a 1.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Scheme 1b). As a fourth substrate we investigated the reactivity of 6,6-dimethylbenzofulvene (16) in the FLP-catalyzed hydrosilylation with 3 (10 mol% 4/1). In accord with the previous examples the hydrosilylation was complete in 24 h in excellent yield (90%, Scheme 2). However, the reaction proceeds with low regioselectivity and a mixture of regioisomers (17:18; 1.2:1 ratio) was obtained.
:1 ratio (Scheme 1b). As a fourth substrate we investigated the reactivity of 6,6-dimethylbenzofulvene (16) in the FLP-catalyzed hydrosilylation with 3 (10 mol% 4/1). In accord with the previous examples the hydrosilylation was complete in 24 h in excellent yield (90%, Scheme 2). However, the reaction proceeds with low regioselectivity and a mixture of regioisomers (17:18; 1.2:1 ratio) was obtained.
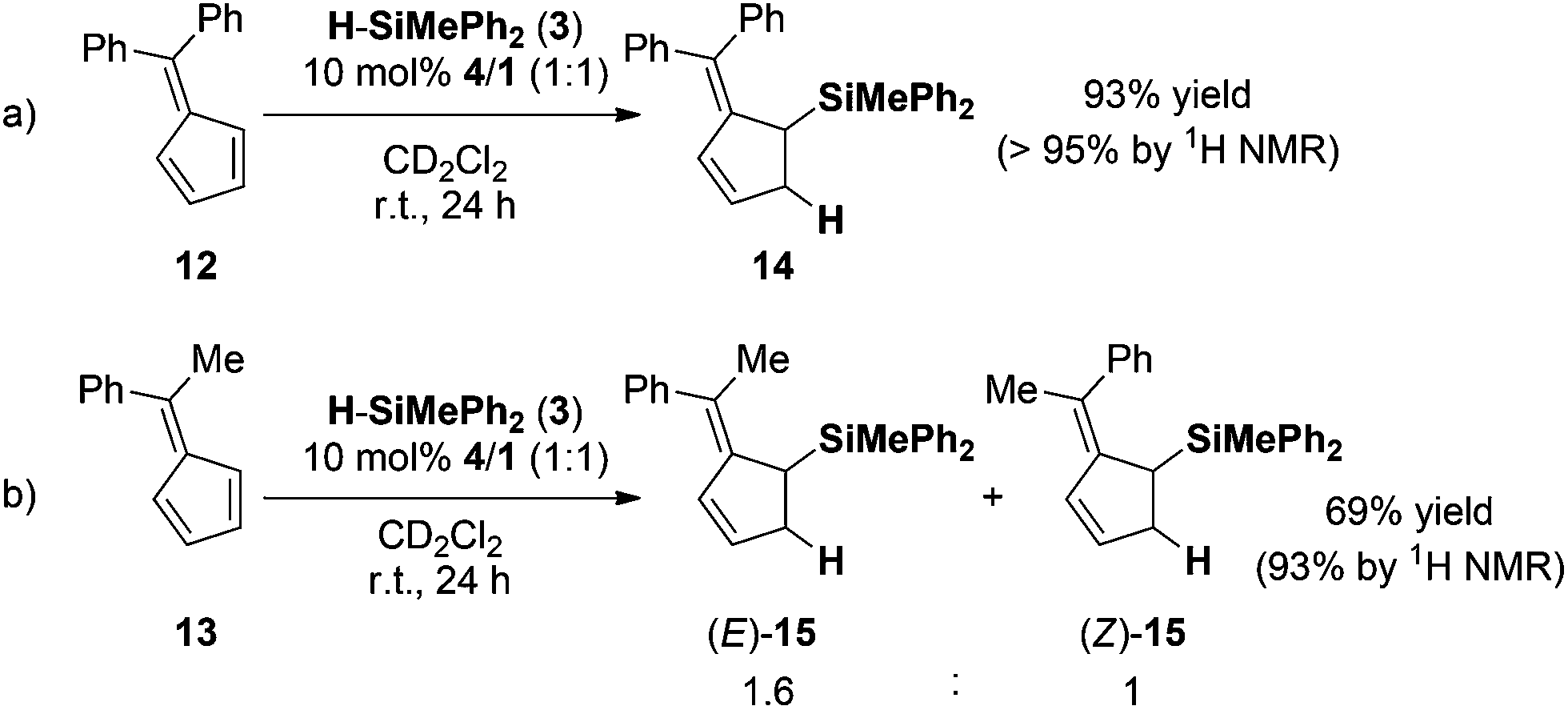 | ||
| Scheme 1 Hydrosilylation of (a) 6,6-diphenylfulvene (12) and (b) 6-phenyl-6-methylfulvene (13). | ||
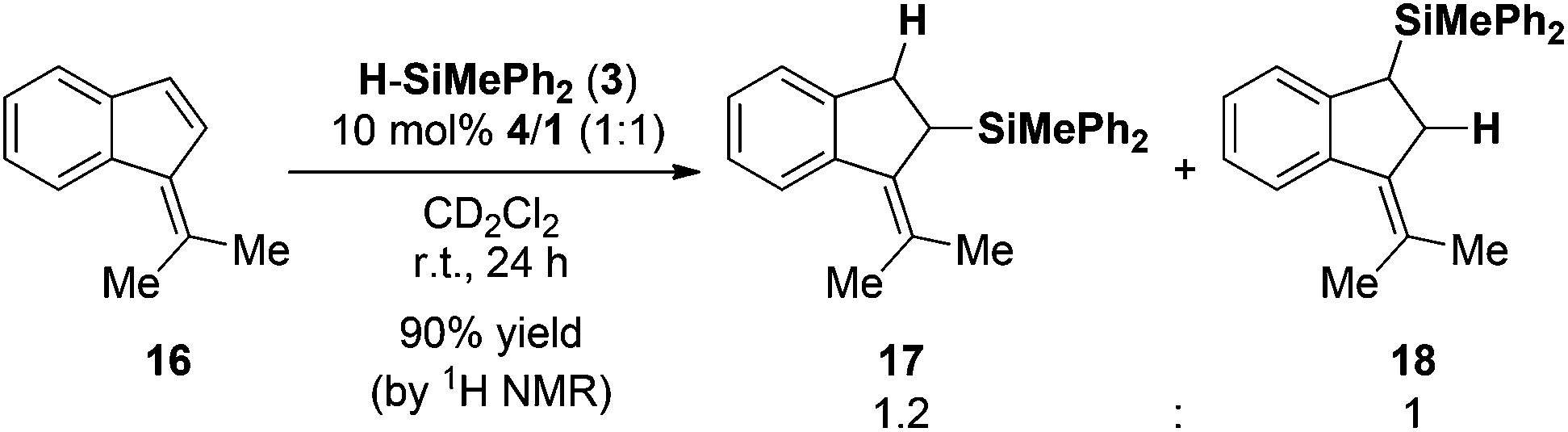 | ||
| Scheme 2 Hydrosilylation of dimethylbenzofulvene (the yield was determined by 1H NMR spectroscopy using the residual solvent signal as an internal standard). | ||
Catalytic hydrogenation
The catalytic hydrosilylation of 6,6-disubstituted fulvenes was achieved using FLPs derived from P(1-naphth)3 (4), P(C6F5)Ph2 (8) and (p-tol)2NMe (11) and B(C6F5)3 (1) as Lewis acids. Our group has shown earlier that the same FLPs were active catalysts for the metal-free hydrogenation of allyl silanes7d,9 giving rise to a hydosilylation/hydrogenation domino-reaction sequence.5a Consequently we first investigated the reduction of the prepared allylic silanes using H2 and 4/1 as catalysts. Indeed, when 5 was subjected to H2-atmosphere in the presence of 4/1 the corresponding cyclopentenylsilane 19 together with small amounts of 1-iso-propylcyclopent-1-ene (20) and hydrosilane 3 as by-products were observed (Scheme 3a).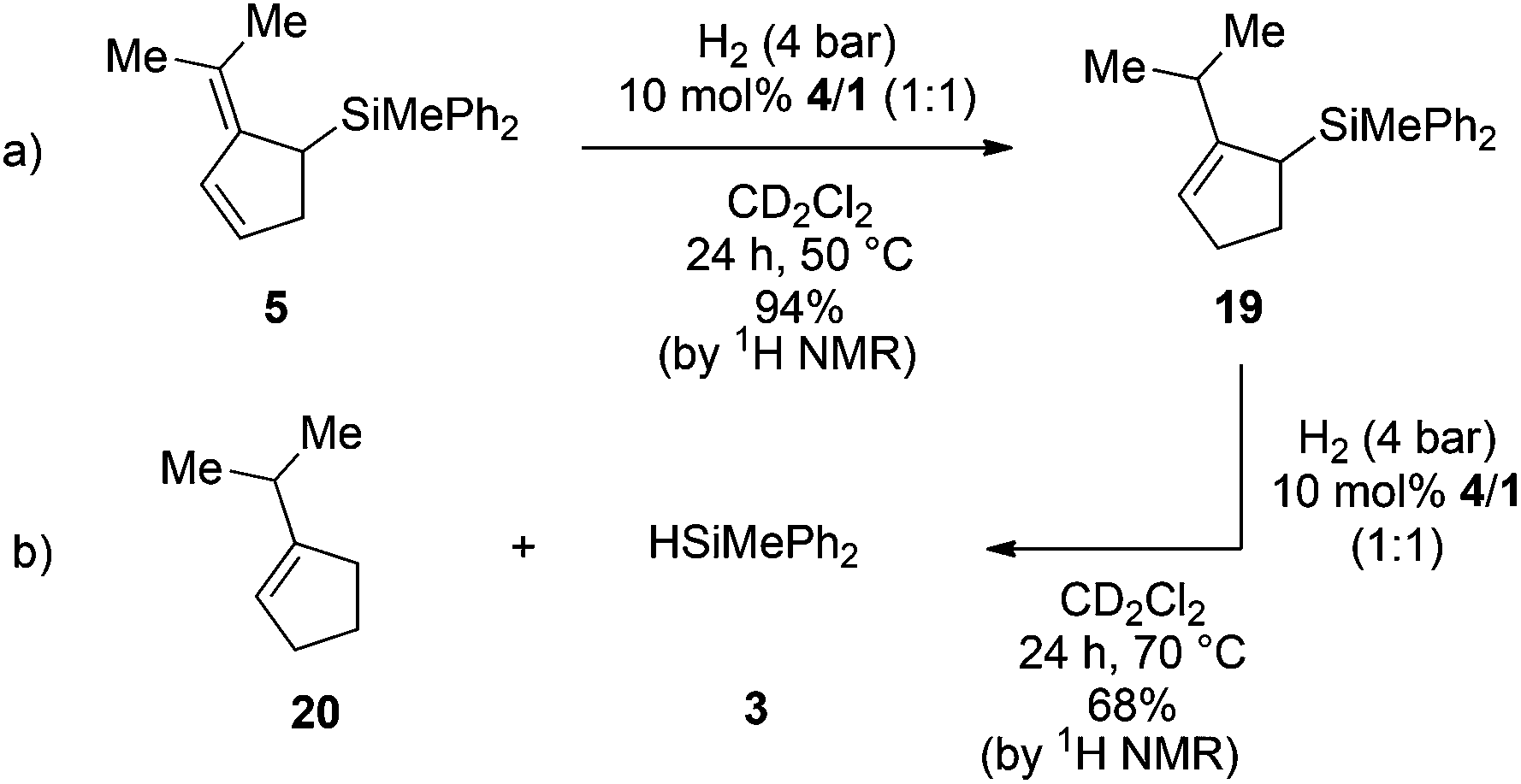 | ||
| Scheme 3 FLP catalyzed hydrogenation of allylsilane 5 (yields were determined by 1H NMR spectroscopy using the residual solvent signal as an internal standard). | ||
Surprisingly the hydrogenation provided (2-iso-propylcyclopent-2-en-1-yl)trimethylsilane (19) as the sole isomer, indicating a double bond migration during the reaction (vide infra for mechanistic details). Since product 19 is once again an allyl silane a second reduction step to the saturated iso-propyl substituted cyclopentylsilane seems feasible. However, prolongation of the reaction time or increase of the reaction temperature (up to 70 °C) did not result in the reduction of the double bond but unexpectedly in the dehydrosilylation of 19 to produce iso-propyl-cyclopentene (20) and the hydrosilane 3 in 68% yield (Scheme 3b). Accordingly the by-product formation in the hydrogenation of 5 is a result of the over-hydrogenation of 19. Such reactivity is known for allyl silanes when treated with strong Brønsted acids.17 However, these reactions usually require nucleophilic oxo-groups in order to cleave the silicon carbon bond. The unexpected protodesilylation prompted us to investigate the reactions in more detail. Therefore hydrogenation experiments of allyl silane 5 were conducted using 10 mol% of FLPs comprising different Lewis bases (Table 2). Lewis pairs consisting of electron rich phosphines P(t-Bu)3 (6)/1 and P(Mes)3 (7)/1 were not reactive (Table 2, entries 1 and 2). FLPs with a less electron releasing phosphine or amine e.g.8 and 9 were able to catalyze the hydrogenation of 5 even at room temperature (entries 4 and 5). Products resulting from protodesilylation were already generated in significant amounts even under these mild reaction conditions. The FLP-catalyzed protodesilylation was not only restricted to 19. Exposure of the mixture of 17 and 18 (1.2:1) to H2-atmosphere in the presence of 4/1 (10 mol%) resulted in the formation of the iso-propyl indene 21. As expected, only the allyl silane 17 underwent protodesilylation (56%) while the homo allyl silane 18 remained unchanged (Scheme 4).
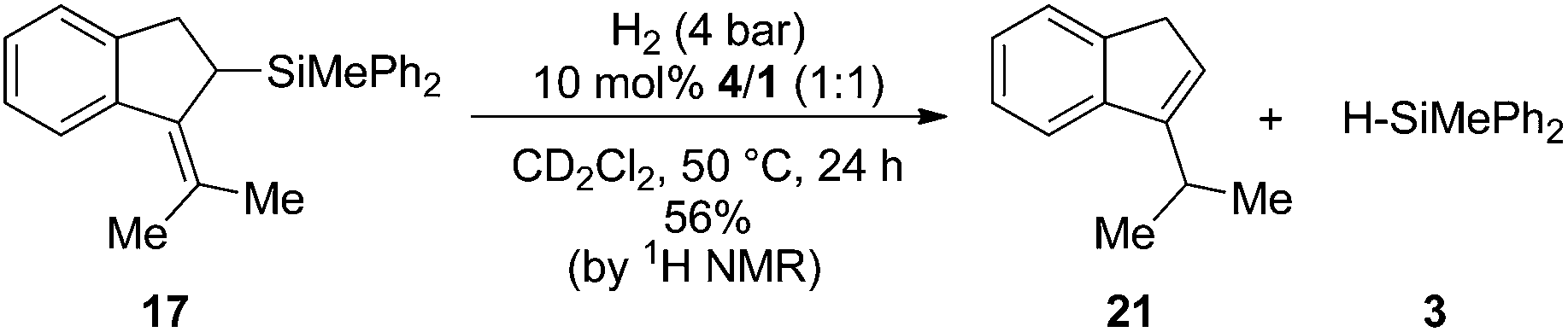 | ||
| Scheme 4 FLP mediated protodesilylation of allylsilane 17 (the yield was determined by 1H NMR spectroscopy using the residual solvent signal as an internal standard). | ||
|
|
||||
|---|---|---|---|---|
| Entry | Lewis base | T [°C] | Conv. [%] | Product ratio 19:20 |
| a Reactions performed on a 0.1 mmol scale in CD2Cl2 (0.5 mL, 0.2 M) at the given temperature using 10 mol% of B(C6F5)3 (1) and Lewis base under 4 bar of hydrogen atmosphere. Yields were determined by 1H NMR spectroscopy after 24 h with the residual solvent signal as an internal standard. | ||||
| 1 | 6 | 70 | 0 | — |
| 2 | 7 | 70 | 0 | — |
| 3 | 4 | 50 | >95 | 10.8:1 |
| 4 | 8 | r.t. | 76 | 5.3:1 |
| 5 | 9 | r.t. | 68 | 7.5:1 |
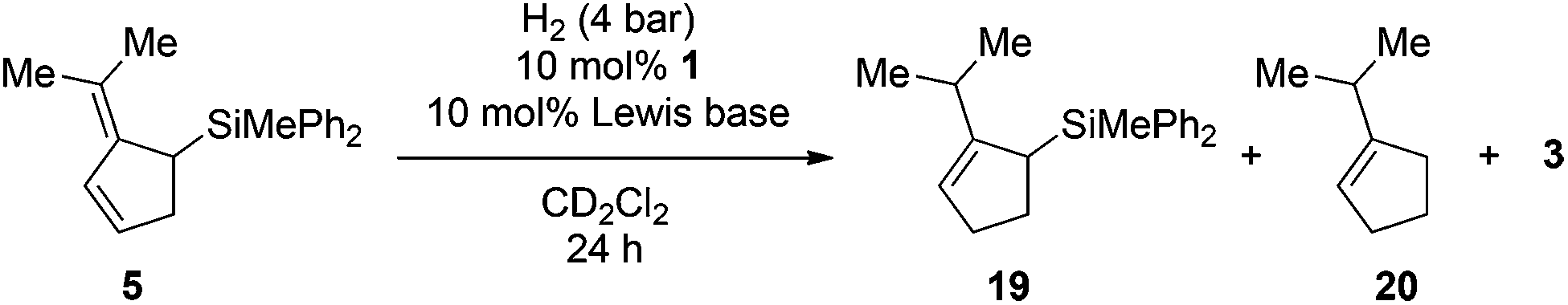
Finally the two allyl silanes 14 and E/Z-15 were subjected to the hydrogenation (10 mol% 4/1), but even after prolonged reaction time (48 h) at elevated temperatures (70 °C) the corresponding hydrogenation products were not identified. However, under the reaction conditions the allyl silanes underwent rearrangement to the corresponding vinyl silanes (compare Table 2). Subsequent control experiments revealed that this rearrangement is catalyzed by B(C6F5)3 (1).18 The symmetrical diphenyl-derivative 14 was converted to the corresponding vinyl silane 22 in quantitative yield after 48 h at 70 °C (Table 3, entry 1 > 95% yield). Interestingly the two diastereomers E/Z-15 displayed different rates for the rearrangement. In the mixture of E/Z-15 (1.6:1) the E-diastereomer underwent the Lewis acid-catalyzed rearrangement more readily (entry 2: 40 °C, 24 h) than Z-15 (entry 2: 70 °C, 92 h).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Starting material | Product | Temp. (° C) | Time (h) | Yield (%) | ||
| R= | R′= | ||||||
| a Reactions performed on a 0.1 mmol scale in CD2Cl2 (0.5 mL, 0.2 M) at the given temperature and reaction time using 10 mol% of B(C6F5)3 (1). Yields were determined by 1H NMR spectroscopy with the residual solvent signal as an internal standard. | |||||||
| 1 | Ph | Ph | 14 | 22 | 70 | 48 | >95 |
| 2 | Ph | Me | (E)-15 | (E)-22 | 40 | 24 | >95 |
| 3 | Me | Ph | (Z)-15 | (Z)-22 | 70 | 92 | 78 |
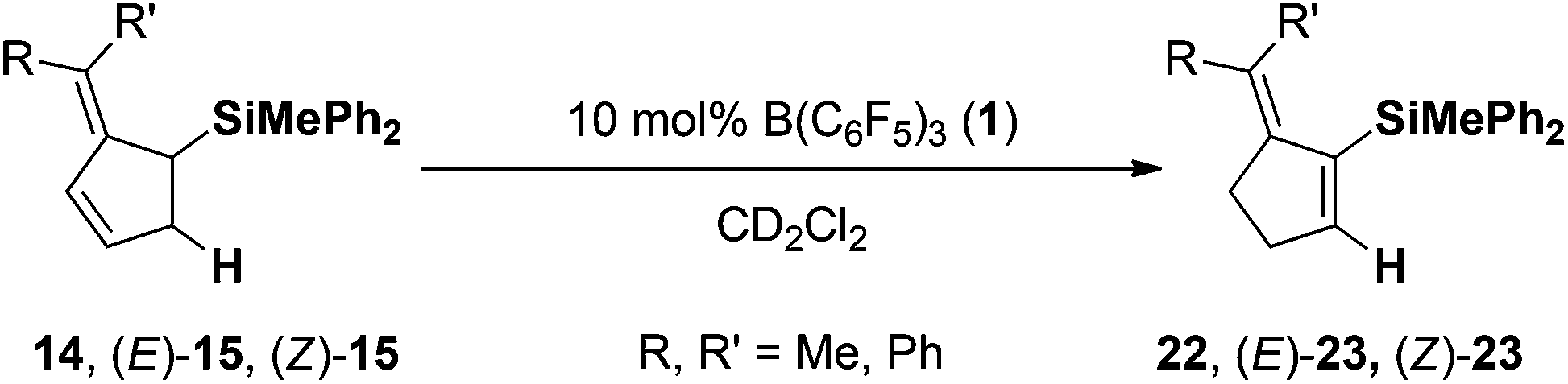
The combined data for the hydrosilylation of 2, hydrogenation of 5 and final protodesilylation of 19 suggest that this sequence may be combined in a triple FLP-catalyzed domino-reaction cascade using hydrosilane 3 as the mediator (Scheme 5).
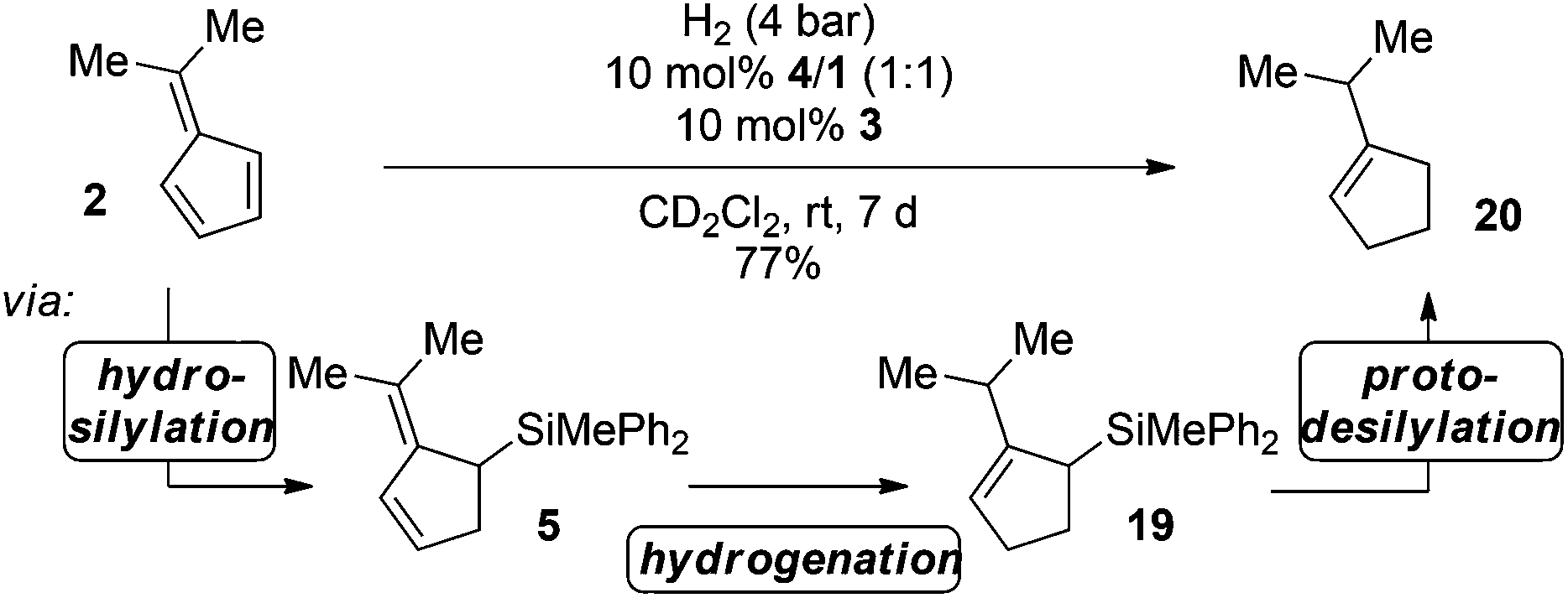 | ||
| Scheme 5 FLP-catalyzed triple reaction cascade for the hydrosilane mediated hydrogenation of fulvene 2 (the yield was determined by 1H NMR spectroscopy using the residual solvent signal as an internal standard). | ||
Indeed, after reaction of 2 with 10 mol% FLP consisting of 4/1, 10 mol% hydrosilane 3 in the presence of H2 (4 bar) for 24 h at room temperature, complete consumption of the starting material was observed, yielding a mixture of cycloaddition and hydrosilylation products. However, after an additional 6 days the mixture was converted to iso-propylcyclopentene (20) in 77% yield. In this sequence the hydrosilane 3 enabled the FLP-catalyzed hydrogenation of 6,6-dimethylfulvene (6), which was not possible by direct metal-free hydrogenation as discussed earlier.
Mechanistic investigations
Finally we investigated the FLP-catalyzed triple cascade in more detail by deuteration experiments (Scheme 6).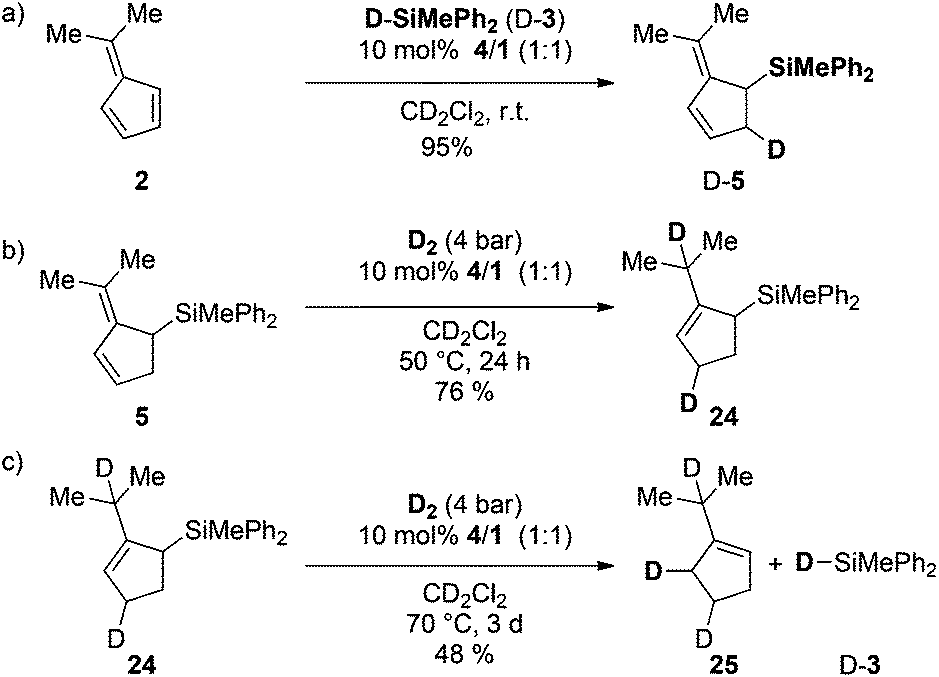 | ||
| Scheme 6 (a) Deuterosilylation of dimethylfulvene (2), (b) deuteration of 5 and (c) deuterodesilylation of allylsilane 24 (yields were determined by 1H NMR spectroscopy with the residual solvent signal as an internal standard). | ||
Treatment of 2 with deuterosilane 3 furnished the 2-(propan-2-ylidene)-5-deutero-cyclopent-3-en-1-yl)silane (D-5) selectively in quantitative yields (>95%, Scheme 6a). The exposure of 5 to D2-atmosphere in the presence of FLP 4/1 provided the 1,4-addition of deuterium to the diene with concomitant double bond migration, supporting a protonation/hydride addition mechanism (Scheme 6b). The resulting 3-cyclopentene 24 underwent deuterodesilylation in 48% yield with selective vicinal deuterium incorporation and formation of equimolar amounts of deuterosilane D-3 (Scheme 6c). Accordingly, protodesilylation occurs via electrophilic attack of the double bond and nucleophilic deuteride-transfer from the borodeuteride to the silyl-group. Based on these observations a catalytic cycle for the FLP-catalyzed triple reaction cascade is proposed (Scheme 7). After the FLP-assisted hydrosilylation of 2 the allyl silane 5 is hydrogenated via metal-free H2-activation to the cyclopentene 19. Finally hydrosilane 3 is regenerated by FLP-mediated H2-activation, and subsequent protodesilylation of 19 closes the catalytic cycle.
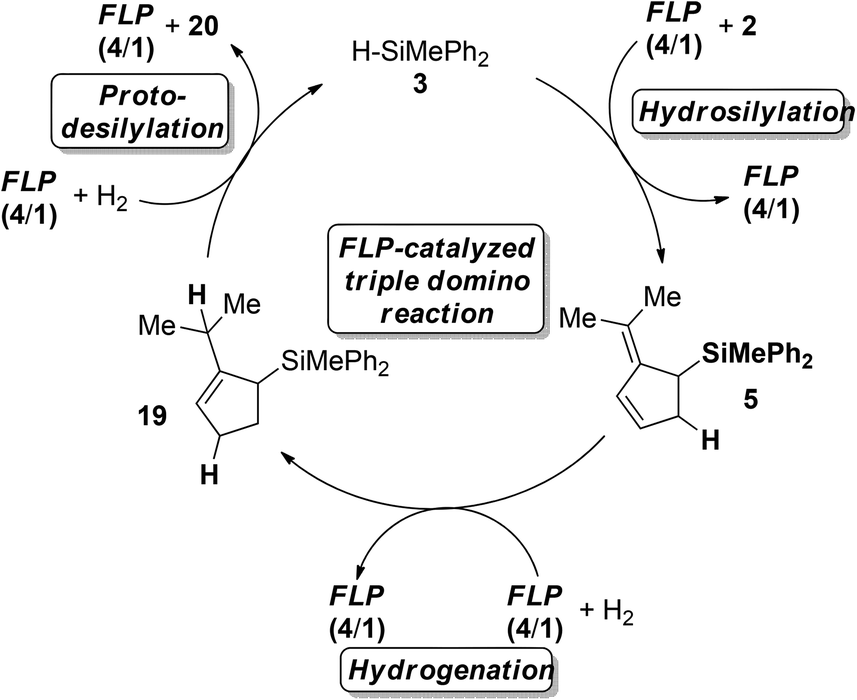 | ||
| Scheme 7 Catalytic cycle for the FLP-catalyzed triple reaction cascade. | ||
Conclusions
In conclusion, we have shown that FLPs are potent catalysts for the hydrosilylation of pentafulvenes in excellent yields. Here the FLPs not only catalyzed the hydrosilylation with remarkable regioselectivity but also suppressed undesired side reactions e.g. the Lewis acid catalyzed oligomerization of fulvenes. Dimethyl-substituted allyl silanes underwent FLP-catalyzed hydrogenation and protodesilylation. The latter observation led to the development of the triple reaction cascade consisting of hydrosilylation/hydrogenation/protodesilylation, allowing for the metal-free hydrogenation of 6,6-dimethyl fulvene to iso-propyl cyclopentene. The mechanisms of the individual reactions were investigated by deuteration experiments.Acknowledgements
The authors acknowledge the Landesgraduiertenförderung of the State of Baden-Württemberg for a Ph.D. grant to S. T. We would also like to acknowledge the German research foundation (DFG) for a Heisenberg fellowship to J. P.Notes and references
- (a) J. G. de Vries and C. J. Elsevier, The handbook of homogeneous hydrogenation, Wiley-VCH, 2007 Search PubMed; (b) I. Ojima, in Organic Silicon Compounds, John Wiley & Sons, Ltd, 1989, p. 1479 Search PubMed.
- (a) M. E. Fasulo, M. C. Lipke and T. D. Tilley, Chem. Sci., 2013, 4, 3882 RSC; (b) C. D. F. Königs, H. F. T. Klare and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 10076 CrossRef PubMed; (c) E. P. Plueddemann, Silane Coupling Agents, Springer, US, 2013 Search PubMed; (d) A. M. Tondreau, C. C. H. Atienza, K. J. Weller, S. A. Nye, K. M. Lewis, J. G. P. Delis and P. J. Chirik, Science, 2012, 335, 567 CrossRef CAS PubMed; (e) D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440 CrossRef CAS PubMed; (f) B. Marciniec, Hydrosilylation: A Comprehensive Review on Recent Advances, Springer, 2010 Search PubMed; (g) A. K. Roy, in Adv. Organomet. Chem, ed. A. F. H. Robert West and J. F. Mark, Academic Press, 2007, vol. 55, p. 1 Search PubMed; (h) M. Oestreich and S. Rendler, Angew. Chem., Int. Ed., 2005, 44, 1661 CrossRef CAS PubMed; (i) P. B. Glaser and T. D. Tilley, J. Am. Chem. Soc., 2003, 125, 13640 CrossRef CAS PubMed; (j) I. E. Markó, S. Stérin, O. Buisine, G. Mignani, P. Branlard, B. Tinant and J.-P. Declercq, Science, 2002, 298, 204 CrossRef PubMed; (k) R. M. Hill, Silicone Surfactants, Taylor & Francis, 1999 Search PubMed; (l) B. Marciniec, Comprehensive handbook on hydrosilylation, Pergamon Press, 1992 Search PubMed; (m) M. Voronkov, L. I. Kopylova, E. Lukevics and V. B. Pukhnarevitch, Perspectives of Hydrosilylation, Institute of Organic Synthesis, Riga, Latvia, 1992 Search PubMed; (n) P. B. Hitchcock, M. F. Lappert and N. J. W. Warhurst, Angew. Chem., Int. Ed., 1991, 30, 438 CrossRef; (o) S. Patai and Z. Rappoport, The chemistry of organic silicon compounds, Wiley, 1989 CrossRef; (p) A. Mortreux, F. Petit and Commission of the European Communities, Industrial Applications of Homogeneous Catalysis, Springer, 1988 Search PubMed; (q) J. L. Speier, in Adv. Organomet. Chem, ed. F. G. A. Stone and W. Robert, Academic Press, 1979, vol. 17, p. 407 Search PubMed.
- (a) J. Hermeke, M. Mewald and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 17537 CrossRef CAS PubMed; (b) W. E. Piers, A. J. V. Marwitz and L. G. Mercier, Inorg. Chem., 2011, 50, 12252 CrossRef CAS PubMed; (c) J. M. Blackwell, D. J. Morrison and W. E. Piers, Tetrahedron, 2002, 58, 8247 CrossRef CAS; (d) J. M. Blackwell, E. R. Sonmor, T. Scoccitti and W. E. Piers, Org. Lett., 2000, 2, 3921 CrossRef CAS PubMed; (e) D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440 CrossRef CAS.
- (a) M. H. Holthausen, M. Mehta and D. W. Stephan, Angew. Chem., Int. Ed., 2014, 53, 6538 CrossRef CAS PubMed; (b) A. Simonneau and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 11905 CrossRef CAS PubMed; (c) M. Pérez, L. J. Hounjet, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 18308 CrossRef PubMed; (d) M. Rubin, T. Schwier and V. Gevorgyan, J. Org. Chem., 2002, 67, 1936 CrossRef CAS PubMed.
- (a) L. Greb, P. Ona-Burgos, A. Kubas, F. C. Falk, F. Breher, K. Fink and J. Paradies, Dalton Trans., 2012, 41, 9056 RSC; (b) L. Greb and J. Paradies, Topics in Current Chemistry: Frustrated Lewis Pairs II: Expanding the Scope, 2013, vol. 334, p. 81 Search PubMed.
- (a) G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124 CrossRef CAS PubMed; (b) P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 8050 ( Angew. Chem., Int. Ed. , 2007 , 119 , 8196–8199 ) CrossRef CAS PubMed; (c) G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880 CrossRef CAS PubMed; (d) D. W. Stephan, S. Greenberg, T. W. Graham, P. Chase, J. J. Hastie, S. J. Geier, J. M. Farrell, C. C. Brown, Z. M. Heiden, G. C. Welch and M. Ullrich, Inorg. Chem., 2011, 50, 12338 CrossRef CAS PubMed; (e) P. Spies, S. Schwendemann, S. Lange, G. Kehr, R. Fröhlich and G. Erker, Angew. Chem., Int. Ed., 2008, 47, 7543 ( Angew. Chem., Int. Ed. , 2008 , 120 , 7654–7657 ) CrossRef CAS PubMed; (f) P. Spies, G. Erker, G. Kehr, K. Bergander, R. Froehlich, S. Grimme and D. W. Stephan, Chem. Commun., 2007, 5072 RSC.
- (a) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS PubMed; (b) D. W. Stephan, Org. Biomol. Chem., 2008, 6, 1535 RSC; (c) D. W. Stephan, Org. Biomol. Chem., 2012, 10, 5740 RSC; (d) J. Paradies, Angew. Chem., Int. Ed., 2014, 53, 3552 CrossRef CAS PubMed; (e) J. Paradies, Synlett, 2013, 777 CrossRef CAS PubMed.
- For the FLP-catalyzed hydrogenation of silylenol ethers see: H. D. Wang, R. Frohlich, G. Kehr and G. Erker, Chem. Commun., 2008, 5966 RSC.
- (a) L. Greb, S. Tussing, B. Schirmer, P. Ona-Burgos, K. Kaupmees, M. Lokov, I. Leito, S. Grimme and J. Paradies, Chem. Sci., 2013, 4, 2788 RSC; (b) L. Greb, P. Oña-Burgos, B. Schirmer, S. Grimme, D. W. Stephan and J. Paradies, Angew. Chem., Int. Ed., 2012, 124, 10311 ( Angew. Chem. Int. Ed. , 2012 , 51 , 10164 ) CrossRef.
- (a) S. S. Bhojgude, T. Kaicharla, A. Bhunia and A. T. Biju, Org. Lett., 2012, 14, 4098 CrossRef CAS PubMed; (b) N. Coskun, J. Ma, S. Azimi, C. Gärtner and I. Erden, Org. Lett., 2011, 13, 5952 CrossRef CAS PubMed; (c) B.-C. Hong, F.-L. Chen, S.-H. Chen, J.-H. Liao and G.-H. Lee, Org. Lett., 2005, 7, 557 CrossRef CAS PubMed; (d) B.-C. Hong, Y.-J. Shr and J.-H. Liao, Org. Lett., 2002, 4, 663 CrossRef CAS PubMed; (e) V. Nair, G. Anilkumar, K. V. Radhakrishnan, M. V. Nandakumar and S. Kumar, Tetrahedron, 1997, 53, 15903 CrossRef CAS; (f) P. Bickert, B. Hildebrandt and K. Hafner, Organometallics, 1984, 3, 653 CrossRef CAS; (g) E. D. Bergmann, Chem. Rev., 1968, 68, 41 CrossRef CAS.
- (a) D. M. Whitehead, P. A. Helliwell, S. C. McKeown and A. Routledge, React. Funct. Polym., 2009, 69, 884 CrossRef CAS PubMed; (b) J. A. Bajgrowicz, T. B. Bourdin and P. Gygax, Eur. Patent, 1067118A1, 2001 Search PubMed; (c) J. E. Nystroem and P. Helquist, J. Org. Chem., 1989, 54, 4695 CrossRef CAS; (d) A. Steinmeyer, W. Schwede and F. Bohlmann, Liebigs Ann. Chem., 1988, 925 CrossRef CAS.
- C. M. Momming, G. Kehr, R. Frohlich and G. Erker, Chem. Commun., 2011, 47, 2006 RSC.
- (a) L. F. Tietze, G. Brasche and K. M. Gericke, Domino Reactions in Organic Sysnthesis, Wiley-VCH Verlag, Weinheim, 2006 Search PubMed; (b) P. von Zezschwitz and A. de Meijere, Top. Organomet. Chem., 2006, 19, 49 CrossRef CAS; (c) A. de Meijere, P. von Zezschwitz and S. Bräse, Acc. Chem. Res., 2005, 38, 413 CrossRef CAS PubMed; (d) A. de Meijere, P. von Zezschwitz, H. Nuske and B. Stulgies, J. Organomet. Chem., 2002, 653, 129 CrossRef CAS; (e) L. F. Tietze, Chem. Rev., 1996, 96, 115 CrossRef CAS PubMed.
- (a) Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307 CAS; (b) Z. Otwinowski, D. Borek, W. Majewski and W. Minor, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2003, 59, 228 CrossRef PubMed; (c) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 467 CrossRef; (d) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed ; CCDC 1008790.
- (a) D. Chen, V. Leich, F. Pan and J. Klankermayer, Chem. – Eur. J., 2012, 18, 5184 CrossRef CAS PubMed; (b) C. A. Reed, Acc. Chem. Res., 1998, 31, 325 CrossRef CAS.
- (a) T. A. Rokob, A. Hamza, A. Stirling and I. Papai, J. Am. Chem. Soc., 2009, 131, 2029 CrossRef CAS PubMed; (b) L. Rocchigiani, G. Ciancaleoni, C. Zuccaccia and A. Macchioni, J. Am. Chem. Soc., 2013, 136, 112 CrossRef PubMed.
- (a) I. Fleming and J. A. Langley, J. Chem. Soc., Perkin Trans. 1, 1981, 1421 RSC; (b) I. Fleming, D. Marchi and S. K. Patel, J. Chem. Soc., Perkin Trans. 1, 1981, 2518 RSC.
- The rearrangement was not observed in the presence of 4/H2, pure 4 or pure H2.
Footnote |
| † Electronic supplementary information (ESI) available: General information; synthetic procedures; NMR spectroscopic data. CCDC 1008790. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob01346h |
| This journal is © The Royal Society of Chemistry 2014 |