
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Annulated and bridged tetrahydrofurans from alkenoxyl radical cyclization†‡
Christine
Schur
a,
Harald
Kelm
a,
Thomas
Gottwald
b,
Arne
Ludwig
b,
Rainer
Kneuer
b and
Jens
Hartung
*a
aFachbereich Chemie, Organische Chemie, Technische Universität Kaiserslautern, Erwin-Schrödinger-Straße, D-67663 Kaiserslautern, Germany. E-mail: hartung@chemie.uni-kl.de; Fax: +49-631-205-3921; Tel: +49-631-205-2431
bInstitut für Organische Chemie, Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 22nd August 2014
Abstract
4-Pentenoxyl radicals sharing two or more carbon atoms with a cycloalkane cyclize in a predictable manner stereoselectively and regioselectively to afford in solutions of bromotrichloromethane cycloalkyl-fused or -bridged 2-bromomethyltetrahydrofurans in up to 95% yield. Stereoselectivity in alkenoxyl radical ring closures arises from cumulative steric effects. The substituent positioned the closest to the alkene carbon, which is being attacked by the oxygen radical, exerts the strongest stereodirecting effect. This principal inductor guides 5-exo-cyclization 2,3-trans- or 2,4-cis-selectively. The substituent located further from the attacked π-bond is the secondary inductor. A secondary inductor in the relative trans-configuration enhances stereodifferentiation by the primary inductor; a cis-configured secondary inductor decreases this effect. A secondary inductor is not able to overrule the guiding effect of a similar sized primary inductor. Intramolecular 4-pentenoxyl radical additions to a cyclohexene-bound exo-methylene group or to endocyclic double bonds proceed cis-specifically, as exemplified by synthesis of a diastereomerically pure bromobicyclo[2.2.1]heptyl-annulated tetrahydrofuran from the verbenylethyloxyl radical. According to theory, the experimental 2,3-cis-specificity in alkoxyl radical cyclization to an endocyclic π-bond arises from strain associated with the 2,3-trans-ring closure.
1. Introduction
4-Pentenoxyl radicals add intramolecularly to the inner alkene carbon with rate constants of 108 per second and above.1–6 Alkyl or ortho-substituted aryl groups in position 1 exert a stereodirecting effect, leading to 2,5-trans-configured tetrahydrofurans as principal products. Carbon substituents at positions 2 and 3 direct 4-pentenoxyl radical cyclization 2,4-cis-and 2,3-trans-selectively.7–9 Stereodifferentiation by alkyl or aryl groups arises from steric effects, which gradually increases as the distance between a controlling substituent and the attacked π-bond shortens, for example from a 15/85-cis/trans-ratio at room temperature to <2/98 by shifting a tert-butyl group from position 1 to position 3.8 In synthesis, 5-exo-cyclized 4-pentenoxyl radicals are preferentially trapped by a heteroatom atom donor,10,11 for introducing halogen,12–14 alkylsulfanyl,15 or other synthetically useful functional groups.16The model to explain stereodifferentiation by a carbon substituent in 4-pentenoxyl radical cyclization predicts that the intramolecular addition proceeds via a distorted twist-conformer of tetrahydrofuran as the favored transition structure (twist-model),8,17 differing from the cyclohexane-based Beckwith–Houk-model for carbon radical cyclization.18,19 Application of the alkoxyl radical approach to synthesis of more demanding targets, for example biologically active terpene-, acetogenin-, and fatty acid-derived cycloalkyl-fused tetrahydrofurans,20–22 requires to extend the model in order to predict the selectivity for constructing bicyclic compounds.7,23 Lessons from carbon radical chemistry have taught that stereodifferentiation in synthesis of bicyclic compounds is difficult to extrapolate by transferring results from monocycle to bicycle formation, since transannular and other strain effects may superimpose in an unpredictable manner.24,25 To find out whether embedding two carbons of a 4-pentenoxyl radical into a cycloaliphatic framework conserves or changes guidelines for stereoselective tetrahydrofuran synthesis, we examined in this study bromocyclization of cis/trans-cycloalkyl-bridged alkenoxyl radicals, having the carbon–carbon double bond located in a conformationally flexible side chain (types A and B), in the exo-position of cyclohexane (type C), or incorporated into an alicyclic core (types D and E, Fig. 1).
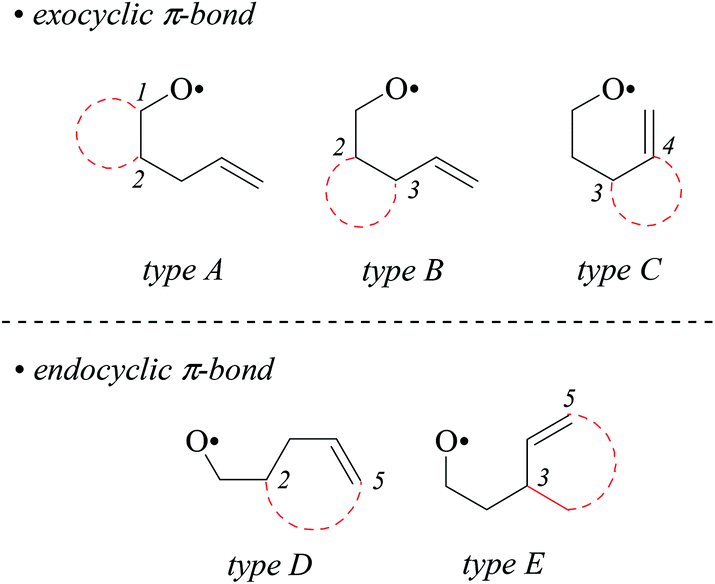 | ||
| Fig. 1 Classification of cycloalkyl-bridged 4-pentenoxyl radicals (type A–C) and alkenoxyl radicals having an endocyclic carbon–carbon double bond (type D–E; dotted red lines symbolize alkyl bridging). | ||
The most important finding from the study shows that cycloalkyl-bridged 4-pentenoxyl radicals cyclize in a predictable manner stereoselectively and regioselectively, to afford in solutions of bromotrichloromethane cycloalkyl-annulated or -bridged bromomethyltetrahydrofurans in up to 95% yield. The principal stereoinductor is the substituent attached the closest to the carbon–carbon double bond, which is being attacked by the radical oxygen. A principal inductor guides 5-exo-cyclization 2,3-trans- or 2,4-cis-selectively. The substituent bound further from the attacked π-bond is the secondary inductor. A trans-arranged secondary inductor enhances stereocontrol of the primary inductor, and a cis-configured secondary inductor decreases this effect. A secondary inductor of similar steric size, located further from the attacked π-bond is not able to overrule the directing effect of the principal inductor. Oxygen radicals attached via a methylene- or an ethylene-spacer to cyclohexene cyclize cis-specifically, as exemplified by synthesis of a diastereomerically pure bromobicyclo[2.2.1]heptyl-annulated tetrahydrofuran from a type-E radical. The propensity of cyclohexenylethyloxyl radicals to cyclize 2,3-cis-specifically arises from strain associated with the 2,3-trans-ring closure, as derived by a Marcus analysis of density functional-calculated reaction energies and barriers.
2. Results and interpretation
2.1 Alkenoxyl radicals from 3-alkenoxy-4-methylthiazole-2(3H)-thiones
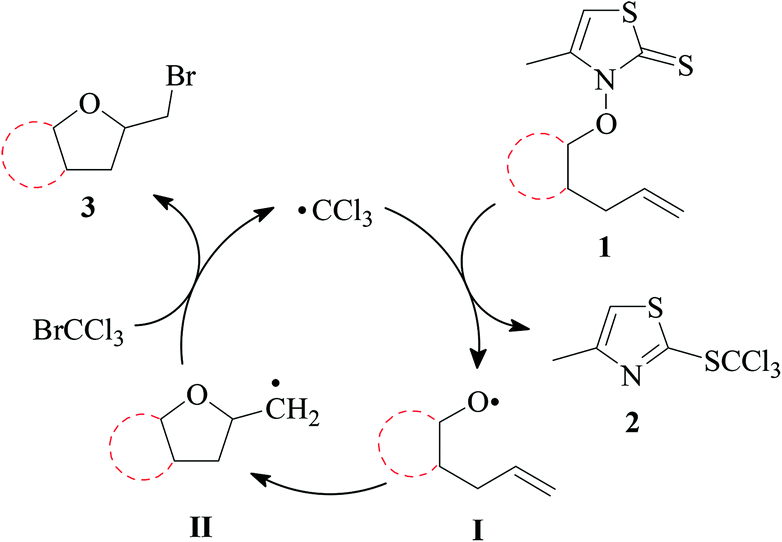
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.17 Trapping of carbon radical II by bromotrichloromethane yields bromomethyltetrahydrofuran 3 as a target product, and the trichloromethyl radical for propagating the chain reaction.
:2.17 Trapping of carbon radical II by bromotrichloromethane yields bromomethyltetrahydrofuran 3 as a target product, and the trichloromethyl radical for propagating the chain reaction.
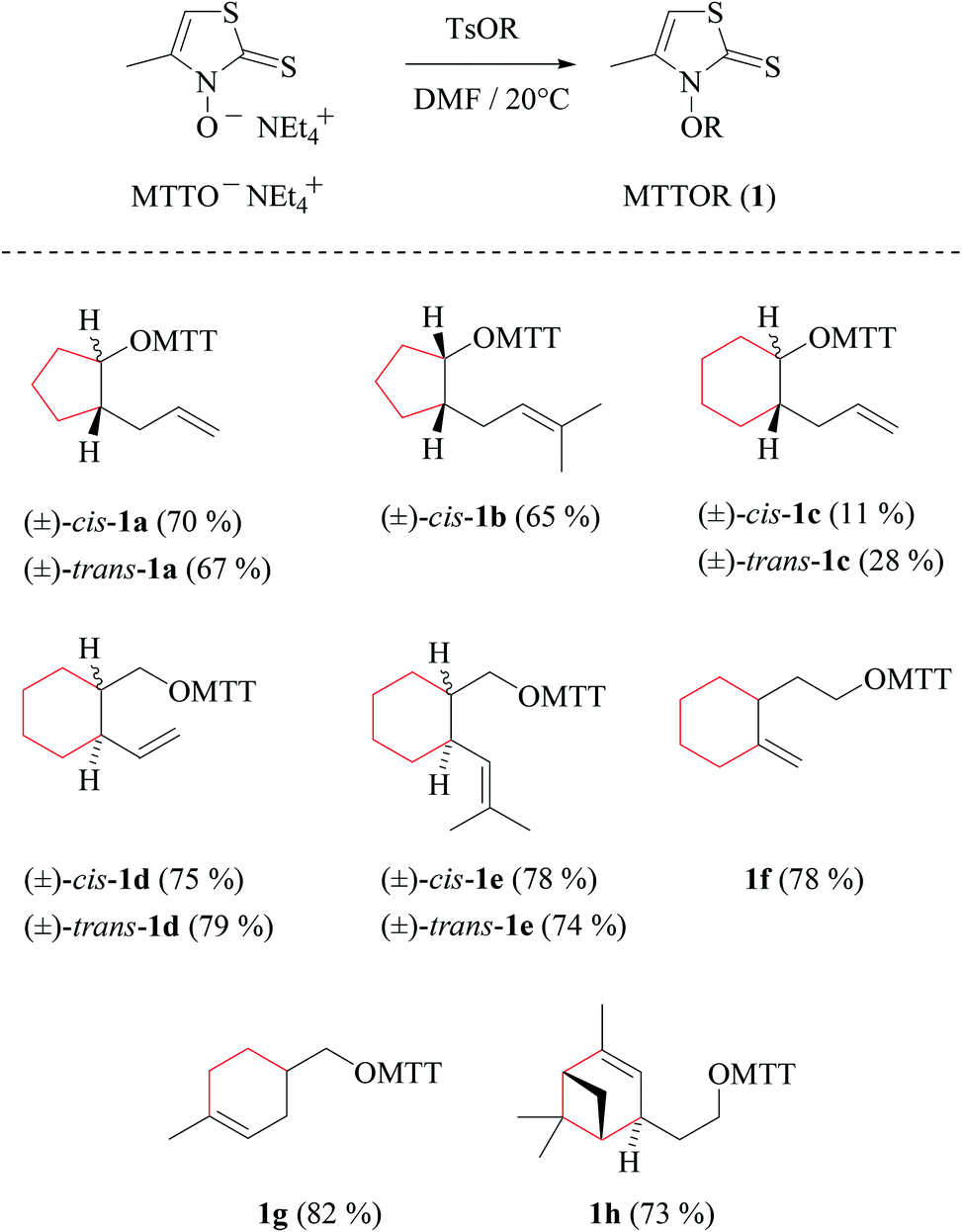 | ||
| Scheme 2 Indexing, yields, and structure formulas of MTTORs 1a–h prepared from O-alkenyl tosylates and 3-hydroxy-4-methylthiazole-2(3H)-thione tetraethylammonium salt (segments drawn in red refer to subunits represented by dotted lines in Fig. 1 and in Scheme 1). | ||
3-Alkenoxyl-4-methylthiazole-2(3H)-thiones obtained as described above are oils (1a–b, cis-1c, 1f) or crystalline solids (trans-1c, 1d–e, 1g, 1h), stable for months when stored in vials at room temperature. Recrystallizing 3-(methylcyclohexenyl)-methyloxy-thiazolethione 1g removed a regioisomer, which had been formed with 5/95-selectivity as a by-product from the [4 + 2]-cycloaddition between isoprene and methyl acrylate, in the first step of the synthesis for constructing the 1,4-disubstituted cyclohexenyl nucleus.37,38
3-Alkenoxy-4-methylthiazole-2(3H)-thiones 1a–h show diagnostic carbon-13 chemical shifts for the thiocarbonyl carbon (180–181 ppm) and the thiohydroxamate-bound carbon (74.8–92.1 ppm). The compounds absorb in solutions of methanol UV-light, leading to absorption-maxima at λ = 316–319 nm (lgε ∼ 3.10–3.19 m2 mol−1). Photoexciting the tail end of this absorption band with 350 nm light causes the nitrogen–oxygen bond in 1 to break homolytically, liberating oxygen radicals without an externally added initiator.39
In solution, 3-alkoxy-4-methylthiazole-2(3H)-thiones show the phenomenon of hindered rotation about the nitrogen–oxygen bond, becoming apparent in nuclear magnetic resonance spectra of, for example, 3-isopropoxy-4-methyl-thiazole-2(3H)-thione by signal coalescence at −60 °C, and a twofold set of resonances below this temperature.40 The lowest in energy conformer has the ester carbon C7 offset by almost 90 degrees from the thiohydroxamate plane, to prevent close contacts with the thione sulfur and methyl group in position 4 of the heterocyclic core.41 The structure of 3-alkenyloxythiazolethione 1g in the solid state corresponds to the predicted minimum conformation of thiazolethione-derived O-alkyl thiohydroxamates in solution and in the gas phase. The crystals available for determining the structure of compound 1g were systematically disordered showing, according to the model used for solving and refining the structure, a 78/22-ratio of diastereomers at crystallographic independent sites.§ The diastereomers differ with respect to the configuration at C8/C8a (ESI‡) and the helicity at the nitrogen–oxygen bond, showing both the characteristic offset of thiohydroxamate bound carbon 7 from the heterocyclic plane [major diastereomer (ds): C2–N3–O1–C7 = 91.0(2)°; minor ds: C2–N3a–O1a–C7a = 59.8(6)°] and bond lengths which are diagnostic for primary O-alkyl thiohydroxamates [major ds (Fig. 2): C2–S2 = 1.666(1) Å, C2–N3 = 1.358(2) Å, N3–O1 = 1.386(2) Å; minor ds (ESI‡): C2–N3a = 1.366(6) Å, N3a–O1a = 1.373(4) Å].42
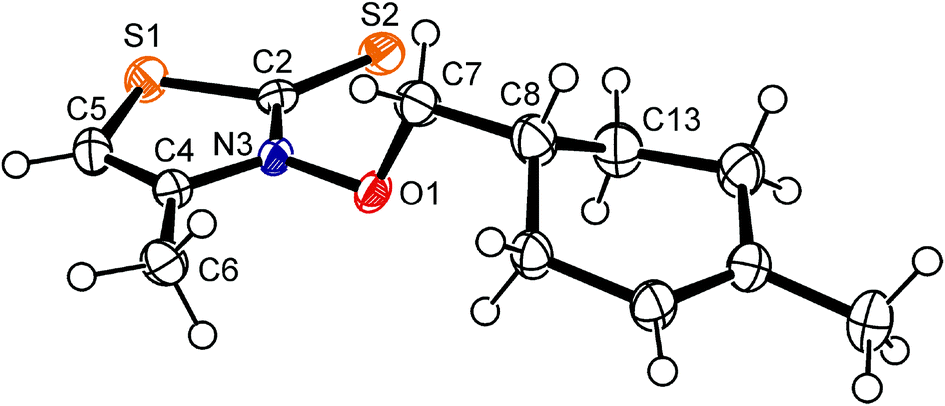 | ||
| Fig. 2 Ellipsoid graphic of 3-[(1-methylcyclohex-1-en-4-yl)-methyloxy]-4-methylthiazole-2(3H)-thione (1g) in the solid state [major diastereomer at 150 K; the (R,P)-isomer was arbitrarily chosen from the racemate (R,P)/(S,M)-1g for presentation (50% probability level); hydrogen atoms are drawn as circles of an arbitrary radius; oxygen is depicted in red, nitrogen in blue, and sulfur in orange; for depiction of the minor diastereomer (S,P)/(R,M)-1g, see the ESI‡]. | ||
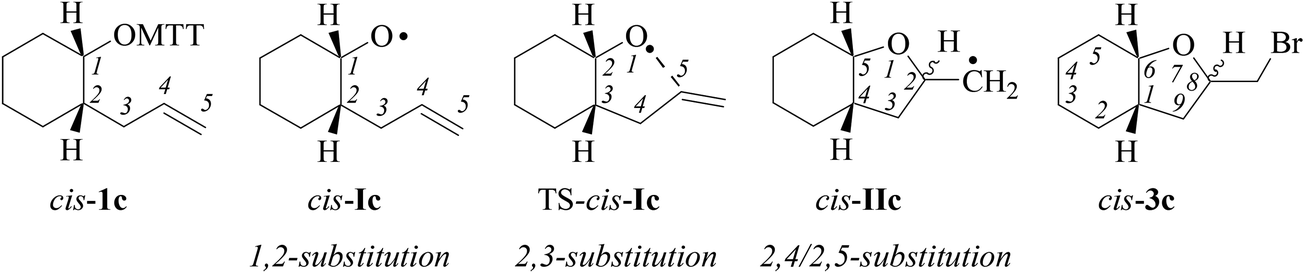 | ||
| Fig. 3 Convention used in this article for numbering atoms in product classes associated with alkoxyl radical reactions, exemplified for alkenoxythiazolethione cis-1c, derived 4-alkenoxyl radical cis-Ic, the transition structure for the 5-exo-cyclization TS-cis-Ic, 5-exo-cyclized carbon radical cis-IIc, and bicyclic bromocyclization product cis-3c. | ||
2.2 Alkenoxyl radical addition to exocyclic double bonds
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | cis-1 | R | Conditions | 2/% |
cis-3a–b/% (1,3-cis:trans) |
4/% |
| 1 | 1a | H | hν/25 °C | 85 |
3a: 10 (70:30) |
4a: 60 |
| 2 | 1a | H | AIBN/80 °C | 87 |
3a: 8 (71:29) |
4a: 54 |
| 3 | 1b | CH3 | hν/25 °C | 73 |
3b: 49 (64:36) |
4b: 31 |
| 4 | 1b | CH3 | AIBN/80 °C | 75 |
3b: 34 (56:44) |
4b: 35 |
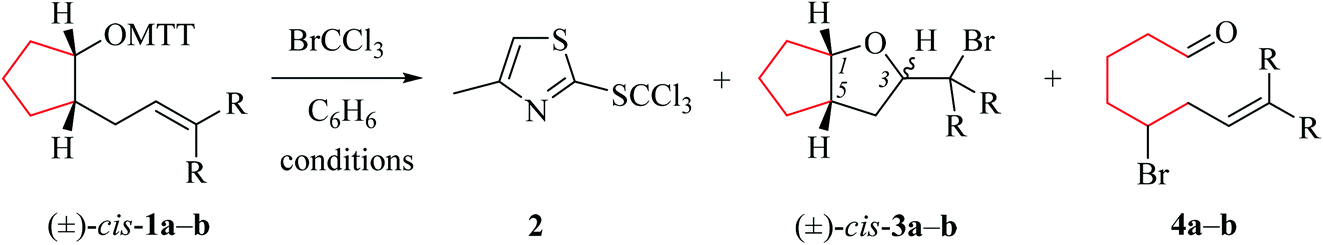
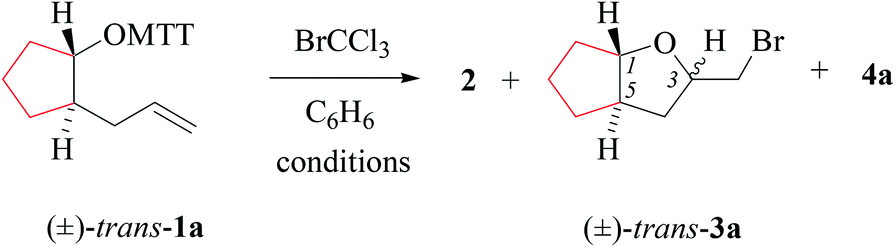
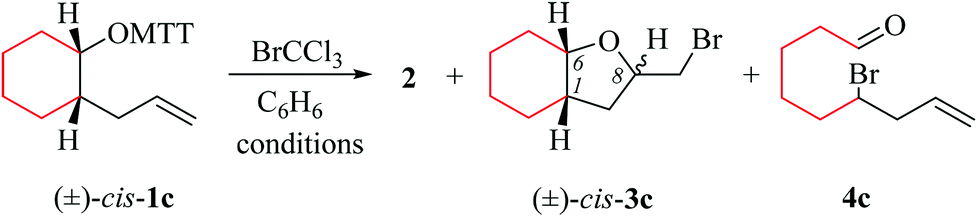
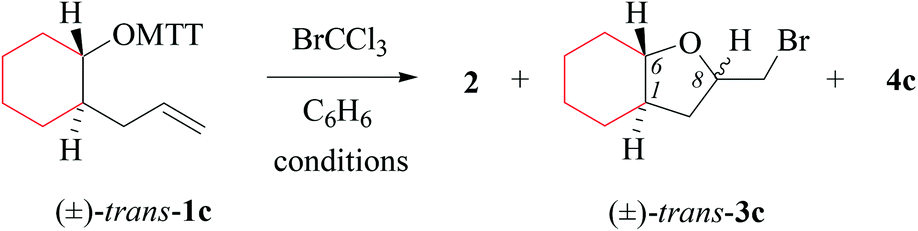
(i) Methods of alkenoxyl radical generation and product analysis. Photolyzing solutions of O-alkenyl thiohydroxamates 1a–c in benzene containing 10 equivalents (1.67 M) of bromotrichloromethane, using Rayonet® chamber apparatus equipped with 350 nm illuminants, quantitatively consume the starting material within 30 minutes, as determined by thin layer chromatography. Reaction mixtures from photochemical experiments tended to turn turbid and yellow. A gas chromatogram (GC) recorded by the end of the reaction time provided information on the original product pattern and distribution. Column chromatography furnished samples of purified 2-(trichloromethylsulfanyl)-4-methyl-1,3-thiazole (2), 5-exo-bromocyclized products 3a–c, and β-fragmented unsaturated 5-bromoaldehydes 4a–b, for collecting analytical data (Tables 1–4, and Experimental). Solutions from thermally initiated reactions were in addition charged with 15 mole percent of azo-α,α-bis-(isobutyronitrile) (AIBN) as the initiator. Such mixtures remained clear but tended to turn yellow by the end of the reaction.
(ii) Product pattern and kinetic interpretation. Reactions between O-(2-allylcycloalkyl) thiohydroxamates cis-1a–c and bromotrichloromethane furnish bromomethyltetrahydrofurans cis-3a–c, with the yields gradually decreasing for thermally initiated reactions from 61% for cis-3c through 34% for cis-3b to 8% for cis-3a (Table 1, entries 2 and 4; Table 3, entry 2). The reactions gave bromoaldehydes 4a–c as co-products in yields increasing from 12% for 4c, through 35% for 4b to 54% to 4a. Photolyzing or heating O-(2-allylcyclopentyl) thiohydroxamate trans-1a in the presence of bromotrichloromethane provided bromooctanal 4a, but no bromomethyltetrahydrofuran trans-3a as secured by independent analysis of an authentic sample of the compound (ESI‡). The ratio of the bromocyclized product trans-3c and bromoaldehyde 4c obtained from O-cyclohexylallyl ester trans-1c is similar to the ratio of cis-3c and 4c obtained from the stereoisomer cis-1c (entry 2 in Tables 3 and 4). The pattern of products obtained from radical reactions conducted at 80 °C in summary is similar, except for bromoaldehyde 4c, which did not form in the room temperature experiments. None of the reactions furnished 2-allylcycloalkanols or 2-allylcycloalkanones in verifiable amounts (GC-MS).
In kinetically controlled reactions, the quotient between bromomethyltetrahydrofuran 3 and bromoaldehyde 4 is equivalent to the relative rate constant for the addition (kadd) versus β-fragmentation (kβ) (Scheme 3). Kinetic control for oxygen radical addition to terminal double bonds is documented.8 For the following reason we suggest that the sequence leading to bromoaldehyde 4 under conditions chosen in this study also is kinetically controlled. In 1.67 molar solution of bromotrichloromethane, the effective rate constant for bromine atom trapping by secondary alkyl radicals, such as III, is approximately 4.3 × 108 s−1, based on kBr for the 6-hepten-2-yl radical (2.6 × 108 M−1 s−1; 26 °C)48 as a reference. The rate constant kadd for the 4-formylbutyl radical 5-exo-cyclization (8.7 × 105 s−1; 80 °C),49 serving as a reference for the reaction III → I, is by almost three orders of magnitude slower than the effective rate of bromine atom transfer from bromotrichloromethane to the secondary carbon radical III.
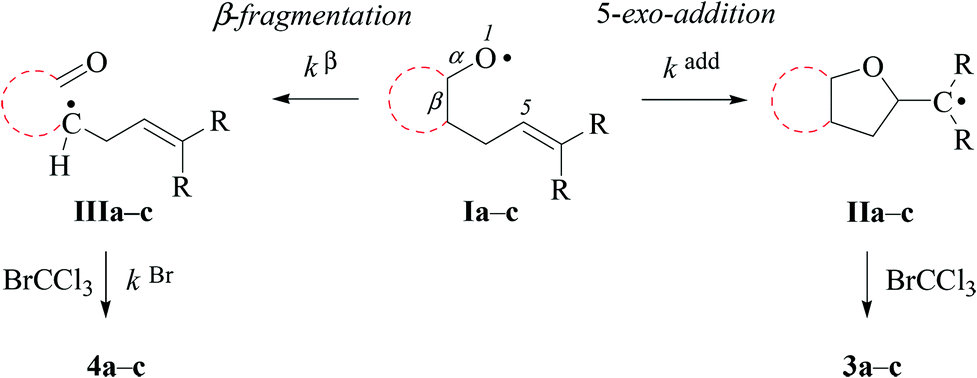 | ||
| Scheme 3 Competing reaction pathways for mechanistically interpreting the origin and yields of 5-exo-cyclization- versus β-fragmentation-products from 2-allylcycloalkyloxyl radicals Ia–c (R = H, CH3; for details associated with rate constants kβ, kBr, and kadd, see the text). | ||
For comparing rates of 5-exo-cyclization to rates of β-fragmentation for intermediates Ia–c, we standardized reactant concentrations and used a tenfold molar excess of bromotrichloromethane. Under such conditions, the ratio of bromide 3 to 4 corresponds to the quotient kadd/kβ, gradually increasing along the series of radicals trans-Ia (krel = 0), cis-Ia (0.2), cis-Ib (1.0) to cis/trans-Ic (krel = 5–9). Dividing kadd for the 4-pentenoxyl radical cyclisation (5.2 × 108 s−1; 26 °C) by kβ for the cyclopentoxyl radical β-fragmentation (4.7 × 108 s−1; 80 °C) for calibrating the competition system with the aid of absolute rate constants leads to a similar order of magnitude for the kadd/kβ ratio.2,49
The propensity of cyclopentane-fused 4-pentenoxyl radicals to provide β-fragmented products, such as bromoaldehydes 4a–b, arises from strain, being ∼20 kJ mol−1 higher for cyclopentane than for cyclohexane.50 Substituting methyl for hydrogen at the terminal alkene carbon increases the fraction of the 5-exo-cyclized product from cis-3a to cis-3b, which we address to a rate enhancing polar effect of the methyl group in oxygen radical additions.51
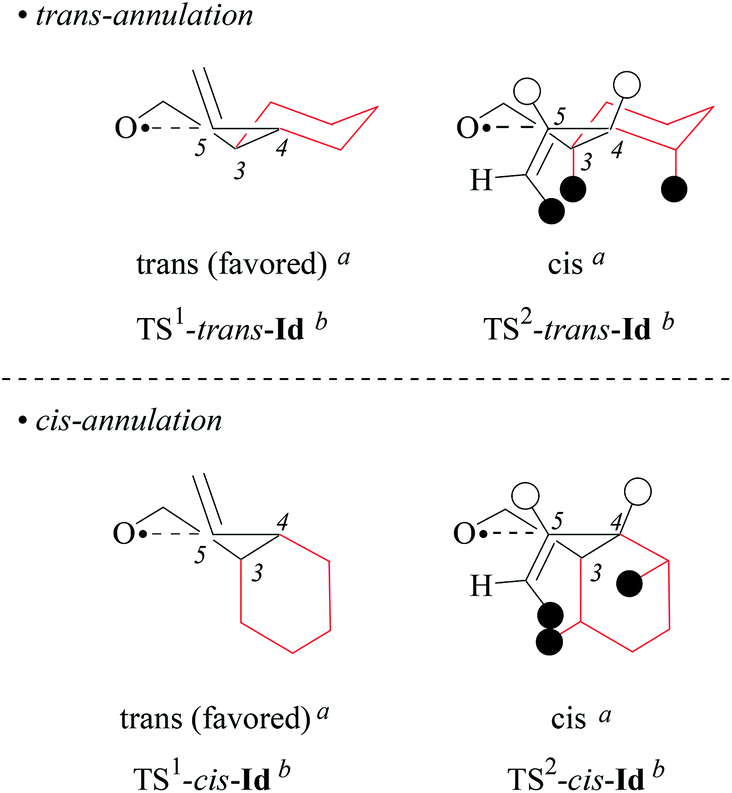
(iii) Stereochemical guidelines. 1,2-Cycloalkyl-bridged 4-pentenoxyl radicals Ia–c cyclize 2,4-cis-selectively showing that the substituent in position 2 is the principal stereoinductor for 5-exo-cyclization of type-A radicals. A trans-arranged secondary inductor in position 1 enhances the directing effect of the principal inductor; a cis-configured secondary inductor decreases this effect.
(iv) On the origin of 2,4-cis-selectivity in 5-exo-cyclization of type-A 4-pentenoxyl radicals. To understand the origin of 2,4-cis-selectivity, we modelled transition structures (TS) of 2-allylcyclohexyl-1-oxyl radical 5-exo-cyclization Ic → IIc, using assessed electronic structure methods.14,52
For stereochemical analysis, we considered transition structures for 2,4-cis-(TS1) and 2,4-trans-cyclization (TS2) of allylcyclohexyloxyl radicals cis/trans-Ic (Fig. 4 and ESI;‡ see also section 2.4). Transition structure searches according to an established methodology17 (ESI‡) led to twist (T)-conformers of tetrahydrofuran (Fig. 4), similar to intermediates modelled for 5-exo-cyclization of monosubstituted 4-pentenoxyl radicals.8 The radical oxygen in transition structures TS1,2-cis/trans-Ic lies for stereoelectronic reasons in a plane defined by inner alkene carbon (C5) and the allylic carbon (C4). Carbons 2 and 3 are offset into opposite directions from this plane, leading to 2T3-(TS2-cis-Ic, TS1-trans-Ic, and TS2-trans-Ic) and 2T3-conformers of tetrahydrofuran (TS1-cis-Ic). cis/trans-Diastereodifferentiation, in this model, occurs by rotating the vinyl substituent by 180 degrees about the bond between carbons 4 and 5. Positioning the vinyl group and carbon 3 on opposite sides of the twist plane prevents eclipsing of hydrogens at carbons 4 and 5, thus favoring transition structures TS1-cis-Ic and TS1-trans-Ic. Favored transition structures furthermore have the cyclohexyl-substituent in position 3 bound equatorially to the distorted tetrahydrofuran nucleus (Fig. 4). Positioning the cyclohexyl-substituent at carbon 3 axially leads to transannular repulsion between the hydrogen attached to carbon 5 and one of the axial cyclohexane hydrogens (TS2-cis-Ic). The lowest in energy transition structure modelled for the 2,4-trans-ring closure of trans-Ic (TS2-trans-Ic; Fig. 4, bottom right) shows an eclipsing of hydrogens at carbons 4 and 5.
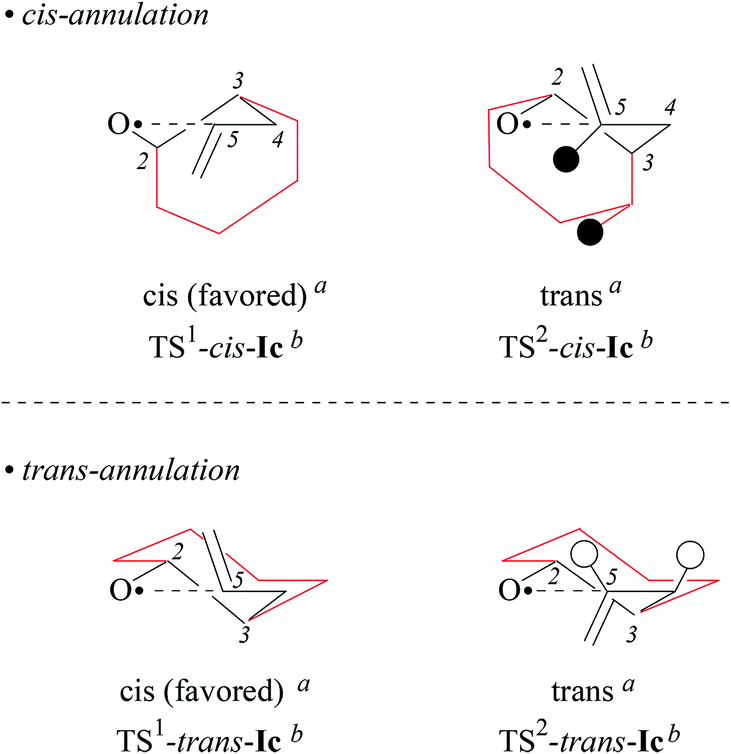 | ||
| Fig. 4 Modelled transition structures for 2,4-cis (TS1) and 2,4-trans (TS2)-cyclization of alkenoxyl radicals cis-Ic (top) and trans-Ic (bottom). Black circles represent hydrogens experiencing close contacts (dH,H = 2.423 Å; B3LYP/6-31+G**), open circles symbolize eclipsed hydrogens (H–C4–C3–H = 2.1°). aStereodescriptor referring to the configuration at carbons 2 and 4 in cyclized radical IIc (cf. Fig. 3). bStereodescriptor referring to the configuration at carbons 2 and 3 in TS-Ic. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1d | Conditions | 2/% |
3d/% (6,7-cis:trans) |
5d/% (1,2-cis:trans) |
| a Stereodescriptor referring to the configuration of bridgehead carbons in products 3d and 5d. | |||||
| 1 | cis | hν/25 °C | 71 | 1,6-cisa: 70 (20:80) |
1,6-cisa: 3 (99:1) |
| 2 | cis | AIBN/80 °C | 88 | 1,6-cisa: 81 (21:79) |
1,6-cisa: 5 (99:1) |
| 3 | trans | hν/25 °C | 78 | 1,6-transa: 57 (7:93) |
1,6-transa: 10 (50:50) |
| 4 | trans | AIBN/80 °C | 97 | 1,6-transa: 70 (10:90) |
1,6-transa: 17 (41:59) |
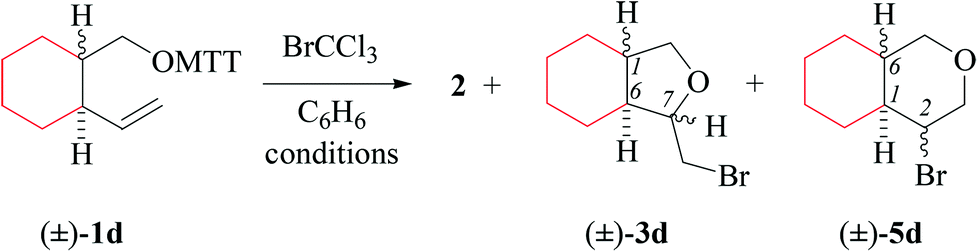
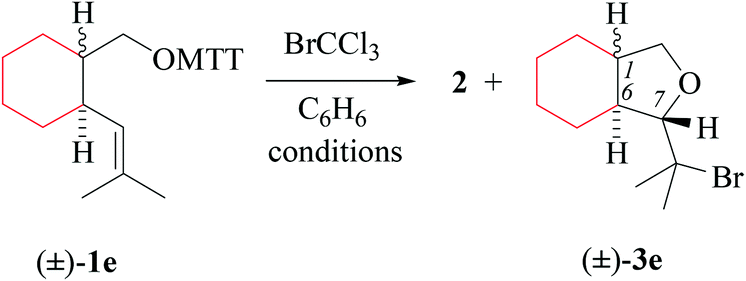
(i) Methods of alkenoxyl radical generation and product analysis. Thermally induced reactions between O-(2-vinylcycloalkyl-1-methyloxy) thiohydroxamates cis-1d/e and bromotrichloromethane furnish 81% of 7-bromomethyltetrahydrofuran cis-3d and 95% of bromoisopropyl derivative cis-3e (entry 2 in Tables 5 and 6). The former reaction provided in addition 5% of the diastereomerically pure 6-endo-cyclized product cis-5d, which was not obtained from dimethylvinyl-congener cis-1e (GC-MS).
Heating O-(2-vinylcycloalkyl-1-methyloxy) thiohydroxamate trans-1d in the presence of bromotrichloromethane furnishes an 80/20-mixture of 5-exo/6-endo-bromocyclized products trans-3d and trans-5d, whereas O-[2-(dimethylvinyl)-cyclohexylmethyl] ester trans-1e affords bromopropyltetrahydrofuran trans-3e as a single diastereomer (Tables 5 and 6, entry 4). Photochemical reactions gave 13–20% lower yields of bromocyclization products 3 and 5 taken together, and 8–19% less thiazole 2, than thermally initiated reactions (entries 1 and 3 in Tables 5 and 6).
(ii) Effect of methyl substitution at the terminal alkene carbon. Substituting two hydrogens at the terminal alkene carbon by methyl improves the stereoselectivity and regioselectivity in cyclization of type-B 4-pentenoxyl radicals (Tables 5 and 6). Terminal methyl groups furthermore improve the regioselectivity of the intramolecular addition, occurring with 80/20-selectivity for trans-Id, 94/6 for cis-Id, and 5-exo-specifically for cis/trans-Ie (GC-MS; Table 5, entries 2 and 4, and Table 6).
(iii) Stereochemical guidelines. 2,3-Cycloalkyl-bridged 4-pentenoxyl radicals Id–e cyclize 2,3-trans-selectively, indicating that the principal stereoinductor in cyclization of type-B radicals is the substituent in position 3 of the radical. Fusing 4-pentenoxyl radicals in relative trans-positions of cyclohexane enhances stereodifferentiation by the principal inductor.
(iv) On the origin of 2,3-trans-selectivity in 5-exo-cyclization of type-B 4-pentenoxyl radicals. Models built as instructed in section 2.1 show that type-B cyclohexyl-bridged 4-pentenoxyl radicals cis/trans-Id–e cyclize 2,3-trans-selectively, because steric constraints disfavor the 2,3-cis-mode of ring closure. In transition structures for 2,3-cis-cyclization, van der Waals repulsion between the (E)-positioned alkene substituent and the axially arranged hydrogens raises conformational free energy. The second aspect raising conformational free energy thus disfavoring a transition structure is eclipsing of hydrogens bound to carbons 4 and 5 (for TS2-trans-Id and TS2-cis-Id; Fig. 5). Extending the size of the (E)-substituent from hydrogen to methyl raises transannular repulsion, explaining the stereodirecting effect of a terminal substituent in cyclization of cis/trans-Ie.
 | ||
| Fig. 5 Transition structure models for explaining the origin of 2,3-trans-stereoselectivity in 5-exo-cyclization type-B pentenoxyl radicals, exemplified by favored intermediates TS1-cis/trans-Id and disfavored intermediates TS2-cis/trans-Id. Hydrogen atoms drawn as black circles give rise to 1,3-diaxial repulsion, ecliptically arranged hydrogens are drawn as open circles. aStereodescriptor referring to the configuration at carbons 2 and 3 in cyclized radical IId (cf. Fig. 3). bStereodescriptor referring to the configuration at carbons 3 and 4 in TS-Id. | ||
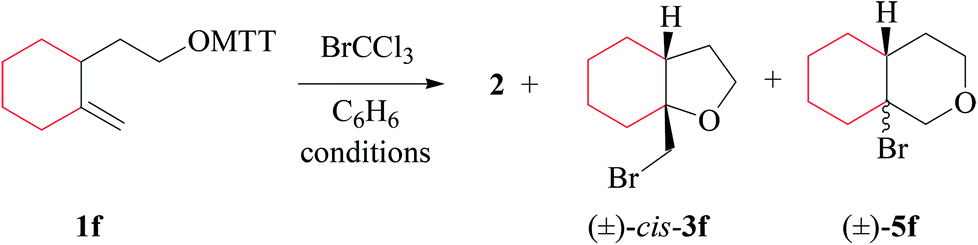
(i) Methods of alkoxyl radical generation and product analysis. 3-[2-(2-methylenecyclohexyl)-ethyloxy]-thiazolethione 1f furnishes the 5-exo-bromocyclized product cis-3f as a single diastereomer, bridgehead brominated oxadecalin 5f, and 2-(trichloromethylsulfanyl)-thiazole 2, when heated in the presence of bromotrichloromethane at 80 °C (Table 7, entry 2). The photochemical reaction provides a similar product pattern, although lower yields of compounds 2 and cis-3f. The fraction of bromotetrahydropyran 5f remained almost unchanged (Table 7, entry 1).
|
|
||||
|---|---|---|---|---|
| Entry | Conditions | 2/% | cis-3f/% |
5f/% (cis:trans) |
| 1 | hν/25 °C | 54 | 24 | 21 (19:81) |
| 2 | AIBN/80 °C | 80 | 35 | 22 (32:68) |
The 5-exo/6-endo-selectivity of radical If (53:47) at room temperature falls below the value reported for the 4-methyl-4-pentenoxyl radical (69:31) and is higher than the regioselectivity determined for the 4-tert-butyl-4-pentenoxyl radical (46:54).17 Regioselectivity in 4-pentenoxyl radical cyclization originates from a balance between FMO attractions, torsional strain, and steric shielding. A carbon substituent in position 4 lowers the barrier for 6-endo-addition based on favorable frontier molecular orbital (FMO) interactions for the C,O-addition to the terminal carbon. Steric blocking of the incoming oxygen radical gradually lowers the rate of 5-exo-addition as the size of the carbon substituent in position 4 increases. The fraction tetrahydropyranyl radical IVf obtained from 6-endo-cyclization of If is in line with the general mechanistic interpretation.17 Homolytic bromination of tetrahydropyranyl radical IVf occurs for steric reasons preferentially from the axial side (Scheme 4), similar to bromination of structurally related cyclohexyl radicals.53
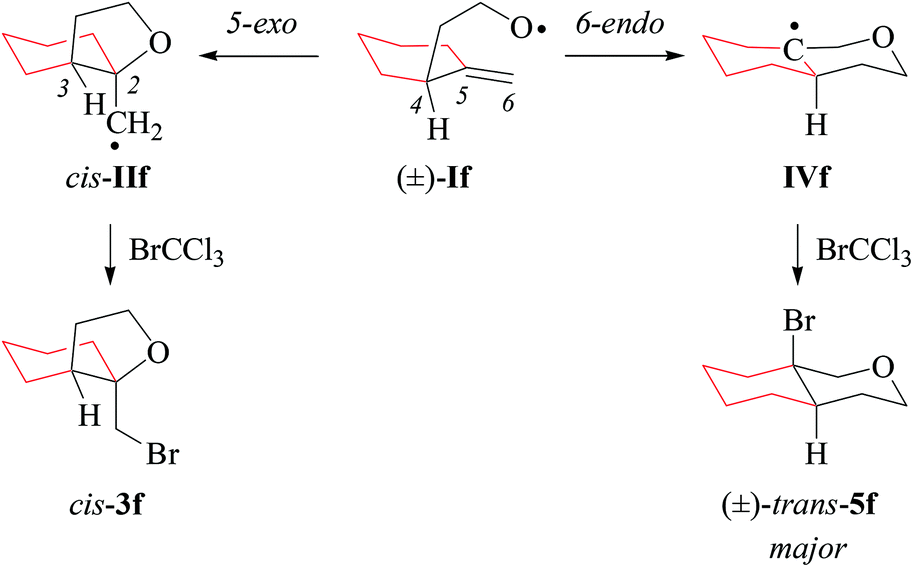 | ||
| Scheme 4 Stereoselectivity in cyclization of the 1-methylenecyclohexyl-2-ethyloxyl radical If (type C). | ||
(ii) Stereochemical guideline. Methylenecyclohexylethoxyl radical If cyclizes 2,3-cis-specifically (Scheme 4).
2.3 Cyclization onto endocyclic double bonds
For investigating the stereoselectivity in intramolecular addition to endocyclic double bonds we examined bromocyclization of 3-[2-(2-methylenecyclohexyl)-ethyloxy]-thiazolethione 1g (type D; section 3.1) and verbenylethanol-derived thiohydroxamate 1h (type E; section 3.2).|
|
|||
|---|---|---|---|
| Entry | Conditions | 2/% |
3g/% (1,2-cis:trans) |
| a Protons experiencing deshielding by changing the position of the carbon–bromine bond from anti to syn, and vice versa, are printed in blue. | |||
| 1 | hν/25 °C | 69 | 64 (28:72) |
| 2 | AIBN/80 °C | 76 | 72 (19:81) |
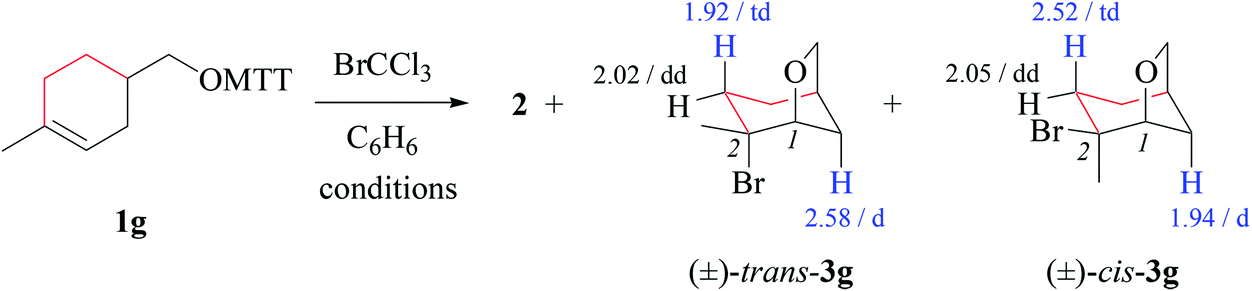
Bicyclic tetrahydrofuran 3g forms at 80 °C as a 19/81-mixture of 1,2-cis/trans-stereoisomers. Resonances of protons in β- and γ-positions to the carbon–bromine bond experience a shift dispersion by ∼0.5 ppm upon changing orientation of the bromosubstituent from anticlinal or antiperiplanar to synclinal (Table 8). We address this phenomenon to magnetic anisotropy induced by the carbon–bromine bond, possibly in combination with three nonbonding electron pairs at bromine.54
(ii) Stereochemical guideline. Cyclohexenylmethyloxyl radical 1g cyclizes 2,4-cis-specifically (Scheme 5).
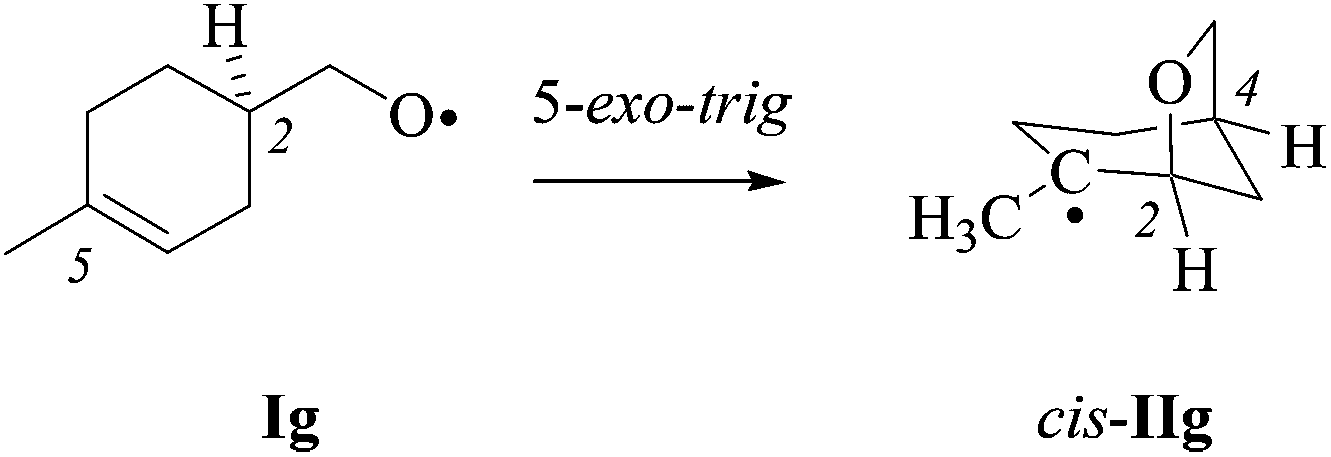 | ||
| Scheme 5 2,4-cis-cyclization of alkenoxyl radical Ig (type D; from 1g). | ||
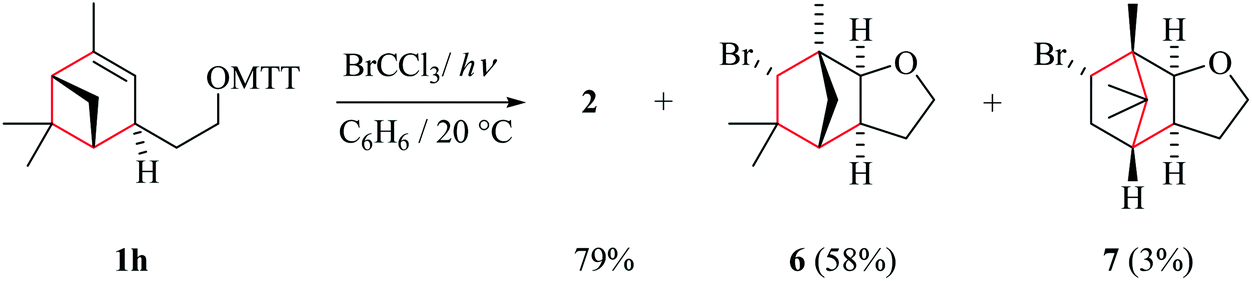 | ||
| Scheme 6 Formation of bicyclic products from 3-(verbenylethyloxy)-thiazole-2(3H)-thione 1h. | ||
(ii) Stereochemical guideline. Cyclohexenylethyloxyl radical Ih cyclizes cis-specifically (Scheme 7).
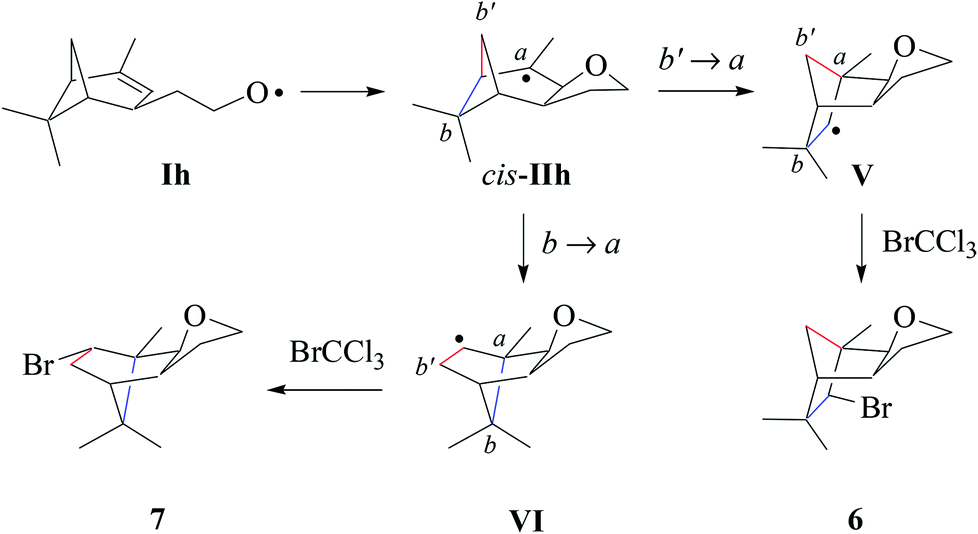 | ||
| Scheme 7 Proposed pathways for product formation from verbenylethyloxyl radical Ih (type E; from 1h). | ||
(iii) Verbenylethyloxyl radical chemistry. In extension to the chemistry summarized in this article, we propose that tricyclic products 6 and 7 arise from a sequence composed of intramolecular addition Ih → cis-IIh, ring-opening of cyclobutylmethyl radical cis-IIh, and bromine atom trapping by rearranged radicals V and VI (Scheme 7). 1,2-Shifting of the methylene bridge releases cyclobutyl strain in radical cis-IIh, leading to the secondary carbon radical V. For steric reasons, we expect trapping of the bicyclic radical V by bromotrichloromethane to occur from the concave face due to shielding of the convex side with the vicinal exo-oriented methyl group. The minor product 7, according to the proposed model, results from 1,2-shifting of the dimethylmethylene bridge cis-IIh → VI and subsequent homolytic bromination.
2.4 Strain effects in alkoxyl radical additions
For estimating differences in energy barriers associated with 2,3-cis- and 2,3-trans-cyclization of type-E alkenoxyl radicals, we modelled energetics associated with 5-exo-cyclization using electronic structure methods.55,56 The 2-(cyclohexen-3-yl)-ethyloxyl radical Ii, in this approach, served as a truncated model for the verbenyl-4-ethyloxyl radical Ih, while the 4-pentenoxyl radical 5-exo-cyclization Ij → IIj and the methoxyl radical addition to the inner carbon of propene served as references (Schemes 8 and 9).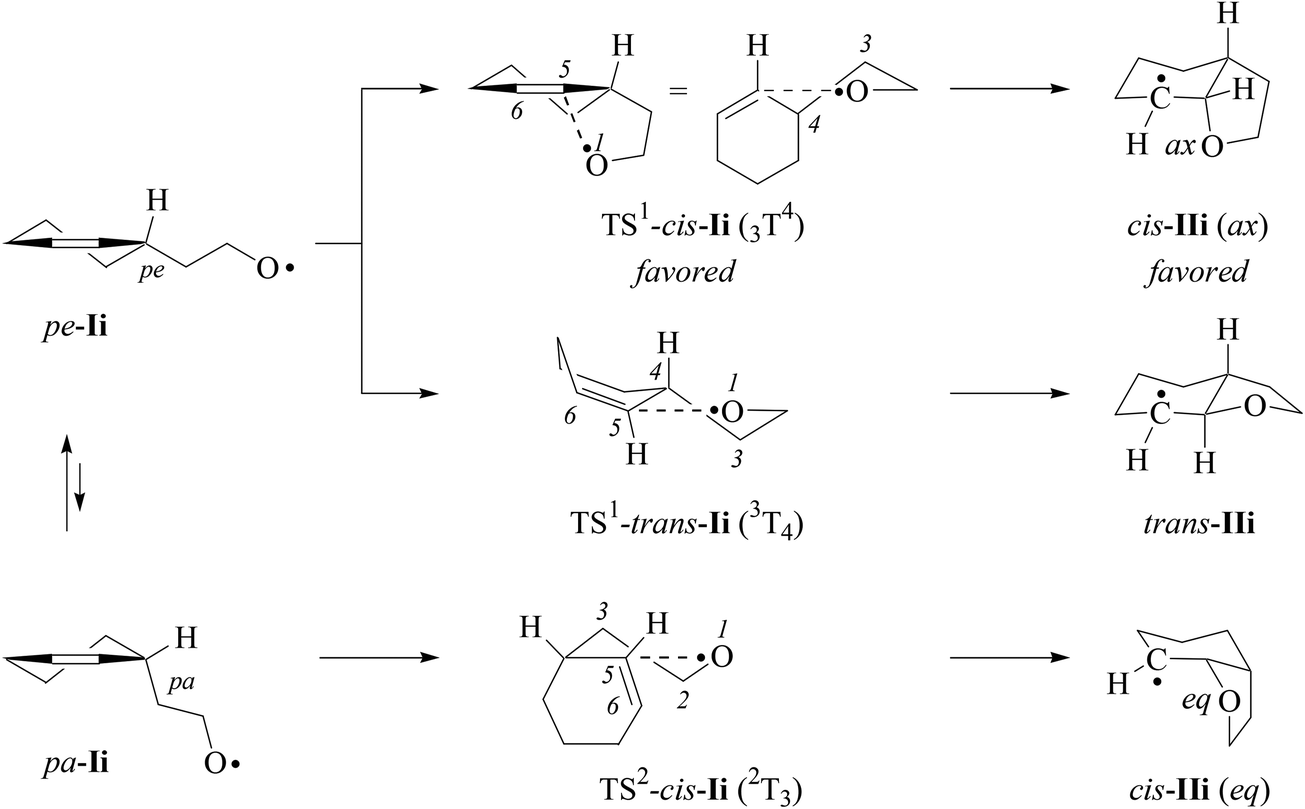 | ||
| Scheme 8 Structure formulas of radicals and intermediates associated with the 1-(cyclohexen-3-yl)-ethyl-2-oxyl radical 5-exo-cyclization (pe = pseudo-equatorial; pa = pseudo-axial; ax = axial; eq = equatorial). | ||
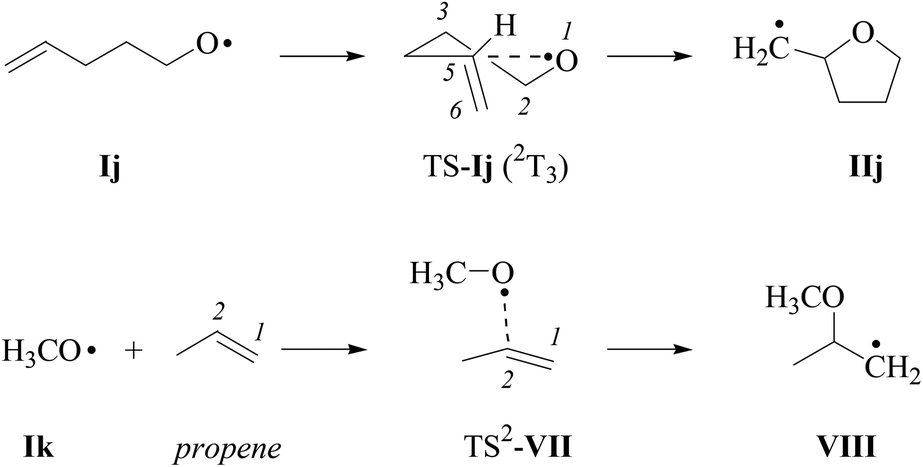 | ||
| Scheme 9 Structure formulas of radicals and intermediates associated with the 4-pentenoxyl radical 5-exo-cyclization (top) and methoxyl radical addition to the inner carbon of propene (bottom; for discussion of TS1-VII, refer to the text and the ESI‡). | ||
(i) Density functional theory. For computing ground state energies of radicals and energies of transition structures, we used Becke's three parameter Lee–Young–Parr-hybrid functional (B3LYP)57,58 and Becke's half and half Lee–Young–Parr hybrid functional (BHandHLYP)59 in combination with 6-31+G**- and 6-311G**-basis sets.56 All selected density functional/basis set-combinations reproduce experimental stereo- and regioselectivity for oxygen radical addition to carbon–carbon double bonds with a precision coming close to the accuracy for determining experimental selectivity.8,14,17,52,60
(ii) Theoretical approach. For calculating equilibrium structures of conformational flexible molecules and transition structures associated with radical addition to carbon–carbon double bonds we used an established strategy.8,14 According to theory, the 2-(cyclohexen-3-yl)-ethyloxyl radical Ii favors pseudo-equatorial (pe) positioning of the ethyloxyl radical side to pseudo-axial (pa), as expressed by a modelled 90/10-mixture of pe/pa-conformers of Ii at 298 K (B3LYP/6-31+G**; ESI‡). Both conformers served as starting points for modeling 5-exo-cyclizations.
Equilibrium structures of propene, alkoxyl radicals Ii–k, cyclized radicals IIi–j, and the addition product VIII lack in negative eigenvalues of second derivatives of energy-minimized wavefunctions. Transition structures TS-I and TS-VII show one imaginary frequency i, describing the trajectory of oxygen radical addition to the inner alkene carbon (Table 9).61 Attempts to localize a transition structure for the trans-5-exo-cyclization of conformer pa-Ii led to TS1-trans-Ii, already available from conformer pe-Ii.
| I | ib/cm−1 | d = O1–C5c/Å | α = O1–C5–C6d/° | ω = H5–C4–C5–C6e/° |
|---|---|---|---|---|
| a B3LYP/6-31+G**-calculated values; numbers in parentheses arise from BHandHLYP/6-31+G**-calculations and values in brackets from BHandHLYP/6-311G**-calculations. b i = imaginary mode of vibration. c O–C2 for TS2-VII. d O–C2–C1 for TS2-VII. e C–C2–H2–C1 for TS2-VII. f For the transition structure TS1-VII, refer to the ESI. | ||||
| TS1-cis-Ii (3T4) | −342 | 2.077 | 103.5 | 161.6 |
| (−496) | (2.024) | (103.9) | (159.4) | |
| [−529] | [2.018] | [103.0] | [158.5] | |
| TS1-trans-Ii (3T4) | −397 | 2.042 | 122.7 | 152.9 |
| (−547) | (2.003) | (121.6) | (152.2) | |
| [−575] | [1.986] | [121.3] | [151.5] | |
| TS2-cis-Ii (2T3) | −353 | 2.058 | 100.8 | −160.5 |
| (−500) | (2.010) | (101.5) | (−158.6) | |
| [−531] | [1.990] | [101.6] | [−157.6] | |
| TS-Ij | −382 | 2.046 | 99.1 | 162.1 |
| (−531) | (1.996) | (100.4) | (159.9) | |
| [−558] | [1.997] | [100.5] | [159.0] | |
| TS2-VIIf | −334 | 2.061 | 98.3 | 161.2 |
| (−489) | (1.999) | (100.6) | (158.6) | |
| [−517] | [1.979] | [100.8] | [157.8] | |
(iii) Quality of the models. Computed wavefunctions characterizing equilibrium structures show expectation values for the spin operator 〈S2〉 close to 0.75 for oxygen and carbon radicals (ESI‡), as expected for doublet states. Wavefunctions describing transition structures show 〈S2〉-values of ∼0.77 for B3LYP-calculated intermediates and 0.82–0.84 for BHandHLYP-calculated transition structures (ESI‡). The effect of spin contamination in BHandHLYP-calculated transition structures was discussed previously, but is not considered relevant for attaining reasonable precision in determining computed relative energies.52
(iv) Methoxyl radical addition to propene. Theory predicts a lower barrier for methoxyl radical addition to the terminal carbon than for addition to the inner carbon of propene (ΔG298 = −5.0 to −8.5 kJ mol−1; ESI‡). The decision to compare structure and energetics from the disfavored mode of addition to data obtained for monocycle and bicycle formation was guided by structural similarity between TS-Ii–j and TS-VII on one side, and derived addition products IIi–j, VIII on the other (Table 9, Schemes 8 and 9).
(v) Thermochemistry. Cyclization of the 2-(cyclohexen-3-yl)-ethyloxyl radical Ii → IIi, according to zero-point energy corrected reaction energies (B3LYP/6-31+G**), is for all considered pathways strongly exothermic (ΔRE = −35 to −47 kJ mol−1), pointing to a notable barrier for the reverse reaction, the β-fragmentation. Computed energetics for the addition Ii → IIi are similar to the values calculated for the 4-pentenoxyl radical ring closure Ij → IIj (ΔRE = −41 kJ mol−1), and are less exothermic than the methoxyl radical addition to the inner carbon of propene (ΔRE = −53 mol−1). BHandHLYP-calculations provide similar trends for reaction energies, except for a stronger driving force for the intermolecular addition (Table 10).
| Reaction | Method | ΔE‡/kJ mol−1 | ΔRE/kJ mol−1 | ΔE‡i/kJ mol−1 | ΔE‡TD/kJ mol−1 | x ‡ |
|---|---|---|---|---|---|---|
| pe-Ii → cis-IIi (ax) | B3LYP/6-31+G** | 16.7 | −44.1 | 35.4 | −18.6 | 0.2 |
| BHandHLYP/6-31+G** | 39.3 | −46.4 | 60.3 | −21.0 | 0.4 | |
| BHandHLYP/6-311G** | 40.6 | −44.7 | 60.9 | −20.3 | 0.4 | |
| pe-Ii → trans-IIi | B3LYP/6-31+G** | 73.1 | −34.5 | 89.5 | −16.4 | 0.4 |
| BHandHLYP/6-31+G** | 99.1 | −35.7 | 116.3 | −17.2 | 0.5 | |
| BHandHLYP/6-311G** | 100.7 | −33.7 | 116.9 | −16.2 | 0.5 | |
| pa-Ii → cis-IIi (eq) | B3LYP/6-31+G** | 17.4 | −46.8 | 37.1 | −19.7 | 0.2 |
| BHandHLYP/6-31+G** | 39.5 | −50.4 | 62.2 | −22.7 | 0.3 | |
| BHandHLYP/6-311G** | 40.9 | −49.0 | 63.1 | −22.1 | 0.4 | |
| Ij → IIj | B3LYP/6-31+G** | 19.8 | −40.5 | 37.3 | −17.5 | 0.2 |
| BHandHLYP/6-31+G** | 41.0 | −43.5 | 60.8 | −19.8 | 0.4 | |
| BHandHLYP/6-311G** | 41.7 | −41.8 | 60.8 | −19.1 | 0.4 | |
| Ik + propene → VIII | B3LYP/6-31+G** | 20.5 | −53.8 | 43.2 | −22.7 | 0.2 |
| BHandHLYP/6-31+G** | 37.3 | −61.2 | 64.2 | −27.0 | 0.3 | |
| BHandHLYP/6-311G** | 36.3 | −61.0 | 63.1 | −26.8 | 0.3 |
(vi) Transition structures. The distance d between the radical oxygen and the attacked carbon, as predicted by B3LYP theory for transition structures of cyclohexenylethyloxyl radical cyclization (2.04–2.08 Å), 4-pentenoxyl radical cyclization (2.05 Å) and methoxyl radical addition to propene (2.06 Å), is marginally wider than those obtained from BHandHLYP-calculations (1.98–2.02 Å; Table 9). Values for the angle α describing oxygen radical attack to the inner alkene carbon are grouped for all calculated transition structures in the range between 98 and 104 degrees, being more acute than the angle calculated for the highest in the energy transition structure TS-trans-Ii (3T4) (121–122 degrees; Table 9). Absolute values of improper torsion angles ω for transition structures TS-Ii, TS-Ij, and TS-VIII, according to B3LYP- and BHandHLYP-theory, are close to 160 degrees, indicating the hybridization change at the attacked carbon from sp2 (ω = 180° for propene) toward sp3 (122° for propane).
Superimposing atomic coordinates of 4-pentenoxyl radical cores illustrates that density functional-calculated favored transition structures for the cyclohexenylethyloxyl radical cyclization and the 4-pentenoxyl radical ring closure are nearly identical (Fig. 6). A 3T4-conformation, as predicted by theory for TS-cis-Ii (3T4), is separated by only 36 degrees from a 2T3-conformer in TS-Ij (2T3) on the pseudorotatory cycle of tetrahydrofuran.62
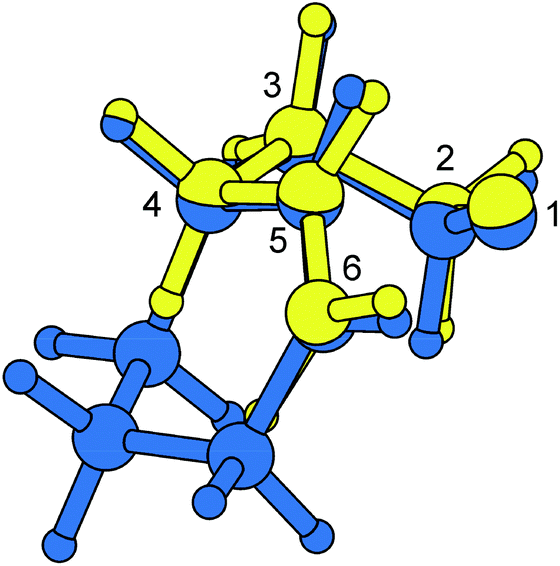 | ||
| Fig. 6 Match plot of B3LYP/6-31+G**-computed transition structures TS-cis-Ii (3T4) (blue) and TS-Ij (2T3) (yellow). | ||
(vii) Energy barriers. The barrier for 2,3-cis-cyclization of cyclohexenylethyloxyl radical Ii (ΔE‡ = 17 kJ mol−1) is similar to the barrier predicted by B3LYP-theory for the 4-pentenoxyl radical 5-exo-cyclization (20 kJ mol−1) and the methoxyl radical addition to propene (21 kJ mol−1). BHandHLYP-computed barriers for 2,3-cis-cyclization of Ii and 5-exo-cyclization of Ij are higher, but generally show the same trends (Table 10).
The computed relative Gibbs free energy of activation for the 2,3-trans-mode of cyclization is 55 kJ mol−1 above the value for the lowest in the energy pathway of 2,3-cis-ring closure (B3LYP; 58 kJ mol−1 for BHandHLYP calculations using either the 6-31+G** or the 6-311G** basis set; ESI‡). A Gibbs free activation energy difference of 55 kJ mol−1 translates for a kinetically controlled reaction and a temperature of 298.15 K into a relative rate constant of 4 × 109 in favor of the 2,3-cis-cyclization. Detecting a 2,3-trans-bromocyclized product with such a precision was beyond the capability of analytic instruments used in the study.
(viii) Marcus analysis. For analyzing strain and electronic effects on barriers of 5-exo-alkenoxyl radical cyclization, we split zero-point vibrational energy-corrected electronic barriers (ΔE‡) into an intrinsic (ΔE‡i) and a thermodynamic term (ΔE‡TD), using Marcus theory (Fig. 7, Table 10, eqn (1)–(3)).63–65 The intrinsic part describes contributions of strain and steric repulsion in a thermoneutral degenerated reaction to the barrier ΔE‡i in a transition structure located half way on the reaction coordinate (x‡ = 0.5) between reactant(s) (x = 0) and product(s) (x = 1; Fig. 7). The thermodynamic part of the barrier ΔE‡TD describes energy changes arising from incipient bond forming and bond breaking in a transition structure.
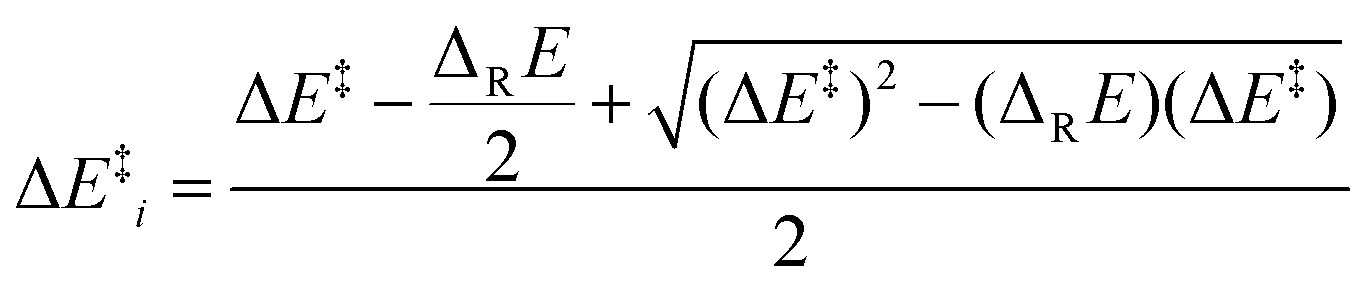 | (1) |
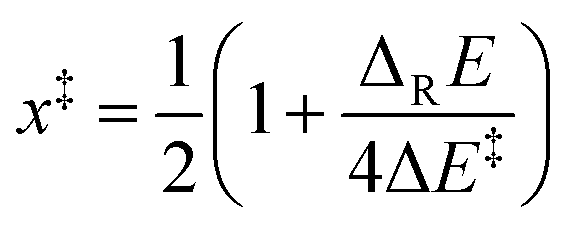 | (2) |
| ΔE‡ = ΔE‡i + ΔE‡TD | (3) |
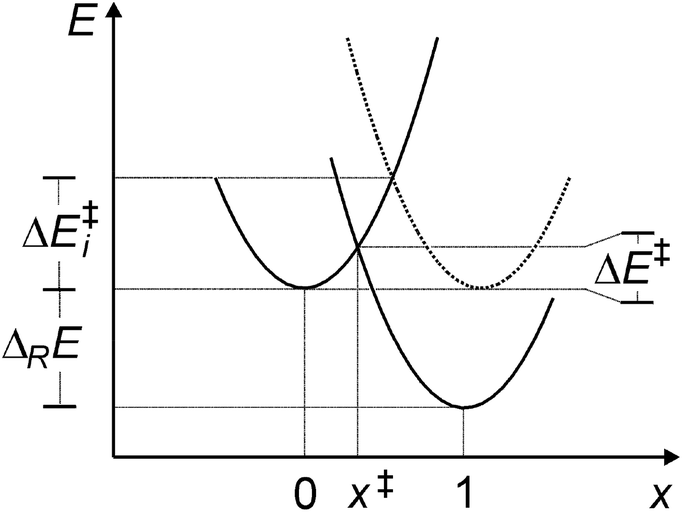 | ||
| Fig. 7 Potential energy curves E(x) for reaction associated with an energy change ΔRE across a barrier ΔE‡, having an intrinsic barrier ΔE‡i, according to Marcus-theory, and harmonic potentials of the identical curvature for the initial (x = 0) and final state (x = 1). | ||
(ix) Localizing transition structures – the x‡-value. According to Marcus theory, reaction energies and barriers obtained from B3LYP-calculations translate into a x‡-value of 0.2 for positioning transition structures on the respective reaction coordinates for C,O-addition (eqn (2)). Transition structures predicted from BHandHLYP-calculated energies are positioned later on the same reaction coordinate (x‡ = 0.3–0.5), due to higher barriers ΔE‡. Considering the magnitude of the experimental rate constant kadd = 5 × 108 s−1 for the 4-pentenoxyl 5-exo-cyclization Ij → IIj2 and computed significant reaction energies, we expect transition structures of alkoxyl radical additions in extension to the Hammond-postulate66 to be localized early on a reaction coordinate, rather than midway between reactant I and product II.
(x) The role of the thermodynamic barrier. In transition structures associated with alkoxyl radical addition to alkenes, incipient carbon–oxygen bond formation and carbon–carbon π-bond breaking in summary is exothermic, lowering the intrinsic barrier by a thermodynamic contribution of −19 to −20 kJ mol−1. This thermodynamic barrier ΔE‡TD is surprisingly similar for 2,3-cis- and 2,3-trans-ring cyclization of cyclohexenylethyloxyl radical Ii (−16 to −20 kJ mol−1), the 4-pentenoxyl radical 5-exo-cyclization Ij → IIj (−18 kJ mol−1; B3LYP/6-31+G**), and the barrier for methoxyl radical addition to the inner carbon of propene (−23 kJ mol−1). BHandHLYP-computed energies lead to more negative thermodynamic barriers, but show otherwise similar trends. From the data we concluded that the thermodynamic barrier is not the key parameter for explaining the experimental 2,3-cis-specificity of verbenylethyloxyl radical cyclization.
(xi) The role of the intrinsic barrier. Intrinsic barriers modelled for 2,3-cis-cyclization of cyclohexenylethyloxyl radical Ii (ΔE‡i = 35–37 kJ mol−1, B3LYP; for BHandHLYP-calculated values, refer to Table 10) and 5-exo-cyclization of 4-pentenoxyl radical Ij (37 kJ mol−1) are marginally smaller than the intrinsic barrier for methoxyl radical addition to the inner carbon of propene (43 kJ mol−1). An intrinsic barrier of 90 kJ mol−1 predicted for 2,3-trans-cyclization of the cyclohexenylethyloxyl radical Ii exceeds the value for the barriers of all other investigated oxygen radical additions in the study by far. From this information we concluded that the experimentally observed 2,3-cis-stereospecificity of the verbenylethyloxyl radical cyclization originates from a large intrinsic barrier associated with the 2,3-trans-ring closure.
3. Concluding remarks
Cycloalkyl-fused and -bridged 4-pentenoxyl radicals provide bicyclic and tricyclic tetrahydrofurans by 5-exo-cyclizations. The selectivity determining step is the intramolecular oxygen radical addition to the carbon–carbon double bond, occurring in most instances with notable stereochemical preference. From the observed stereoselectivities we concluded that a system exists, which can be summed up by two new directives for predicting the stereochemical outcome of similar cyclizations not exemplified in this article. The new guidelines supplement the set of existing directives, developed for predicting the major products in synthesis of monocyclic disubstituted tetrahydrofurans by the oxygen radical method.7,14,67The first of the new guidelines ranks the hierarchy of two similarly sized stereoinductors by the distance between the alkyl group and the alkene carbon which is being approached by the oxygen radical. This guideline states that the substituent positioned the closest to the attacked alkene carbon is the principal (primary) inductor, guiding 5-exo-cyclization 2,3-trans- and 2,4-cis-selectively. The substituent bound further from the attacked π-bond is the secondary inductor, enhancing stereodifferentiation exerted by the principal inductor in the case of the trans-configuration, and decreasing this effect in the case of the cis-configuration. A secondary inductor is not able to overrule the guiding effect of a similarly sized primary inductor. The first guideline applies to 5-exo-cyclization of type-A and type-B 4-pentenoxyl radicals (Fig. 1).
The second new directive states that 4-pentenoxyl radical 5-exo-cyclization to a cyclohexene-bound exo-methylene group or an endocyclic double bond occurs cis-specifically. The second guideline refers to intramolecular addition of type-C–E radicals (Fig. 1).
From the hierarchy of similar-sized inductors we expect a substituent located in position 3 to also control the stereoselectivity in 5-exo-cyclization of 4-pentenoxyl radicals having similar sized substituents attached to carbons 1, 2, and 3. According to the first new guideline, a group in position 2 will be secondary and a group in position 1 the least effective, the tertiary inductor. From today's point of view we expect the stereoisomer of a 1,2,3-substituted 4-pentenoxyl radical corresponding to an all-trans-configured type-A and type-B radical to cyclize with notable 2,3-trans-, 2,4-cis-, and 2,5-trans-selectivity, possibly providing a single diastereomer. In the same model, a sterically more demanding substituent in position 2, for example tert-butyl, should be able to overrule the effect of a smaller group in position 3, such as methyl. Stereochemical questions of this kind attracted our attention and are being pursued at the moment in our laboratory, with the aim to provide new solutions to synthesis of functionalized ethers from oxygen radical addition to alkenes.
4. Experimental
4.1. General
For general laboratory practice and instrumentation see ref. 42 and the ESI.‡4.2. General methods
4.3 3-Alkenyloxy-4-methylthiazole-2(3H)-thiones
:1 (v/v), Rf = 0.51. Yield: 2.67 g (10.5 mmol, 70%), colorless oil.361H-NMR (CDCl3, 600 MHz) δ 1.60–1.75 (m, 3 H), 1.80–1.84 (m, 1 H), 1.87–1.93 (m, 2 H), 2.03–2.06 (m, 1 H), 2.22 (d, J = 1.2 Hz, 3 H), 2.22–2.27 (m, 1 H), 2.67–2.71 (m, 1 H), 4.99–5.01 (m, 1 H), 5.08 (dq, Jd = 17.0, Jq = 1.7 Hz, 1 H), 5.74–5.77 (m, 1 H), 5.98–6.05 (m, 1 H), 6.16 (q, J = 0.9 Hz, 1 H). 13C-NMR (CDCl3, 150 MHz) δ 13.8, 21.8, 29.2, 29.3, 30.1, 45.2, 88.2, 102.9, 115.4, 137.9, 139.2, 181.0. UV (methanol): λmax (lgε/m2 mol−1) 319 nm (3.14), 210 nm (3.05). Anal. Calcd for C12H17NOS2 (255.40): C, 56.43; H, 6.71; N, 5.48; S, 25.11; Found: C, 56.48; H, 6.56; N, 5.68; S, 25.27.
:1 (v/v), Rf = 0.38. Yield: 1.13 g (4.42 mmol, 67%), yellow oil.361H-NMR (CDCl3, 400 MHz) δ 1.28–1.37 (m, 1 H), 1.67–1.84 (m, 3 H), 1.86–1.93 (m, 1 H), 1.98–2.09 (m, 1 H), 2.21–2.30 (m, 2 H), 2.24 (d, J = 1.2 Hz, 3 H), 2.22–2.27 (m, 1 H), 4.97–5.05 (m, 2 H), 5.43–5.46 (m, 1 H), 5.78 (ddt, Jd = 17.0, 10.2, Jt = 6.8 Hz, 1 H), 6.16 (q, J = 1.4 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 14.1, 23.2, 30.4, 30.6, 37.6, 43.7, 92.1, 102.9, 116.3, 136.4, 138.8, 181.0. UV (methanol): λmax (lgε/m2 mol−1) 319 nm (3.17), 208 nm (3.09). Anal. Calcd for C12H17NOS2 (255.40): C, 56.43; H, 6.71; N, 5.48; S, 25.11; Found: C, 56.49; H, 6.85; N, 5.46; S, 24.96.
:2 (v/v), Rf = 0.30. Yield: 1.49 g (5.26 mmol, 65%), colorless oil. 1H-NMR (CDCl3, 400 MHz) δ 1.59–1.76 (m, 3 H), 1.64 (s, 3 H), 1.71 (s, 3 H), 1.77–2.03 (m, 4 H), 2.18–2.30 (m, 1 H), 2.23 (d, J = 1.4 Hz, 3 H), 2.47–2.59 (m, 1 H), 5.33 (ddt, Jd = 7.9, 6.5, Jt = 1.5 Hz, 1 H), 5.72 (td, Jt = 4.5, Jd = 1.7 Hz, 1 H), 6.15 (d, J = 1.4 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 13.8, 17.9, 21.9, 25.8, 27.1, 29.1, 29.4, 46.0, 88.5, 102.8, 123.5, 132.0, 139.2, 181.0. UV (methanol): λmax (lgε/m2 mol−1) 318 nm (3.14), 205 nm (3.16). Anal. Calcd for C14H21NOS2 (283.45): C, 59.32; H, 7.47; N, 4.94; S, 22.62; Found: C, 59.33; H, 7.38; N, 5.03; S, 22.47.
:2 (v/v), Rf = 0.28. Conversion 84%. Yield: 637 mg (2.36 mmol, 11%), colorless oil. 1H-NMR (CDCl3, 600 MHz) δ 1.23–1.49 (m, 4 H), 1.54–1.83 (m, 4 H), 2.09–2.38 (m, 2 H), 2.25 (s, 3 H), 2.55–2.62 (m, 1 H), 4.99–5.08 (m, 2 H), 5.25–5.32 (m, 1 H), 5.73–5.84 (m, 1 H), 6.16 (d, J = 1.2 Hz, 1 H). 13C-NMR (CDCl3, 150 MHz) δ 14.2, 20.4, 23.8, 26.1, 26.7, 30.5, 37.5, 85.2, 102.8, 116.1, 137.2, 139.1, 180.7. UV (methanol): λmax (lgε/m2 mol−1) 318 nm (3.18), 210 nm (3.12). Anal. Calcd for C13H19NOS2 (269.42): C, 57.96; H, 7.11; N, 5.20; S, 23.80; Found: C, 57.94; H, 7.34; N, 5.47; S, 23.73.
:2 (v/v), Rf = 0.32. Yield: 412 mg (1.53 mmol, 28%), colorless oil, which crystallizes on standing at 20 °C. M.p. = 62–63 °C. 1H-NMR (CDCl3, 400 MHz) δ 1.03–1.13 (m, 1 H), 1.14–1.30 (m, 2 H), 1.32–1.44 (m, 1 H), 1.61–1.73 (m, 2 H), 1.73–1.84 (m, 2 H), 1.87–1.96 (m, 1 H), 2.13 (dt, Jd = 14.3, Jt = 8.3 Hz, 1 H), 2.24 (d, J = 1.2 Hz, 3 H), 2.83 (dddd, J = 14.1, 4.8, 3.4, 1.9 Hz, 1 H), 4.94–5.15 (m, 3 H), 5.79–5.97 (m, 1 H), 6.15 (d, J = 1.2 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 14.1, 24.2, 25.0, 30.19, 30.23, 36.4, 41.8, 86.0, 102.8, 116.3, 136.8, 139.2, 180.7. UV (methanol): λmax (lgε/m2 mol−1) 318 nm (3.14), 211 nm (3.04). Anal. Calcd for C13H19NOS2 (269.42): C, 57.96; H, 7.11; N, 5.20; S, 23.80; Found: C, 58.16; H, 7.16; N, 5.19; S, 23.67.
:2 (v/v), Rf = 0.33. Yield: 1.03 g (3.81 mmol, 75%), colorless oil, which crystallizes on standing at −18 °C. M.p. = 61–62 °C. 1H-NMR (CDCl3, 400 MHz) δ 1.34–1.65 (m, 6 H), 1.67–1.79 (m, 2 H), 2.16–2.22 (m, 1 H), 2.23 (d, J = 1.2 Hz, 3 H), 2.50 (dq, Jd = 8.3, Jq = 4.1 Hz, 1 H), 4.18 (dt, Jd = 23.0, Jt = 7.1 Hz, 2 H), 4.98–5.12 (m, 2 H), 6.02 (ddd, J = 17.1, 10.4, 8.4 Hz, 1 H), 6.14 (d, J = 1.2 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 13.5, 22.2, 24.3, 25.5, 30.5, 38.5, 40.9, 78.2, 102.6, 115.8, 137.7, 138.4, 180.2. UV (methanol): λmax (lgε/m2 mol−1) 316 nm (3.19), 206 nm (3.11). Anal. Calcd for C13H19NOS2 (269.42): C, 57.96; H, 7.11; N, 5.20; S, 23.80; Found: C, 57.89; H, 6.94; N, 5.20; S, 23.44.
:2 (v/v), Rf = 0.33. Yield: 466 mg (1.73 mmol, 79%), yellow oil, which crystallizes from diethyl ether to afford colorless crystals. M.p. = 51–52 °C. 1H-NMR (CDCl3, 400 MHz) δ 1.14–1.41 (m, 4 H), 1.65–1.82 (m, 4 H), 1.84–1.96 (m, 1 H), 2.14–2.29 (m, 1 H), 2.24 (d, J = 1.0 Hz, 3 H), 4.10 (t, J = 7.6 Hz, 1 H), 4.41 (dd, J = 7.3, 3.4 Hz, 1 H), 4.97–5.04 (m, 2 H), 5.71 (ddd, J = 17.2, 10.0, 9.1 Hz, 1 H), 6.13 (d, J = 0.7 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 13.5, 25.4, 25.6, 29.5, 33.5, 41.0, 44.6, 79.3, 102.6, 114.9, 137.7, 142.2, 180.3. UV (methanol): λmax (lgε/m2 mol−1) 317 nm (3.13), 208 nm (3.05). Anal. Calcd for C13H19NOS2 (269.42): C, 57.96; H, 7.11; N, 5.20; S, 23.80; Found: C, 57.92; H, 7.05; N, 5.20; S, 23.70.
:2 (v/v), Rf = 0.31. Yield: 761 mg (2.56 mmol, 78%), colorless oil, colorless crystals on standing at 20 °C. M.p. = 78 °C. 1H-NMR (CDCl3, 400 MHz) δ 1.29–1.56 (m, 6 H), 1.62 (d, J = 1.5 Hz, 3 H) 1.67–1.81 (m, 2 H), 1.71 (d, J = 1.5 Hz, 3 H), 2.11–2.25 (m, 1 H), 2.21 (d, J = 1.4 Hz, 3 H), 2.71–2.80 (m, 1 H), 4.15 (d, J = 6.8 Hz, 2 H), 5.35 (ddt, Jd = 9.8, 3.0, Jt = 1.5 Hz, 1 H), 6.13 (q, J = 1.4 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 13.1, 18.0, 21.9, 24.8, 25.0, 26.2, 31.8, 34.3, 38.9, 78.9, 102.5, 123.5, 132.9, 137.8, 180.3. UV (methanol): λmax (lgε/m2 mol−1) 316 nm (3.10), 204 nm (3.18). Anal. Calcd for C15H23NOS2 (297.48): C, 60.56; H, 7.79; N, 4.71; S, 21.55; Found: C, 60.43; H, 7.68; N, 4.76; S, 21.43.
:2 (v/v), Rf = 0.31. Yield: 1.86 g (6.25 mmol, 74%), yellow oil, which was crystallized from ethyl acetate to afford a colorless solid. M.p. = 47–48 °C. 1H-NMR (CDCl3, 400 MHz) δ 1.01–1.15 (m, 1 H), 1.20–1.40 (m, 3 H), 1.54–1.86 (m, 4 H), 1.57 (d, J = 1.0 Hz, 3 H), 1.67 (d, J = 0.7 Hz, 3 H), 1.98–2.11 (m, 1 H), 2.14–2.29 (m, 1 H), 2.22 (d, J = 1.2 Hz, 3 H), 4.07 (t, J = 7.6 Hz, 1 H), 4.35 (dd, J = 7.3, 3.2 Hz, 1 H), 4.97 (dt, Jd = 9.3, Jt = 1.5 Hz, 1 H), 6.12 (d, J = 1.5 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 13.2, 18.1, 25.6, 25.7, 25.8, 29.9, 33.4, 38.7, 42.1, 79.5, 102.5, 128.8, 131.7, 137.8, 180.2. UV (methanol): λmax (lgε/m2 mol−1) 317 nm (3.13), 209 nm (3.02). Anal. Calcd for C15H23NOS2 (297.48): C, 60.56; H, 7.79; N, 4.71; S, 21.55; Found: C, 60.41; H, 7.73; N, 4.74; S, 21.47.
:1 (v/v), Rf = 0.39. Yield: 2.57 g (9.54 mmol, 78%), yellow oil. 1H-NMR (CDCl3, 400 MHz) δ 1.25–1.38 (m, 1 H), 1.42–1.55 (m, 2 H), 1.56–1.74 (m, 2 H), 1.76–1.89 (m, 2 H), 2.03 (ddd, J = 13.0, 8.5, 4.5 Hz, 1 H), 2.14 (td, Jt = 14.1, Jd = 7.2 Hz, 1 H), 2.20–2.39 (m, 2 H), 2.27 (d, J = 1.2 Hz, 3 H), 4.38 (q, J = 7.3 Hz, 1 H), 4.45 (q, J = 7.3 Hz, 1 H), 4.60 (s, 1 H), 4.70 (s, 1 H), 6.15 (d, J = 1.2 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 13.4, 24.1, 28.6, 30.0, 34.0, 34.7, 39.4, 74.8, 102.6, 106.1, 137.7, 151.6, 180.3. UV (methanol): λmax (lgε/m2 mol−1) 316 nm (3.18), 206 nm (3.10). Anal. Calcd for C13H19NOS2 (269.42): C, 57.96; H, 7.11; N, 5.20; S, 23.80. Found: C, 57.74; H, 7.12; N, 5.14; S, 23.93.
ε/m2 mol−1) 317 nm (3.11), 206 nm (3.06). Anal. Calc. for C12H17NOS2 (255.39): C, 56.43; H, 6.71; N, 5.49; S, 25.11; Found: C, 56.34; H, 6.69; N, 5.46; S, 25.26. X-ray crystallography. T = 150(2) K, λ = 0.71073 Å, monoclinic, P21/n, a = 9.1980(3) Å, b = 13.3225(3) Å, c = 10.6301(3) Å, β = 100.318(3)°, Z = 4, µ = 0.395 mm−1, completeness 99.6% (2θ = 57.0), goodness-of-fit on F2 = 1.009; final R indices [I > 2σ(I)]: R1 = 0.0373, wR2 = 0.0891.
:2 (v/v), Rf = 0.28. Yield: 463 mg (1.50 mmol, 73%) colorless oil, which crystallizes on standing at 20 °C. [α]25D = −52.9 (c = 1.02/ethanol). 1H-NMR (CDCl3, 600 MHz) δ 0.86 (s, 3 H), 1.15 (d, J = 9.0 Hz, 1 H), 1.29 (s, 3 H), 1.67 (t, J = 1.7 Hz, 3 H), 1.79 (dq, Jd = 14.0, Jq = 7.0 Hz, 1 H), 1.89 (dq, Jd = 13.8 Hz, Jq = 6.9 Hz, 1 H), 1.95–2.05 (m, 2 H), 2.21 (dt, Jd = 8.8, Jt = 5.6 Hz, 1 H), 2.28 (d, J = 1.0 Hz, 3 H), 2.48–2.57 (m, 1 H), 4.41–4.52 (m, 2 H), 5.19 (d, J = 0.8 Hz, 1 H), 6.15 (d, J = 1.0 Hz, 1 H). 13C-NMR (CDCl3, 150 MHz) δ 13.5, 20.4, 22.9, 26.5, 27.8, 31.6, 36.4, 40.8, 45.1, 47.6, 75.0, 102.7, 119.3, 137.7, 145.5, 180.4. Anal. Calcd for C16H23NOS2 (309.49): C, 62.09; H, 7.49; N, 4.53; S, 20.72; Found: C, 61.81; H, 7.51; N, 4.56; S, 21.09.
4.4 Alkoxyl radical reactions
:5 (v/v). 3-Bromomethyl-2-oxabicyclo[3.3.0]octane cis-(3a). Yield: 42.3 mg (206 µmol, 10%), yellow liquid, 70/30-mixture of 1,3-cis/trans-isomers, i.e. rel-(1S,3S,5S)-3a/rel-(1S,3R,5S)-3a. Rf = 0.48 for diethyl ether–pentane = 1:5 (v/v). Anal. Calcd for C8H13BrO (205.09): C, 46.85; H, 6.39; Found: C, 46.73; H, 6.40. rel-(1S,3S,5S)-3a: 1H-NMR (CDCl3, 400 MHz) δ 1.37–1.47 (m, 1 H), 1.50–1.68 (m, 3 H), 1.69–1.92 (m, 3 H), 1.98 (ddd, J = 12.8, 8.7, 7.2 Hz, 1 H), 2.68–2.75 (m, 1 H), 3.29–3.36 (m, 1 H), 3.36–3.42 (m, 1 H), 4.23 (quin, J = 6.2 Hz, 1 H), 4.60 (t, J = 4.8 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz): δ 24.6, 32.8, 34.7, 35.5, 38.4, 42.8, 78.8, 85.7. rel-(1S,3R,5S)-3a: 1H-NMR (CDCl3, 400 MHz) δ 1.16–1.30 (m, 1 H), 1.37–1.47 (m, 1 H), 1.50–1.68 (m, 3 H), 1.69–1.92 (m, 2 H), 2.22–2.35 (m, 1 H), 2.65–2.78 (m, 1 H), 3.44 (d, J = 5.5 Hz, 2 H), 3.89 (dq, Jd = 10.3, Jq = 5.3 Hz, 1 H), 4.45 (t, J = 6.3 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz). δ 23.3, 33.2, 33.8, 34.6, 39.3, 42.9, 78.8, 85.8. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 438 mg (1.76 mmol, 85%), yellow liquid. Rf = 0.40 for diethyl ether–pentane = 1:5 (v/v). 1H-NMR (CDCl3, 400 MHz) δ 2.57 (s, 3 H), 7.30 (s, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 17.3, 96.9, 122.6, 153.2, 155.8. Anal. Calcd for C5H4NCl3S2 (248.57): C, 24.16; H, 1.62; N, 5.64; S, 25.80. Found: C, 24.17; H, 1.82; N, 5.70; S, 25.50. 5-Bromoct-7-enal (4a). Yield: 253.6 mg (1.24 mmol, 60%), yellow liquid. Rf = 0.28 for diethyl ether–pentane = 1:5 (v/v). 1H-NMR (CDCl3, 600 MHz) δ 1.71–1.97 (m, 4 H), 2.43–2.54 (m, 2 H), 2.62 (t, J = 6.6 Hz, 2 H), 3.91–4.11 (m, 1 H), 5.10–5.18 (m, 2 H), 5.84 (ddt, Jd = 17.0, 10.3, Jt = 6.9 Hz, 1 H), 9.78 (t, J = 1.6 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 20.1, 37.5, 43.0, 43.2, 55.3, 118.1, 134.5, 201.8. HRMS (EI+) m/z 204.0149/206.0124 (M+); calculated mass for C8H13OBr+: 204.0150/206.0129. Thermal reaction. cis-1a 526 mg (2.06 mmol). Eluent used for column chromatography: diethyl ether–pentane = 1:5 (v/v). 3-Bromomethyl-2-oxabicyclo[3.3.0]octane cis-(3a). Yield: 33.4 mg (163 µmol, 8%), yellow liquid, 71/29-mixture of 1,3-cis/trans-isomers, i.e. rel-(1S,3S,5S)-3a/rel-(1S,3R,5S)-3a. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 446 mg (1.80 mmol, 87%), yellow liquid. 5-Bromoct-7-enal (4a). Yield: 230 mg (1.12 mmol, 54%), yellow liquid.
:5 (v/v). 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 110 mg (443 μmol, 56%), yellow liquid. 5-Bromoct-7-enal (4a). Yield: 72.5 mg (354 μmol, 44%), yellow liquid. Unlike-5,7-dibromo-9,9,9-trichlorononanal. Yield: 9.3 mg (23.1 μmol, 3%), yellow liquid. Rf = 0.22 for diethyl ether–pentane = 1:5 (v/v). 1H-NMR (CDCl3, 400 MHz) δ 1.72–2.05 (m, 4 H), 2.40–2.67 (m, 4 H), 3.23 (dd, J = 15.9, 4.8 Hz, 1 H), 3.47 (dd, J = 16.0, 5.3 Hz, 1 H), 4.18–4.29 (m, 1 H), 4.37 (ddt, Jd = 8.0, 6.6, Jt = 5.1 Hz, 1 H), 9.80 (t, J = 1.5 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 19.6, 35.8, 42.9, 44.8, 48.3, 51.9, 62.3, 96.6, 201.5. HRMS (EI+) m/z 400.8288/402.8250 (M − H); calculated mass for C9H12OCl3Br2+: 400.8291/402.8261. Like-5,7-dibromo-9,9,9-trichlorononanal. Yield: 36.8 mg (91.2 μmol, 11%), yellow liquid. Rf = 0.20 for diethyl ether–pentane = 1:5 (v/v). 1H-NMR (CDCl3, 600 MHz) δ 1.75–2.01 (m, 4 H), 2.23 (ddd, J = 15.3, 11.6, 2.1 Hz, 1 H), 2.46–2.61 (m, 3 H), 3.24 (dd, J = 15.7, 6.2 Hz, 1 H), 3.57 (dd, J = 15.7, 4.6 Hz, 1 H), 4.21–4.35 (m, 1 H), 4.70 (dddd, J = 11.2, 6.5, 4.4, 2.4 Hz, 1 H), 9.80 (t, J = 1.2 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 20.0, 38.2, 42.9, 47.5, 48.0, 54.4, 62.6, 96.6, 201.5. HRMS (EI+) m/z 400.8314/402.8297/404.8233 (M − H); calculated mass for C9H12OCl3Br2+: 400.8300/402.8312/404.8232. Thermal reaction. trans-1a 250 mg (979 µmol); eluent used for column chromatography: diethyl ether–pentane = 1:5 (v/v). 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 208 mg (836 μmol, 85%), yellow liquid. 5-Bromoct-7-enal (4a). Yield: 145.5 mg (709 μmol, 73%), yellow liquid. Unlike-5,7-dibromo-9,9,9-trichlorononanal. Yield: 19.2 mg (47.6 μmol, 5%), yellow liquid. Like-5,7-dibromo-9,9,9-trichlorononanal. Yield: 34.1 mg (84.5 μmol, 9%), yellow liquid.
:5 (v/v). 3-(2-Bromoprop-2-yl)-2-oxabicyclo[3.3.0]octane cis-3b. Yield: 119 mg (510 μmol, 49%), 64/36-mixture of 1,3-cis/trans-isomers, i.e. rel-(1S,3S,5S)-3b/rel-(1S,3R,5S)-3b, pale yellow oil. Rf = 0.56 for diethyl ether–pentane = 1:5 (v/v). HRMS (EI+) m/z 231.0399/233.0372 (M − H); calculated mass for C10H16OBr+: 231.0385/233.0364. rel-(1S,3S,5S)-3b: 1H-NMR (CDCl3, 400 MHz) δ 1.35–1.51 (m, 2 H), 1.51–1.67 (m, 3 H), 1.71 (s, 3 H), 1.75 (s, 3 H), 1.78–1.86 (m, 1 H), 1.91 (dd, J = 13.1, 6.0 Hz, 1 H), 2.21 (ddd, J = 12.3, 9.5, 5.5 Hz, 1 H), 2.63–2.75 (m, 1 H), 3.62 (dd, J = 10.5, 5.4 Hz, 1 H), 4.45 (t, J = 6.3, Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 23.5, 30.3, 30.6, 33.4, 33.9, 37.1, 42.4, 67.0, 85.3, 86.7. rel-(1S,3R,5S)-3b: 1H-NMR (CDCl3, 400 MHz) δ 1.35–1.51 (m, 1 H), 1.51–1.76 (m, 6 H), 1.69 (s, 3 H), 1.73 (s, 3 H), 2.12 (dt, Jd = 12.9, Jt = 8.5 Hz, 1 H), 2.63–2.75 (m, 1 H), 3.91 (dd, J = 8.0, 6.6 Hz, 1 H), 4.61–4.67 (m, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 24.9, 30.1, 30.8, 33.0, 34.9, 36.8, 43.5, 69.3, 86.7, 87.2. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 190 mg (764 μmol, 73%), pale yellow liquid. 5-Brom-8-methylnon-7-enal (4b). Yield: 75.6 mg (324 μmol, 31%), pale yellow liquid. Rf = 0.25 for diethyl ether–pentane = 1:5 (v/v). 1H-NMR (CDCl3, 400 MHz) δ 1.62 (s, 3 H), 1.72 (s, 3 H), 1.76–2.02 (m, 4 H), 2.37–2.68 (m, 4 H), 3.93–4.03 (m, 1 H), 5.17 (t, J = 7.0 Hz, 1 H), 9.78 (s, 1 H). 13C-NMR (CDCl3, 150 MHz) δ 18.1, 20.3, 25.8, 37.5, 37.8, 43.1, 56.9, 120.5, 134.8, 201.9. HRMS (EI+) m/z 232.0463/234.0452 (M+); calculated mass for C10H17OBr+: 232.0463/234.0442. Thermal reaction. cis-1b 292 mg (1.03 mmol); eluent used for column chromatography: diethyl ether–pentane = 1:5 (v/v). 3-(2-Bromoprop-2-yl)-2-oxoabicyclo[3.3.0]octane cis-3b. Yield: 81.8 mg (351 μmol, 34%), 56/44-mixture of 1,3-cis/trans-isomers, i.e. rel-(1S,3S,5S)-3b/rel-(1S,3R,5S)-3b, yellow oil. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 193 mg (776 μmol, 75%), yellow liquid. 5-Brom-8-methylnon-7-enal (4b). Yield: 84.2 mg (361 μmol, 35%), yellow liquid.
:10 (v/v). 8-(Bromomethyl)-7-oxabicyclo[4.3.0]nonane cis-(3c). Yield: 50.2 mg (229 μmol, 45%), 89/11-mixture of 6,8-cis/trans-isomers i.e. rel-(1S,6S,8S)-3c/rel-(1S,6S,8R)-3c, colorless liquid. Rf = 0.44 for diethyl ether–pentane = 1:10 (v/v). Anal. Calcd for C9H15BrO (219.12): C, 49.33; H, 6.90. Found: C, 49.46; H, 6.80. rel-(1S,6S,8S)-3c. 1H-NMR (CDCl3, 400 MHz) δ 1.21–1.29 (m, 1 H), 1.31–1.37 (m, 1 H), 1.38–1.45 (m, 1 H), 1.47–1.56 (m, 3 H), 1.57–1.70 (m, 2 H), 1.76–1.85 (m, 1 H), 2.12–2.20 (m, 2 H), 3.40 (dd, J = 10.0, 6.7 Hz, 1 H), 3.53 (dd, J = 10.0, 5.9 Hz, 1 H), 3.91 (q, J = 5.0 Hz, 1 H), 4.15 (quin, J = 6.7 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 21.4, 23.5, 28.5, 29.0, 36.4, 36.5, 37.6, 77.8, 78.3. rel-(1S,6S,8R)-3c. 1H-NMR (CDCl3, 400 MHz) δ 1.17–1.70 (m, 7 H), 1.76–1.85 (m, 1 H), 1.87–1.94 (m, 2 H), 2.06–2.12 (m, 1 H), 3.39 (dd, J = 6.6, 10.0 Hz, 1 H), 3.47 (dd, J = 10.0, 5.0 Hz, 1 H), 4.05 (q, J = 3.8 Hz, 1 H), 4.37–4.43 (m, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 20.5, 24.0, 27.6, 28.1, 37.2, 37.8, 38.4, 76.4, 77.7. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 92.8 mg (373 μmol, 73%), colorless liquid. Rf = 0.36 for diethyl ether–pentane = 1:10 (v/v). Thermal reaction. cis-1c 81.6 mg (303 μmol); eluent used for chromatography: diethyl ether–pentane = 1:10 (v/v). 8-(Bromomethyl)-7-oxabicyclo[4.3.0]nonane cis-(3c). Yield: 40.6 mg (185 μmol, 61%), 68/32-mixture of 6,8-cis/6,8-trans-isomers, i.e. rel-(1S,6S,8S)-3c/rel-(1S,6S,8R)-3c, colorless liquid. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 55.9 mg (225 μmol, 74%), colorless liquid. 6-Bromo-8-nonenal (4c). Yield: 7.8 mg (36.0 μmol, 12%), yellow oil.
:1 (v/v). 8-(Bromomethyl)-7-oxabicyclo[4.3.0]nonane28trans-(3c). Yield: 55.0 mg (251 μmol, 49%), 8/92-mixture of 6,8-cis/6,8-trans-isomers, i.e. rel-(1R,6S,8S)-3c/rel-(1R,6S,8R)-3c, colorless liquid. Rf = 0.50 for diethyl ether–pentane = 1:5 (v/v). rel-(1R,6S,8S)-3c: 1H-NMR (CDCl3, 400 MHz) δ 1.04–1.58 (m, 6 H), 1.67–2.13 (m, 5 H), 3.11 (dt, Jd = 3.7 Hz, Jt = 10.4 Hz, 1 H), 3.34 (dd, J = 6.7, 10.1 Hz, 1 H), 3.42 (dd, J = 4.9, 10.1 Hz, 1 H), 4.20–4.24 (m, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 24.2, 25.7, 28.9, 31.2, 35.3, 36.3, 43.9, 76.7, 84.5. rel-(1R,6S,8R)-3c: 1H-NMR (CDCl3, 400 MHz) δ 1.08–1.53 (m, 6 H), 2.18–2.24 (m, 1 H), 1.69–2.12 (m, 4 H), 3.23 (dt, Jd = 4.0 Hz, Jt = 10.4 Hz, 1 H), 3.41 (dd, J = 6.5, 10.1 Hz, 1 H), 3.50 (dd, J = 5.3, 10.1 Hz, 1 H), 4.24–4.30 (m, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 24.2, 25.5, 28.8, 31.3, 36.7, 37.1, 46.3, 77.3, 83.3. Thermal reaction. cis-1c 111 mg (412 μmol); eluent used for chromatography: diethyl ether–pentane = 1:5 (v/v). 8-(Bromomethyl)-7-oxabicyclo[4.3.0]nonane trans-(3c). Yield: 63.1 mg (288 μmol, 70%), 13/87-mixture of 6,8-cis/trans-isomers, i.e. rel-(1R,6S,8S)-3c/rel-(1R,6S,8R)-3c, pale yellow liquid. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 81.8 mg (329 μmol, 80%), yellow liquid. 6-Bromo-8-nonenal (4c). Yield: 7.4 mg (33.8 μmol, 8%), yellow oil. 1H-NMR (CDCl3, 400 MHz) δ 1.36–1.52 (m, 1 H), 1.54–1.74 (m, 3 H), 1.76–1.90 (m, 2 H), 2.47 (dt, Jd = 7.1, Jt = 1.8 Hz, 2 H), 2.61 (tt, J = 6.7, 1.3 Hz, 2 H), 4.02 (dtd, Jd = 8.2, 4.9, Jt = 6.5, Hz, 1 H), 5.09–5.19 (m, 2 H), 5.75–5.93 (m, 1 H), 9.78 (t, J = 1.6 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 21.4, 27.1, 38.0, 43.3, 43.7, 55.8, 118.0, 134.7, 202.3. HRMS (EI+) m/z 218.0296/220.0282 (M+); calculated mass for C9H15OBr+: 218.0306/220.0286.
:10 (v/v). 2-Bromo-4-oxabicyclo[4.4.0]decane cis-(5d). Yield: 3.5 mg (16.0 μmol, 3%), i.e. rel-(1S,2S,6R)-5d, yellow liquid. Rf = 0.46 for diethyl ether–pentane = 1:10 (v/v). 1H-NMR (CDCl3, 600 MHz) δ 1.18–1.53 (m, 6 H), 1.63–1.89 (m, 2 H), 1.97–2.07 (m, 1 H), 2.09–2.23 (m, 1 H), 3.47 (t, J = 10.8 Hz, 1 H), 3.57–3.65 (m, 1 H), 3.67–3.76 (m, 1 H), 4.14 (dd, J = 11.1, 4.7 Hz, 1 H), 4.34 (td, Jt = 10.6, Jd = 4.5 Hz, 1 H). 13C-NMR (CDCl3, 150 MHz, −37.3 °C) δ 19.7, 25.1, 25.9, 28.5, 39.4, 42.2, 49.5, 73.3, 73.6. HRMS (EI+) m/z 218.0307/220.0286 (M+); calculated mass for C9H15OBr+: 218.0306/220.0286. 7-Bromomethyl-8-oxabicyclo[4.3.0]nonane cis-(3d). Yield: 75.1 mg (343 μmol, 70%), colorless liquid, 20/80-mixture of 6,7-cis/6,7-trans-isomers, i.e. rel-(1R,6S,7R)-3d/rel-(1R,6S,7S)-3d. Rf = 0.31 for diethyl ether–pentane = 1:10 (v/v). Anal. Calcd for C9H15BrO (219.12): C, 49.33; H, 6.90. Found: C, 49.24; H, 6.88. rel-(1R,6S,7R)-3d. 1H-NMR (CDCl3, 400 MHz) δ 1.13–1.22 (m, 2 H), 1.23–1.39 (m, 1 H), 1.46–1.56 (m, 1 H), 1.57–1.67 (m, 3 H), 1.72–1.80 (m, 1 H), 2.12 (dq, Jd = 10.7, Jq = 5.4 Hz, 1 H), 2.48–2.60 (m, 1 H), 3.30–3.37 (m, 1 H) 3.37–3.46 (m, 1 H), 3.80 (dd, J = 11.1, 7.8 Hz, 1 H), 3.87–3.93 (m, 1 H), 4.15 (td, Jt = 7.2, Jd = 4.2 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 20.7, 20.8, 23.6, 24.4, 30.8, 38.1, 40.4, 69.4, 82.7. rel-(1R,6S,7S)-3d. 1H-NMR (CDCl3, 400 MHz) δ 1.23–1.36 (m, 1 H), 1.39–1.49 (m, 3 H), 1.52–1.60 (m, 2 H), 1.61–1.70 (m, 2 H), 2.14 (quin, J = 6.0 Hz, 1 H), 2.20–2.30 (m, 1 H), 3.33–3.40 (m, 1 H), 3.42–3.49 (m, 1 H), 3.66 (dd, J = 8.1, 4.4 Hz, 1 H) 3.89 (dd, J = 8.0, 5.9 Hz, 1 H) 3.98–4.06 (m, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 22.4, 23.5, 25.4, 25.9, 35.9, 38.4, 41.8, 72.8, 80.6. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 85.3 mg (343 μmol, 71%), colorless liquid. Thermal reaction. cis-1d 136 mg (505 μmol); eluent used for chromatography: diethyl ether–pentane = 1:10 (v/v). 2-Bromo-4-oxabicyclo[4.4.0]decane cis-(5d). Yield: 5.6 mg (25.6 μmol, 5%), i.e. rel-(1S,2S,6R)-5d, yellow liquid. 6,7-cis-7-Bromomethyl-8-oxabicyclo[4.3.0]nonane cis-(3d). Yield: 19.0 mg (86.7 μmol, 17%), rel-(1R,6S,7R)-3d colorless liquid. 6,7-trans-7-Bromomethyl-8-oxabicyclo[4.3.0]nonane cis-(3d). Yield: 70.5 mg (322 μmol, 64%), i.e. rel-(1R,6S,7S)-3d colorless liquid. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 111 mg (446 μmol, 88%), colorless liquid.
:10 (v/v). 2-Bromo-4-oxabicyclo[4.4.0]decane trans-(5d). Yield: 11.8 mg (53.9 μmol, 5%), rel-(1S,2S,6S)-5d colorless liquid. Rf = 0.46 for diethyl ether–pentane = 1:10 (v/v). 1H-NMR (CDCl3, 400 MHz) δ 0.84–1.04 (m, 2 H), 1.24–1.42 (m, 3 H), 1.44–1.55 (m, 2 H), 1.73–1.84 (m, 2 H), 2.13–2.23 (m, 1 H), 3.13 (t, J = 10.9 Hz, 1 H), 3.43–3.57 (m, 1 H), 3.70–3.88 (m, 2 H), 4.12 (dd, J = 11.1, 4.7 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 25.75, 25.83, 27.6, 30.7, 44.1, 49.7, 54.4, 73.2, 73.7. Anal. Calcd for C9H15BrO (219.12): C, 49.33; H, 6.90. Found: C, 48.88; H, 6.86. HRMS (EI+) m/z 218.0298/220.0277 (M+); calculated mass for C9H15OBr+: 218.0306/220.0286. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 199 mg (801 μmol, 78%), colorless liquid. 93/7-mixture of exo/endo-isomers 7-Bromomethyl-8-oxabicyclo[4.3.0]nonane trans-(3d) and 2-Bromo-4-oxabicyclo-[4.4.0]decane trans-(5d). Yield: 137 mg (624 μmol, 61%), 7/93-mixture of 6,7-cis/trans-isomers, i.e. rel-(1S,6S,7R)-3d/rel-(1S,6S,7S)-3d and rel-(1S,2R,6S)-5d, colorless liquid. Rf = 0.25 for diethyl ether–pentane = 1:10 (v/v). Anal. Calcd for C9H15BrO (219.12): C, 49.33; H, 6.90. Found: C, 49.04; H, 6.93. rel-(1S,6S,7S)-3d. 1H-NMR (CDCl3, 400 MHz) δ 1.03–1.19 (m, 2 H), 1.20–1.38 (m, 3 H), 1.66–1.97 (m, 5 H), 3.36–3.45 (m, 2 H), 3.52–3.58 (m, 1 H), 3.71 (ddd, J = 9.7, 5.5, 4.2 Hz, 1 H), 3.96 (t, J = 7.3 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 25.3, 25.5, 27.4, 27.7, 35.3, 46.3, 49.7, 72.3, 81.6. rel-(1S,6S,7R)-3d. 1H-NMR (CDCl3, 400 MHz) δ 0.98–1.11 (m, 1 H), 1.14–1.28 (m, 3 H), 1.62–1.72 (m, 2 H), 1.75–1.97 (m, 4 H), 3.15–3.24 (m, 1 H), 3.25–3.34 (m, 2 H), 3.97–4.04 (m, 1 H), 4.21–4.29 (m, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 25.4, 26.0, 26.2, 27.8, 33.9, 42.7, 48.0, 72.7, 79.6. HRMS (EI+) m/z 125.0940 (M − CH2Br); calculated mass for C8H13O+: 125.0966. rel-(1S,2R,6S)-5d. 1H-NMR (CDCl3, 400 MHz) δ 1.03–1.38 (m, 3 H), 1.49–1.56 (m, 2 H), 1.66–1.97 (m, 5 H), 3.05 (t, J = 11.1 Hz, 1 H), 3.80–3.86 (m, 2 H), 4.11–4.16 (m, 2 H). 13C-NMR (CDCl3, 100 MHz) δ 25.2, 25.7, 27.5, 31.4, 36.4, 45.4, 57.0, 73.5, 73.8. HRMS (EI+) m/z 218.0304/220.0287 (M+); calculated mass for C9H15OBr+: 218.0306/220.0286. Thermal reaction. cis-1d 270 mg (1.00 μmol); eluent used for chromatography: diethyl ether–pentane = 1:10 (v/v). 2-Bromo-4-oxabicyclo[4.4.0]decane trans-(5d). Yield: 21.7 mg (99.0 μmol, 10%), rel-(1S,2S,6S)-5d yellow liquid. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 243 mg (977 μmol, 98%), colorless liquid. 91/9-mixture of exo/endo-isomers 7-Bromomethyl-8-oxabicyclo[4.3.0]nonane trans-(3d) and 2-Bromo-4-oxabicyclo[4.4.0]decane trans-(5d). Yield: 168 mg (767 μmol, 77%), 10/90-mixture of 6,7-cis/6,7-trans-isomers, i.e. rel-(1S,6S,7R)-3d/rel-(1S,6S,7S)-3d and rel-(1S,2R,6S)-5d, pale yellow liquid.
:10 (v/v). 7-(2-Bromoprop-2-yl)-8-oxabicyclo[4.3.0]nonane cis-(3e). Yield: 107 mg (433 μmol, 80%), i.e. rel-(1R,6S,7S)-3e, pale yellow liquid. Rf = 0.48 for diethyl ether–pentane = 1:10 (v/v). 1H-NMR (CDCl3, 600 MHz) δ 1.32–1.43 (m, 2 H) 1.45–1.55 (m, 3 H) 1.59–1.67 (m, 2 H) 1.68–1.76 (m, 7 H) 2.25–2.33 (m, 2 H) 3.63–3.69 (m, 2 H) 3.91 (dd, J = 8.2, 5.9 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 23.1 (2C, HMQC), 25.2, 28.1, 30.1, 30.9, 38.6, 40.3, 69.9, 72.6, 90.1. Anal. Calcd for C11H19BrO (247.17): C, 53.45; H, 7.75; Found: C, 53.32; H, 7.59. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 110 mg (443 μmol, 82%), pale yellow liquid. Thermal reaction: cis-1e 151 mg (508 μmol); eluent used for chromatography: diethyl ether–pentane = 1:10 (v/v). 7-(2-Bromoprop-2-yl)-8-oxabicyclo[4.3.0]nonane cis-(3e). Yield: 119 mg (481 μmol, 95%), i.e. rel-(1R,6S,7S)-3e, pale yellow liquid. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 114 mg (460 μmol, 90%), colorless liquid.
:10 (v/v). 7-(2-Bromoprop-2-yl)-8-oxabicyclo[4.3.0]nonane trans-(3e). Yield: 94.4 mg (382 μmol, 81%), i.e. rel-(1S,6S,7S)-3e, colorless liquid. Rf = 0.39 for diethyl ether–pentane = 1:10 (v/v). 1H-NMR (CDCl3, 400 MHz) δ ppm 1.04–1.35 (m, 4 H) 1.46–1.58 (m, 1 H) 1.68–1.91 (m, 4 H), 1.74 (s, 3 H), 1.77 (s, 3 H), 2.06–2.14 (m, 1 H), 3.37 (dd, J = 11.3, 7.4 Hz, 1 H), 3.44 (d, J = 9.2 Hz, 1 H), 3.92 (t, J = 7.1 Hz, 1 H). 13C-NMR (CDCl3, 150 MHz) δ 25.3, 25.9, 27.3, 30.5, 30.7, 31.0, 47.2, 47.8, 69.3, 72.1, 89.4. Anal. Calcd for C11H19BrO (247.17): C, 53.45; H, 7.75; Found: C, 53.62; H, 7.82. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 92.4 mg (372 μmol, 79%), colorless liquid. Thermal reaction. trans-1e 147 mg (495 μmol); eluent used for chromatography: diethyl ether–pentane = 1:10 (v/v). 7-(2-Bromoprop-2-yl)-8-oxabicyclo[4.3.0]nonane trans-(3e). Yield: 115 mg (465 μmol, 94%), i.e. rel-(1S,6S,7S)-3e, colorless liquid. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 120 mg (483 μmol, 98%), colorless liquid.
:10 (v/v). cis-(1-Bromo)-3-oxabicyclo[4.4.0]decane cis-(5f). Yield: 8.2 mg (37.4 μmol, 4%), yellow liquid. Rf = 0.46 for diethyl ether–pentane = 1:10 (v/v). 1H-NMR (CDCl3, 400 MHz, 22.2 °C) δ 1.31–1.59 (m, 4 H), 1.62–1.90 (m, 3 H), 1.96–2.25 (m, 3 H), 2.34 (ddd, J = 14.7, 12.1, 4.2 Hz, 1 H), 3.51 (br. t, J = 10.0 Hz, 1 H), 3.78 (d, J = 11.4 Hz, 1 H), 3.92 (d, J = 11.6 Hz, 1 H), 4.01 (br. d, J = 10.0 Hz, 1 H). 1H-NMR (CDCl3, 600 MHz, −39.7 °C) δ 1.35–1.45 (m, 3 H), 1.46–1.53 (m, 1 H), 1.65–1.72 (m, 2 H), 1.73–1.82 (m, 1 H), 2.03–2.13 (m, 2 H), 2.15–2.21 (m, 1 H), 2.26–2.33 (m, 1 H), 3.46–3.52 (m, 1 H), 3.78 (d, J = 11.2 Hz, 1 H), 3.91 (d, J = 11.2 Hz, 1 H), 4.03 (dd, J = 11.3, 4.8 Hz, 1 H). 13C-NMR (CDCl3, 150 MHz, −39.7 °C) δ 19.3, 22.7, 27.3, 29.4, 32.1, 42.0, 68.7, 72.9, 78.4. Anal. Calcd for C9H15BrO (219.12): C, 49.33; H, 6.90; Found: C, 49.58; H, 6.83. cis-(1-bromomethyl)-9-oxabicyclo[4.3.0]nonane cis-(3f). Yield: 53.6 mg (245 μmol, 24%), pale yellow liquid. Rf = 0.36 for diethyl ether–pentane = 1:10 (v/v). 1H-NMR (CDCl3, 600 MHz) δ 1.27–1.33 (m, 1 H), 1.34–1.44 (m, 2 H), 1.47–1.56 (m, 2 H), 1.61 (m, 1 H), 1.73 (m, 1 H), 1.78–1.85 (m, 2 H), 2.09 (m, 1 H), 2.26 (quin, J = 6.6 Hz, 1 H), 3.35 (d, J = 10.6 Hz, 1 H), 3.46 (d, J = 10.6 Hz, 1 H), 3.90 (td, Jt = 8.7, Jd = 5.6 Hz, 1 H), 3.97 (td, Jt = 8.5, Jd = 6.5 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 22.0, 22.5, 27.3, 30.8, 30.9, 39.2, 39.8, 65.4, 81.6. Anal. Calcd for C9H15BrO (219.12): C, 49.33; H, 6.90; Found: C, 49.17; H, 6.79. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 137 mg (551 μmol, 54%), pale yellow liquid. trans-(1-Bromo)-3-oxabicyclo[4.4.0]decane trans-(5f). Yield: 37.1 mg (169 μmol, 17%), yellow liquid. Rf = 0.28 for diethyl ether–pentane = 1:10 (v/v). 1H-NMR (CDCl3, 400 MHz) δ 1.12 (tt, J = 11.4, 3.6 Hz, 1 H), 1.22–1.36 (m, 2 H), 1.37–1.51 (m, 3 H), 1.63–1.71 (m, 1 H), 1.73–1.87 (m, 2 H), 1.90–2.06 (m, 2 H), 3.35 (d, J = 12.1 Hz, 1 H), 3.50 (td, Jt = 12.0, Jd = 2.5 Hz, 1 H), 3.98 (d, J = 12.3 Hz, 1 H), 4.07 (dd, J = 11.5, 4.7 Hz, 1 H). 13C-NMR (CDCl3, 150 MHz) δ 21.8, 25.6, 29.0, 29.8, 36.3, 45.2, 68.8, 76.9, 78.4. Anal. Calcd for C9H15BrO (219.12): C, 49.33; H, 6.90; Found: C, 49.33; H, 7.02. Thermal reaction. 1f 270 mg (1.00 mmol); eluent used for column chromatography: diethyl ether–pentane = 1:10 (v/v). cis-1-Bromo-3-oxabicyclo[4.4.0]decane cis-(5f). Yield: 14.8 mg (67.5 μmol, 7%), yellow liquid. cis-(1-Bromomethyl)-9-oxabicyclo[4.3.0]nonane cis-(3f). Yield: 76.7 mg (350 μmol, 35%), pale yellow liquid. trans-1-Bromo-3-oxabicyclo[4.4.0]decane trans-(5f). Yield: 33.5 mg (153 μmol, 15%), yellow liquid. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 199 mg (801 μmol, 80%), pale yellow liquid.
:1:15 (v/v/v). rel-(1R,4R,5R)-4-Bromo-4-methyl-6-oxabicyclo[3.2.1]octane trans-(3g). Yield: 213 mg (1.04 mmol, 50%), pale yellow liquid. Rf = 0.46 for diethyl ether–acetone–pentane = 1:1:15 (v/v/v). 1H-NMR (CDCl3, 400 MHz) δ 1.48–1.57 (m, 1 H), 1.77 (s, 3 H), 1.79–1.89 (m, 2 H), 1.90–1.96 (m, 1 H), 1.97–2.06 (m, 1 H), 2.33–2.39 (m, 1 H), 2.58 (d, J = 11.9 Hz, 1 H), 3.73–3.88 (m, 2 H), 4.10 (d, J = 5.8 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz) δ 26.8, 32.3, 33.9, 35.4, 36.4, 67.9, 72.3, 83.0. Anal. Calcd for C8H13BrO (205.09): C, 46.85; H, 6.39; Found: C, 46.74; H, 6.36. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 358 mg (1.44 mmol, 69%), pale yellow liquid. Rf = 0.35 for diethyl ether–acetone–pentane = 1:1:15 (v/v/v). rel-(1R,4S,5R)-4-Bromo-4-methyl-6-oxabicyclo[3.2.1]octane cis-(3g). Yield: 62.0 mg (302 μmol, 14%), pale yellow liquid. Rf = 0.22 for diethyl ether–acetone–pentane = 1:1:15 (v/v/v) 1H-NMR (CDCl3, 600 MHz) δ 1.51–1.62 (m, 2 H), 1.72–1.82 (m, 1 H), 1.78 (s, 3 H), 1.94 (d, J = 12.0 Hz, 1 H), 2.05 (dd, J = 13.6, 5.9 Hz, 1 H), 2.42–2.46 (m, 1 H), 2.51 (td, Jt = 13.2, Jd = 7.0 Hz, 1 H), 3.85 (ddd, J = 8.1, 4.4, 1.4 Hz, 1 H), 3.93 (d, J = 7.9 Hz, 1 H), 4.31 (d, J = 6.1 Hz, 1 H). 13C-NMR (CDCl3, 100 MHz): δ 28.7 (2C, HMQC), 34.1, 34.4, 37.5, 71.4, 71.5, 84.9. Anal. Calcd for C8H13BrO (205.09): C, 46.85; H, 6.39. Found: C, 46.87; H, 6.37. Thermal reaction. 1g 513 mg (2.00 mmol); eluent used for column chromatography: diethyl ether–acetone–pentane = 1:1:15 (v/v/v). rel-(1R,4R,5R)-4-Bromo-4-methyl-6-oxabicyclo[3.2.1]octane trans-(3g). Yield: 238 mg (1.16 mmol, 58%), pale yellow liquid. 4-Methyl-2-(trichloromethylsulfanyl)-thiazole (2). Yield: 377 mg (1.52 mmol, 76%), pale yellow liquid. rel-(1R,4S,5R)-4-Bromo-4-methyl-6-oxabicyclo[3.2.1]octane cis-(3g). Yield: 57.2 mg (279 μmol, 14%), pale yellow liquid.
:4 (v/v). rel-(1R,2S,6S,7S,9S)-9-bromo-1,8,8-trimethyl-3-oxatricyclo[5.2.1.02,6]-decane (6). Yield: 148.2 mg (58%), yellow liquid. [α]25D = 36.6 (c = 0.85/ethanol). 1H-NMR (CDCl3, 600 MHz): δ 1.08 (s, 3 H), 1.10 (s, 3 H), 1.15 (s, 3 H), 1.33 (dd, J = 11.1, 1.7 Hz, 1 H), 1.35–1.41 (m, 1 H), 1.55 (dd, J = 11.3, 1.3 Hz, 1 H), 1.60 (s, 1 H), 2.04 (dddd, J = 12.1, 9.5, 5.8, 1.3 Hz, 1 H), 2.60–2.66 (m, 1 H), 3.40 (ddd, J = 11.1, 8.6, 5.9 Hz, 1 H), 3.79 (s, 1 H), 3.88–3.97 (m, 2 H). 13C-NMR (CDCl3, 100 MHz): δ 15.4, 25.9, 30.3, 31.8, 34.9, 40.5, 41.8, 50.9, 53.4, 67.9, 72.1, 83.4. HRMS (EI+) m/z 258.0612 (M+); calculated mass for C12H19OBr+: 258.0442 260. 4-Methyl-2-(trichloromethylsulfanyl)-thiazol (2). Yield: 193 mg (774 μmol, 79%), colorless liquid. rel-(1S,2S,6S,7S,9S)-9-bromo-1,10,10-trimethyl-3-oxatricyclo[5.2.1.02,6]-decane (7). Yield: 7.7 mg (3%). 1H-NMR (CDCl3, 400 MHz) δ 0.93 (s, 3 H), 1.05 (s, 6 H), 1.81–2.04 (m, 4 H), 2.32–2.42 (m, 1 H), 3.01 (ttd, Jt = 9.7, 4.7, Jd = 1.4 Hz, 1 H), 3.85–3.93 (m, 1 H), 4.30 (ddd, J = 11.2, 5.8, 1.7 Hz, 1 H), 4.35–4.44 (m, 2 H). 13C-NMR (CDCl3, 100 MHz) δ 13.8, 20.4, 20.5, 26.0, 31.2, 43.0, 47.2, 51.8, 53.2, 53.8, 71.7, 91.2. HRMS (EI+) m/z 258.0612/260.0612 (M+); calculated mass for C12H19OBr+: 258.0619/260.0599.
Acknowledgements
This work is part of Ph.D. theses of C. S., T. G., and R. K., and an undergraduate research project of A. L. We express our gratitude to the State Rheinland-Pfalz (scholarship for C. S. and equipment for computational chemistry), and the Deutsche Forschungsgemeinschaft (grant Ha1705/5-2) for financial support.Notes and references
- A. L. J. Beckwith and B. P. Hay, J. Am. Chem. Soc., 1988, 110, 4415–4416 CrossRef CAS.
- J. Hartung and F. Gallou, J. Org. Chem., 1995, 60, 6706–6716 CrossRef CAS.
- M. Rueda-Becerril, J. C. T. Leung, C. R. Dunbar and G. M. Sammis, J. Org. Chem., 2011, 76, 7720–7729 CrossRef CAS PubMed.
- J. M. Surzur, M. P. Bertrand and R. Nougier, Tetrahedron Lett., 1969, 4197–4200 CrossRef CAS.
- J. M. Surzur and M. P. Bertrand, Bull. Soc. Chim. Fr., 1973, 1861 CAS.
- R. D. Rieke and N. A. Moore, J. Org. Chem., 1972, 37, 413–418 CrossRef CAS.
- J. Hartung, T. Gottwald and K. Špehar, Synthesis, 2002, 1469–1498 CrossRef CAS PubMed.
- J. Hartung, K. Daniel, C. Rummey and G. Bringmann, Org. Biomol. Chem., 2006, 4, 4089–4100 CAS.
- For stereodirecting effects of amino- or silyloxy groups see: M. Zlotorzynska, H. Zhai and G. M. Sammis, J. Org. Chem., 2010, 75, 864–872 CrossRef CAS PubMed.
- J. Hartung, R. Kneuer, S. Laug, P. Schmidt, K. Špehar, I. Svoboda and H. Fuess, Eur. J. Org. Chem., 2003, 4033–4052 CrossRef CAS PubMed.
- J. Hartung, T. Gottwald and M. Greb, Synlett, 2004, 65–68 CrossRef.
- J. Hartung and R. Kneuer, Tetrahedron: Asymmetry, 2003, 14, 3019–3031 CrossRef CAS PubMed.
- N. Schneiders, T. Gottwald and J. Hartung, Eur. J. Org. Chem., 2009, 799–801 Search PubMed.
- C. Schur, N. Becker, U. Bergsträßer, T. Gottwald and J. Hartung, Tetrahedron, 2011, 67, 2338–2347 CrossRef CAS PubMed.
- P. Fries, M. K. Müller and J. Hartung, Org. Biomol. Chem., 2013, 11, 2630–2637 CAS.
- P. Fries and J. Hartung, J. Am. Chem. Soc., 2011, 133, 3906–3912 CrossRef CAS PubMed.
- J. Hartung, R. Kneuer, C. Rummey and G. Bringmann, J. Am. Chem. Soc., 2004, 126, 12121–12129 CrossRef CAS PubMed.
- A. L. J. Beckwith and C. H. Schiesser, Tetrahedron, 1985, 41, 3925–3941 CrossRef CAS.
- K. N. Houk, M. N. Paddon-Row, D. C. Spellmeyer, N. G. Rondan and S. Nagase, J. Org. Chem., 1986, 51, 2874–2879 CrossRef CAS; D. C. Spellmeyer and K. N. Houk, J. Org. Chem., 1987, 52, 959–974 CrossRef.
- M. Stoll and M. Hinder, Helv. Chim. Acta, 1950, 33, 1251–1261 CrossRef CAS PubMed; J. M. Castro, S. Salido, J. Altarejos, M. Nogueras and A. Sanchez, Tetrahedron, 2002, 58, 5941–5949 CrossRef.
- R. R. Gadikota, A. I. Keller, C. S. Callam and T. L. Lowary, Tetrahedron: Asymmetry, 2003, 14, 737–742 CrossRef CAS.
- W. Francke, S. Schulz, V. Sinnwell, W. A. König and Y. Roisin, Liebigs Ann. Chem., 1989, 1195–1201 CrossRef CAS PubMed; H.-L. Wang, C.-H. Zhao, G. Szöcs, S. P. Chinta, S. Schulz and C. O. Brumata, J. Chem. Ecol., 2013, 39, 790–796 CrossRef PubMed.
- J. Hartung, T. M. Kopf, R. Kneuer and P. Schmidt, C. R. Acad. Sci., Paris, Chim./Chem., 2001, 4, 649–666 CAS.
- D. P. Curran, N. A. Porter and B. Giese, in Stereochemistry of Radical Reactions: Concepts, Guidelines, and Synthetic Applications, Wiley, Weinheim, 1995, vol. X, ch. X, pp. 27–115 Search PubMed.
- B. Giese, B. Koppling, T. Göbel, J. Dickhaut, G. Thoma, K. J. Kulicke and F. Trach, in Organic Reactions, ed. L. A. Paquette, Wiley, Weinheim, 1996, vol. 48, ch. 2, pp. 301–856 Search PubMed.
- J. Hartung, C. Schur, I. Kempter and T. Gottwald, Tetrahedron, 2010, 66, 1365–1374 CrossRef CAS PubMed.
- T. Gottwald, M. Greb and J. Hartung, Synlett, 2004, 65–68 CAS.
- For 2-(prop-2-en-1-yl)cyclopentan-1-ol: S. Baskaran, I. Islam and S. Chandrasekaran, J. Org. Chem., 1990, 55, 891–895 CrossRef CAS; D. P. Curran and H. J. Liu, J. Chem. Soc., Perkin Trans. 1, 1994, 1377–1393 RSC; L. S. Hegedus and J. M. McKearin, J. Am. Chem. Soc., 1982, 104, 2444–2451 CrossRef.
- For 2-(3-methylbut-2-en-1-yl)-cyclopentan-1-ol: L. Streinz and M. Romaňuk, Collect. Czech. Chem. Commun., 1978, 43, 647–654 CrossRef CAS.
- For 2-(prop-2-en-1-yl)-cyclohexan-1-ol: S. Baskaran, I. Islam and S. Chandrasekaran, J. Org. Chem., 1990, 55, 891–895 CrossRef CAS; J. M. Schomaker, B. R. Travis and B. Borhan, Org. Lett., 2003, 5, 3089–3092 CrossRef PubMed; V. Spezialé, M. Armat and A. Lattes, J. Heterocycl. Chem., 1976, 13, 349–356 CrossRef PubMed.
- For 2-(1-methylenecyclohex-2-yl)-ethan-1-ol: A. Segre, R. Viterbo and G. Parisi, J. Am. Chem. Soc., 1957, 79, 3503–3505 CrossRef CAS; G. E. Gream, A. K. Serelis and T. I. Stoneman, Aust. J. Chem., 1974, 27, 1711–1729 CrossRef.
- J. Hartung, S. Hünig, R. Kneuer, M. Schwarz and H. Wenner, Synthesis, 1997, 1433–1438 CrossRef CAS PubMed.
- For cis-[2-(ethyen)-cyclohex-1-yl]-methanol and cis-[2-(methylprop-1-en-1-yl)-cyclohex-1-yl]-methanol: B. D. Kelly, J. M. Allen, R. E. Tundel and T. H. Lambert, Org. Lett., 2009, 11, 1381–1383 CrossRef CAS PubMed.
- For trans-[2-(ethylen)-cyclohex-1-yl]-methanol and trans-[2-(methylprop-1-en-1-yl)-cyclohex-1-yl]-methanol: R. Kuhn and I. Butula, Liebigs Ann. Chem., 1968, 718, 50–77 CrossRef CAS PubMed; I. J. Jakovac, H. B. Goodbrand, K. P. Lok and J. B. Jones, J. Am. Chem. Soc., 1982, 104, 4659–4665 CrossRef; R. M. Borzilleri and S. M. Weinreb, J. Am. Chem. Soc., 1994, 116, 9789–9790 CrossRef; R. M. Borzilleri, S. M. Weinreb and M. Parvez, J. Am. Chem. Soc., 1995, 117, 10905–10913 CrossRef; B. D. Kelly, J. M. Allen, R. E. Tundel and T. H. Lambert, Org. Lett., 2009, 11, 1381–1383 CrossRef PubMed.
- For 2-{(1S,4S,5R)-2,6,6-trimethylbicyclo[3.1.1]hept-2-en-4-yl}ethanol: R. K. Guy and R. A. DiPietro, Synth. Commun., 1992, 22, 687–692 CrossRef CAS; X. Wei, J. C. Lorenz, S. Kapadia, A. Saha, N. Haddad, C. A. Busacca and C. H. Senanayake, J. Org. Chem., 2007, 72, 4250–4253 CrossRef PubMed; P. A. Grieco, J. D. Clark and C. T. Jagoe, J. Am. Chem. Soc., 1991, 113, 5488–5489 CrossRef; M. T. Reetz and A. Gansäuer, Tetrahedron, 1993, 49, 6025–6030 CrossRef.
- J. Hartung, I. Kempter, T. Gottwald, M. Schwarz and R. Kneuer, Tetrahedron: Asymmetry, 2009, 20, 2097–2104 CrossRef CAS PubMed.
- For (1-methylcyclohex-1-en-4-yl)-methanol: T. Inukai and M. Kasai, J. Org. Chem., 1965, 30, 3567–3569 CrossRef CAS.
- S. A. Monti and G. L. White, J. Org. Chem., 1975, 40, 215–217 CrossRef CAS.
- M. Arnone, J. Hartung and B. Engels, J. Phys. Chem. A, 2005, 109, 5943–5950 CrossRef CAS PubMed.
- J. Hartung, R. Kneuer, M. Schwarz and M. Heubes, Eur. J. Org. Chem., 2001, 4733–4736 CrossRef CAS.
- J. Hartung, S. Altermann, U. Bergsträßer, T. Gottwald, I. Kempter, C. Schur and M. Heubes, Tetrahedron, 2009, 65, 7527–7532 CrossRef CAS PubMed.
- J. Hartung, U. Bergsträsser, K. Daniel, N. Schneiders, I. Svoboda and H. Fuess, Tetrahedron, 2009, 65, 2567–2573 CrossRef CAS PubMed.
- U. Bünzli-Trepp, Handbuch für die systematische Nomenklatur der Organischen Chemie, Metallorganischen Chemie und Koordinationschemie, Logos-Verlag, Berlin, 2001, pp. 15–56 Search PubMed.
- A. Hantzsch and J. H. Weber, Ber. Dtsch. Chem. Ges., 1887, 20, 3118–3132 CrossRef PubMed.
- O. J. Widman, Prakt. Chem., 1888, 38, 185–201 CrossRef PubMed.
- G. P. Moss, Pure Appl. Chem., 1999, 71, 513–529 CAS.
- A. Baeyer, Ber. Dtsch. Chem. Ges., 1900, 33, 3771–3775 CrossRef PubMed.
- J. Hartung, B. Hertel and F. Trach, Chem. Ber., 1993, 126, 1187–1191 CrossRef CAS PubMed.
- A. L. J. Beckwith and B. P. Hay, J. Am. Chem. Soc., 1989, 111, 230–234 CrossRef CAS.
- K. S. Pitzer and W. E. Donath, J. Am. Chem. Soc., 1959, 81, 3213–3218 CrossRef CAS.
- J. Hartung, T. Gottwald and R. Kneuer, Synlett, 2001, 749–752 CrossRef CAS.
- J. C. Tripp, C. H. Schiesser and D. P. Curran, J. Am. Chem. Soc., 2005, 127, 5518–5527 CrossRef CAS PubMed.
- W. Damm, B. Giese, J. Hartung, T. Hasskerl, K. N. Houk, O. Hueter and H. Zipse, J. Am. Chem. Soc., 1992, 114, 4067–4079 CrossRef CAS.
- R. J. Abraham, M. A. Warne and L. Griffiths, J. Chem. Soc., Perkin Trans. 2, 1997, 2151–2160 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03 (Revision E.01), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- J. B. Foresman and Æ. Frisch, Exploring Chemistry with Electronic Structure Methods, Gaussian Inc., Pittsburgh, PA, 2nd edn, 1996 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- I. V. Alabugin and M. Manoharan, J. Am. Chem. Soc., 2005, 127, 12583–12594 CrossRef CAS PubMed.
- G. Schaftenaar and J. H. Noordik, Comput.-Aided Mol. Des., 2000, 14, 123–134 CrossRef CAS.
- B. Fuchs, in Topics in Stereochemistry, ed. E. L. Eliel and N. L. Allinger, Wiley, New York, 1978, vol. 10, pp. 1–94 Search PubMed.
- R. A. Marcus, J. Phys. Chem., 1968, 72, 891–899 CrossRef CAS.
- P. Gisdakis and N. Rösch, J. Am. Chem. Soc., 2001, 123, 697–701 CrossRef CAS PubMed.
- C. W. Wu and J.-J. Ho, J. Org. Chem., 2006, 71, 9595–9601 CrossRef CAS PubMed.
- G. S. Hammond, J. Am. Chem. Soc., 1955, 77, 334–338 CrossRef CAS.
- I. Kempter, B. Frensch, T. Kopf, R. Kluge, R. Csuk, I. Svoboda, H. Fuess and J. Hartung, Tetrahedron, 2014, 70, 1918–1927 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to the memory of Athelstan (Athel) L. J. Beckwith in recognition of his pioneering and creative contributions to the chemistry of free radicals in general and to free radical cyclizations in particular. We will miss his humor and his intellectual approach to chemical science. |
| ‡ Electronic supplementary information (ESI) available: Standard instrumentation, the protocol for ESI containing instrumentation, synthesis of 4-pentenols, O-pentenyl tosylates, carbon-13 spectra of selected compounds, calculated atomic coordinates and energies of transition structures and radicals. CCDC 1008593. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob01266f |
| § Crystallographic data (excluding structure factors) for the structure in this paper are deposited with the Cambridge Crystallographic Data Centre as supplementary publication [CCDC 1008593 (compound 1g)]. |
| This journal is © The Royal Society of Chemistry 2014 |