
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Monitoring penetratin interactions with lipid membranes and cell internalization using a new hydration-sensitive fluorescent probe†
Oleksandr M.
Zamotaiev
ab,
Viktoriia Y.
Postupalenko
a,
Volodymyr V.
Shvadchak
a,
Vasyl G.
Pivovarenko
*ab,
Andrey S.
Klymchenko
*a and
Yves
Mély
a
aLaboratoire de Biophotonique et Pharmacologie, UMR 7213 du CNRS, Université de Strasbourg, Faculté de Pharmacie, 74 route du Rhin, 67401 Illkirch, France. E-mail: andrey.klymchenko@unistra.fr; Fax: +33 368 85 43 13; Tel: +33 368 85 42 55
bOrganic Chemistry Chair, Chemistry Faculty, Taras Shevchenko National University of Kyiv, 01601 Kyiv, Ukraine. E-mail: pvg_org@ukr.net; Fax: +38044-2898391; Tel: +38044-2393312
First published on 15th July 2014
Abstract
A new fluorescent label N-[4′-(dimethylamino)-3-hydroxyflavone-7-yl]-N-methyl-β-alanine (7AF) was synthesized. Due to two electron donor groups at the opposite ends of the chromophore, an excited state intramolecular proton transfer (ESIPT) resulting in a dual emission was observed even in highly polar media and its fluorescence quantum yield was found to be remarkably high in a broad range of solvents including water. As a consequence, this label exhibits a remarkable sensitivity to the hydration of its environment, which is observed as a color switch between the emission of the ESIPT product (T* form) and that of the normal N* form. The 7AF label was coupled to the N-terminus of penetratin, a cell penetrating peptide, in order to study its interactions with lipid membranes and internalization inside the cells. As expected, the binding of penetratin to lipid membranes resulted in a dramatic switch in the relative intensity of its two emission bands as compared to its emission in buffer. Our studies with different lipid compositions confirmed the preference of penetratin to lipid membranes of the liquid disordered phase. After incubation of low concentrations of labeled penetratin with living cells, ratiometric imaging revealed, in addition to membrane-bound species, a significant fraction of free peptide in cytosol showing the characteristic emission from aqueous medium. At higher concentrations of penetratin, mainly peptides bound to cell membrane structures were observed. These observations confirmed the ability of penetratin to enter the cytosol by direct translocation through the cell plasma membrane, in addition to the classical entry by endocytosis. The present probe constitutes thus a powerful tool to study the interaction of peptides with living cells and their internalization mechanisms.
Introduction
Despite the ever growing development of fluorescent probes and instrumental techniques for tracking the localization of biomolecules in living cells,1–3 it remains a challenge to monitor directly their molecular environment and interactions with their cellular targets.4,5 Förster resonance energy transfer (FRET), being the most commonly used approach, is limited because it requires double labeling. In this respect, the use of environment-sensitive probes, which change their emission color or intensity as a function of their environment properties (polarity, hydration, viscosity, etc.),6–9 appears as a promising and simple alternative. These probes should sensitively report on changes in the environment of a given biomolecule from the highly polar environment of the cytoplasm to the apolar environment in cellular lipid compartments.10Most of the polarity sensors developed in the last decade for peptide labeling were based on popular solvatochromic fluorescent dyes undergoing intramolecular charge transfer, namely Prodan derivatives,11–13 dimethylaminophthalimide,14 and others.15–17 They were applied to investigate protein–protein interactions and protein conformational transitions.11–14,16,18,19 Though effective enough in the mentioned studies, these probes show limited spectroscopic properties and sensitivity to binding events. Most of them are limited to applications in a relatively non-polar environment, because in polar media, especially in water, their fluorescence is quenched15 or is poorly sensitive to the environment.
3-Hydroxyflavones (3HFs) are fluorescent dyes exhibiting strong sensitivity of their dual emission to the environment as a result of an excited-state intramolecular proton transfer (ESIPT) reaction20 (Scheme 1). The ratio of emission of the normal (N*) to the photo-tautomer (T*) species provides a concentration-independent response to the environment polarity and H-bonding interactions.21–26 Both the mentioned processes inhibit the ESIPT reaction and thus decrease the relative intensity of the T* band. Modification of the structure of 3HFs gives the possibility of sensing separately the hydration27 (H-bond donating property28–30) and H-bond accepting property27,30 of the surroundings. 2-Furyl-3-hydroxychromones31,32 and 4′-methoxy-3HFs26,27 present optimal two-band ratiometric response to the environment in polar media, which was successfully used to study peptide–peptide and peptide–nucleic acid interactions. However, their application for microscopy techniques is limited due to their absorption in the UV region. On the other hand, 4′-(dialkylamino)-3HFs exhibit absorption and emission in the visible spectral region, but due to much higher dipole moments,33 their dual emission is optimally sensing low- and middle-polar media, while in water their T* band is poorly resolved. They were successfully applied to the study of model and cellular membranes6,34,35 and protein–lipid interactions,24,36 but they are strongly quenched in aqueous media. Recently, 4′-(dialkylamino)-3HFs bearing an electron donating amino group in 7-position were shown to exhibit higher emission of the T* form with better resolution of N* and T* bands,37 giving thus the possibility of ratiometric sensing in highly polar media. Therefore, we hypothesized that 7-substituted 4′-(dialkylamino)-3HFs could be a promising environment-sensitive label that combines absorption in the visible region with sensitivity in polar (aqueous) media.
 | ||
| Scheme 1 ESIPT reaction scheme in 3HF dyes. Structure of the 7AF label and the sequence of penetratin. | ||
To validate our hypothesis, we synthesized the N-[4′-(dimethylamino)-3-hydroxyflavone-7-yl]-N-methyl-β-alanine (7AF) probe and characterized its spectroscopic properties. The 7AF probe was found to be highly sensitive to the local concentration of water and could thus be used to quantify it. Moreover, we coupled 7AF to the N-terminus of the cell penetrating peptide (CPP) penetratin, a cationic peptide containing 16 amino acids, 6 of which being lipophilic. Penetratin binds lipid membranes, preferentially to their fluid phases,38 and enters into cells by different mechanisms,38–42 showing a strong potential for applications as a vector for the intracellular delivery of hydrophilic biomolecules and drugs.43,44 The labeled peptide was used to sensitively monitor its binding to lipid vesicles of various compositions and lipid phases. Moreover, the new hydration-sensitive label allowed us to monitor the internalization of penetratin into cells and distinguish for the first time two of its internalization pathways by ratiometric imaging.
Results and discussion
Label synthesis and characterization
The synthesis of the new label (7AF) was performed in five steps, through the preparation of substituted 4-amino-2-hydroxyacetophenone (3). It was converted into 3-hydroxyflavone (4) by condensation with 4-N,N-dimethylaminobenzaldehyde and a subsequent oxidative heterocyclization (Scheme 2). Then, the target product 7AF was obtained through carboxyethylation of the 7-amino group by a Michael reaction (see Materials and methods and ESI†).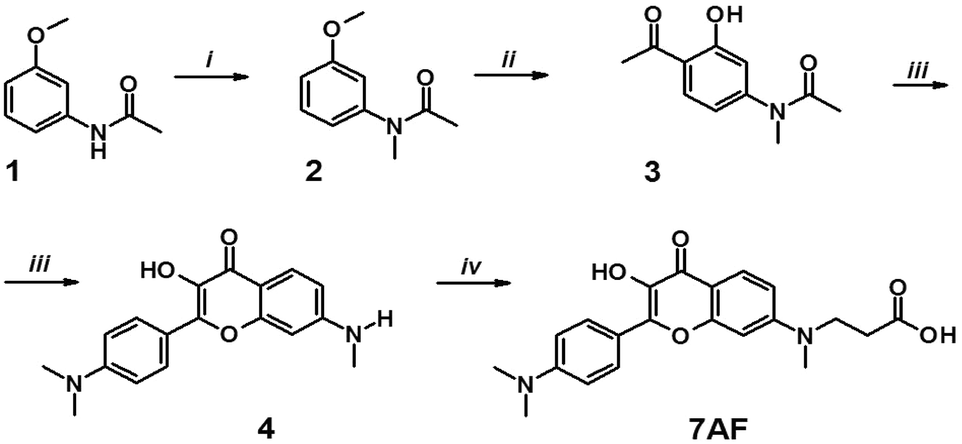 | ||
| Scheme 2 Synthesis of the 7AF label: (i) NaH, CH3I, THF; (ii) AcCl, AlCl3; (iii) MeONa, DMF, p-Me2N–C6H4–CHO, then MeONa, H2O2; (iv) AcOH, acrylic acid. | ||
Spectroscopic properties
The absorption spectrum of 7AF in different solvents consists of one band centered at about 400 nm, similar to its parent 4′-(dialkylamino)-3HF analogues.15,23,25,34,35 In organic solvents, 7AF shows two well-separated emission bands corresponding to the normal (N*) and tautomer (T*) forms of the dye24 (Fig. 1A, S1A and Table S1†). The position of the N* emission band strongly shifts to the red upon an increase in solvent polarity (Table S1†), like for other 4′-dialkylaminoflavones.6,23,35,45,46 Remarkably, the relative intensity of the N* band in aprotic solvents is very small even for highly polar solvents, which is a clear difference with the parent 4′-dialkylamino-3HFs (Table S1,†Fig. 1A and ref. 6,23,35,45). In protic solvents, the N* band relative intensity increases drastically and in water it becomes dominant (Fig. 1A). Thus, from polar aprotic DMF to polar water solvent, 7AF undergoes a complete switch from the red-shifted emission of the T* form to the blue-shifted emission of the N* form (Fig. 1B). This is a clear advantage in terms of environment-sensitivity compared to other 4′-(dialkylamino)-3HF, which showed a relatively strong N* band emission in polar aprotic solvents.24 In line with previous reports,27 this dramatic effect of protic solvents is likely due to the inhibition of the ESIPT reaction by H-bonding of the dye with the solvent. Moreover, measurements of the ratio of the emission band intensities (IN*/IT*) for the 7AF probe in DMF–water binary mixtures show the possibility to quantify the water concentration. Indeed, the logarithm of IN*/IT* changes linearly with the total concentration of water (Fig. 1B inset, Fig. S3†). Significant deviations from the linearity are observed only at high water content (≥70%) that we attribute to preferential solvation effects.27 Importantly, the 7AF emission is not sensitive to pH in the range of 6–9 (pKa = 10.5, Fig. S2†) and to ionic strength in aqueous solutions (Fig. S1B†). The IN*/IT* ratio is also poorly sensitive to basic solvents, since the intensity of the N* form is negligible in DMF (Fig. 1A). All mentioned features allow the application of the log(IN*/IT*) fluorescence parameter for estimating the local water concentration CL27 or, alternatively, the partial volume WA of water in the probe surroundings (relative local water access), defined as WA = 100% CL/55.5. | ||
| Fig. 1 (A) Fluorescence spectra of the 7AF label in water, methanol, ethanol, tetrahydrofuran and N,N-dimethylformamide, normalized at their maxima. Excitation wavelength was at the absorption maximum. (B) Fluorescence spectra of 7AF in mixtures of DMF and water. Inset: plot of the logarithm of fluorescence intensity ratio log(IN*/IT*) versus total concentration of water in the mixture. The solid line represents the fit with the equation: log(IN*/IT*) = 0.0377Cwater − 0.975 (R2 = 0.97). | ||
In contrast to most solvatochromic fluorophores including 3HFs, the 7AF label shows rather high fluorescence quantum yields (11–44%) in all types of solvents, including water. Moreover, emission of N* and T* bands is better separated than for other dialkylamino derivatives of 3HFs (Fig. 1A).24 Thus, in line with our initial aim, the new label shows both absorption in the visible region and highly sensitive dual emission, which allows quantitative estimation of the probe hydration.
Applications of 7AF for investigating penetratin–membrane interactions
The penetratin peptide labeled by 7AF at its N-terminus (Scheme 1) was prepared by solid-phase synthesis. In aqueous buffer, the labeled peptide shows a single emission band close to that of the label alone, but with a slightly higher fluorescence quantum yield (Fig. 2A, Table 1). The shoulder of the T*-band emission at 597 nm (Fig. 2A) is more pronounced than for the free label, pointing to a slight decrease in hydration due to the proximity of the peptide backbone. | ||
| Fig. 2 Fluorescence spectra of 7AF-labeled penetratin alone and in the presence of lipid vesicles: (A) normalized spectra of the 7AF label and 7AF-labeled penetratin in 20 mM phosphate buffer, 150 mM NaCl (pH = 7.4) at 20 °C. Deconvolution of their fluorescence spectra into the N* and T* forms. (B) Fluorescence spectra of 7AF-labeled penetratin in the presence of DOPC LUVs at different lipid/peptide (L/P) molar ratios. (C) Changes of the N*/T* ratio and corresponding local hydration of 7AF–penetratin bound to lipid vesicles at different L/P ratios. | ||
| Media | λ ABS | λ N* | λ T* | I N*/IT* | QY (%) | W A (%) |
|---|---|---|---|---|---|---|
| a λ ABS – position of absorption maxima (nm); λN* and λT* – positions of the fluorescence maxima of N* and T* forms, respectively. IN*/IT* – ratio of integral intensities of the two emission bands (errors are ±2%). The presented fluorescence data were obtained from deconvolution of the fluorescence spectra, as shown in Fig. 2A. QY (%), fluorescence quantum yield (errors are ±5%), measured using FE in EtOH (QY = 51%51) as a reference; WA – calculated water access (%): WA = 100% CW/55.5 mol L−1 where the concentration of water CW was obtained from the calibration curve in Fig. 1B. Lipid/peptide ratio is 1000. b Data for the 7AF probe alone. | ||||||
| 7AF in buffer (25 °C)b | 411 | 534 | 593 | 13 | 11 | 100 |
| Buffer (25 °C) | 414 | 535 | 597 | 6.4 | 14 | 85 |
| DOPC (25 °C) | 411 | 497 | 584 | 0.66 | 40 | 39 |
DOPC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ch (6:4) :Ch (6:4) |
413 | 514 | 583 | 0.52 | 41 | 34 |
| DPPC (25 °C) | 411 | 497 | 586 | 2.2 | 23 | 40 |
| DPPC (45 °C) | — | 506 | 584 | 0.54 | 40 | 33 |
| SM (25 °C) | 417 | 490 | 585 | 1.7 | 31 | 55 |
| SM (50 °C) | — | 496 | 583 | 1.1 | 50 | 44 |
| SM:Ch (6:4, 25 °C) |
— | 533 | 583 | 2.8 | 18 | 58 |
| SM:Ch (6:4, 50 °C) |
— | 513 | 583 | 0.92 | 46 | 44 |
Next, we investigated the interaction of the labeled penetratin with model lipid membranes. Penetratin is known to induce aggregation of zwitterionic lipid vesicles in a large range of lipid to peptide (L/P) ratios.39 This makes problematic the application of intensiometric probes for investigating peptide–membrane interactions. The ratiometric response of the 7AF probe overcomes this problem, since the ratiometric signal, having internal calibration, is independent of the total fluorescence intensity.
Fluorescence spectra of penetratin in the presence of lipid vesicles depend strongly on the L/P ratio (Fig. 2B). At low L/P ratio (0–20), the spectra are close to that in buffer, indicating a large fraction of free peptide. However, we cannot exclude the presence of penetratin aggregates47 at the membrane surface, but they are likely poorly fluorescent. Starting from a L/P ratio of 40 (Fig. 2B and S4†), which corresponds to a dense membrane surface covering (up to 1 peptide per 5 nm2),39,48 the shape of the spectra changes drastically, so that the fluorescence intensity grows and the IN*/IT* ratio drops. These changes suggest a shift of the label from water to the poorly hydrated lipid membranes, confirming the efficient binding of the peptide to lipid membranes. Low IN*/IT* ratios were also observed for other lipid compositions presenting the fluid phase, such as DOPC:Ch, DPPC (45 °C) and SM:Ch (45 °C) at L/P ratios >100. In contrast, for lipid vesicles presenting the gel phase (DPPC and SM at 25 °C) or the liquid ordered phase (SM:Ch at 25 °C), the IN*/IT* ratio and the shape of the spectra are closer to those in aqueous buffer, even for high L/P ratios. Therefore, the binding of penetratin to these lipid phases appears much less efficient, so that a large fraction of the peptide is in the free form with possibly some poorly fluorescent aggregated species at the lipid membranes. The observed higher affinity of penetratin for fluid phases is in line with previous reports.38 Using the IN*/IT* ratio of the integral band intensities, we estimated the decrease in water access from 85% for the peptide in buffer to 33–50% in lipid membranes of the fluid phase. These last values correspond to the water access values of the interfacial region of the lipid bilayer, determined with other methods.49,50
The effect of the phase preference can be further seen in the temperature-dependence of the emission spectra of 7AF–penetratin in the presence of LUVs composed of DPPC (Fig. 3A and B). Below the phase transition temperature, the fluorescence spectra show a large fraction of free peptide, characterized by a high IN*/IT* ratio. Further increase in the temperature close to the phase transition (41 °C) leads to a sharp decrease in the N*/T* ratio accompanied by an increase in the total fluorescence intensity. This shows that the gel to fluid phase transition results in efficient binding and insertion of penetratin into the lipid membranes. Remarkably, with lipid vesicles presenting the liquid ordered phase (SM:Ch), the observed temperature-dependent decrease in the N*/T* ratio was continuous without a sharp phase transition (Fig. 3B), in line with the relatively broad temperature range for its phase transition. Thus our data indicate that penetratin avoids the liquid ordered phase similarly to the gel phase.
 | ||
| Fig. 3 Temperature dependence of the emission spectra of 7AF–penetratin in the presence of lipid vesicles. (A) Fluorescence spectra of 7AF–penetratin in the presence of DPPC LUVs at various temperatures. (B) Temperature-dependence of the local hydration and N*/T* ratio of 7AF–penetratin in the presence of DPPC or SM-cholesterol (6:4) LUVs. The lipid/peptide ratio is 250 in all cases. | ||
Monitoring interactions with living cells
Understanding the mechanisms of internalization of cell penetrating peptides is of key importance for the development of new efficient delivery systems.52 Due to strong sensitivity of 7AF label to hydration and its high fluorescence quantum yield in both aqueous and lipid media, we used 7AF–penetratin to monitor by fluorescence microscopy internalization of the peptide into the living cells. Two mechanisms of penetratin entry into cells are described, namely endocytosis and direct translocation of the peptide through the plasma membrane.40 Since endocytosis proceeds through binding of the peptide to the plasma membrane, we expect this pathway to be associated with low N*/T* ratios. In contrast, translocation is thought to drive the peptide into the cytosol, so that this pathway should be associated with high N*/T* ratios due to the aqueous nature of the cytosol. In addition, as the quantum yield of the probe is comparable in aqueous and lipid media (Table 1), we should be able to visualize the two pathways simultaneously. Since the balance between the two mechanisms was reported to depend on the extracellular concentration of the peptide, we monitored the internalization of 7AF-labeled penetratin at 2 μM and 10 μM, where translocation and endocytosis are, respectively, the major modes of internalization.53 Moreover, the images were recorded at different incubation times (5, 10 and 30 min), to take into account that penetratin internalization is time dependent.53At 2 μM concentration, the rather slow internalization of penetratin allowed us during the first minutes to clearly distinguish two populations of 7AF-labeled penetratin molecules (Fig. 4A–D). The first and most important population is observed as a diffuse fluorescence in the cytosol, and is characterized by a high intensity ratio of blue/red (B/R) channels in the ratiometric images. This high ratio is in line with the high N*/T* ratio observed in aqueous media, indicating a high level of probe hydration. This diffuse fluorescence in the cytosol is consistent with penetratin peptides that have been internalized by the translocation mechanism. The second population is associated with cell plasma membrane and intracellular dots, and is characterized by a low B/R intensity ratio, in line with the low N*/T* ratio observed on model membranes. This second population could be ascribed to penetratin molecules that are endocytosed by binding first to the plasma membrane and then being internalized in early endosomes formed by invaginations of the cell plasma membranes. After 30 min of incubation, all penetratin molecules were found to exhibit low B/R ratio values, suggesting their association with intracellular lipid compartments (Fig. 4D). This redistribution of the peptides likely results from the peptide partition coefficient that favors the lipid over the aqueous phase.
 | ||
| Fig. 4 Fluorescence microscopy imaging of HeLa cells stained with 2 μM (A–D) and 10 μM (E–F) of 7AF–penetratin after 5 (A, C), 10 (B, E) and 30 (D, F) min of incubation. The color of each pixel represents the value of the intensity ratio of blue/red channels, while the pixel intensity corresponds to the total intensity at both channels. The two-photon excitation wavelength was at 830 nm. | ||
At 10 μM concentration, penetratin internalizes rapidly, so that within 10 min most of the fluorescence is observed inside the cells (Fig. 4E), including some fraction in the nucleus. In the cytoplasm, the labeled penetratin molecules are associated with intracellular dots and exhibit mainly low B/R ratios, in full line with an accumulation of penetratin in endosomal compartments, and thus, with endocytosis being the major internalization pathway. Rare areas with high B/R ratios were also observed, suggesting that a minor population of penetratin may enter through translocation, but the signal associated with this population is probably hidden by the signal of the major population in the lipid compartments. In the nucleus, penetratin is located in a highly hydrated medium (high B/R values), with preferential binding to nucleoli. Thus, penetratin after entering into the cytosol could cross the nuclear membrane and then, due to its numerous positively charged amino acids, it could bind to the negatively charged RNAs concentrated in the nucleoli. At 30 min, the images were very similar to those at 10 min, suggesting that no major redistribution of the penetratin molecules occurs in this time frame (Fig. 4F).
Taken together, our data confirm the two different internalization modes of penetratin and their dependence on the external concentration of penetratin. Due to the dependence of its N*/T* ratio on the hydration of its environment and its comparable quantum yields in hydrated and non-hydrated media, this probe provides the possibility for the first time to distinguish the two internalization pathways by imaging techniques.
Conclusions
A fluorescent label based on 3-hydroxyflavone bearing electron donor groups at 4′ and 7-positions (7AF) was synthesized. The new label showed selective response to the local concentration of water by the intensity ratio of the normal (N*) form to the ESIPT product (T*) form and good quantum yields both in water and lipids. It was grafted to the N-terminus of the cell penetrating peptide penetratin in order to monitor its local hydration, which is expected to change after peptide interaction with lipid membranes. It was found that the probe hydration estimated from its dual emission showed a significant decrease after binding to lipid membranes and the affinity of the peptide to lipid membranes depended strongly on their phase state. Then, we monitored the internalization of the labeled penetratin into living cells with the aim to image its intracellular distribution as well as to distinguish the free peptide species from those bound to cell membranes. Using ratiometric imaging, we evidenced that at low penetratin concentrations, a significant fraction of the peptide is localized in the cytosol without binding to membrane components, suggesting that the peptide is mainly internalized through direct translocation into the cytosol. At high concentrations, endocytosis becomes the major internalization mode, so that the labeled peptides are mainly associated with the plasma membrane and endosomes, as indicated by the characteristic fluorescence intensity ratio of the label corresponding to the lipid environment. Thus, the new label can reveal the internalization mechanisms of a cell penetrating peptide into the living cells.Materials and methods
Reagents and solvents
All the reagents were purchased from Sigma-Aldrich Chemical Company. Solvents for synthesis were of reagent quality and were appropriately dried if necessary. For absorption and fluorescence studies, the solvents were of spectroscopic grade.Synthesis
:20). After the reaction was complete, the mixture was evaporated in vacuo. The obtained oil crystallized after adding 20 mL of heptane. The gray precipitate was filtered and washed with heptane. Yield 12.9 g (87.8%). 1H-NMR (300 MHz, CDCl3): δ 2.09 (s, 3H, COCH3), 3.72 (s, 3H, OCH3), 6.59 (dd, 1H, J = 2 Hz, J = 8.3 Hz, ArH), 6.89 (d, 1H, J = 2 Hz, J = 8.3 Hz, ArH), 7.13 (t, 1H, J = 8.3 Hz, ArH), 7.20 (t, 1H, J = 2 Hz, ArH), 7.24 (br. s, 1H, NH).
:5). After evaporation of the solvent in vacuo, the residue was dissolved in methylene chloride (100 mL), filtered, washed twice with water and dried over MgSO4. After evaporation of the solvent, 7.91 g of yellow oil was obtained. The substance was pure enough for its usage in the next step. Yield 73%. 1H-NMR (300 MHz, CDCl3): δ 1.90 (s, 3H, NCOCH3), 3.25 (s, 3H, NCH3), 3.82 (s, 3H, OCH3), 6.72 (t, 1H, J = 1.9 Hz, ArH), 6.77 (dd, 1H, J = 8.1 Hz, J = 1.9 Hz, ArH), 6.87 (dd, 1H, J = 8.1 Hz, J = 1.9 Hz, ArH), 7.31 (t, 1H, J = 8.1 Hz, ArH).
:20). Then the reaction mixture was poured onto ice-water, the organic layer was separated and washed twice with water, dried with MgSO4 and evaporated. The obtained yellow oil was purified by repeated recrystallization from heptane. Yield 1.73 g (19.4%), white crystals. 1H-NMR (300 MHz, CDCl3): δ 2.01 (s, 3H, NCOCH3), 2.65 (s, 3H, ArCOCH3), 3.29 (s, 3H, NCH3), 6.77 (dd, 1H, ArH, J = 8.4 Hz, J = 2.2 Hz), 6.82 (d, 1H, ArH, J = 2.2 Hz), 7.79 (d, 1H, ArH, J = 8.4 Hz), 12.38 (s, 1H, ArOH).
:5). As a result, 150 mg (20%) of yellow powder was obtained. 1H-NMR (300 MHz, CDCl3): δ 2.97 (s, 3H, NHCH3), 3.08 (s, 6H, NMe2), 6.52 (d, 1H, ArH, J = 1.9 Hz), 6.63 (dd, 1H, ArH, J = 9.1 Hz, J = 1.9 Hz), 6.88 (d, 2H, ArH, J = 9.1 Hz), 7.98 (d, 1H, ArH, J = 9.1 Hz), 8.16 (d, 2H, ArH, J = 9.1 Hz).
:8). Then the mixture was cooled and evaporated in vacuo. To the obtained brown oil 15 mL of water was added. A yellow precipitate was collected, and washed twice with water. The crude product was purified by preparative TLC (CH2Cl2–MeOH, 85:15). Yield 10 mg (16.2%). 1H-NMR (300 MHz, DMSO-d6): δ 2.50 (t, 2H, J = 7 Hz, CH2), 3.00 (s, 6H, NMe2), 3.03 (s, 3H, NMe), 3.71 (t, 2H, J = 7 Hz, NCH2), 6.72 (d, 1H, J = 2.5 Hz, ArH), 6.82 (d, 2H, J = 9.3 Hz, ArH), 6.84 (dd, 1H, J = 9 Hz, J = 2,5 Hz, ArH), 7.82 (d, 1H, J = 9 Hz, ArH), 8.07 (d, 2H, J = 9.3 Hz, ArH), 8.72 (br. s, 1H, OH).
Peptide synthesis, labeling, and purification
Peptides were synthesized by solid phase peptide synthesis on a 433A synthesizer (ABI, Foster City, CA). The synthesis was performed on a 0.1 mmol scale using standard side-chain protected fluorenylmethoxycarbonyl (Fmoc)-amino acids and the HBTU/HOBt coupling protocol. LL-HMP resin (ABI, 0.44 mmol g−1 reactive group concentrations) was used as a solid support. At the end of the synthesis, peptidyl-resin was isolated and washed twice with NMP.Two equivalents (0.15 mmol) of the label were dissolved in 1 mL of NMP mixed with two eq. of HBTU/HOBt coupling solution (in DMF) and added to Fmoc-deprotected peptidylresin (0.075 mmol) swelled in 1 mL of NMP. After a few minutes of shaking, five eq. of DIEA solution were added. Then, the reaction mixture was shaken overnight at 40 °C. Resin was filtered and washed with NMP, methanol and DCM.
Cleavage and deprotection of the peptidylresin were performed for 2 h using a 10 mL trifluoroacetic acid (TFA) solution containing water (5%, v/v), TIS (iPr3SiH, 2.5%, v/v), phenol (1%, w/v), thioanisole (5%, v/v) and ethanedithiol (2.5%, v/v). The solution was concentrated in vacuo and the peptide was precipitated using ice-cold diethyl ether and then pelleted by centrifugation. The pellet was washed with diethyl ether and dried. The peptide was solubilized with aqueous TFA (0.05%, v/v). HPLC purification was carried out on a C8 column (uptisphere 300A, 5 μm; 250 × 10, Interchim, France) in a water–acetonitrile mixture containing 0.05% TFA with a linear gradient (20–60% of acetonitrile for 30 min) and monitored at 210 nm (detection of non-labeled peptide) and 370 nm (detection of labeled peptide). The molecular mass obtained by ion spray mass spectrometry was 2610.4 for 7AF–penetratin (in agreement with the expected theoretical mass of 2610.7). Prior to use, the peptide was dissolved in distilled water, aliquoted and stored at −20 °C. The concentration of the labelled peptide was determined from the label absorbance at 400 nm using ε = 25000 M−1 cm−1.
Preparation of liposomes
All lipidic components were purchased in lyophilized form or in CHCl3 solution from Avanti Polar Lipids (Birmingham, USA). Stock solutions of 2 mM were prepared in CHCl3 and stored in glass vials at −20 °C. Aliquots of the appropriate amounts of stock solutions were mixed in glass flasks. The CHCl3 was removed under vacuum for at least 1 h, and then lipids were rehydrated in 25 mM phosphate buffer, pH 6.5, 150 mM NaCl, to a final concentration of 1 mM lipid in the form of multi-lamellar vesicles (MLVs). Large unilamellar vesicles (LUVs) were prepared by extrusion through polycarbonate films (Nucleopore) in a handheld extruder (Avanti Polar Lipids). The MLV suspension was first reduced in size by 7 passages through a large pore size (1 μm) filter. The suspension was then passed 10 times through a filter with 0.1 μm pore size. The LUVs were prepared at temperatures 5–15 °C above the transition points. The size of all vesicles was determined by DLS using a Zetasizer Nano ZS equipped with a 633 nm laser (Malvern Instruments). The measured diameter of all LUVs was 110 ± 10 nm. The vesicles were used within 5 days of preparation (stored at 4 °C). For vesicles composed of more than one type of lipid, all given percentages are based on relative molar composition.Instrumentation
Proton NMR spectra were recorded on a 300 MHz Bruker spectrometer and mass spectra were recorded on a Mariner System 5155 mass spectrometer using the electrospray ionization (ESI) method. All column chromatography experiments were performed on silica gel (Merck, Kieselgel 60H, Art 7736). Absorption and fluorescence spectra were recorded on a Cary 400 spectrophotometer (Varian) and a FluoroMax 3.0 spectrofluorimeter (Jobin Yvon, Horiba), respectively. For fluorescence studies, the dyes were used at 0.5 to 1 μM concentrations. All measurements were performed in 20 mM phosphate buffer, containing 150 mM NaCl (pH = 7.4) at 20 °C. The excitation wavelength was 400 nm for labeled peptides and 408 nm for the free 7AF label. All the spectra were corrected for Raman scattering and background fluorescence measured before the addition of the labelled peptide. Fluorescence quantum yields were determined using 3-hydroxy-4′-diethylaminoflavone as a reference (quantum yield, φ = 51%51). For the experiments in water, 10 mM phosphate buffer, containing 30 mM NaCl (pH 7.0), was used systematically.Deconvolution of spectra into two bands and calculations of polarity and hydration parameters
Deconvolution of the fluorescence spectra into N* and T* bands was performed using the Siano software kindly provided by Prof. A.O. Doroshenko (Kharkiv, Ukraine), as previously described.54 Then, the fluorescence parameters (cited in Table 1 or applied in figure drawings) were derived from the separated N* and T* bands. The local water concentration of the probe environment was calculated using the equation C = [log(IN*/IT*) + 0.975]/0.0377 (Fig. 1B, inset, Fig. S3†). Then, this concentration was converted into the water access coefficient WA by the equation WA (%) = 100[H2O]L/55.5.Fluorescence microscopy experiments
Fluorescence microscopy experiments were performed using a home-built two-photon laser scanning setup based on an Olympus IX70 inverted microscope with an Olympus 60×1.2 NA water immersion objective.55 Two-photon excitation was provided by a titanium-sapphire laser (Tsunami, Spectra Physics), and photons were detected with Avalanche Photodiodes (APD SPCM-AQR-14-FC, Perkin-Elmer) connected to a counter/timer PCI board (PCI6602, National Instrument). Imaging was carried out using two fast galvo-mirrors in the descanned fluorescence collection mode. Sizes of the images were 70 × 70 μm, resolution 512 × 512 pixel. Images corresponding to the blue and red channels were recorded simultaneously using a dichroic mirror (Beamsplitter 585 DCXR) and two band-pass filters (Brightline HC 520/20 and HQ 585/40). The images were processed with a home-made program under LabView that generates a ratiometric image by dividing the image of the blue channel by that of the red channel. For each pixel, a pseudocolor scale is used for coding the ratio, while the intensity is defined by the integrated intensity recorded for both channels at the corresponding pixel.56 The sensitivity of the two detectors was calibrated using solutions of the 7AF dye in DMF and EtOH.Cell preparation and staining
HeLa cells were cultured in Dulbecco's modified Eagle medium (DMEM, high glucose, Gibco-Invitrogen) supplemented with 10% (v/v) fetal bovine serum (FBS, Lonza), 1% antibiotic solution (penicillin–streptomycin, Gibco-Invitrogen) in a humidified incubator with a 5% CO2 atmosphere at 37 °C. A cell concentration of 5–10 × 104 cells mL−1 was maintained by removal of a portion of the culture and replacing with fresh medium 3 times per week. For microscopy studies, cells were seeded onto a chambered coverglass (IBiDi) at a density of 5 × 104 cells per IBiDi. After washing the cells with Opti-MEM, a solution of 7AF–penetratin diluted in 1 mL of Opti-MEM was added to the cells to final concentrations of 2 μM and 10 μM and immediately measured.Abbreviations
| LUVs | Large unilamellar vesicles |
| DOPC | 1,2-Dioleoyl-sn-glycero-3-phosphatidylcholine |
| DPPC | 1,2-Dipalmitoyl-sn-glycero-3-phosphatidylcholine |
| Ch | Cholesterol |
| SM | Brain sphingomyelin |
Acknowledgements
O.M.Z, V.Y.P., V.G.P., and this work were supported by ANR Fluoaant, ANR Femtostack, ANR Fluometadn, ANR Cellmemprobes, CNRS, and Université de Strasbourg.References
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS PubMed.
- A. S. Klymchenko and R. Kreder, Chem. Biol., 2014, 21, 97–113 CrossRef CAS PubMed.
- J. Zhang, R. E. Campbell, A. Y. Ting and R. Y. Tsien, Nat. Rev. Mol. Cell Biol., 2002, 3, 906–918 CrossRef CAS PubMed.
- C. M. Cooley, K. S. Hettie, J. L. Klockow, S. Garrison and T. E. Glass, Org. Biomol. Chem., 2013, 11, 7387–7392 CAS.
- N. Luisier, A. Ruggi, S. N. Steinmann, L. Favre, N. Gaeng, C. Corminboeuf and K. Severin, Org. Biomol. Chem., 2012, 10, 7487–7490 CAS.
- A. P. Demchenko, Y. Mely, G. Duportail and A. S. Klymchenko, Biophys. J., 2009, 96, 3461–3470 CrossRef CAS PubMed.
- A. S. Klymchenko and Y. Mely, Fluorescence-Based Biosensors: From Concepts to Applications, 2013, vol. 113, pp. 35–58 Search PubMed.
- G. S. Loving, M. Sainlos and B. Imperiali, Trends Biotechnol., 2010, 28, 73–83 CrossRef CAS PubMed.
- A. R. Katritzky and T. Narindoshvili, Org. Biomol. Chem., 2009, 7, 627–634 CAS.
- V. Y. Postupalenko, O. M. Zamotaiev, V. V. Shvadchak, A. V. Strizhak, V. G. Pivovarenko, A. S. Klymchenko and Y. Mely, Bioconjugate Chem., 2013, 24, 1998–2007 CrossRef CAS PubMed.
- B. E. Cohen, T. B. McAnaney, E. S. Park, Y. N. Jan, S. G. Boxer and L. Y. Jan, Science, 2002, 296, 1700–1703 CrossRef CAS PubMed.
- M. Nitz, A. R. Mezo, M. H. Ali and B. Imperiali, Chem. Commun., 2002, 1912–1913 RSC.
- M. E. Vazquez, M. Nitz, J. Stehn, M. B. Yaffe and B. Imperiali, J. Am. Chem. Soc., 2003, 125, 10150–10151 CrossRef CAS PubMed.
- M. Eugenio Vazquez, D. M. Rothman and B. Imperiali, Org. Biomol. Chem., 2004, 2, 1965–1966 CAS.
- G. Loving and B. Imperiali, J. Am. Chem. Soc., 2008, 130, 13630–13638 CrossRef CAS PubMed.
- B. E. Cohen, A. Pralle, X. Yao, G. Swaminath, C. S. Gandhi, Y. N. Jan, B. K. Kobilka, E. Y. Isacoff and L. Y. Jan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 965–970 CrossRef CAS PubMed.
- M. E. Vazquez, J. B. Blanco and B. Imperiali, J. Am. Chem. Soc., 2005, 127, 1300–1306 CrossRef CAS PubMed.
- P. Venkatraman, T. T. Nguyen, M. Sainlos, O. Bilsel, S. Chitta, B. Imperiali and L. J. Stern, Nat. Chem. Biol., 2007, 3, 222–228 CrossRef CAS PubMed.
- K. G. Harikumar, D. I. Pinon, W. S. Wessels, F. G. Prendergast and L. J. Miller, J. Biol. Chem., 2002, 277, 18552–18560 CrossRef CAS PubMed.
- P. K. Sengupta and M. Kasha, Chem. Phys. Lett., 1979, 68, 382–385 CrossRef CAS.
- A. S. Klymchenko, Y. Mely, A. P. Demchenko and G. Duportail, Biochim. Biophys. Acta, 2004, 1665, 6–19 CrossRef CAS PubMed.
- G. M'Baye, Y. Mely, G. Duportail and A. S. Klymchenko, Biophys. J., 2008, 95, 1217–1225 CrossRef PubMed.
- S. Oncul, A. S. Klymchenko, O. A. Kucherak, A. P. Demchenko, S. Martin, M. Dontenwill, Y. Arntz, P. Didier, G. Duportail and Y. Mely, Biochim. Biophys. Acta, 2010, 1798, 1436–1443 CAS.
- V. Y. Postupalenko, V. V. Shvadchak, G. Duportail, V. G. Pivovarenko, A. S. Klymchenko and Y. Mely, Biochim. Biophys. Acta, 2011, 1808, 424–432 CrossRef CAS PubMed.
- V. V. Shvadchak, A. S. Klymchenko, H. de Rocquigny and Y. Mely, Nucleic Acids Res., 2009, 37, e25 CrossRef PubMed.
- O. M. Zamotaiev, V. Y. Postupalenko, V. V. Shvadchak, V. G. Pivovarenko, A. S. Klymchenko and Y. Mely, Bioconjugate Chem., 2011, 22, 101–107 CrossRef CAS PubMed.
- V. G. Pivovarenko, O. M. Zamotaiev, V. V. Shvadchak, V. Y. Postupalenko, A. S. Klymchenko and Y. Mely, J. Phys. Chem. A, 2012, 116, 3103–3109 CrossRef CAS PubMed.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- M. D. Bilokin, V. V. Shvadchak, D. A. Yushchenko, A. S. Klymchenko, G. Duportail, Y. Mely and V. G. Pivovarenko, Tetrahedron Lett., 2009, 50, 4714–4719 CrossRef CAS PubMed.
- M. D. Bilokin’, V. V. Shvadchak, D. A. Yushchenko, G. Duportail, Y. Mely and V. G. Pivovarenko, J. Fluoresc., 2009, 19, 545–553 CrossRef PubMed.
- V. V. Shvadchak, A. S. Klymchenko, H. de Rocquigny and Y. Mely, Nucleic Acids Res., 2009, 37, e25 CrossRef PubMed.
- A. V. Strizhak, V. Y. Postupalenko, V. V. Shvadchak, N. Morellet, E. Guittet, V. G. Pivovarenko, A. S. Klymchenko and Y. Mely, Bioconjugate Chem., 2012, 23, 2434–2443 CrossRef CAS PubMed.
- N. A. Nemkovich, W. Baumann and V. G. Pivovarenko, J. Photochem. Photobiol., A, 2002, 153, 19–24 CrossRef CAS.
- A. S. Klymchenko, S. Oncul, P. Didier, E. Schaub, L. Bagatolli, G. Duportail and Y. Mely, Biochim. Biophys. Acta, 2009, 1788, 495–499 CrossRef CAS PubMed.
- V. V. Shynkar, A. S. Klymchenko, C. Kunzelmann, G. Duportail, C. D. Muller, A. P. Demchenko, J. M. Freyssinet and Y. Mely, J. Am. Chem. Soc., 2007, 129, 2187–2193 CrossRef CAS PubMed.
- V. V. Shvadchak, L. J. Falomir-Lockhart, D. A. Yushchenko and T. M. Jovin, J. Biol. Chem., 2011, 286, 13023–13032 CrossRef CAS PubMed.
- P. T. Chou, C. H. Huang, S. C. Pu, Y. M. Cheng, Y. H. Liu, Y. Wang and C. T. Chen, J. Phys. Chem. A, 2004, 108, 6452–6454 CrossRef CAS.
- A. Lamaziere, O. Maniti, C. Wolf, O. Lambert, G. Chassaing, G. Trugnan and J. Ayala-Sanmartin, Biochim. Biophys. Acta, 2010, 1798, 2223–2230 CrossRef CAS PubMed.
- O. Maniti, I. Alves, G. Trugnan and J. Ayala-Sanmartin, PLoS One, 2010, 5, e15819 CAS.
- I. D. Alves, C. Y. Jiao, S. Aubry, B. Aussedat, F. Burlina, G. Chassaing and S. Sagan, Biochim. Biophys. Acta, 2010, 1798, 2231–2239 CAS.
- Y. Su, R. Mani and M. Hong, J. Am. Chem. Soc., 2008, 130, 8856–8864 CrossRef CAS PubMed.
- T. Letoha, S. Gaal, C. Somlai, Z. Venkei, H. Glavinas, E. Kusz, E. Duda, A. Czajlik, F. Petak and B. Penke, J. Pept. Sci., 2005, 11, 805–811 CrossRef CAS PubMed.
- D. M. Copolovici, K. Langel, E. Eriste and U. Langel, ACS Nano, 2014, 8, 1972–1994 CrossRef CAS PubMed.
- J. H. Jeong, H. Mok, Y. K. Oh and T. G. Park, Bioconjugate Chem., 2009, 20, 5–14 CrossRef CAS PubMed.
- G. M'Baye, Y. Mely, G. Duportail and A. S. Klymchenko, Biophys. J., 2008, 95, 1217–1225 CrossRef PubMed.
- L. Giordano, V. V. Shvadchak, J. A. Fauerbach, E. A. Jares-Erijman and T. M. Jovin, J. Phys. Chem. Lett., 2012, 3, 1011–1016 CrossRef CAS.
- C. C. Lee, Y. Sun and H. W. Huang, Biophys. J., 2010, 98, 2236–2245 CrossRef CAS PubMed.
- I. D. Alves, I. Correia, C. Y. Jiao, E. Sachon, S. Sagan, S. Lavielle, G. Tollin and G. Chassaing, J. Pept. Sci., 2009, 15, 200–209 CrossRef CAS PubMed.
- A. L. Lomize, I. D. Pogozheva and H. I. Mosberg, J. Chem. Inf. Model, 2011, 51, 930–946 CrossRef CAS PubMed.
- N. Kucerka, J. F. Nagle, J. N. Sachs, S. E. Feller, J. Pencer, A. Jackson and J. Katsaras, Biophys. J., 2008, 95, 2356–2367 CrossRef CAS PubMed.
- P. T. Chou, M. L. Martinez and J. H. Clements, J. Phys. Chem., 1993, 97, 2618–2622 CrossRef CAS.
- F. Heitz, M. C. Morris and G. Divita, Br. J. Pharmacol., 2009, 157, 195–206 CrossRef CAS PubMed.
- C. Y. Jiao, D. Delaroche, F. Burlina, I. D. Alves, G. Chassaing and S. Sagan, J. Biol. Chem., 2009, 284, 33957–33965 CrossRef CAS PubMed.
- A. O. Doroshenko, L. B. Sychevskaya, A. V. Grygorovych and V. G. Pivovarenko, J. Fluoresc., 2002, 12, 455–464 CrossRef CAS.
- J. P. Clamme, J. Azoulay and Y. Mely, Biophys. J., 2003, 84, 1960–1968 CrossRef CAS.
- A. S. Klymchenko, S. Oncul, P. Didier, E. Schaub, L. Bagatolli, G. Duportail and Y. Mely, Biochim. Biophys. Acta, 2009, 1788, 495–499 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob01242a |
| This journal is © The Royal Society of Chemistry 2014 |