
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recognition of double-stranded DNA using energetically activated duplexes with interstrand zippers of 1-, 2- or 4-pyrenyl-functionalized O2′-alkylated RNA monomers†
Saswata
Karmakar
a,
Andreas S.
Madsen
b,
Dale C.
Guenther
a,
Bradley C.
Gibbons
ac and
Patrick J.
Hrdlicka
*a
aDepartment of Chemistry, University of Idaho, Moscow, ID 83844, USA. E-mail: hrdlicka@uidaho.edu; Fax: (+1) 208 885 6173; Tel: (+1) 208 885 0108
bDepartment of Chemistry, Technical University of Denmark, Kemitorvet 207, Kgs. Lyngby, DK-2800, Denmark
cDepartment of Chemistry, Brigham Young University-Idaho, ID 83440, USA
First published on 13th August 2014
Abstract
Despite advances with triplex-forming oligonucleotides, peptide nucleic acids, polyamides and – more recently – engineered proteins, there remains an urgent need for synthetic ligands that enable specific recognition of double-stranded (ds) DNA to accelerate studies aiming at detecting, regulating and modifying genes. Invaders, i.e., energetically activated DNA duplexes with interstrand zipper arrangements of intercalator-functionalized nucleotides, are emerging as an attractive approach toward this goal. Here, we characterize and compare Invaders based on 1-, 2- and 4-pyrenyl-functionalized O2′-alkylated uridine monomers X–Z by means of thermal denaturation experiments, optical spectroscopy, force-field simulations and recognition experiments using DNA hairpins as model targets. We demonstrate that Invaders with +1 interstrand zippers of X or Y monomers efficiently recognize mixed-sequence DNA hairpins with single nucleotide fidelity. Intercalator-mediated unwinding and activation of the double-stranded probe, coupled with extraordinary stabilization of probe–target duplexes (ΔTm/modification up to +14.0 °C), provides the driving force for dsDNA recognition. In contrast, Z-modified Invaders show much lower dsDNA recognition efficiency. Thus, even very conservative changes in the chemical makeup of the intercalator-functionalized nucleotides used to activate Invader duplexes, affects dsDNA-recognition efficiency of the probes, which highlights the importance of systematic structure–property studies. The insight from this study will guide future design of Invaders for applications in molecular biology and nucleic acid diagnostics.
Introduction
The right-handed DNA helix is one of the most fundamental structures in Nature due to its role as the carrier of genetic information. The two strands comprising the helix are held together through π–π stacking interactions between neighboring nucleobases and hydrogen bonding between base pairs.1 Development of synthetic ligands that are capable of decoding the sequence information contained within double-stranded DNA (dsDNA) has proven very challenging as the Watson–Crick base pairs are buried deeply within the duplex core and not readily accessible to exogenous agents. The most established dsDNA-targeting agents, i.e., pyrrole–imidazole polyamides,2 engineered proteins3 and triplex-forming oligonucleotides4 or peptide nucleic acids (PNAs),5 accordingly recognize chemical features that are accessible via one of the grooves instead.6 While these strategies are attractive, they are not without limitations. For example, PNA-based approaches normally require low salinity conditions, while triplex-based approaches require targets with polypurine tracts. Approaches with more relaxed sequence requirements have been developed but they still require the presence of purine stretches, a circumstance that may not be met at a target of interest.7 Other strategies allow recognition of mixed-sequence target regions that are unusually accessible to exogenous agents, such as AT-rich cruciforms or transcription bubbles.8Synthetic ligands that recognize mixed-sequence B-DNA via duplex invasion are attractive due to the predictability of Watson–Crick base-pairing rules. However, these approaches must overcome a steep energetic penalty since pre-existing base pairs of B-DNA must be broken prior to probe binding. Pseudocomplementary DNA (pcDNA), i.e., DNA duplexes featuring modified nucleobases that form weak base pairs with each other, while allowing hybridization to complementary DNA, recognize mixed-sequence target regions at B-DNA termini.9 This strategy has been successfully extended to pcPNA, which recognize internal regions of mixed-sequence dsDNA.10 However, the self-inhibitory effects observed at high pcPNA concentrations and the requirement for low salinity, pose potential limitations for pcPNA-mediated strand invasion in biological media.11 γ-PNA, i.e., single-stranded probes that are fully modified with conformationally pre-organized PNA building blocks, are another interesting class of dsDNA-targeting probes capable of recognizing mixed-sequence dsDNA targets. However, they too require non-physiological salinity for optimal strand invasion.12 In summary, oligonucleotide-based probes that recognize mixed-sequence dsDNA targets at physiologic conditions remain largely elusive.
We have recently introduced Invaders as an alternative strategy toward mixed-sequence dsDNA recognition.13 These double-stranded probes are activated for dsDNA recognition through modification with +1 interstrand zippers of intercalator-functionalized nucleotides (for an illustration, see Fig. 1; for a formal definition of the zipper nomenclature, see the Experimental section). This structural motif results in a locally perturbed and destabilized region in the probe duplex since the motif represents a violation of the ‘nearest neighbor exclusion principle’, which states that intercalators, at most, bind to every second base pair of a DNA duplex due to limitations in local helix expandability.14 On the other hand, the two strands comprising an Invader probe display exceptionally strong affinity toward complementary single-stranded DNA (cDNA), since duplex formation results in strongly stabilizing pyrene–nucleobase stacking interactions (Fig. 1). In a key proof-of-concept study, we harnessed the energy difference between reactants (i.e., Invader probes and target duplexes) and products (i.e., probe–target duplexes) to drive recognition of chromosomal DYZ-1 satellite DNA in male bovine kidney cells at non-denaturing conditions.13
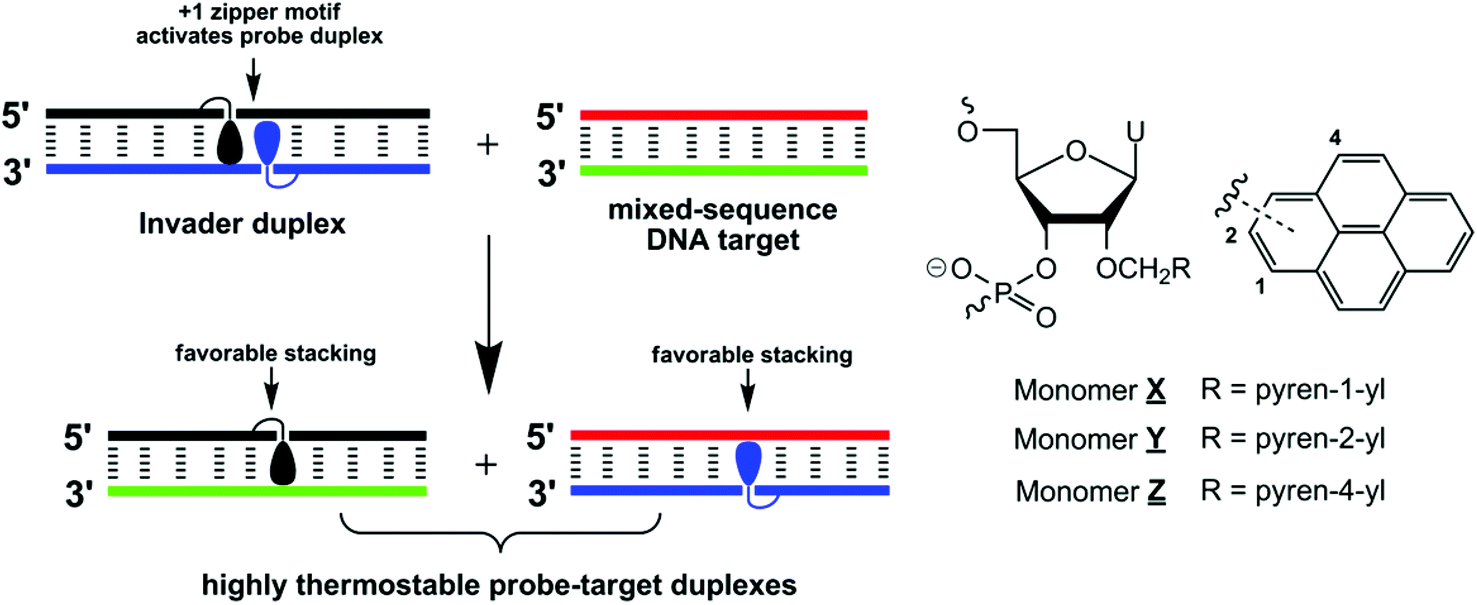 | ||
| Fig. 1 Illustration of the Invader approach for recognition of mixed-sequence DNA and structures of monomers used herein. Droplets denote pyrene moieties. | ||
We originally used 2′-N-(pyren-1-yl)methyl-2′-amino-α-L-LNA (locked nucleic acid) nucleotides as the key activating components of Invader probes,15 but recently discovered that they can be replaced by 2′-O-(pyren-1-yl)methyl-ribonucleotides (Fig. 1).16 The resulting probes display similar dsDNA-recognition efficiency and are much easier to synthesize.17,18 Identification of these simple building blocks allowed us to initiate systematic structure–property relationship studies with the goal of gaining additional insights into the structural determinants governing Invader recognition efficiency. For example, we demonstrated that all four canonical 2′-O-(pyren-1-yl)methyl-RNA monomers can be used to construct dsDNA-binding Invader probes. However, probes using the pyrimidine monomers are particularly efficient.19
In the present study, we wanted to determine if the relative orientation between the pyrene intercalator and sugar skeleton has any impact on Invader-mediated recognition of mixed-sequence dsDNA. Toward this end, the corresponding 2- and 4-pyrenyl functionalized uridines were synthesized and incorporated into oligodeoxyribonucleotides (ONs), which were then characterized with respect to thermal denaturation, thermodynamic, UV-Vis absorption and fluorescence properties (Fig. 1). This was then followed by biophysical characterization of double-stranded probes with different interstrand arrangements of these monomers and recognition experiments using DNA hairpins as model targets.
Results and discussion
Synthesis of O2′-pyrene-functionalized uridine phosphoramidites
Phosphoramidites 4Y and 4Z were prepared in a similar manner as the 2′-O-(pyren-1-yl)methyl-uridine analogue 4X.17b Thus, treatment of O2,O2′-anhydrouridine 120 with tris(pyren-2-yl)methyl borate or tris(pyren-4-yl)methyl borate – generated in situ21via addition of 2-pyrenemethanol22 or 4-pyrenemethanol23 to borane – afforded 2Y and 2Z in modest but acceptable yields (30% and 20% respectively, Scheme 1). Subsequent O5′-dimethoxytritylation gave nucleosides 3Y and 3Z, which upon treatment with 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (PCl reagent) and Hünig's base provided target phosphoramidites 4Y and 4Z in excellent yield.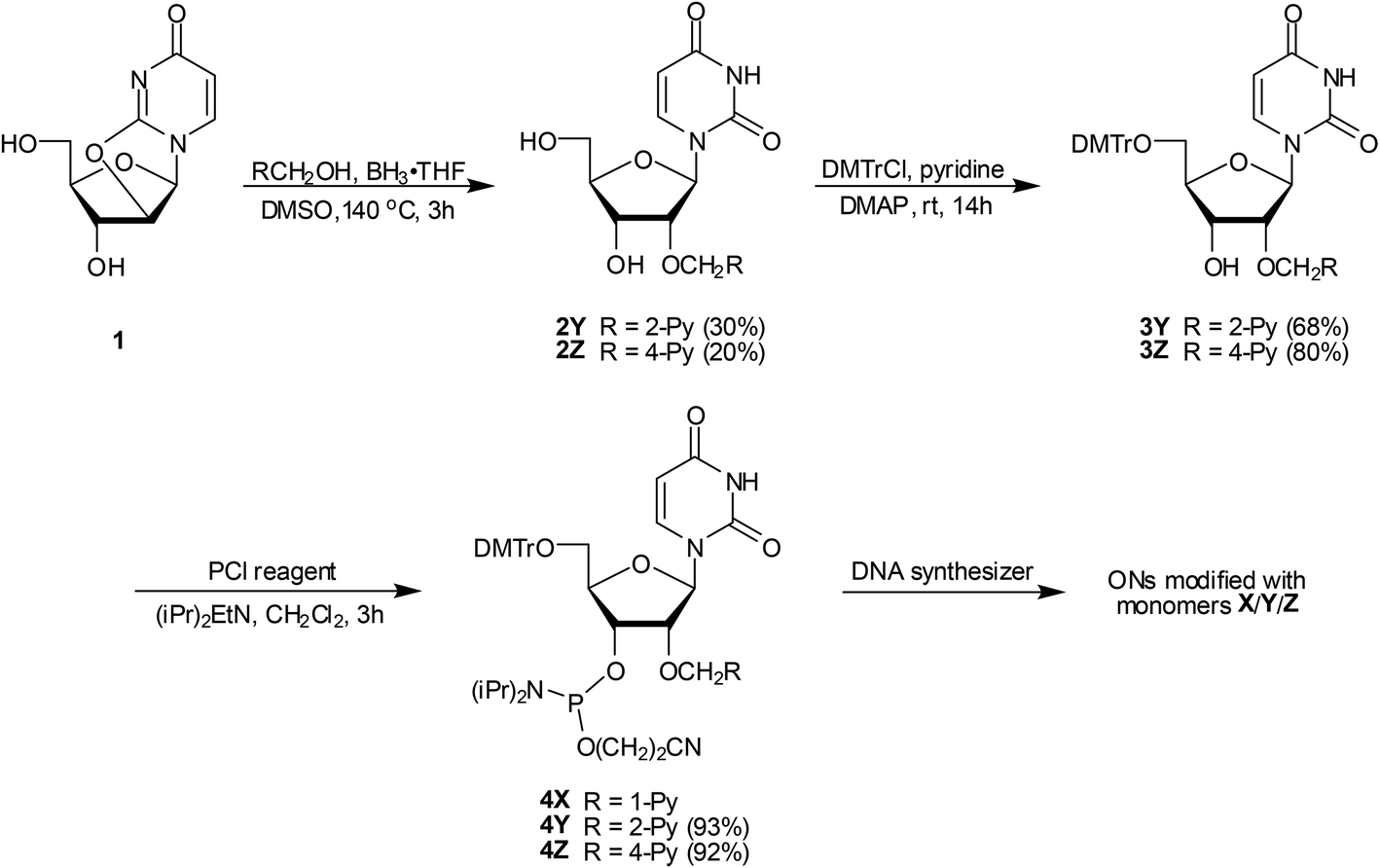 | ||
| Scheme 1 Synthesis of O2′-pyrene-functionalized uridine phosphoramidites 4Y and 4Z. DMTr = 4,4′-dimethoxytrityl; PCl reagent = 2-cyanoethyl-N,N‘-diisopropylchlorophosphoramidite; 1-Py = pyren-1-yl; 2-Py = pyren-2-yl; 4-Py = pyren-4-yl. | ||
Synthesis of modified ONs and experimental design
Phosphoramidites 4X, 4Y and 4Z were used in machine-assisted solid-phase DNA synthesis to incorporate monomers X–Z into ONs using 4,5-dicyanoimidazole as an activator and extended hand-coupling (15 min), which resulted in stepwise coupling yields of ∼99%/∼99%/∼98% for 4X/4Y/4Z, respectively. The identity and purity of the modified ONs was established via MALDI-TOF (Table S1 in the ESI†) and ion-pair reverse phase HPLC (>85% purity), respectively.Monomers X–Z were studied in 9-mer sequence contexts, which we have previously used to screen and identify potential Invader building blocks.16 ONs containing a single incorporation in the 5′-GBG ATA TGC context are denoted X1, Y1, and Z1. Similar conventions apply for the B2–B6 series (Table 1). Reference DNA and RNA strands are denoted D1/D4 and R1/R4, respectively (see footnote, Table 1).
| T m (ΔTm/mod) [°C] | ||||||||
|---|---|---|---|---|---|---|---|---|
| +cDNA | +cRNA | |||||||
| ON | Sequence | B = | X | Y | Z | X | Y | Z |
| a ΔTm = change in Tm relative to reference duplexes D1:D4 (Tm ≡ 29.5 °C), D1:R4 (Tm ≡ 26.5 °C) or R1:D4 (Tm ≡ 24.5 °C), where D1: 5′-GTGATATGC, D4: 3′-CACTATACG, R1: 5′-GUGAUAUGC and R4: 3′-CACUAUACG; Tm's are determined as the maximum of the first derivative of melting curves (A260vs. T) recorded in medium salt buffer ([Na+] = 110 mM, [Cl−] = 100 mM, pH 7.0 (NaH2PO4/Na2HPO4)), using 1.0 μM of each strand. Tm's are averages of at least two measurements within 1.0 °C; A = adenin-9-yl DNA monomer, C = cytosin-1-yl DNA monomer, G = guanin-9-yl DNA monomer, T = thymin-1-yl DNA monomer. For structures of monomers X–Z see Fig. 1. b From ref. 17b. | ||||||||
| B1 | 5′-G![[B with combining low line]](https://www.rsc.org/images/entities/b_char_0042_0332.gif) G ATA TGC G ATA TGC |
34.5 [+5.0] | 35.5 [+6.0] | 29.5 [±0.0] | 24.5 [−2.0] | 25.5 [−1.5] | 20.5 [−6.0] | |
| B2 | 5′-GTG AA TGC |
42.5 [+13.0] | 43.5 [+14.0] | 37.5 [+8.0] | 30.5 [+4.0] | 34.0 [+7.5] | 27.5 [+1.0] | |
| B3 | 5′-GTG ATA GC |
37.5 [+8.0] | 39.0 [+9.5] | 33.5 [+4.0] | 26.5 [±0.0] | 28.0 [+1.5] | 21.5 [−5.0] | |
| B4 | 3′-CAC AT ACG |
33.0 [+3.5] | 35.5 [+6.0] | 26.5 [−3.0] | 20.5 [−4.5] | 23.0 [−1.5] | 16.5 [−8.0] | |
| B5 | 3′-CAC TA ACG |
42.5 [+13.0] | 43.5 [+14.0] | 38.5 [+9.0] | 27.5 [+2.5] | 32.0 [+7.5] | 26.5 [+2.0] | |
| B6 | 3′-CAC A ACG |
43.5 [+7.0] | 45.5 [+8.0] | 32.5 [+1.5] | 24.0 [−0.3] | 28.5 [+2.0] | 16.5 [−4.0] | |
Thermostability of duplexes with complementary DNA/RNA
First, the thermostabilities of duplexes between B1–B6 and complementary DNA or RNA targets were determined from thermal denaturation experiments performed in medium salt phosphate buffer ([Na+] = 110 mM, pH 7.0). The resulting denaturation curves display the expected monophasic sigmoidal transitions (Fig. S1 in the ESI†). Interestingly, Y-modified ONs form much more thermostable duplexes with cDNA than unmodified ONs (ΔTm/mod = +6.0 to +14.0 °C, Table 1). In fact, the resulting duplexes are even more thermostable than X-modified duplexes, suggesting that the 2-pyrenyl moiety of monomer Y is very well accommodated in DNA duplexes. In contrast, Z-modified ONs display considerably lower affinity toward cDNA (ΔTm/mod = −3.0 to +9.0 °C, Table 1). The thermostability trends are sequence-dependent. Thus, ONs in which the pyrene-functionalized monomers are flanked by 3′-purines, induce greater stabilization than ONs with 3′-flanking pyrimidines (e.g., compare ΔTm/mod values for B2- and B4-series, Table 1). This is indicative of 3′-intercalative binding modes of the pyrene moieties,19,24 as 3′-flanking purines would provide larger π-stacking surfaces than 3′-pyrimidines. Less stable duplexes are formed with cRNA (ΔTm/mod = −8.0 to +7.5 °C, Table 1; trend: Y > X > Z), which also points to intercalative pyrene binding modes,18,24a,25 as intercalators generally favor the less compressed B-type helix geometry of DNA:DNA duplexes.26Binding specificity
Next, the binding specificity of singly modified ONs (B2-series) was studied using DNA targets with mismatched nucleotides opposite of the pyrene-functionalized monomer (Table 2). X2/Y2/Z2 discriminate mismatched DNA targets less efficiently than reference strand D1, especially when a mismatched T is opposite of the modification. This is in agreement with other studies in which reduced binding specificity of intercalator-modified ONs was observed.26 Interestingly, the specificity trends mirror the affinity trends. Thus, Y2 discriminates mismatched target more efficiently than X2, which, conversely, discriminates mismatched targets more efficiently than Z2. Additional specificity data are discussed in the ESI (Tables S3 and S4†).| DNA: 3′-CAC TT ACG |
||||||
|---|---|---|---|---|---|---|
| T m [°C] | ΔTm [°C] | |||||
| ON | Sequence |
= |
A | C | G | T |
| a For conditions of thermal denaturation experiments, see Table 1. Tm's of fully matched duplexes are shown in bold. ΔTm = change in Tm relative to fully matched duplex. b From ref. 17b. | ||||||
| D1 | 5′-GTG ATA TGC | 29.5 | −16.5 | −9.5 | −17.0 | |
| X2 | 5′-GTG A![[X with combining low line]](https://www.rsc.org/images/entities/b_char_0058_0332.gif) A TGC A TGC |
42.5 | −13.5 | −5.5 | −7.0 | |
| Y2 | 5′-GTG A![[Y with combining low line]](https://www.rsc.org/images/entities/b_char_0059_0332.gif) A TGC A TGC |
43.5 | −16.5 | −6.5 | −9.0 | |
| Z2 | 5′-GTG A![[Z with combining low line]](https://www.rsc.org/images/entities/b_char_005a_0332.gif) A TGC A TGC |
37.5 | −10.0 | −4.0 | −3.0 | |
Optical spectroscopy
UV-Vis absorption and steady-state fluorescence emission spectra of X/Y/Z-modified ONs in the presence or absence of complementary DNA/RNA targets were recorded next to further ascertain intercalative binding modes of the pyrene moieties, as intercalation is known to induce bathochromic shifts of pyrene absorption peaks27 and nucleobase-mediated quenching of pyrene fluorescence.27a,28 Indeed, X/Y/Z-modified ONs generally display hypochromic and bathochromic shifts in the pyrene absorption spectra upon hybridization with DNA and RNA targets (Δλmax = 0–5 nm, Table 3; Fig. S2–S4 in the ESI†). Slightly greater bathochromic shifts are generally observed upon hybridization with DNA than RNA targets, which again may reflect the preference of intercalators for the less compressed geometry of B-DNA.| λ max [Δλmax]/nm | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| X | Y | Z | |||||||||
| ON | Sequence |
= |
SSP | +cDNA | +cRNA | SSP | +cDNA | +cRNA | SSP | +cDNA | +cRNA |
| a Measurements were performed at 5 °C using a spectrophotometer and quartz optical cells with 1.0 cm path lengths. Buffer conditions are as for thermal denaturation experiments. b From ref. 17b. | |||||||||||
| B1 | 5′-GG ATA TGC |
350 | 353[+3] | 352[+2] | 344 | 346[+2] | 345[+1] | 343 | 344[+1] | 343[±0] | |
| B2 | 5′-GTG AA TGC |
348 | 353[+5] | 352[+4] | 342 | 345[+3] | 345[+3] | 340 | 343[+3] | 343[+3] | |
| B3 | 5′-GTG ATA GC |
350 | 353[+3] | 352[+2] | 344 | 346[+2] | 345[+1] | 343 | 344[+1] | 343[±0] | |
| B4 | 3′-CAC AT ACG |
350 | 352[+2] | 352[+2] | 344 | 345[+1] | 345[+1] | 342 | 344[+2] | 343[+1] | |
| B5 | 3′-CAC TA ACG |
349 | 353[+4] | 352[+3] | 343 | 345[+2] | 345[+2] | 342 | 344[+2] | 344[+2] | |
Steady-state fluorescence emission spectra of X/Y/Z-modified ONs and the corresponding duplexes with cDNA/cRNA display two vibronic bands at λem = 382 ± 3 nm and 402 ± 3 nm, respectively, as well as a small shoulder at ∼420 nm. As anticipated, the fluorescence intensity typically decreases upon hybridization with DNA/RNA targets, with greater decreases typically being observed upon DNA binding (Fig. 2; Fig. S5–S7 in the ESI†). Interestingly, duplexes modified with monomer Z are noticeably less fluorescent than X- or Y-modified duplexes.
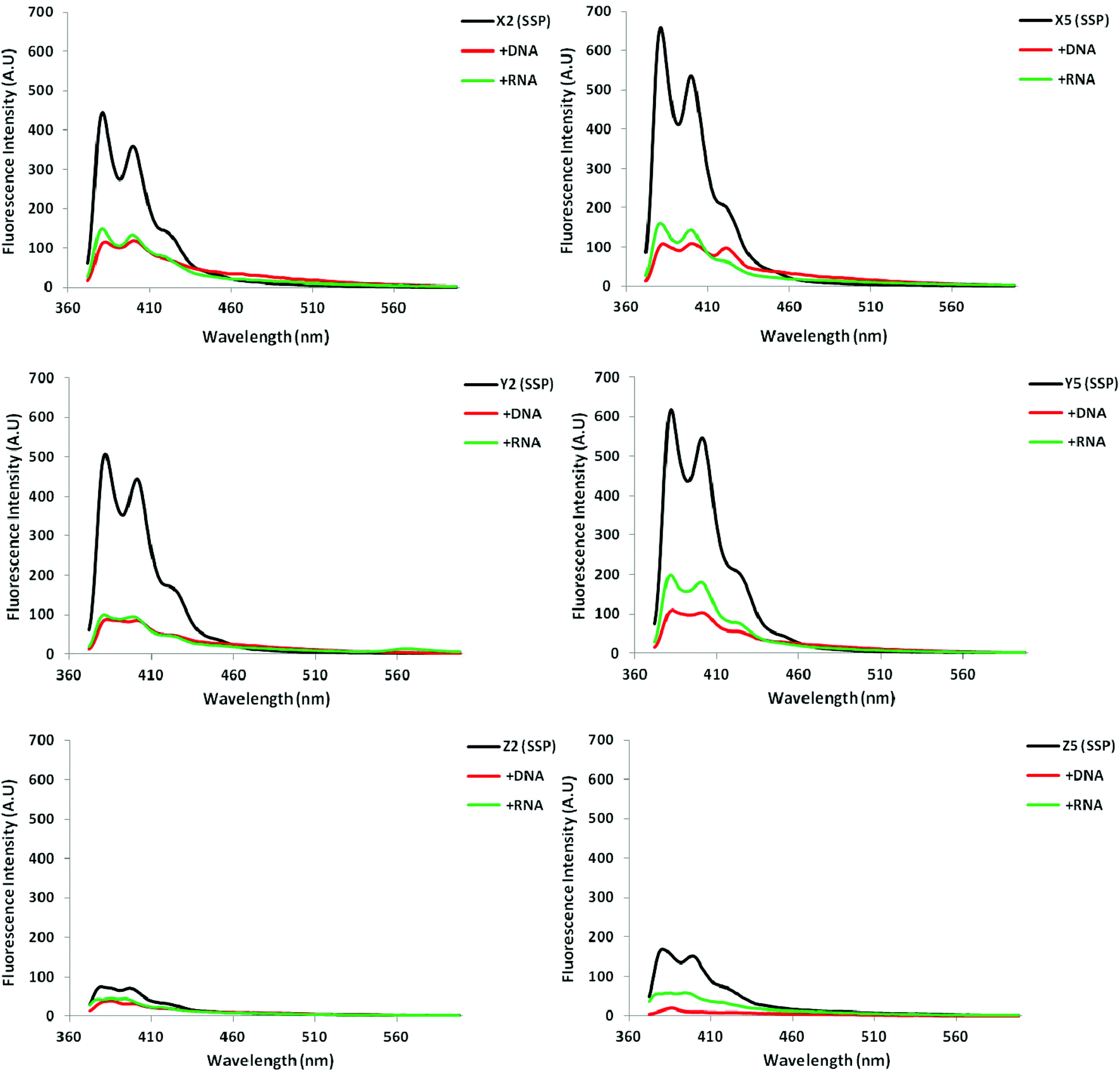 | ||
| Fig. 2 Steady-state fluorescence emission spectra of select X/Y/Z-modified ONs and corresponding duplexes with DNA/RNA targets. Spectra were recorded at T = 5 °C using λex = 350, 345 and 340 nm for X, Y and Z-modified ONs, respectively. Each strand was used at 1.0 μM concentration in Tm buffer. | ||
Molecular modeling of probe–target duplexes
To rationalize the observed biophysical trends, we performed force-field calculations on duplexes between X2/Y2/Z2 and complementary DNA (residue numbering: 5′-G1T2G3A4B5A6T7G8C9:3′-C18A17C16T15A14T13A12C11G10). The complete data set of structural furanose, base pair and dinucleotide step parameters29 is provided in the ESI (Tables S7–S8 and Fig. S9–S14†).In agreement with the photophysical data presented above, the pyrene moieties remain stably intercalated in the duplex core throughout the stochastic dynamics simulations (Fig. 3 top – for details of calculation protocol, see Experimental section). Inspection of the structures reveals that the right-handed duplexes are elongated relative to the corresponding B-DNA reference duplex (e.g., rise increases from ∼3.3 Å to ∼7.3 Å across the B5A6:A14T13 dinucleotide step, Fig. S10 in the ESI†). The overlap between the pyrene and neighboring nucleobases is strongly influenced by the substitution pattern of the pyrene, which explains the observed thermostability trends (Tm decreases Y ≥ X > Z). The pyrene of monomer X stacks with the nucleobase moieties of X5, A6 and A13, while the pyrene of monomer Y is positioned in a manner that facilitates stacking with all four neighboring nucleobase moieties (Fig. 3 bottom). Conversely, the pyrene of monomer Z stacks only with the flanking nucleobases on its own strand, i.e., Z5 and A6. The poorer fit of the pyrene of monomer Z is also reflected in greater perturbation across the B5A6:A14T13 dinucleotide step, i.e., more pronounced buckle (∼6° and ∼−4° in D1:D4, ∼13° and ∼−10° in X2:D4, ∼15° and ∼−11° in Y2:D4, and ∼21° and ∼−19° in Z2:D4, Fig. S9 in the ESI†) and decreased roll (∼12° in D1:D4, ∼−10° in X2:D4, ∼−11° in Y2:D4, and ∼−19° in Z2:D4, Fig. S10 in the ESI†). Another noteworthy observation is that the furanose rings of nucleotides X5A6, Y5A6 and Z5A6 generally exhibit increased South type character relative to the reference duplex (P = 124–171° vs. 102–129°), presumably as a consequence of π–π-stacking between the pyrene and flanking nucleobases (Table S7 in the ESI†).
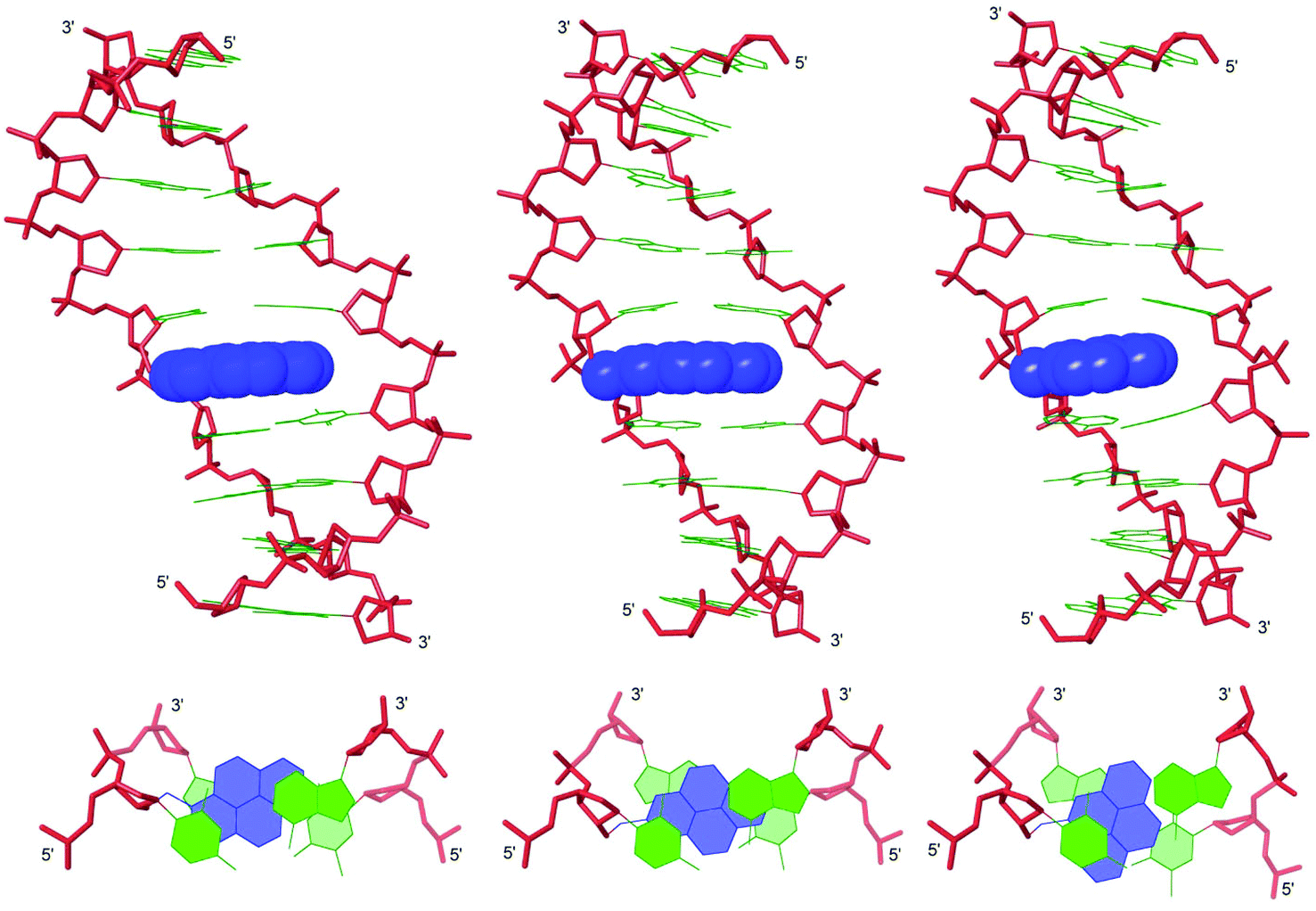 | ||
| Fig. 3 Lowest energy structures of X2:D4 (left), Y2:D4 (middle) and Z2:D4 (right). Upper: side view of duplex; lower: top view of the central duplex region. Color code: sugar phosphate backbone (red); pyrene moieties (blue) and nucleobases (green). Hydrogen atoms, sodium ions and bond orders are omitted for clarity. | ||
To sum up, the molecular modeling structures are useful in explaining the observed thermostability trends and photophysical properties of X/Y/Z-modified duplexes.
Biophysical properties of duplexes with interstrand zippers of X/Y/Z-monomers
Next, we determined the thermostability of DNA duplexes with different interstrand zipper arrangements of X/Y/Z monomers to identify monomers and probe architectures that show the greatest potential for dsDNA-recognition. The impact on duplex thermostability upon incorporation of a second monomer can be additive, greater-than-additive or less-than-additive relative to the corresponding singly modified duplex. The impact is conveniently approximated in terms of Tm's by the term ‘deviation from additivity’ (DA) defined as: DAONA:ONB ≡ ΔTm (ONA:ONB) − [ΔTm (ONA:cDNA) + ΔTm (cDNA:ONB)], where ONA:ONB is a duplex with an interstrand arrangement of monomers. DA also serves as an indicator for dsDNA-recognition potential. Probes with strongly negative DA values are likely to be activated for recognition of iso-sequential dsDNA via the process depicted in Fig. 1, since the products of the recognition process (i.e., probe–target duplexes) are more thermostable than the reactants (i.e., probe duplexes and target duplexes). A more rigorous approach based on differences in ΔG values of probe–target and Invader duplexes is discussed shortly.As expected,13,15,16 duplexes with +1 interstrand monomer arrangements are less thermostable and more strongly activated for dsDNA-recognition than duplexes with other arrangements (compare Tm's and DA values for B2:B5 relative to other probe duplexes, Table 4). Clearly the two pyrene-functionalized monomers interact with each other in an energetically unfavorable manner when placed in this motif. Interestingly, there is little difference in the dsDNA-targeting potential of X2:X5, Y2:Y5 and Z2:Z5 as judged by the DA values; the lower thermostability of Z-modified probe–target duplexes is compensated by an equivalent destabilization of the probe duplexes (compare DA values for X2:X5, Y2:Y5 and Z2:Z5, Table 4).
| ΔG293 [ΔΔG293] (kJ mol−1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| ON | ZP | Sequence | T m (°C) | DA (°C) | Upper ON vs. cDNA | Lower ON vs. cDNA | Probe duplex | ΔG293rec (kJ mol−1) | λ max (nm) |
| a ZP = zipper. For conditions of thermal denaturation and absorption experiments, see Tables 1 and 3, respectively. DA = ΔTm (Invader) − (ΔTm (upper strand vs. cDNA) + ΔTm (lower strand vs. cDNA)). ΔΔG293 is measured relative to ΔG293 for D1:D4 = −41 kJ mol−1. ΔG293rec = ΔG293 (upper strand vs. cDNA) + ΔG293 (lower strand vs. cDNA) − ΔG293 (probe duplex) − ΔG293 (dsDNA target). “±” denotes standard deviation. N/A = the absence of a clear lower base line prevented determination of this value. Tm's for X-modified duplexes have been previously published in ref. 17b but are included to facilitate direct comparison. | |||||||||
| X1 | +4 | 5′-GG ATA TGC |
47.5 | ±0.0 | −46 ± 0 [−5] | −52 ± 0 [−11] | −55 ± 1 [−14] | −2 | 352 |
| X5 | 3′-CAC TA ACG |
||||||||
| X1 | +2 | 5′-GG ATA TGC |
31.5 | −6.5 | −46 ± 0 [−5] | −46 ± 0 [−5] | −45 ± 1 [−4] | −6 | 350 |
| X4 | 3′-CAC AT ACG |
||||||||
| X2 | +1 | 5′-GTG AA TGC |
26.5 | −29.0 | −55 ± 1 [−14] | −53 ± 1 [−12] | −38 ± 1 [+3] | −29 | 348 |
| X5 | 3′-CAC TA ACG |
||||||||
| X2 | −1 | 5′-GTG AA TGC |
39.5 | −6.5 | −54 ± 0 [−13] | −46 ± 0 [−5] | −51 ± 1 [−10] | −8 | 351 |
| X4 | 3′-CAC AT ACG |
||||||||
| X3 | −1 | 5′-GTG ATA GC |
45.5 | −5.0 | −50 ± 0 [−9] | −52 ± 0 [−11] | −55 ± 1 [−14] | −6 | 352 |
| X5 | 3′-CAC TA ACG |
||||||||
| X3 | −3 | 5′-GTG ATA GC |
40.5 | −0.5 | −50 ± 0 [−7] | −46 ± 0 [−3] | −53 ± 1 [−10] | ±0 | 352 |
| X4 | 3′-CAC AT ACG |
||||||||
| Y1 | +4 | 5′-GG ATA TGC |
48.5 | −1.0 | −48 ± 0 [−7] | −55 ± 1 [−14] | −56 ± 1 [−15] | −6 | 346 |
| Y5 | 3′-CAC TA ACG |
||||||||
| Y1 | +2 | 5′-GG ATA TGC |
35.5 | −6.0 | −48 ± 0 [−7] | −46 ± 0 [−5] | −45 ± 1 [−4] | −8 | 346 |
| Y4 | 3′-CAC AT ACG |
||||||||
| Y2 | +1 | 5′-GTG AA TGC |
28.5 | −29.5 | −56 ± 0 [−15] | −55 ± 1 [−14] | −43 ± 1 [−2] | −27 | 339 |
| Y5 | 3′-CAC TA ACG |
||||||||
| Y2 | −1 | 5′-GTG AA TGC |
43.5 | −6.0 | −56 ± 0 [−15] | −46 ± 0 [−5] | −55 ± 1 [−14] | −6 | 346 |
| Y4 | 3′-CAC AT ACG |
||||||||
| Y3 | −1 | 5′-GTG ATA GC |
47.5 | −5.5 | −51 ± 1 [−10] | −55 ± 1 [−14] | −58 ± 1 [−17] | −7 | 346 |
| Y5 | 3′-CAC TA ACG |
||||||||
| Y3 | −3 | 5′-GTG ATA GC |
43.0 | −2.0 | −51 ± 1 [−10] | −46 ± 0 [−5] | −53 ± 1 [−12] | −3 | 346 |
| Y4 | 3′-CAC AT ACG |
||||||||
| Z1 | +4 | 5′-GG ATA TGC |
38.5 | ±0.0 | −44 ± 0 [−3] | −48 ± 1 [−7] | −49 ± 1 [−8] | −2 | 345 |
| Z5 | 3′-CAC TA ACG |
||||||||
| Z1 | +2 | 5′-GG ATA TGC |
27.5 | +1.0 | −44 ± 0 [−3] | −41 ± 0 [±0] | −41 ± 1 [±0] | −3 | 342 |
| Z4 | 3′-CAC AT ACG |
||||||||
| Z2 | +1 | 5′-GTG AA TGC |
19.5 | −27.0 | −48 ± 1 [−7] | −48 ± 1 [−7] | N/A | N/A | 339 |
| Z5 | 3′-CAC TA ACG |
||||||||
| Z2 | −1 | 5′-GTG AA TGC |
30.5 | −4.0 | −48 ± 1 [−7] | −41 ± 0 [±0] | −41 ± 1 [±0] | −7 | 344 |
| Z4 | 3′-CAC AT ACG |
||||||||
| Z3 | −1 | 5′-GTG ATA GC |
37.0 | −5.5 | −46 ± 0 [−5] | −48 ± 1 [−7] | −46 ± 1 [−5] | −7 | 345 |
| Z5 | 3′-CAC TA ACG |
||||||||
| Z3 | −3 | 5′-GTG ATA GC |
32.5 | +2.0 | −46 ± 0 [−5] | −41 ± 0 [±0] | −42 ± 1 [−1] | −4 | 345 |
| Z4 | 3′-CAC AT ACG |
||||||||
The above Tm-based conclusions were corroborated through analysis of thermodynamic parameters for duplex formation, which were derived through fitting of thermal denaturation curves.30 Thus, formation of probe–target duplexes is highly favorable relative to unmodified duplexes, with Y-modified and Z-modified ONs leading to the most and least stable duplexes with cDNA, respectively (see the first two ΔΔG293 columns, Table 4). Stabilization of the probe–target duplexes is largely a consequence of increased entropy (Tables S5 and S6 in the ESI†). On the other hand, B2:B5 duplexes are much less stable (see the third ΔΔG293 column, Table 4) due to strongly increased enthalpy (Tables S5 and S6 in the ESI†). Consequentially, B2:B5 probes display favorable binding free energy for recognition of iso-sequential dsDNA targets as given by ΔG293rec (ONA:ONB) = ΔG293 (ONA:cDNA) + ΔG293 (cDNA:ONB) − ΔG293 (ONA:ONB) − ΔG293 (dsDNA) (i.e., ΔG293rec for B2:B5 ≪ 0 kJ mol−1, Table 4). X2:X5 and Y2:Y5 display very similar ΔG293rec values. Unfortunately, the absence of a clear lower base line in the thermal denaturation curve of Z2:Z5 precluded determination of ΔG293rec for this duplex. Probes with other interstrand arrangements of X/Y/Z-monomers are much more stable and display much less favorable binding free energy for recognition of iso-sequential dsDNA targets (ΔG293rec between−8 and 0 kJ mol−1, Table 4), presumably since the two monomers act independently from each other.
The unique characteristics of duplexes with +1 interstrand monomer arrangements relative to other probe duplexes are also reflected in the blue-shifted pyrene absorption peaks (Fig. S8 in the ESI†), which are indicative of reduced pyrene–nucleobase interactions (compare λmax for B2:B5 with λmax for other probe duplexes (Table 4) or probe–target duplexes (Table 3)).
All probe duplexes exhibit vibronic peaks at λem = 380 ± 2 nm, 400 ± 3 nm and ∼420 ± 3 nm in their steady-state fluorescence emission spectrum (Fig. 4). In addition, duplexes with +2 interstrand monomer arrangements display prominent and unstructured emission centered at λem ∼ 490 nm, which is consistent with pyrene excimer emission (Fig. 4).15,16,19,31 The presence of these signals shows that the pyrene moieties are stacking with an interplanar separation of ∼3.4 Å. We speculate that stacking occurs in the major groove in a similar manner as previously suggested for duplexes with +2 zipper arrangements of pyrene-functionalized ara-uridine monomers.31b,32 Duplexes with +1 interstrand monomer arrangements also display excimer emission albeit of much weaker intensity. In this case, the two pyrene moieties most likely stack inside the duplex core in a similar manner as recently suggested for duplexes modified with 2′-N-(pyren-1-yl)methyl-2′-amino-α-L-LNA monomers; the spatial separation for pyrene–pyrene stacking in the major groove is too large.15a,16 Dual intercalation would also explain the energetic lability of the B2:B5 duplexes, as a localized region with one intercalator per base pair would ensue, which represents a violation of the ‘nearest neighbor exclusion principle’.14
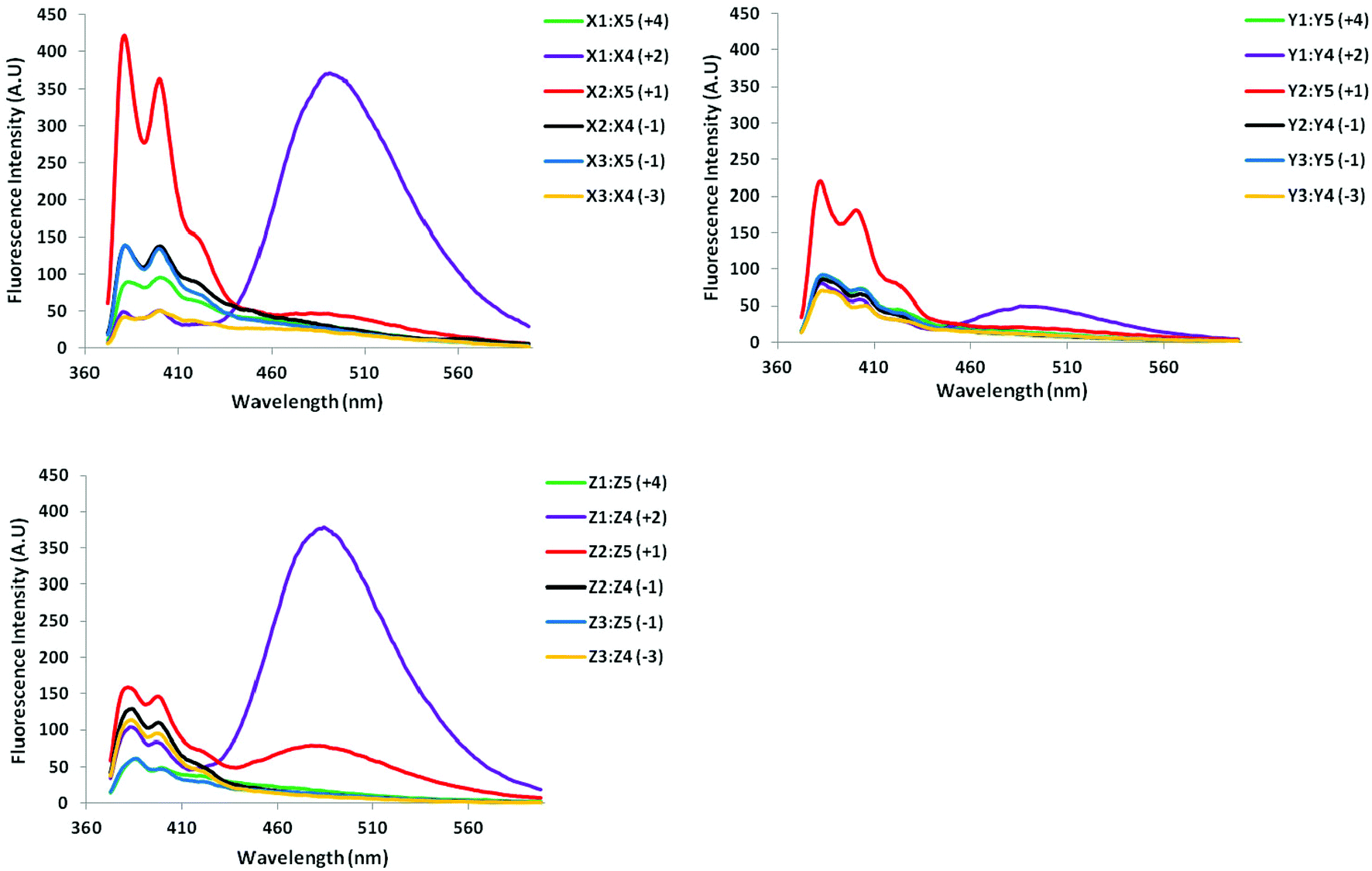 | ||
| Fig. 4 Steady-state fluorescence emission spectra of duplexes with different interstrand monomer arrangements of X, Y, and Z (zipper type indicated in parenthesis). For experimental conditions, see Fig. 2. Spectra for X-modified duplexes were previously reported in ref. 16 but are included for comparison. | ||
Molecular modeling of B2:B5 duplexes
Force-field based simulations of B2:B5 duplexes were performed to gain additional insight into the binding mode of the pyrene moieties. The results of these simulations point toward two possible binding modes, i.e., Type A in which both pyrene moieties intercalate into the duplex core (Fig. 5) and Type B in which one pyrene is intercalating and the other is extruded into the major groove (Fig. S15 in the ESI†). The presence of excimer signals in the steady state emission spectra of X2:X5 and Z2:Z5 (Fig. 4), suggests that these duplexes are capable of adopting Type A conformations. However, the weak intensity of the excimer signals – especially with Y2:Y5 – suggests that B2:B5 duplexes equilibrate between Type A and Type B conformations. Type A structures are characterized by significant elongation (rise ∼ 10 Å) and local perturbation (buckle ∼ 30° and ∼−30° at B5A6 and A14B13, respectively, and roll ∼ −30° across X5A6:X14B13 and Y5A6:Y14B13 and ∼−18° across Z5A6:A14) (Fig. S11–S12 in the ESI†). Type B structures are less elongated (rise ∼ 7.5 Å) and less perturbed (buckle ∼ 20° and ∼−20° at B5A6 and A14B13, respectively, and roll ∼ −5°) (Fig. S13–S14 in the ESI†). Both conformations can account for the lability of the B2:B5 duplexes; base pairing in the vicinity of the interstrand zipper is distorted in both structures and the presence of a large hydrophobic pyrene in the major grove (Type B) will almost certainly perturb the stabilizing hydration layer or cause unfavorable steric interactions.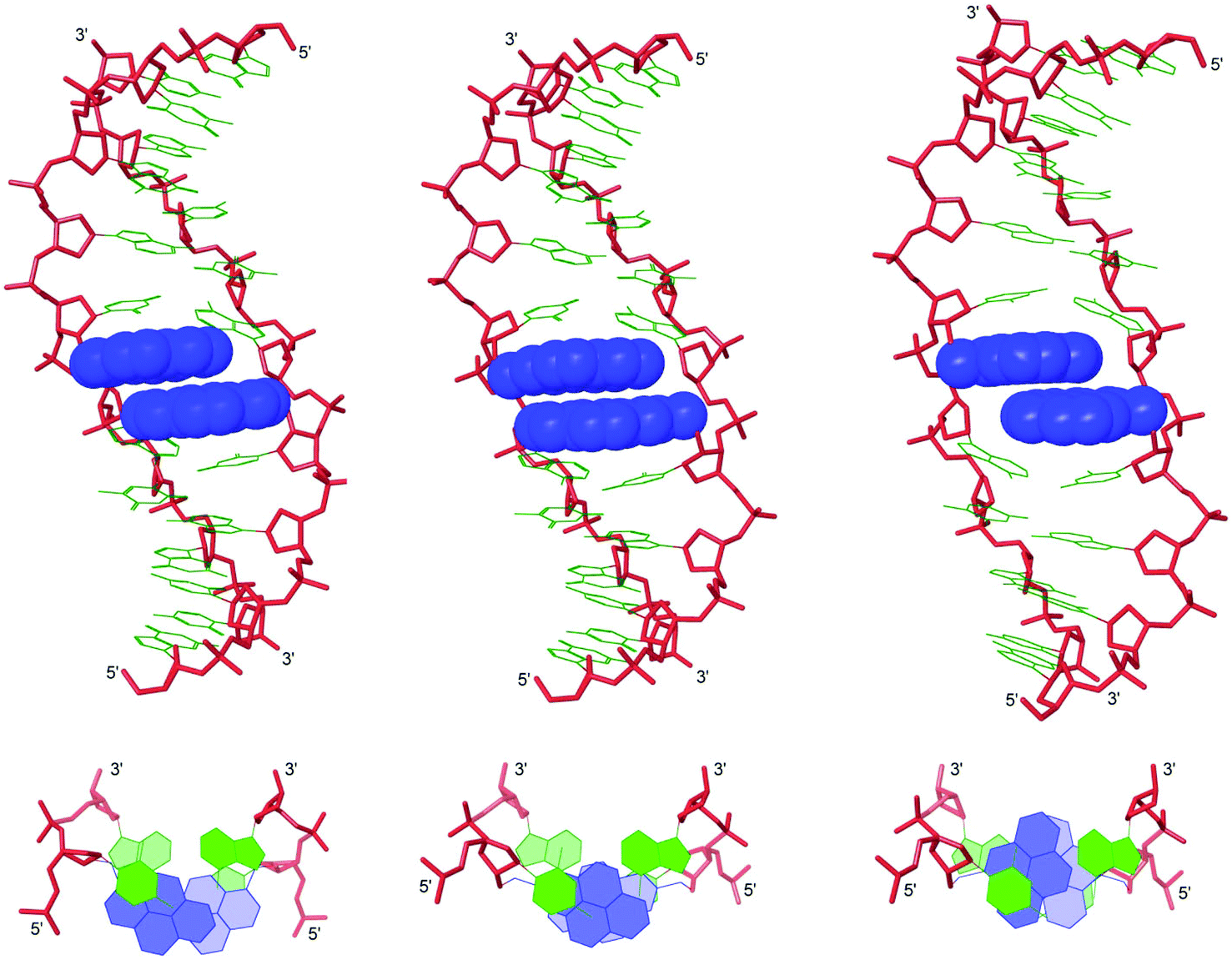 | ||
| Fig. 5 Lowest energy structures of X2:X5 (left), Y2:Y5 (middle) and Z2:Z5 (right) in Type A conformation. Top: side view of duplex; bottom: top view of the central duplex region. For color code, see Fig. 3. | ||
Recognition of DNA hairpins using activated probe duplexes
The ΔG293rec values indicate that duplexes with +1 interstrand zippers of X/Y/Z monomers are the most strongly activated probes for dsDNA-recognition (Table 4). We therefore decided to evaluate these probes in greater detail using a gel shift assay that we have used to screen other potential Invader monomers with.13,16 Thus, a digoxigenin (DIG) labeled DNA hairpin (DH) – comprised of a 9-mer double-stranded mixed-sequence stem that is linked by a T10 loop – was used as a model target (Fig. 6a).33 As expected, the unimolecular nature of DH1 strongly stabilizes the stem region as seen from the significantly higher Tm relative to the corresponding linear DNA duplex (Tm = 57.0 °C vs. 29.5 °C, respectively; Fig. 6b and Table 1). Incubation of DH1 with X2:X5 or Y2:Y5 in a HEPES buffer for 15 h at ambient temperature resulted in dose-dependent recognition as evidenced by the emergence of a slower migrating band on non-denaturing PAGE gels (Fig. 6c).33 It is particularly noteworthy that as little as 1.0 molar equivalent of Y2:Y5 resulted in ∼50% dsDNA-recognition (Fig. 6d). In contrast, very little recognition was observed even at 500-fold molar excess of Z2:Z5 (Fig. 6c and 6d). This was surprising to us considering the very similar DA values of the B2:B5 duplexes (Table 4). To determine if the poor dsDNA-recognition efficiency of Z2:Z5 was due to insufficient stability of the recognition complex and/or slow reaction kinetics, we performed separate experiments in which DH1 was annealed in the presence of 500-fold molar excess of X2:X5 or Z2:Z5 (i.e., heated to 95 °C for 2 min, cooled to 8° over 3 h, then analyzed on non-denaturing PAGE gels). The resulting gel electrophoretograms (results not shown) were essentially identical to those from the room temperature incubation experiments discussed above, which suggests that the recognition complex between Z2:Z5 and DH1 is not sufficiently stable at the experimental conditions of the assay.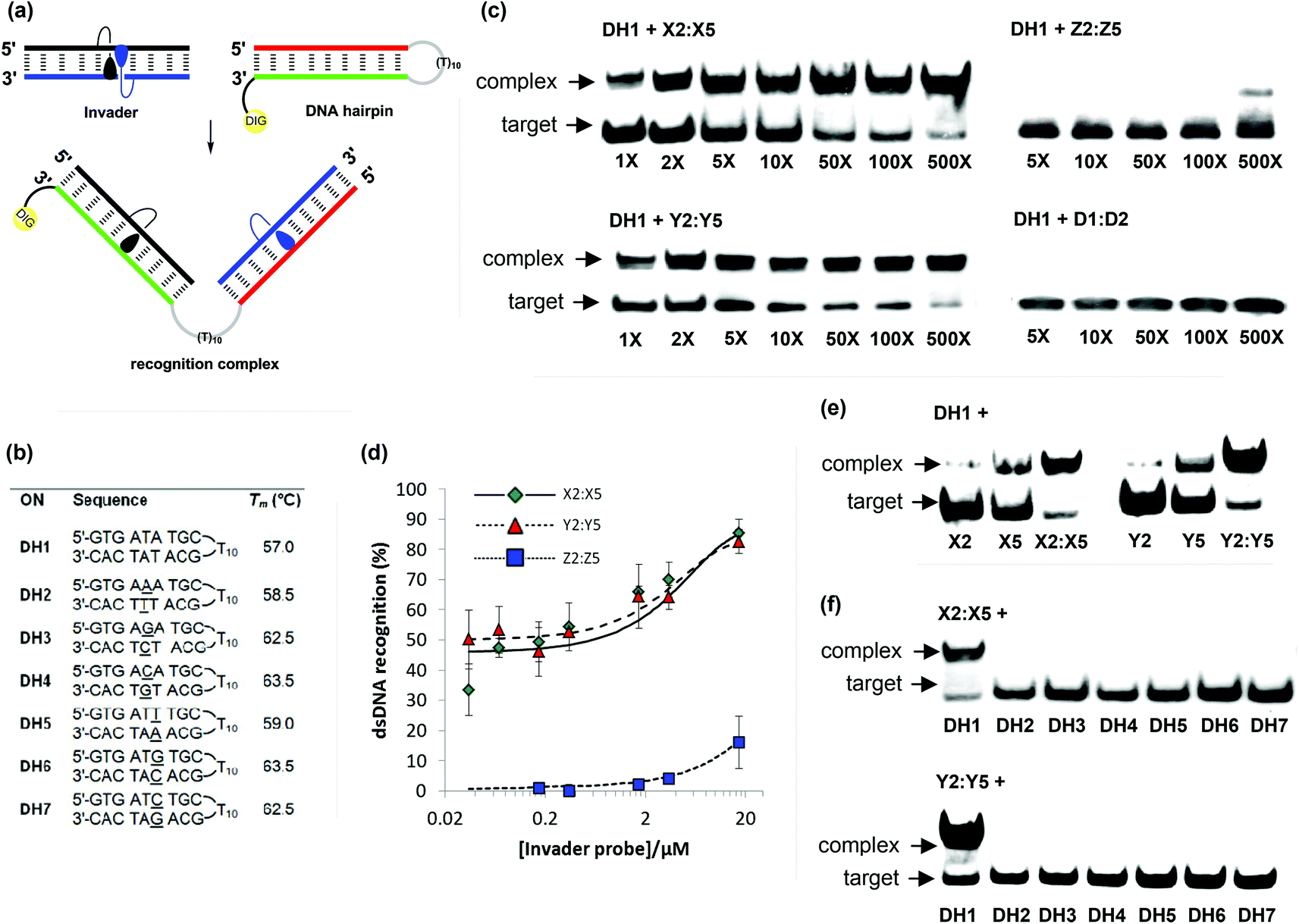 | ||
| Fig. 6 Recognition of model dsDNA targets using activated probe duplexes. (a) Illustration of recognition process; (b) sequences and intramolecular Tm's of DNA hairpins with isosequential (DH1) or mismatched stems (DH2-DH7) relative to B2:B5 probes (for conditions of thermal denaturation experiments, see Table 1); (c) representative gel electrophoretograms illustrating recognition of DH1 using 1- to 500-fold molar excess of X2:X5, Y2:Y5, Z2:Z5, or unmodified D1:D4; (d) dose-response curves (average of three independent experiments; error bars represent standard deviation); (e) incubation of DH1 with 500-fold molar excess of single-stranded X2, X5, Y2 or Y5 or double-stranded X2:X5 or Y2:Y5; (f) gel electrophoretograms illustrating incubation of DH1–DH7 with 500-fold molar excess of X2:X5 or Y2:Y5. Experimental conditions for electrophoretic mobility shift assay: separately pre-annealed targets (34.4 nM) and probes (variable concentration) were incubated for 15 h at ambient temperature in 1× HEPES buffer (50 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 10% sucrose, 1.4 mM spermine tetrahydrochloride, pH 7.2) and then run on 16% non-denaturing PAGE (performed at 70 V, 2 h, ∼4 °C) using 0.5× TBE as a running buffer (45 mM Tris, 45 mM boric acid, 1 mM EDTA); DIG: digoxigenin. | ||
As expected, the unmodified control duplex D1:D4 did not result in formation of recognition complexes even when used at 500-fold molar excess, as there is no driving force to overcome the energetic penalty of opening DH1 (Fig. 6c). The use of 500-fold molar excess of single-stranded X2/X5/Y2/Y5 only resulted in 15–40% recognition of DH1 (Fig. 6e), which demonstrates that both probe strands are necessary for efficient dsDNA-recognition.
Finally, the binding specificity of X2:X5 and Y2:Y5 was examined in detail by incubating the probe duplexes with DNA hairpins DH2–DH7, which are fully base-paired but singly mismatched relative to the Invader probes (underlined residues indicate position of sequence deviation, Fig. 6b). Importantly, neither a 500-fold molar excess of X2:X5 nor Y2:Y5 resulted in recognition of the singly mismatched DNA hairpins, which demonstrates that hairpin recognition is highly specific (Fig. 6f).
Conclusion
Short synthetic routes to suitably protected 2′-O-(pyren-2-yl)-methyluridine and 2′-O-(pyren-4-yl)methyluridine have been developed. ONs modified with 2′-O-(pyren-2-yl)methyluridines display greater affinity toward complementary DNA and better mismatch discrimination than ONs modified with the corresponding 1-pyrenyl or 4-pyrenyl analogues. Molecular modeling suggests this to be a consequence of more efficient π–π-stacking between the intercalating pyrene moiety and neighboring base pairs.DNA duplexes with +1 interstrand arrangements of these pyrene-functionalized monomers display lower thermostability, more blue-shifted pyrene absorption maxima, more distinctive fluorescence emission profiles and greater potential for dsDNA-recognition than duplexes with other monomer arrangements. Force field simulations suggest that the +1 zipper motif results in significant perturbation of the duplex. Although the chemical differences between the three studied monomers are relatively minor, significant variations in the dsDNA-recognition efficiencies of the resulting probes are observed. Thus, probes with +1 interstrand zippers of 2′-O-(pyren-2-yl)methyluridines recognize mixed-sequence DNA hairpins very efficiently, whereas probes constructed with the corresponding 2′-O-(pyren-4-yl)methyluridines do not. Remarkably, Invaders based on 2′-O-(pyren-2-yl)methyluridines result in ∼50% recognition of dsDNA when used in equimolar quantities. These findings encourage additional structure–property relationship studies based on the 2-pyrenyl scaffolds with the goal of further increasing the dsDNA recognition efficiency of Invader probes, especially considering that detection of chromosomal DNA using Invaders based on the slightly less efficient 2′-O-(pyren-1-yl)-RNA monomers already has been demonstrated.13 Proof-of-concept studies aiming at expanding the repertoire of applications for Invader probes in molecular biology, nucleic acid diagnostics and medicinal chemistry are ongoing and will be reported in due course.
Experimental
Preparation of 2-pyrenemethanol
2-Pyrenemethanol was obtained from 4,5,9,10-tetrahydropyrene by combining known literature protocols.22 First, a solution of bromine (1.24 mL, 24.2 mmol) in anhydrous DMF (40 mL) was added dropwise over 2 h to a room temperature solution of 4,5,9,10-tetrahydropyrene22a (5.00 g, 24.2 mmol) in anhydrous DMF (40 mL). The mixture was stirred for 4 h at rt after completed addition, poured into cold water and stirred overnight. The resulting precipitate was filtered, washed with water, dried and purified by silica gel column chromatography (petroleum ether) to provide an off-white solid material (5.50 g), which was used in the next step without further purification.The solid material was dissolved in anhydrous benzene (400 mL) along with DDQ (14.4 g, 63.6 mmol), and the solution was refluxed under an argon atmosphere for 4 h. The resulting slurry was filtered through a pad of celite, which was thoroughly washed with benzene. The filtrate was washed with aq. NaOH (10% v/v, 3 × 100 mL) and water (3 × 100 mL). The organic phase was evaporated to dryness and the resulting residue purified by silica gel column chromatography (petroleum ether) to afford a pale yellow solid material (4.60 g), which was used in the next step without further purification.
Next, n-butyllithium (9.82 mL, 24.5 mmol, 2.5 M in hexane) was added slowly to a −78 °C solution of the solid material in anhydrous ether–THF (350 mL, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v). The brightly red solution was stirred at −78 °C for 2 h and then slowly treated with anhydrous DMF (2.6 mL, 33.5 mmol). The reaction mixture was stirred at −30 to −50 °C for 1 h and then at rt for an additional 15 h. At this point, the reaction mixture was poured into ice-cold water (∼100 mL) and the aqueous phase was extracted with EtOAc (3 × 100 mL). The combined organic layers were evaporated to near dryness and the resulting residue was purified by silica gel column chromatography (0–3% CH2Cl2 in petroleum ether, v/v) to afford a brightly yellow solid material (3.50 g), which was used in the next step without further purification.
:1 v/v). The brightly red solution was stirred at −78 °C for 2 h and then slowly treated with anhydrous DMF (2.6 mL, 33.5 mmol). The reaction mixture was stirred at −30 to −50 °C for 1 h and then at rt for an additional 15 h. At this point, the reaction mixture was poured into ice-cold water (∼100 mL) and the aqueous phase was extracted with EtOAc (3 × 100 mL). The combined organic layers were evaporated to near dryness and the resulting residue was purified by silica gel column chromatography (0–3% CH2Cl2 in petroleum ether, v/v) to afford a brightly yellow solid material (3.50 g), which was used in the next step without further purification.
Lastly, sodium borohydride (0.69 g, 18.2 mmol) was added to a solution of this solid material in anhydrous THF (120 mL) and the reaction mixture was stirred overnight at rt. The reaction mixture was cooled on an ice bath and aqueous NaHCO3 (10% v/v, 50 mL) was added carefully. The aqueous layer was extracted with EtOAc (3 × 100 mL) and the combined organic layers were evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–1% MeOH in CH2Cl2, v/v) to afford 2-pyrenemethanol22c (3.45 g, 62% over four steps) as a white solid material. Rf = 0.5 (10% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 232.0875 ([M]+, C17H12O, Calc. 232.0888); 1H NMR (DMSO-d6): δ 8.28 (d, 2H, J = 7.8 Hz, H6/H8), 8.25 (s, 2H, H1/H3), 8.17 (s, 4H, H4/H5/H9/H10), 8.03–8.06 (ap t, 1H, J = 7.5 Hz, H7), 5.52 (t, 1H, ex, J = 5.7 Hz, OH), 4.97 (d, 2H, J = 5.7 Hz, CH2OH); 13C NMR (DMSO-d6) δ 140.7 (C2), 130.5, 130.4, 127.31 (C4/C5),34 127.29 (C9/C10), 125.9 (C7), 124.9 (C6/C8), 123.8, 123.0 (C1/C3), 122.9, 63.2 (CH2Py).
Preparation of 4-pyrenemethanol
4-Pyrenemethanol was obtained from 4,5,9,10-tetrahydropyrene by combining known literature protocols23 and protocols for the synthesis of 2-pyrenemethanol. First, a solution of bromine (0.62 mL, 12.0 mmol) in glacial acetic acid (25 mL) was added dropwise over 1 h to a room temperature solution of 4,5,9,10-tetrahydropyrene 322a (2.50 g, 12.0 mmol) in glacial acetic acid (25 mL). After ended addition, the reaction mixture was heated to 80 °C for 30 min, and then slowly cooled to rt, leading to the formation of a precipitate, which was isolated to provide a white solid material (2.90 g, 10.1 mmol) which used in the next step without further purification.The solid material and DDQ (7.56 g, 33.3 mmol) were dissolved in anhydrous benzene (300 mL) and refluxed for 4 h. The resulting slurry was filtered through a celite pad, which was thoroughly washed with benzene. The filtrate was washed with aq. NaOH (10% v/v, 3 × 80 mL) and water (3 × 80 mL). The organic phase was evaporated to dryness and the resulting residue purified via silica gel column chromatography (petroleum ether) to obtain a pale yellow solid (2.00 g), which was used in the next step without further purification.
Next, n-butyllithium (4.27 mL, 10.7 mmol, 2.5 M in hexane) was added slowly to a −78 °C solution of this solid material in anhydrous ether–THF (170 mL, 1:1 v/v). The brightly red solution was stirred at −78 °C for 2 h and then treated slowly with anhydrous DMF (1.12 mL, 14.6 mmol). The reaction mixture was stirred at −30 to −50 °C for 1 h and then at rt for additional 15 h. The reaction mixture was poured into ice-cold water (∼50 mL), the aqueous phase was extracted with EtOAc (3 × 50 mL), and the combined organic phase evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–3% CH2Cl2 in pet ether, v/v) to afford a brightly yellow solid material (1.20 g), which was used in the next step without further purification.
Finally, NaBH4 (0.24 g, 6.25 mmol) was added to a solution of the solid material in anhydrous THF (80 mL) and the reaction mixture was stirred overnight at rt. The reaction mixture was cooled on an ice-bath and aqueous NaHCO3 (10% v/v, 20 mL) was added carefully. The aqueous layer was extracted with EtOAc (3 × 80 mL) and the combined organic layers were evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–1% MeOH in CH2Cl2, v/v) to afford 4-pyrenemethanol (1.15 g, 41% over four steps) as a white solid material. Rf = 0.5 (10% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 232.0881 ([M]+, C17H12O, Calc. 232.0888); 1H NMR (DMSO-d6): δ 8.39–8.41 (dd, 1H, J = 7.4 Hz, 0.8 Hz, H3), 8.26–8.32 (m, 3H, H1/H6/H8), 8.24 (s, 1H, H5), 8.17–8.21 (2d, 2H, J = 8.8 Hz, H9/H10), 8.05–8.11 (m, 2H, H2/H7), 5.53 (t, 1H, ex, J = 5.5 Hz, OH), 5.19–5.21 (dd, 2H, J = 5.5 Hz, 0.8 Hz, CH2Py); 13C NMR (DMSO-d6) δ 137.1, 130.9, 130.4, 130.3, 128.9, 127.4 (C9/C10), 127.1 (C9/C10), 126.2 (C2/C7), 125.9 (C2/C7), 125.1 (C1/C6/C8), 125.0 (C1/C6/C8), 124.8 (C1/C6/C8), 124.3 (C5), 124.0, 123.2, 121.2 (C3), 61.4 (CH2Py).
General O2′-alkylation protocol for the preparation of 2Y/2Z (description for ∼4.4 mmol scale)
The appropriate pyrenemethanol, solid NaHCO3 and borane (1.0 M solution in THF) were placed in a pressure tube, suspended in anhydrous DMSO and stirred under an argon atmosphere at rt until effervescence ceased (∼10 min). At this point, O2,O2′-anhydrouridine 120 was added (specific quantities of substrates and reagents are given below), the pressure tube was purged with argon and sealed, and the mixture was heated at ∼140 °C until analytical TLC indicated full conversion (∼3 h). After cooling to rt, the reaction mixture was poured into water (∼50 mL), stirred for 30 min and diluted with EtOAc (∼100 mL). The organic phase was washed with water (4 × 50 mL), evaporated to dryness and the resulting crude purified by silica gel column chromatography (2–4%, MeOH in CH2Cl2, v/v) to afford a residue, which was further purified through precipitation from refluxing methanol (30 min) to afford nucleoside 2 (yield specified below).2′-O-(Pyren-2-yl)methyl-uridine (2Y)
O2,O2′-Anhydrouridine 1 (1.00 g, 4.42 mmol), 2-pyrenemethanol (2.05 g, 8.84 mmol), NaHCO3 (74.3 mg. 0.88 mmol), BH3 in THF (2.21 mL, 2.21 mmol) and anhydrous DMSO (8 mL) were mixed, reacted, worked up, and purified as described above to afford nucleoside 2Y (0.60 g, 30%) as a white solid material. Rf: 0.4 (10% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 481.1386 ([M + Na]+, C26H22N2O6·Na+, Calc. 481.1370; 1H NMR (DMSO-d6):35δ 11.38 (s, 1H, ex, NH), 8.28–8.30 (d, 2H, J = 7.8 Hz, H6Py, H8Py), 8.23 (s, 2H, H1Py, H3Py), 8.17–8.20 (d, 2H, J = 8.8 Hz, H4Py, H10Py), 8.10–8.13 (d, 2H, J = 8.8 Hz, H5Py, H9Py), 8.07 (t, 1H, J = 7.8 Hz, H7Py), 7.85 (d, 1H, J = 8.0 Hz, H6), 6.09 (d, 1H, J = 4.9 Hz, H1′), 5.51 (d, 1H, J = 8.0 Hz, H5), 5.35 (d, 1H, ex, J = 5.7 Hz, 3′-OH), 5.16–5.19 (d, 1H, J = 12.7 Hz, CH2Py), 5.11 (t, 1H, ex, J = 4.9 Hz, 5′-OH), 5.03–5.06 (d, 1H, J = 12.7 Hz, CH2Py), 4.23–4.26 (m, 1H, H3′), 4.12–4.15 (m, 1H, H2′), 3.98–4.00 (m, 1H, H4′), 3.65–3.69 (m, 1H, H5′), 3.57–3.62 (m, 1H, H5′); 13C NMR (DMSO-d6): δ 163.0, 150.6, 140.2 (C6), 136.3, 130.54, 130.50, 127.5 (C4Py, C10Py), 127.2 (C5Py, C9Py), 126.1 (C7Py), 125.1 (C6Py, C8Py), 123.7 (C1Py, C3Py), 123.2, 101.8 (C5), 86.4 (C1′), 85.2 (C4′), 80.8 (C2′), 71.3 (CH2Py), 68.4 (C3′), 60.5 (C5′).2′-O-(Pyren-4-yl)methyl-uridine (2Z)
O2,O2′-Anhydrouridine 1 (0.50 g, 2.21 mmol), 4-pyrenemethanol (1.03 g, 4.42 mmol), NaHCO3 (37.1 mg. 0.44 mmol), BH3 in THF (1.10 mL, 1.10 mmol) and anhydrous DMSO (5 mL) were mixed, reacted, worked up, and purified as described above to afford nucleoside 2Y (200 mg, 20%) as a white solid. Rf: 0.4 (10% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 481.1399 ([M + Na]+, C26H22N2O6·Na+, Calc. 481.1370; 1H NMR (DMSO-d6): δ 11.28 (s, 1H, ex, NH), 8.42 (d, 1H, J = 7.8 Hz, Py), 8.29–8.31 (m, 2H, Py), 8.24–8.26 (d, 1H, J = 7.3 Hz, Py), 8.18–8.22 (m, 3H, Py), 8.07 (t, 1H, J = 7.5 Hz, Py), 8.03 (dd, 1H, J = 7.8 Hz, 7.5 Hz, Py), 7.77 (d, 1H, J = 8.0 Hz, H6), 6.08 (d, 1H, J = 5.4 Hz, H1′), 5.39–5.43 (m, 3H, 1 ex, 3′-OH, H5 and CH2Py), 5.21–5.24 (d, 1H, J = 12.5 Hz, CH2Py), 5.09 (t, 1H, ex, 5′-OH), 4.29–4.33 (m, 1H, H3′), 4.20–4.23 (m, 1H, H2′), 3.97–4.00 (m, 1H, H4′), 3.63–3.68 (m, 1H, H5′), 3.57–3.62 (m, 1H, H5′); 13C NMR (DMSO-d6): δ 162.8, 150.5, 140.0 (C6), 132.8, 130.8, 130.5, 129.9, 129.0, 127.5 (Py), 127.0 (Py), 126.9 (Py), 126.3 (Py), 125.9 (Py), 125.3 (Py), 125.2 (Py), 124.1, 123.5, 121.7 (Py), 101.6 (C5), 86.0 (C1′), 85.4 (C4′), 80.7 (C2′), 70.2 (CH2Py), 68.4 (C3′), 60.6 (C5′).General O5′-DMTr-protection protocol for the preparation of 3Y/3Z (description for ∼1 mmol scale)
The appropriate nucleoside 2 (specific quantities given below) was co-evaporated twice with anhydrous pyridine and redissolved in anhydrous pyridine. To this was added 4,4′-dimethoxytritylchloride (DMTrCl) and catalytic N,N-dimethyl-4-aminopyridine (DMAP), and the reaction mixture was stirred at rt under an argon atmosphere until TLC indicated complete conversion (∼14 h). The reaction mixture was diluted with CH2Cl2 (40 mL) and the organic phase was sequentially washed with water (2 × 30 mL) and sat. aq. NaHCO3 (2 × 50 mL). The organic phase was evaporated to near dryness and the resulting crude co-evaporated with abs. EtOH and toluene (2:1, v/v, 3 × 3 mL) and purified by silica gel column chromatography (0–5%, MeOH in CH2Cl2, v/v) to afford nucleoside 3 (yield specified below).
5′-O-(4,4′-Dimethoxytrityl)-2′-O-(pyren-2-yl)methyl-uridine (3Y)
Nucleoside 2Y (500 mg, 1.09 mmol), DMTrCl (0.64 g, 1.63 mmol) and DMAP (∼9 mg) in anhydrous pyridine (10 mL) were mixed, reacted, worked up and purified as described above to afford 3Y (0.57 g, 68%) as a pale yellow foam. Rf: 0.6 (5% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 783.2706 ([M + Na]+, C47H40N2O8·Na+, Calc. 783.2677); 1H NMR (DMSO-d6):35δ 11.43 (s, 1H, ex, NH), 8.27–8.31 (m, 4H, H1Py, H3Py, H6Py, H8Py), 8.17–8.20 (d, 2H, J = 8.9 Hz, H4Py, H10Py), 8.11–8.13 (d, 2H, J = 8.9 Hz, H5Py, H9Py), 8.07 (t, 1H, J = 7.5 Hz, H7Py), 7.67 (d, 1H, J = 8.0 Hz, H6), 7.31–7.33 (m, 2H, DMTr), 7.22–7.26 (m, 2H, DMTr), 7.15–7.20 (m, 5H, DMTr), 6.78–6.84 (m, 4H, DMTr), 6.07–6.08 (d, 1H, J = 3.9 Hz, H1′), 5.45 (d, 1H, ex, J = 6.7 Hz, 3′-OH), 5.13–5.23 (m, 3H, H5, CH2Py), 4.31–4.35 (m, 1H, H3′), 4.18–4.20 (m, 1H, H2′), 4.09–4.12 (m, 1H, H4′), 3.68 (s, 3H, CH3O), 3.65 (s, 3H, CH3O), 3.30–3.34 (m, 1H, H5′; overlap with H2O), 3.23–3.26 (m, 1H, H5′); 13C NMR (DMSO-d6): δ 162.9, 158.1, 158.0, 150.4, 144.4, 140.1 (C6), 136.2, 135.3, 135.0, 130.6, 130.5, 129.7 (DMTr), 129.6 (DMTr), 127.8 (DMTr), 127.6 (DMTr), 127.5 (Py), 127.2 (Py), 126.7 (Py), 126.1 (Py), 125.1 (Py), 123.68, 123.66 (Py), 123.2, 113.2 (DMTr), 113.1 (DMTr), 101.5 (C5), 87.2 (C1′), 85.9, 82.9 (C4′), 80.6 (C2′), 71.3 (CH2Py), 68.7 (C3′), 62.7 (C5′), 54.94 (CH3O), 54.92 (CH3O). A minor impurity of chloroform at 79.1 ppm was identified in the 13C NMR.5′-O-(4,4′-Dimethoxytrityl)-2′-O-(pyren-4-yl)methyl-uridine (3Z)
Nucleoside 2Z (150 mg, 0.33 mmol), DMTrCl (191 mg, 0.49 mmol) and DMAP (∼4 mg) in anhydrous pyridine (5 mL) were mixed, reacted, worked up and purified as described above to afford 3Z (200 mg, 80%) as a pale yellow foam. Rf: 0.6 (5% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 783.2695 ([M + Na]+, C47H40N2O8·Na+, Calc. 783.2677); 1H NMR (DMSO-d6):36δ 11.36 (s, 1H, ex, NH), 8.45 (d, 1H, J = 7.8 Hz, Py), 8.17–8.33 (m, 6H, Py), 8.05 (t, 2H, H2Py, H7Py), 7.60 (d, 1H, J = 8.2 Hz, H6), 7.30–7.32 (m, 2H, DMTr), 7.17–7.25 (m, 7H, DMTr), 6.78–6.84 (m, 4H, DMTr), 6.07 (d, 1H, J = 4.1 Hz, H1′), 5.49 (d, 1H, J = 6.2 Hz, ex, 3′-OH), 5.44–5.46 (m, 1H, J = 12.7 Hz, CH2Py), 5.30–5.34 (d, 1H, J = 12.7 Hz, CH2Py), 5.08 (d, 1H, J = 8.2 Hz, H5), 4.38–4.42 (m, 1H, H3′), 4.26–4.28 (m, 1H, H2′), 4.09–4.12 (m, 1H, H4′), 3.71 (s, 3H, CH3O), 3.69 (s, 3H, CH3O), 3.30–3.34 (m, 1H, H5′ – partial overlap with H2O), 3.22–3.25 (m, 1H, H5′); 13C NMR (DMSO-d6): δ 162.8, 158.1, 158.0, 150.4, 144.4, 140.0 (C6), 135.3, 135.0, 132.7, 130.9, 130.5, 129.9, 129.7 (DMTr), 129.6 (DMTr), 129.0, 127.8 (DMTr), 127.6 (DMTr), 127.5 (Py), 127.1 (Py), 126.8 (Py), 126.7 (DMTr), 126.3 (C2Py), 126.0 (C7Py), 125.29 (Py), 125.25 (Py), 124.1, 123.5, 121.7 (Py), 113.2 (DMTr), 113.1 (DMTr), 101.4 (C5), 86.9 (C1′), 85.9, 83.2 (C4′), 80.5 (C2′), 70.2 (CH2Py), 68.7 (C3′), 62.8 (C5′), 55.0 (CH3O).General O3′-phosphitylation protocol for the preparation of 4Y and 4Z (description for ∼0.25 mmol scale)
The appropriate nucleoside 3 (specific quantities given below) was co-evaporated with anhydrous 1,2-dichloroethane and redissolved in anhydrous CH2Cl2. To this was added N,N-diisopropylethylamine (DIPEA) and 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (PCl-reagent) and the reaction mixture was stirred at rt under an argon atmosphere until TLC indicated complete conversion (∼3 h), whereupon abs. EtOH (0.3 mL) and CH2Cl2 (5 mL) were sequentially added. The organic phase was washed with sat. aq. NaHCO3 (2 mL), evaporated to near dryness, and the resulting residue purified by silica gel column chromatography (40–70% EtOAc in petroleum ether, v/v) to afford phosphoramidite 4 (yield specified below).3′-O-(2-Cyanoethoxy(diisopropylamino)phosphinyl))-5′-O-(4,4′-dimethoxytrityl)-2′-O-(pyren-2-yl)methyl-uridine (4Y)
Nucleoside 3Y (145 mg, 0.19 mmol), DIPEA (136 μL, 0.76 mmol) and PCl-reagent (85 μL, 0.38 mmol) in anhydrous CH2Cl2 (5 mL) were mixed, reacted, worked up and purified as described above to afford phosphoramidite 4Y (170 mg, 93%) as a white foam. Rf: 0.8 (5% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 983.3746 ([M + Na]+, C56H57N4O9P·Na+, Calc. 983.3755); 31P NMR (CDCl3): δ 150.5, 150.2.3′-O-(2-Cyanoethoxy(diisopropylamino)phosphinyl))-5′-O-(4,4′-dimethoxytrityl)-2′-O-(pyren-4-yl)methyl-uridine (4Z)
Nucleoside 3Z (190 mg, 0.25 mmol), DIPEA (178 μL, 0.99 mmol) and PCl-reagent (111 μL, 0.49 mmol) in anhydrous CH2Cl2 (5 mL) were mixed, reacted, worked up and purified as described above to afford phosphoramidite 4Z (220 mg, 92%) as a white foam. Rf: 0.8 (5% MeOH in CH2Cl2, v/v); MALDI-HRMS m/z 983.3766 ([M + Na]+, C56H57N4O9P·Na+, Calc. 983.3755); 31P NMR (CDCl3) δ 150.1, 150.0.Protocol – synthesis and purification of ONs
Synthesis of Y/Z-modified ONs was performed on an automated DNA synthesizer using 0.2 μmol scale succinyl linked LCAA-CPG (long chain alkyl amine controlled pore glass) columns with a pore size of 500 Å. Standard protocols for incorporation of DNA phosphoramidites were used. A ∼50-fold molar excess of modified phosphoramidites in anhydrous acetonitrile (at 0.05 M), along with extended oxidation (45 s) and coupling times (activator: 0.01 M 4,5-dicyanoimidazole, 15 min) was used during hand-couplings, which resulted in stepwise coupling yields of 4Y and 4Z of ∼99% and ∼98%, respectively. Cleavage from solid support and removal of protecting groups was accomplished upon treatment with 32% aq. ammonia (55 °C, 24 h). The crude ONs were purified via ion-pair reverse phase HPLC (XTerra MS C18 column) using 0.05 M triethylammonium acetate–water/acetonitrile gradient, followed by detritylation (80% aq. AcOH, 20 min), and precipitation (NaOAc–NaClO4–acetone, −18 °C for 12–16 h). The identity of synthesized ONs was established through MALDI-MS analysis (Table S1†) recorded in positive ions mode on a Quadrupole Time-Of-Flight Tandem Mass Spectrometer (Q-TOF Premiere) equipped with a MALDI source (Waters Micromass LTD, U.K) using anthranilic acid as a matrix, while purity (>85%) was verified by ion-pair reverse phase HPLC running in analytical mode.Protocol – thermal denaturation studies
Concentrations of ONs were estimated using the following extinction coefficients for DNA (OD μmol−1): G (12.01), A (15.20), T (8.40), C (7.05); for RNA (OD μmol−1): G (13.70), A (15.40), U (10.00), C (9.00); for pyrene (OD μmol−1): (22.4)31 Strands were thoroughly mixed and denatured by heating to 70–85 °C, followed by cooling to the starting temperature of the experiment. Quartz optical cells with a path length of 1.0 cm were used. Thermal denaturation temperatures (Tm's) of duplexes (1.0 μM final concentration of each strand) were measured on a Cary 100 UV/VIS spectrophotometer equipped with a 12-cell Peltier temperature controller and determined as the maximum of the first derivative of the thermal denaturation curve (A260vs. T) recorded in medium salt buffer (Tm buffer: 100 mM NaCl, 0.1 mM EDTA, and pH 7.0 adjusted with 10 mM Na2HPO4 and 5 mM Na2HPO4). The temperature of the denaturation experiments ranged from at least 15 °C below Tm to 20 °C above Tm (although not below 3 °C). A temperature ramp of 0.5 °C min−1 was used in all experiments. Reported Tm-values are averages of two experiments within ±1.0 °C.Protocol – determination of thermodynamic parameters
Thermodynamic parameters for duplex formation were determined through baseline fitting of denaturation curves (van't Hoff analysis) using software provided with the UV/VIS spectrometer. Bimolecular reactions, two-state melting behavior, and a heat capacity change of ΔCp = 0 upon hybridization were assumed.30 A minimum of two experimental denaturation curves were each analyzed at least three times to minimize errors arising from baseline choice. Averages and standard deviations are listed.Protocol – absorption spectra
UV-vis absorption spectra (range: 300–400 nm) were recorded at 5 °C using the same samples and instrumentation as in the thermal denaturation experiments.Protocol – steady-state fluorescence emission spectra
Steady-state fluorescence emission spectra of ONs modified with pyrene-functionalized monomers X–Z and the corresponding duplexes with complementary DNA/RNA targets, were recorded in non-deoxygenated thermal denaturation buffer (each strand at 1.0 μM concentration) and obtained as an average of five scans using an excitation wavelength of λex = 350, 345 or 340 nm for X-, Y- or Z-modified ONs, excitation slit 5.0 nm, emission slit 2.5 nm and a scan speed of 600 nm min−1. Experiments were determined at 5 °C to ascertain maximal hybridization of probes to DNA/RNA targets (a stream of nitrogen was blown in to the chamber to prevent condensation).Protocol – molecular modeling
The initial structures of the X/Y/Z-modified DNA duplexes were generated by building and modifying a standard B-type DNA duplex using MacroModel v9.8.37 The charge of the phosphodiester backbone was neutralized with sodium ions, which were placed 3.0 Å from non-bridging oxygen atoms and restrained to this distance by a force constant of 100 kJ mol−1 Å−2. A preliminary minimization was carried out using the Polak–Ribiere conjugate gradient method (convergence criteria 0.1 kJ mol−1 Å−1), the AMBER94 force field38 with the improved parambsc0 parameter set,39 and the implicit GB/SA continuum solvation model40 as implemented in MacroModel v9.8; all atoms were frozen except those in the O2′-substituent of the modified monomers. Non-bonded interactions were treated with extended cut-offs (van der Waals 8.0 Å and electrostatics 20.0 Å).The seed structure for X2:D4 was generated by taking previously reported NMR data on X-modified DNA duplexes into account.31 Thus, distances between atoms for which NOE contacts have been reported were constrained to 3.0–5.0 Å by a force constant of 100 kJ mol−1 Å−2 (i.e., distances between (i) pyrene and H5 of the modified uridine, (ii) pyrene and H2/H8 of the 3′-flanking adenosine, and (iii) pyrene and H6 of the thymine opposite of the 3′-flanking adenosine). The structure was minimized as described above with the following considerations: (i) all atoms were allowed to move freely during minimization, (ii) sodium ions were restrained as above, and (iii) hydrogen bonding of the outermost base pairs were restrained by a force constant of 100 kJ mol−1 Å−2 ((C)2–O⋯HN–2(G), distance 1.99 Å, (C)N3⋯H–N1(G), distance 1.90 Å and (C)4–NH⋯O–6(G), distance 1.69 Å). The resulting lowest energy structure formed the basis for construction of the seed structures for all other studied duplexes. Pyrene moieties of the other duplexes were manually adjusted to intercalated positions and the duplexes minimized as described for X2:D4. Seed structures were then submitted to 5 ns of stochastic dynamics (simulation temperature 300 K, time step 2.2 fs, SHAKE all bonds to hydrogen) using the same constraints as employed above (except for NOE constraints), during which 1000 structures were sampled and subsequently minimized. The resulting structures were analyzed using MacroModel v9.8 and the 3DNA web server.41
Protocol – electrophoretic mobility shift assay
This assay was performed, essentially as previously described.13 Unmodified DNA hairpins DH1–DH7 were obtained from commercial sources and used without further purification. The DNA hairpins were 3′-DIG-labeled using the 2nd generation DIG Gel Shift Kit (Roche Applied Bioscience) as recommended by the manufacturer. DIG-labeled ONs obtained in this manner, were diluted and used without further purification in the recognition experiments. Pre-annealed probes (90 °C for 10 min, cooled to room temperature over 15 min) and DIG-labeled DNA hairpins (34.4 nM) were mixed and incubated in HEPES buffer (50 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 10% sucrose, 1.44 mM spermine tetrahydrochloride, pH 7.2) for 15 h at ambient temperature (∼21 ± 3 °C). The reaction mixtures were then diluted with 6x DNA loading dye (Fermentas) and loaded onto a 16% non-denaturing polyacrylamide gel. Electrophoresis was performed using a constant voltage of 70 V for 2 h at ∼4 °C. Gels were blotted onto a positively charged nylon membrane (Roche Applied Bioscience) using constant voltage with external cooling (100 V, ∼4 °C). The membranes were exposed to anti-digoxigenin-AP Fab fragments as recommend by the manufacturer of the DIG Gel Shift Kit, transferred to a hybridization jacket, and incubated with the substrate (CSPD) in detection buffer for 10 min at 37 °C. The chemiluminescence of the formed product was captured on X-ray film, which was developed using an X-Omatic 1000A X-ray film developer (Kodak). The resulting bands were quantified using a Fluor-S MultiImager (Bio-Rad, Hercules, CA) equipped with Quantity One software. Invasion efficiency was determined as the intensity ratio between the recognition complex band and the total lane. An average of three independent experiments is reported along with standard deviations.Definition of zipper nomenclature
The following nomenclature describes the relative arrangement between two monomers positioned on opposing strands in a duplex. The number n describes the distance measured in number of base pairs and has a positive value if a monomer is shifted toward the 5′-side of its own strand relative to a second reference monomer on the other strand. Conversely, n has a negative value if a monomer is shifted toward the 3′-side of its own strand relative to a second reference monomer on the other strand.Acknowledgements
This study was supported by Award Number GM088697 from the National Institute of General Medical Sciences, National Institutes of Health; Awards IF13-001 and IF14-012 from the Higher Education Research Council, Idaho State Board of Education; The Office of Naval Research (N00014-10-1-0282); INBRE Program, NIH grant no. P20 RR016454 (National Center for Research Resources) and P20 GM103408 (National Institute of General Medical Sciences); and Minitube of America. A.S.M. is a Sapere Aude Postdoctoral Fellow (The Danish Council for Independent Research). We thank Dr. Lee Deobald (EBI Murdock Mass Spectrometry Center, Univ. Idaho) for assistance with mass spectrometric analysis, Prof. Carolyn Bohach (Food Science, Univ. Idaho) for access to gel documentation stations, and Prof. Jean'ne Shreeve (Dept. Chemistry, Univ. Idaho) for access to Parr reactors.Notes and references
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737–738 CrossRef CAS PubMed.
- (a) P. B. Dervan and B. S. Edelson, Curr. Opin. Struct. Biol., 2003, 13, 284–299 CrossRef CAS PubMed; (b) M. S. Blackledge and C. Melander, Bioorg. Med. Chem., 2003, 21, 6101–6114 CrossRef PubMed.
- (a) I. Ghosh, C. I. Stains, A. T. Ooi and D. J. Segal, Mol. BioSyst., 2006, 2, 551–560 RSC; (b) A. J. Bogdanove and D. F. Voytas, Science, 2011, 333, 1843–1846 CrossRef CAS PubMed; (c) T. Gaj, C. A. Gersbach and C. F. Barbas III, Trends Biotechnol., 2013, 31, 397–405 CrossRef CAS PubMed.
- M. Duca, P. Vekhoff, K. Oussedik, L. Halby and P. B. Arimondo, Nucleic Acids Res., 2008, 36, 5123–5138 CrossRef CAS PubMed.
- (a) P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CAS; (b) T. Bentin and P. E. Nielsen, Biochemistry, 1996, 35, 8863–8869 CrossRef CAS PubMed; (c) K. Kaihatsu, D. A. Braasch, A. Cansizoglu and D. R. Corey, Biochemistry, 2002, 41, 11118–11125 CrossRef CAS PubMed; (d) K. Kaihatsu, B. A. Janowski and D. R. Corey, Chem. Biol., 2004, 11, 749–758 CrossRef CAS PubMed.
- For other groove-binding strategies see e.g.: (a) W. C. Tse and D. L. Boger, Chem. Biol., 2004, 11, 1607–1617 CrossRef CAS PubMed; (b) P. L. Hamilton and D. P. Arya, Nat. Prod. Rep., 2012, 29, 134–143 RSC.
- (a) T. Bentin, H. J. Larsen and P. E. Nielsen, Biochemistry, 2003, 42, 13987–13995 CrossRef CAS PubMed; (b) K. Kaihatsu, R. H. Shah, X. Zhao and D. R. Corey, Biochemistry, 2003, 42, 13996–14003 CrossRef CAS PubMed; (c) D. A. Horne and P. B. Dervan, J. Am. Chem. Soc., 1990, 112, 2435–2437 CrossRef CAS; (d) V. V. Filichev, C. M. Nielsen, N. Bomholt, C. H. Jessen and E. B. Pedersen, Angew. Chem., Int. Ed., 2006, 45, 5311–5315 CrossRef CAS PubMed; (e) D. A. Rusling, V. E. C. Powers, R. T. Ranasinghe, Y. Wang, S. D. Osborne, T. Brown and K. Fox, Nucleic Acids Res., 2005, 33, 3025–3032 CrossRef CAS PubMed; (f) Y. Hari, S. Obika and T. Imanishi, Eur. J. Org. Chem., 2012, 2875–2887 CrossRef CAS.
- (a) T. Ishihara and D. R. Corey, J. Am. Chem. Soc., 1999, 121, 2012–2020 CrossRef CAS; (b) L. Milne, Y. Xu, D. M. Perrin and D. S. Sigman, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3136–3141 CrossRef CAS PubMed; (c) V. V. Filichev, B. Vester, L. H. Hansen and E. B. Pedersen, Nucleic Acids Res., 2005, 33, 7129–7137 CrossRef CAS PubMed; (d) B. A. Janowski, K. Kaihatsu, K. E. Huffman, J. C. Schwartz, R. Ram, D. Hardy, C. R. Mendelson and D. R. Corey, Nat. Chem. Biol., 2005, 1, 210–215 CrossRef CAS PubMed; (e) R. Beane, S. Gabillet, C. Montaillier, K. Arar and D. R. Corey, Biochemistry, 2008, 47, 13147–13149 CrossRef CAS PubMed.
- (a) I. V. Kutyavin, R. L. Rhinehart, E. A. Lukhtanov, V. V. Gorn, R. B. Meyer Jr. and H. B. Gamper Jr., Biochemistry, 1996, 35, 11170–11176 CrossRef CAS PubMed; (b) I. V. Smolina and V. V. Demidov, Chem. Biol., 2003, 10, 591–595 CrossRef CAS PubMed.
- (a) J. Lohse, O. Dahl and P. E. Nielsen, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11804–11808 CrossRef CAS PubMed; (b) T. Ishizuka, J. Yoshida, Y. Yamamoto, J. Sumaoka, T. Tedeschi, R. Corradini, S. Sforza and M. Komiyama, Nucleic Acids Res., 2008, 36, 1464–1471 CrossRef CAS PubMed.
- V. V. Demidov, E. Protozanova, K. I. Izvolsky, C. Price, P. E. Nielsen and M. D. Frank-Kamenetskii, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5953–5958 CrossRef CAS PubMed.
- (a) S. Rapireddy, R. Bahal and D. H. Ly, Biochemistry, 2011, 50, 3913–3918 CrossRef CAS PubMed; (b) R. Bahal, B. Sahu, S. Rapireddy, C.-M. Lee and D. H. Ly, ChemBioChem, 2012, 13, 56–60 CrossRef CAS PubMed.
- B. A. Didion, S. Karmakar, D. C. Guenther, S. Sau, J. P. Verstegen and P. J. Hrdlicka, ChemBioChem, 2013, 14, 1534–1538 CrossRef CAS PubMed.
- D. M. Crothers, Biopolymers, 1968, 6, 575–584 CrossRef CAS PubMed.
- (a) P. J. Hrdlicka, T. S. Kumar and J. Wengel, Chem. Commun., 2005, 4279–4281 RSC; (b) S. P. Sau, T. S. Kumar and P. J. Hrdlicka, Org. Biomol. Chem., 2010, 8, 2028–2036 RSC.
- S. P. Sau, A. S. Madsen, P. Podbevsek, N. K. Andersen, T. S. Kumar, S. Andersen, R. L. Rathje, B. A. Anderson, D. C. Guenther, S. Karmakar, P. Kumar, J. Plavec, J. Wengel and P. J. Hrdlicka, J. Org. Chem., 2013, 78, 9560–9570 CrossRef CAS PubMed.
- (a) M. Nakamura, Y. Shimomura, Y. Ohtoshi, K. Sasa, H. Hayashi, H. Nakano and K. Yamana, Org. Biomol. Chem., 2007, 5, 1945–1951 RSC; (b) S. Karmakar, B. A. Anderson, R. L. Rathje, S. Andersen, T. Jensen, P. Nielsen and P. J. Hrdlicka, J. Org. Chem., 2011, 76, 7119–7131 CrossRef CAS PubMed.
- (a) T. S. Kumar, A. S. Madsen, M. E. Østergaard, S. P. Sau, J. Wengel and P. J. Hrdlicka, J. Org. Chem., 2009, 74, 1070–1081 CrossRef CAS PubMed; (b) N. K. Andersen, B. A. Anderson, J. Wengel and P. J. Hrdlicka, J. Org. Chem., 2013, 78, 12690–12702 CrossRef CAS PubMed.
- S. Karmakar, D. C. Guenther and P. J. Hrdlicka, J. Org. Chem., 2013, 78, 12040–12048 CrossRef CAS PubMed.
- S. K. Roy and J. Y. Tang, Org. Process Res. Dev., 2000, 4, 170–171 CrossRef CAS.
- B. S. Ross, R. H. Springer, Z. Tortorici and S. Dimock, Nucleosides Nucleotides, 1997, 16, 1641–1643 CAS.
- 2-Pyrenemethanol was obtained from pyrene according to: (a) V. V. Filichev, I. V. Astakhova, A. D. Malakhov, V. A. Korshun and E. B. Pedersen, Chem. – Eur. J., 2008, 14, 9968–9980 CrossRef CAS PubMed; (b) R. G. Harvey, S. Schmolka, C. Cortez and H. Lee, Synth. Commun., 1988, 18, 2207–2209 CrossRef CAS; (c) K. K. Laali and P. E. Hansen, J. Org. Chem., 1997, 62, 5804–5810 CrossRef CAS . See Scheme S1† and the ESI† for further details.
- 4-Pyrenemethanol was obtained from pyrene according to ref. 22a and (a) A. Streitwieser Jr., R. G. Lawler and D. Schwaab, J. Org. Chem., 1965, 30, 1470–1473 CrossRef; (b) M. Konieczny and R. G. Harvey, J. Org. Chem., 1979, 44, 2158–2160 CrossRef CAS; (c) R. G. Harvey, M. Konieczny and J. Pataki, J. Org. Chem., 1983, 48, 2930–2932 CrossRef CAS . See Scheme S2† and the ESI† for further details.
- (a) K. Yamana, R. Iwase, S. Furutani, H. Tsuchida, H. Zako, T. Yamaoka and A. Murakami, Nucleic Acids Res., 1999, 27, 2387–2392 CrossRef CAS PubMed; (b) M. Nakamura, Y. Fukunaga, K. Sasa, Y. Ohtoshi, K. Kanaori, H. Hayashi, H. Nakano and K. Yamana, Nucleic Acids Res., 2005, 33, 5887–5895 CrossRef CAS PubMed.
- (a) U. B. Christensen and E. B. Pedersen, Nucleic Acids Res., 2002, 30, 4918–4925 CrossRef CAS PubMed; (b) T. Bryld, T. Højland and J. Wengel, Chem. Commun., 2004, 1064–1065 RSC.
- (a) V. A. Korshun, D. A. Stetsenko and M. J. Gait, J. Chem. Soc., Perkin Trans. 1, 2002, 1092–1104 RSC; (b) C. Dohno and I. Saito, ChemBioChem, 2005, 6, 1075–1081 CrossRef CAS PubMed.
- (a) G. Dougherty and J. R. Pilbrow, Int. J. Biochem., 1984, 16, 1179–1192 CrossRef CAS PubMed; (b) H. Asanuma, T. Fujii, T. Kato and H. Kashida, J. Photochem. Photobiol., C, 2012, 13, 124–135 CrossRef CAS.
- (a) M. Manoharan, K. L. Tivel, M. Zhao, K. Nafisi and T. L. Netzel, J. Phys. Chem., 1995, 99, 17461–17472 CrossRef CAS; (b) Y. J. Seo, J. H. Ryu and B. H. Kim, Org. Lett., 2005, 7, 4931–4933 CrossRef CAS PubMed; (c) J. N. Wilson, Y. Cho, S. Tan, A. Cuppoletti and E. T. Kool, ChemBioChem, 2008, 9, 279–285 CrossRef CAS PubMed.
- For an overview of nucleic acid parameters, see: W. K. Olson, M. Bansal, S. K. Burley, R. E. Dickerson, M. Gerstein, S. C. Harvey, U. Heinemann, X.-J. Lu, S. Neidle, Z. Shakked, H. Sklenar, M. Suzuki, C.-S. Tung, E. Westhof, C. Wolberger and H. M. Berman, J. Mol. Biol., 2001, 313, 229–237 CrossRef CAS PubMed.
- J. L. Mergny and L. Lacroix, Oligonucleotides, 2003, 13, 515–537 CrossRef CAS PubMed.
- (a) F. M. Winnik, Chem. Rev., 1993, 93, 587–614 CrossRef CAS; (b) N. N. Dioubankova, A. D. Malakhov, D. A. Stetsenko, M. J. Gait, P. E. Volynsky, R. G. Efremov and V. A. Korshun, ChemBioChem, 2003, 4, 841–847 CrossRef CAS PubMed; (c) P. J. Hrdlicka, B. R. Babu, M. D. Sørensen and J. Wengel, Chem. Commun., 2004, 1478–1479 RSC; (d) F. Seela and S. A. Ingale, J. Org. Chem., 2010, 75, 284–295 CrossRef CAS PubMed; (e) R. Haner, F. Garo, D. Wenger and V. L. Malinovskii, J. Am. Chem. Soc., 2010, 132, 7466–7471 CrossRef CAS PubMed; (f) F. Wojciechowski, J. Lietard and C. J. Leumann, Org. Lett., 2012, 14, 5176–5179 CrossRef CAS PubMed.
- (a) I. V. Astakhova, A. D. Malakhov, I. A. Stepanova, A. V. Ustinov, S. L. Bondarev, A. S. Paramonov and V. A. Korshun, Bioconjugate Chem., 2007, 18, 1972–1980 CrossRef CAS PubMed; (b) I. V. Astakhova, A. V. Ustinov, V. A. Korshun and J. Wengel, Bioconjugate Chem., 2011, 22, 533–539 CrossRef CAS PubMed.
- We use the DNA hairpin assay in lieu of footprinting experiments to avoid reliance on 32P-labelled targets. Moreover, linearized plasmids were not used as targets due to insufficient mobility differences on non-denaturing PAGE gels between recognition complexes and free linearized plasmids. We have previously demonstrated that the recognition complex between Invaders and DNA hairpins indeed is comprised of three strands as depicted in Fig. 6a. Long incubation periods were chosen to ensure that equilibrium was reached. However, shorter incubation periods (<3 h) can be used for biological applications see ref. 13.
- The assignment of C4/C5 and C9/C10 is interchangeable.
- The assignments of H4Py/H10Py and H5Py/H9Py (and the corresponding 13C NMR signals) are interchangeable.
- The assignments of H2Py/H7Py (and the corresponding 13C NMR signals) are interchangeable.
- MacroModel, version 9.8, Schrödinger, LLC, New York, NY, 2010 Search PubMed.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- A. Pérez, I. Marchán, D. Svozil, J. Sponer, T. E. Cheatham, C. A. Laughton and M. Orozco, Biophys. J., 2007, 92, 3817–3829 CrossRef PubMed.
- W. C. Still, A. Tempczyk, R. C. Hawley and T. Hendrickson, J. Am. Chem. Soc., 1990, 112, 6127–6129 CrossRef CAS.
- G. Zheng, X.-J. Lu and W. K. Olson, Nucleic Acids Res., 2009, 37, W240–W246 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental section; MS-data for modified ONs; representative thermal denaturation curves; additional denaturation and thermodynamic data; additional absorption and steady-state fluorescence emission spectra; complete data set of structural parameters from modeling studies; lowest energy representations of Type B structures; NMR spectra for nucleosides 2Y–4Z. See DOI: 10.1039/c4ob01183j |
| This journal is © The Royal Society of Chemistry 2014 |