
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel ruthenium-catalyst for hydroesterification of olefins with formates†
Irina
Profir
a,
Matthias
Beller
*a and
Ivana
Fleischer
*ab
aLeibniz Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein-Strasse 29a, 18059 Rostock, Germany
bInstitut für organische Chemie, Universität Regensburg, Universitätsstrasse 31, 93040 Regensburg, Germany. E-mail: ivana.fleischer@chemie.uni-regensburg.de
First published on 30th July 2014
Abstract
An alternative ruthenium-based catalyst for the hydroesterification of olefins with formates is reported. The good activity of our system is ensured by the use of a bidentate P,N-ligand and ruthenium dodecacarbonyl. A range of formates can be used for selective alkoxycarbonylation of aromatic olefins. In addition, the synthesis of selected aliphatic esters is realized. The proposed active ruthenium complex has been isolated and characterized.
The conversion of alkenes to esters by carbonylation is a broadly applied and commercially relevant process. It is conventionally performed by the reaction of an alkene with CO gas and corresponding alcohols.1 However, due to the toxicity and the intricate handling of CO, there is an increasing interest in applying alternative carbonyl sources in such transformations.2
As an example, hydroesterification with formates proves to be an attractive route to esters. Since Sneeden and co-workers discovered the ruthenium-catalyzed formation of methyl propionate from ethylene with methyl formate,3 much effort has been put into improving the efficiency and applicability of this reaction. Hence, significantly milder reaction conditions have been developed in the course of the past two decades. Beside ruthenium, researchers are also striving to design hydroesterification catalysts based on other transition metals such as rhodium,4 palladium5 or cobalt.6 Still, the restricted substrate scope as well as the often irreversible decomposition of the formates remain major challenges.
In the past, the most active catalyst systems for simple substrates such as ethylene and methyl formate have been based on ruthenium precursors containing PPN, halide and CO ligands.7 Additionally, the coordination of polar solvents such as DMF resulted in increased activity of the tested catalysts.8 Later, it was reported that ancillary PCy3 not only enabled a broader substrate scope but also allowed high conversion and good yields of the desired products without supplementary gas pressure derived from CO and N2 gas, respectively.9
In 2004, pyridylmethyl formate has been successfully utilized in hydroesterification reactions as CO source.10 Chang and co-workers investigated the positive ligand effects on the reactions with this chelating formate which allowed the conversion of a variety of alkenes in excellent yields using different ruthenium sources as catalyst precursors.11 The addition of catalytic amounts of Bu4NI made the reduction of the working temperature to 70 °C possible.12 These conditions were also successively applied in the formation of amides based on formamide derivatives.13
More recently, efforts regarding a wider range of applicable formates have been reported by Manabe and co-workers when introducing imidazole derivatives as potent additives.14 With respect to the most effective ligands they postulated bidentate coordination to ruthenium forming a five membered metallacycle that would activate the formate C–H bond and yield the desired product without precedent decarbonylation. Despite the improvements achieved by this approach, the amount of ruthenium used for this conversion and related reactions remained high with 15 mol%.
Herein, we present an improved catalyst system composed of Ru3(CO)12 and a phosphine substituted imidazole derivative (Scheme 1).15 The in situ generated ruthenium complex (2.5 mol%) successfully catalyzes the formation of esters in yields up to 95% at 135 °C. Although the formate undergoes decarbonylation, the reaction can be performed in glass pressure tubes (3 mmol scale). Dimethyl formamide proved to be the most suitable solvent with respect to both yield and purity of the chromatographically isolated products.
 | ||
| Scheme 1 Ruthenium-catalyzed olefin hydroesterification. | ||
Employing Ru3(CO)12 and ligand 4a in our model reaction (Table 1) at 135 °C yielded the ester in 89% with a linear to branched ratio of 67![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :33. In comparison, the absence of any additive to Ru3(CO)12 yielded in only 2% and 10% product at 135 °C and 150 °C, respectively (Table 1, entries 1 and 2).
:33. In comparison, the absence of any additive to Ru3(CO)12 yielded in only 2% and 10% product at 135 °C and 150 °C, respectively (Table 1, entries 1 and 2).
|
|
|||
|---|---|---|---|
| Entry | Additive | Yield of 3a (%) |
3aa:3′aab |
| Conditions: 3.0 mmol benzyl formate, 4.5 mmol styrene, 0.83 mol% Ru3(CO)12, 2.5 mol% additive, 1.5 ml DMF, 135 °C, 24 h.a Isolated yield.b Determined by 1H NMR.c The data in parentheses represents results obtained at 150 °C.d Reaction was carried out at 150 °C under otherwise identical conditions.e 5.0 mol% of LiCl. | |||
| 1 | 4a | 89 (82)c | 67:33 (69:31)c |
| 2 | — | 2 (10) | 65:35 (77:23) |
| 3 | 4e | 21 (40) | 76:24 (80:20) |
| 4 | PCy3 | 6 | 67:33 |
| 5 | 4e + PCy3 | 10 | 72:28 |
| 6d | 4b | Traces | — |
| 7 | 4c | 87 | 78:22 |
| 8 | 4d | 28 | 77:23 |
| 9d | 4f | Traces | — |
| 10d | 5a | 7 | 76:24 |
| 11d | 5b | Traces | — |
| 12d | 5c | 10 | 82:18 |
| 13 | Pyridine | Traces | — |
| 14 | TMEDA | No conversion | — |
| 15 | PPh3 | Traces | — |
| 16 | Bu4NI | 22 | 72:28 |
| 17 | Bu4NBr | 30 | 83:17 |
| 18 | 4a + Bu4NBr | 61 | 62:38 |
| 19 | LiCle | 57 | 75:25 |
| 20 | 4a + LiCle | 41 | 51:49 |
A number of other additives has been tested under the optimized conditions (3.0 mmol benzyl formate, 4.5 mmol styrene, 0.83 mol% Ru3(CO)12, 2.5 mol% additive, 1.5 ml DMF, 135 °C, 24 h). To confirm that the reactivity of the system derives from the bidentate P,N-coordination of ligand 4a to the ruthenium center, experiments investigating the individual impact of the phosphine and imidazole moiety have been conducted. Notably, the presence of 1,2-dimethyl imidazole (4e) increased the yield of product (Table 1, entry 3) in comparison to the additive-free system, whereas PCy3 alone had a negligible influence on the outcome of this conversion (Table 1, entry 4). The combination of 4e and PCy3 seemed to inhibit the reaction rather than promoting it (Table 1, entry 5) compared to the results of utilizing 4a.
While the diphenyl phosphine analogue 4b showed no activity at all, the isopropylphosphine substituted imidazole 4c exhibited good activity and selectivity, but was rather difficult to handle due to its low melting point and severe air sensitivity (Table 1, entries 6 and 7). Ligand 4f, which was successfully applied in ruthenium-catalyzed hydroformylation of olefins by our group,16 was inactive in the model system. Also, the lutidine-based phosphine ligands175 that would supposedly form a five-membered ring with ruthenium as observed with 4a, gave unsatisfactory yields up to 10% at 150 °C (Table 1, entries 10–12). Using Bu4NI, Bu4NBr and LiCl as halide sources both in combination with and without ligand 4a had a decreasing effect on the yield (Table 1, entries 16–20).
With the exception of 4c, none of the ligands came close to the activity of 4a. Analyzing the reaction mixtures via GC usually revealed the presence of a small amount of benzyl alcohol and a major amount of benzyl formate after 24 h. In most cases increasing the temperature to 150 °C did not improve the conversion to a significant extent.
As far as formates are concerned, we were delighted to discover a broader applicability among both aliphatic and aromatic derivatives. Although undergoing decarbonylation, yields up to 95% have been obtained. As depicted in Table 2, steric properties of the utilized formates play a pivotal role for successful product formation. While ethyl formate and n-propyl formate showed excellent conversion resulting in 95% and 94% of the corresponding esters, the yield dropped dramatically to 35% when isopropyl formate was applied under identical conditions. tert-Butyl formate showed no product generation at all (Table 2, entry 6). On the other hand, steric hindrance in reactive formates had a positive impact on the linear to branched product ratio, as is shown for isopropyl and phenyl formate (Table 2, entries 4, 5 and 7).
|
|
||||
|---|---|---|---|---|
| Entry | R (1x) | Yielda (%) |
3xa:3′xab |
|
| Conditions: 3.0 mmol formate, 4.5 mmol styrene, 0.83 mol% Ru3(CO)12, 2.5 mol% ligand 4a, 1.5 ml DMF, 135 °C, 24 h.a Isolated yield.b Determined by 1H NMR.c Reaction was carried out at 150 °C under otherwise identical conditions. | ||||
| 1 | Me | 1b | 71 | 52:48 |
| 2 | Et | 1c | 95 | 66:34 |
| 3 | n Pr | 1d | 94 | 64:36 |
| 4 | iPr | 1e | 35 | 83:17 |
| 5c | iPr | 1e | 50 | 81:19 |
| 6 | t Bu | 1f | — | — |
| 7 | Ph | 1g | 56 | 96:4 |

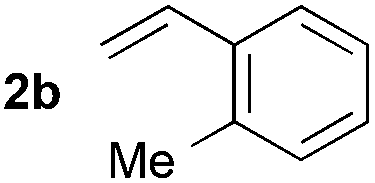
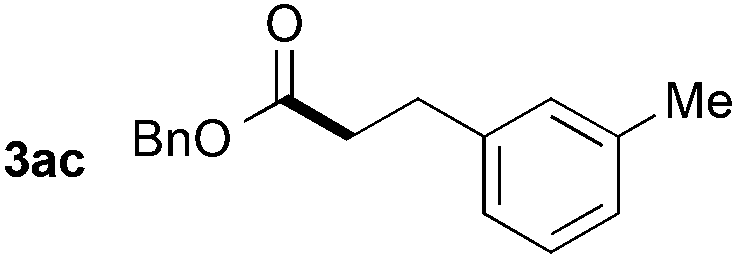
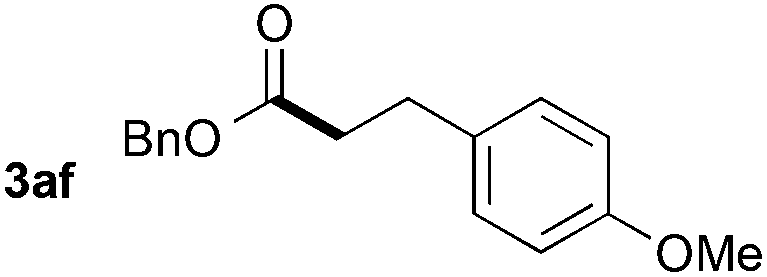
Utilizing different alkenes illustrates the scope and limitations of the presented system (Table 3). In this context, benzyl formate was chosen as substrate for analytical reasons. The degree of decarbonylation and the conversion of the resulting alcohol to the desired product could be observed via GC analysis of the reaction mixtures. Additionally, the combination of yield and regioselectivity obtained with this formate fulfilled the requirements the best in our opinion. While styrene derivatives have been converted successfully at 135 °C (Table 3, entries 1–5), aliphatic alkenes proved to be more challenging. Notably, allylbenzene (2g) and β-methyl styrene (2′g) gave mixtures of the same products 3ag, 3′ag and 3′′ag (Table 3, entries 7 and 8) and the linear product 3ag was the main component. α-Substituted styrenes, such as 2h (Table 3, entry 8), could only be converted to a low amount of product and α-phenyl styrene did not react at all.
|
|
|||
|---|---|---|---|
| Entry | Alkene | Producta | Yieldb (%) (3ay:3′ay)c |
| Conditions: 3.0 mmol benzyl formate, 4.5 mmol alkene, 0.83 mol% Ru3(CO)12, 2.5 mol% ligand 4a, 1.5 ml DMF, 135 °C, 24 h.a Only linear products are displayed in the table. For the structures of the branched products please check the electronic supplementary information (ESI).b Isolated yield.c Ratio determined by 1H NMR.d Reaction was carried out at 150 °C.e 1.7 mol% Ru3(CO)12, 5.0 mol% ligand 4a.f Extended reaction time of 65 h. | |||
| 1 |
|

|
88 (76:24) |
| 2 |

|
|
85 (51:49) |
| 3 |

|

|
72 (64:36) |
| 4 |

|

|
86 (56:44) |
| 5 |

|
|
57 (51:49) |
| 6d,e |

|

|
42 (59:22:18) |
| 7d,e |

|
31 (51:29:20) |
|
| 8d,e |
|

|
10 (>99:1) |
| 9d |
|

|
35 |
| 10d |

|

|
11 (>99:1) 72f (>99:1) |
| 11d |

|

|
17 (76:24) |
| 12d,e |

|

|
21 (76:24) 44f (70:30) |
| 13d |

|
40f (67:33) |
|
| 14 |

|

|
73 (82:18) |

|
|||
Aliphatic alkenes react slower than aromatic alkenes, even at 150 °C. This is shown exemplarily when using 3,3-dimethyl 1-butene (2j) which yielded the product in 11% after 24 h and 72% after 65 h (Table 3, entry 10). Similar to β-methyl styrene, 2-octene underwent isomerization of the double bond and yielded the same products 3al and 3′al as 1-octene (Table 3, entries 12 and 13). At this point, we expected more isomers to be in the product mixture but identifying them by 1H NMR was impossible due to their small concentration and overlap of the signals with those of the main products.
When applying isoprene (2m) under the milder conditions (135 °C), the unsubstituted double bond was converted selectively to the linear product. The sterically hindered double bond was isomerized without further conversion resulting in the β,γ- and α,β-unsaturated carbonyl compounds 3am and 3′am, respectively, at a ratio of 82:18 and an overall yield of 73% (Table 3, entry 14).
In our attempt to identify the active catalytic species, we observed the coordination of ligand 4a to the ruthenium cluster by applying different ratios and analyzing the resulting solutions by NMR spectroscopy (Fig. 1). The metal to ligand ratios [Ru]/4a 1:1 and 1:2 show one main phosphorous species each, indicating complete coordination of the ligand to the metal in both cases. In contrast, applying the metal in excess gives another compound in addition to the 1:1 moiety. We assume that the coordination of the ligand initiates the stepwise dissolution of the [Ru3] cluster, resulting in complexes [Ru3]-4a and [Ru2]-4a at 66 ppm and 53 ppm, respectively, whereas single [Ru] units are coordinated by two ligands at first (59 ppm). Thus, these intermediate species can be observed as minor signals in the 1:1 spectrum. When applying [Ru]/4a 1:3, double coordination of 4a to ruthenium is observed along with uncoordinated ligand. Transferring these ratios to our model system, [Ru]/4a 2:1 gave lower yield but increased linear to branched selectivity compared to 1:1, whereas [Ru]/4a 1:2 showed no conversion of the formate at all.
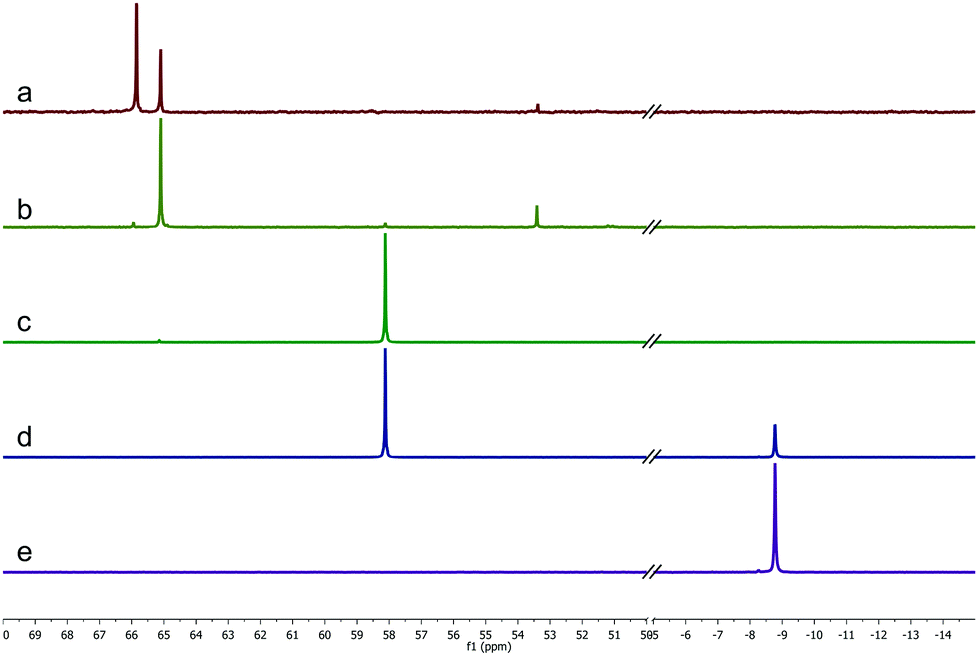 | ||
| Fig. 1
31P NMR spectra of different [Ru]/4a ratios in toluene-d8; (a) 2:1, (b) 1:1, (c) 1:2, (d) 1:3, (e) ligand without ruthenium. | ||
In addition we successfully isolated complex 6 from the 1:1 mixture of ruthenium and ligand 4a (Fig. 2) as dark orange crystals. In contrast to the in situ catalyst system, immediate gas evolution was observed in solution at ambient temperature when applying the complex in the model reaction.
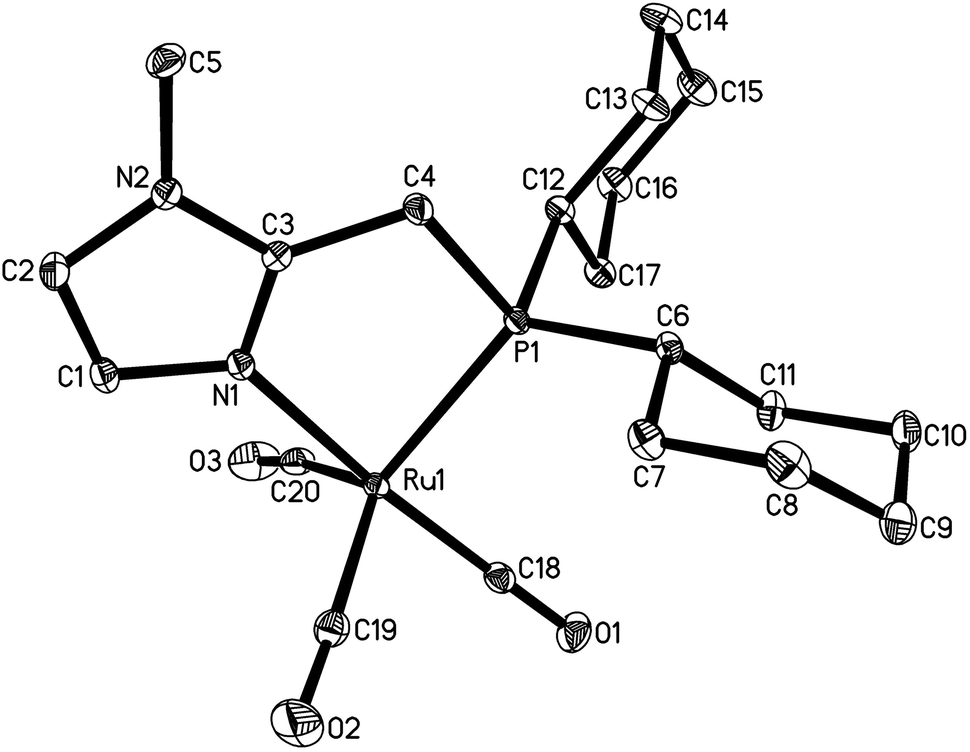 | ||
| Fig. 2 X-Ray structure of complex 6 generated from Ru3(CO)12 and ligand 4a at a ratio of 1:1. | ||
Analyzing the possible coordination of both the formate and the alkene in the course of the reaction has also been attempted in NMR tubes. There was no change in the NMR shifts of the substrates or the complex at ambient temperature. However, at 135 °C partial decomposition of the formate was observed. These results led us to believe that the gas evolution mentioned above derived from substitution of CO ligands by solvent molecules in the complex rather than from decomposition of benzyl formate. By application of only 1.5 mol% of 6 in model reaction 77% of product with a linear to branched ratio of 69:31 were obtained after 24 h at 135 °C. For comparison, experiments have been carried out at 100 °C with both the in situ system [Ru]/4a and the isolated complex 6, respectively. GC analysis of the reaction mixtures after 24 h revealed only low conversion of the preliminary generated benzyl alcohol and yields below 10% of the desired product.
The recently proposed mechanism for the ruthenium-catalyzed hydroesterification of alkenes with formates by Manabe and co-workers is in agreement with our considerations (Scheme 2).18 The reaction starts with the generation of complex 6, though coordination of solvent molecules to the ruthenium center in addition to or in lieu of CO is possible. [Ru] then inserts into the C–H bond of the formate (I). The path following the decarbonylation of the formate (II), coordination of the alkene (III) and carbonylation of the alkoxide moiety (IV) seems more plausible in our case due to the observed accumulation of benzyl alcohol and CO pressure in the model system. On the other hand C–H activation and direct coordination of the alkene to form species IV before the product is released and the active ruthenium complex is regenerated may occur to a minor extent.
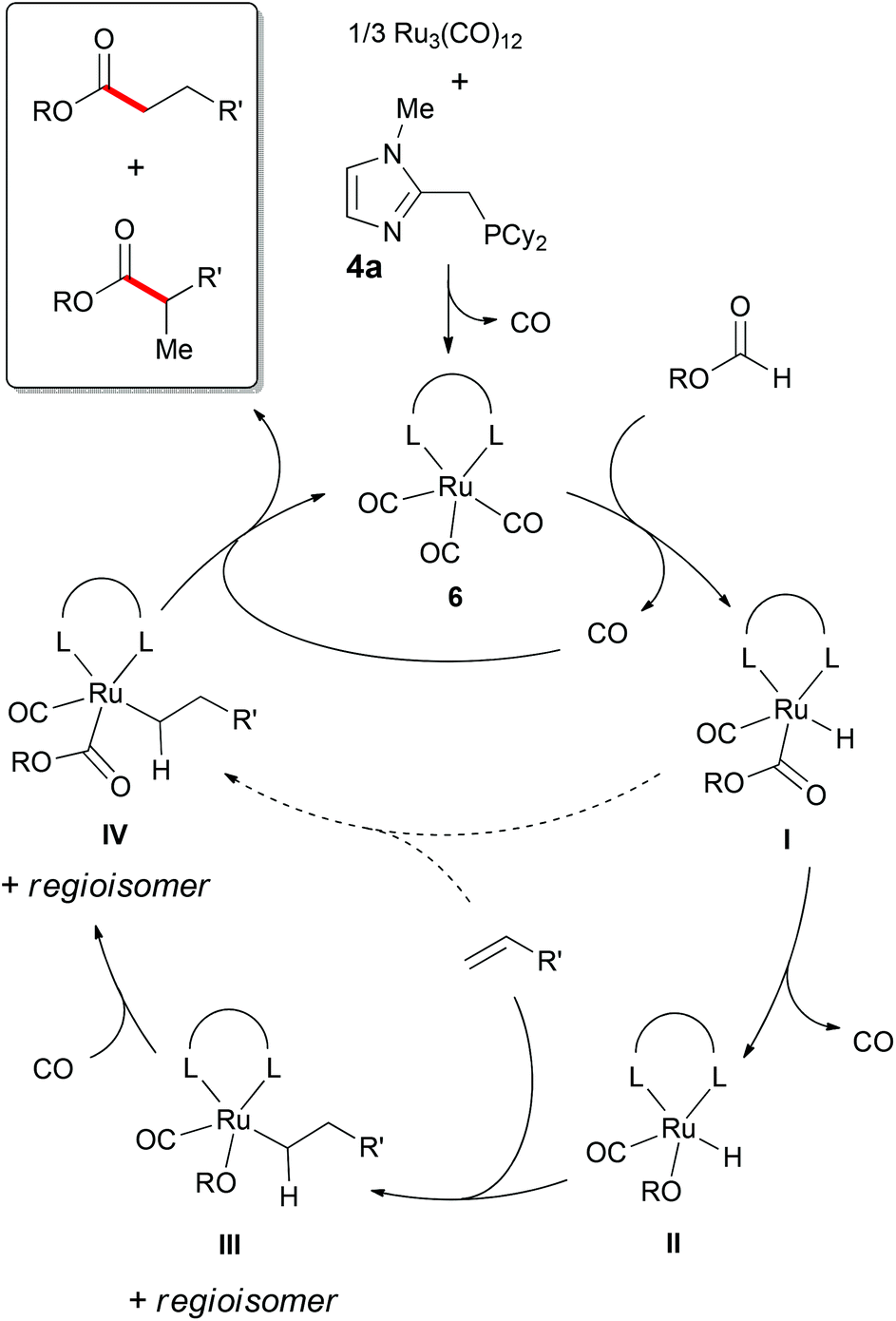 | ||
| Scheme 2 Proposed mechanism for the hydroesterification of alkenes with formates.18 | ||
Conclusions
In summary, the employment of bidentate imidazole-based phosphine ligands provided the most active Ru catalyst system for the alkoxycarbonylation of styrene derivatives with formates. Both aliphatic and aromatic formates were successfully converted to the corresponding esters. Steric hindrance in both alkene and formate led to improved regioselectivity. Aromatic olefins can be carbonylated under comparably milder reaction conditions. In addition, for the first time we were able to isolate and employ a well-defined Ru-complex in this reaction, which provides opportunities for tuning of its catalytic activity and further mechanistic studies.We are grateful to the State of Mecklenburg-Western Pomerania, the Deutsche Forschungsgemeinschaft (Leibniz-price) and the Fonds der Chemischen Industrie (Liebig Fellowship for I.F.) for the financial support. We thank Dr K. Junge, B. Wendt and Dr A. Pews-Davtyan for the provision of ligands and Dr A. Spannenberg for crystallographic analysis of the isolated complex. We further thank the analytical department, workshop and glassblower of the Leibniz Institute of Catalysis for their support.
Notes and references
- For reviews see: (a) G. Kiss, Chem. Rev., 2001, 101, 3435–3456 CrossRef CAS PubMed; (b) B. El Ali, H. Alper, M. Beller and C. Bolm, Transition Metals for Organic Synthesis, Wiley-VCH, Weinheim, 2008, 49–67 Search PubMed; (c) A. Brennführer, H. Neumann and M. Beller, ChemCatChem, 2009, 1, 28–41 CrossRef PubMed.
- T. Morimoto and K. Kakiuchi, Angew. Chem., Int. Ed., 2004, 43, 5580–5588 CrossRef CAS PubMed.
- P. Isnard, B. Denise, R. P. A. Sneeden, J. M. Cognion and P. Durual, J. Organomet. Chem., 1983, 256, 134–139 CrossRef.
- J.-P. Simonato, J. Mol. Catal. A: Chem., 2003, 197, 61–64 CrossRef CAS.
- (a) J. Grévin and P. Kalck, J. Organomet. Chem., 1994, 476, C23–C24 CrossRef; (b) I. Fleischer, R. Jennerjahn, D. Cozzula, R. Jackstell, R. Franke and M. Beller, ChemSusChem, 2013, 6, 417–420 CrossRef CAS PubMed; (c) H. Wang, B. Dong, Y. Wang, J. Li and Y. Shi, Org. Lett., 2014, 16, 186–189 CrossRef CAS PubMed.
- Y. N. Cui, J. M. Yin, D. B. Gao, Y. P. Jia, G. Y. Zhou and S. M. Li, Chin. Chem. Lett., 2007, 18, 17–20 CrossRef CAS PubMed.
- (a) G. Lavigne, N. Lugan, P. Kalck, J. M. Soulié, O. Lerouge, J. Y. Saillard and J. F. Halet, J. Am. Chem. Soc., 1992, 114, 10669–10670 CrossRef CAS; (b) S. Fabre, P. Kalck and G. Lavigne, Angew. Chem., Int. Ed. Engl., 1997, 36, 1092–1095 CrossRef CAS PubMed.
- L. Noël, G. Lavigne, J. M. Soulié, S. Fabre and P. Kalck, Organometallics, 1995, 14, 1712–1731 CrossRef.
- T. Kondo, T. Okada and T. Mitsudo, Organometallics, 1999, 18, 4123–4127 CrossRef CAS.
- L. Wang and P. E. Floreancig, Org. Lett., 2004, 6, 4207–4210 CrossRef CAS PubMed.
- (a) S. Ko, Y. Na and S. Chang, J. Am. Chem. Soc., 2002, 124, 750–751 CrossRef CAS PubMed; (b) B. Li, S. Lee, K. Shin and S. Chang, Org. Lett., 2014, 16, 2010–2013 CrossRef CAS PubMed.
- (a) E. J. Park, J. M. Lee, H. Han and S. Chang, Org. Lett., 2006, 8, 4355–4358 CrossRef CAS PubMed; (b) N. Armanino, M. Lafrance and E. M. Carreira, Org. Lett., 2014, 16, 572–575 CrossRef CAS PubMed.
- (a) S. Ko, H. Han and S. Chang, Org. Lett., 2003, 5, 2687–2690 CrossRef CAS PubMed; (b) N. Armanino and E. M. Carreira, J. Am. Chem. Soc., 2013, 135, 6814–6817 CrossRef CAS PubMed; (c) B. Li, Y. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 1125–1131 CrossRef CAS PubMed.
- H. Konishi, T. Ueda, T. Muto and K. Manabe, Org. Lett., 2012, 14, 4722–4725 CrossRef CAS PubMed.
- K. Junge, B. Wendt, F. A. Westerhaus, A. Spannenberg, H. Jiao and M. Beller, Chem. – Eur. J., 2012, 18, 9011–9018 CrossRef CAS PubMed.
- (a) I. Fleischer, K. M. Dyballa, R. Jennerjahn, R. Jackstell, R. Franke, A. Spannenberg and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 2949–2953 CrossRef CAS PubMed; (b) I. Fleischer, L. Wu, I. Profir, R. Jackstell, R. Franke and M. Beller, Chem. – Eur. J., 2013, 32, 10589–10594 CrossRef PubMed.
- A. Pews-Davtyan, X. Fang, R. Jackstell, A. Spannenberg, W. Baumann, R. Franke and M. Beller, Chem. – Asian. J., 2014, 9, 1168–1174 CrossRef CAS PubMed.
- H. Konishi and K. Manabe, Synlett, 2014 DOI:10.1055/s-0033-1339136.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1003596. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob01246a |
| This journal is © The Royal Society of Chemistry 2014 |