 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total synthesis of the cyclic monoterpenoid pyrano[3,2-a]carbazole alkaloids derived from 2-hydroxy-6-methylcarbazole†‡
Cemena
Gassner
,
Ronny
Hesse
,
Arndt W.
Schmidt
and
Hans-Joachim
Knölker
*
Department Chemie, Technische Universität Dresden, Bergstrasse 66, 01069 Dresden, Germany. E-mail: hans-joachim.knoelker@tu-dresden.de; Fax: +49 351 463-37030
First published on 8th July 2014
Abstract
The synthesis of seven pyrano[3,2-a]carbazole alkaloids has been achieved using their putative biogenetic precursor 2-hydroxy-6-methylcarbazole as key intermediate.
Introduction
The research groups of Chakraborty, Furukawa, Ito and Wu have isolated a wide range of biologically active carbazole alkaloids from plants of the family Rutaceae (genera Murraya, Clausena and Glycosmis).1,2 The pyrano[3,2-a]carbazoles, e.g.1–9, are an important subgroup of carbazole alkaloids which exhibit diverse structural features (Fig. 1).2 In 1964, girinimbine (1) was among the first carbazole alkaloids which have been isolated by Chakraborty et al. from terrestrial plants.3 Only two years later, the same group described the isolation of the corresponding prenyl-substituted homologue mahanimbine (2).4 Biogenetically, both compounds derive from 2-hydroxy-3-methylcarbazole by fusion with either prenyl or geranyl diphosphate.2 Originally, girinimbine was erroneously assigned as structure 3,3a but subsequently it had to be reassigned as 1.3b,c,d Isogirinimbine (3) biogenetically could have been formed from 2-hydroxy-6-methylcarbazole (10) and prenyl diphosphate as C5 building block (Scheme 1). Interestingly, although isogirinimbine (3) has not been found in nature so far, the corresponding carbazole alkaloids 4–9 resulting from fusion of 2-hydroxy-6-methylcarbazole (10) and geranyl diphosphate were isolated from natural sources.2 In 1970, Kapil et al. isolated mahanimbicine [(+)-4] and bicyclomahanimbicine (6) from Murraya koenigii.5 Subsequently, Crombie and Whiting et al. proposed the correct structure for bicyclomahanimbicine (6).6 It is interesting to note that also in 1970, Joshi et al. obtained (−)-4 from the leaves of the same plant and named it isomahanimbine,7 however, the absolute configuration was not determined. The structures of 4 and 6 were supported by synthesis.4c,d,5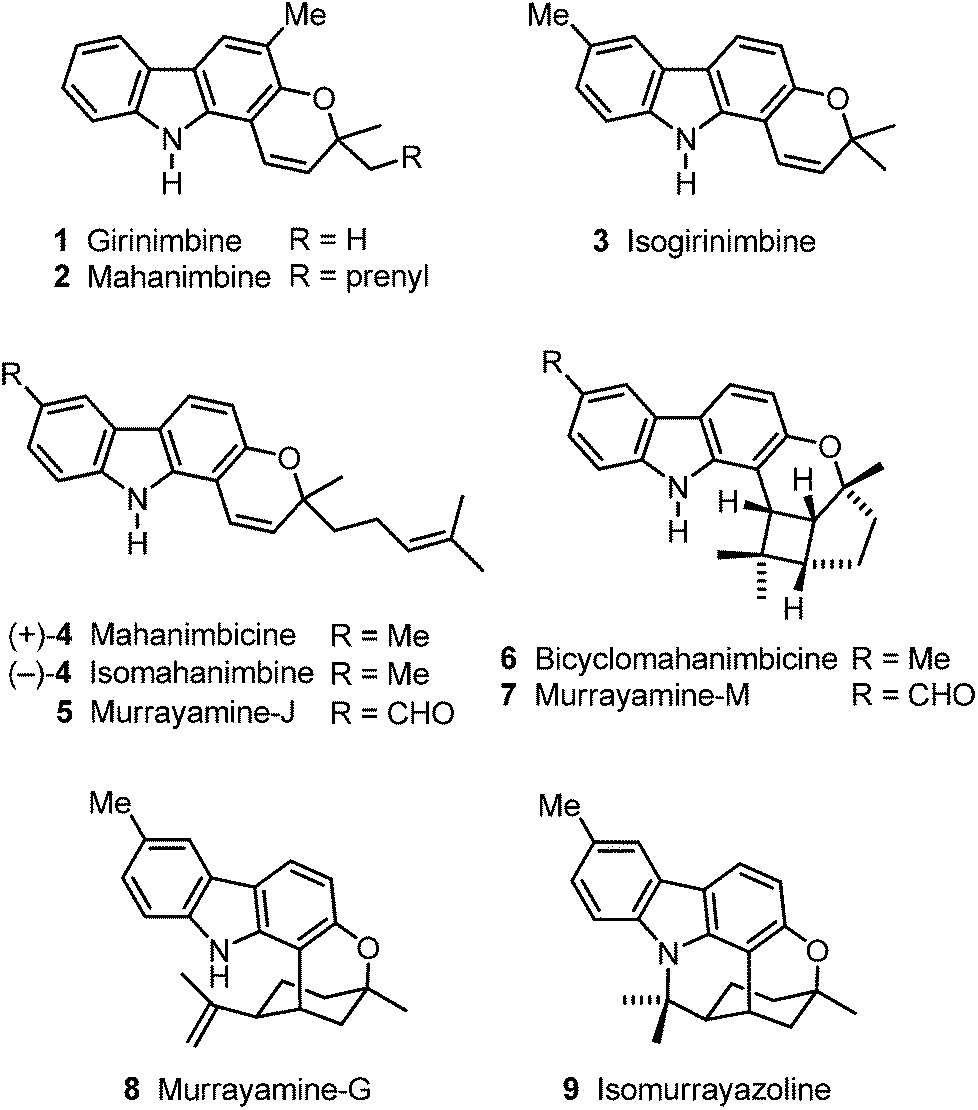 | ||
| Fig. 1 Naturally occurring pyrano[3,2-a]carbazole alkaloids 1–9. | ||
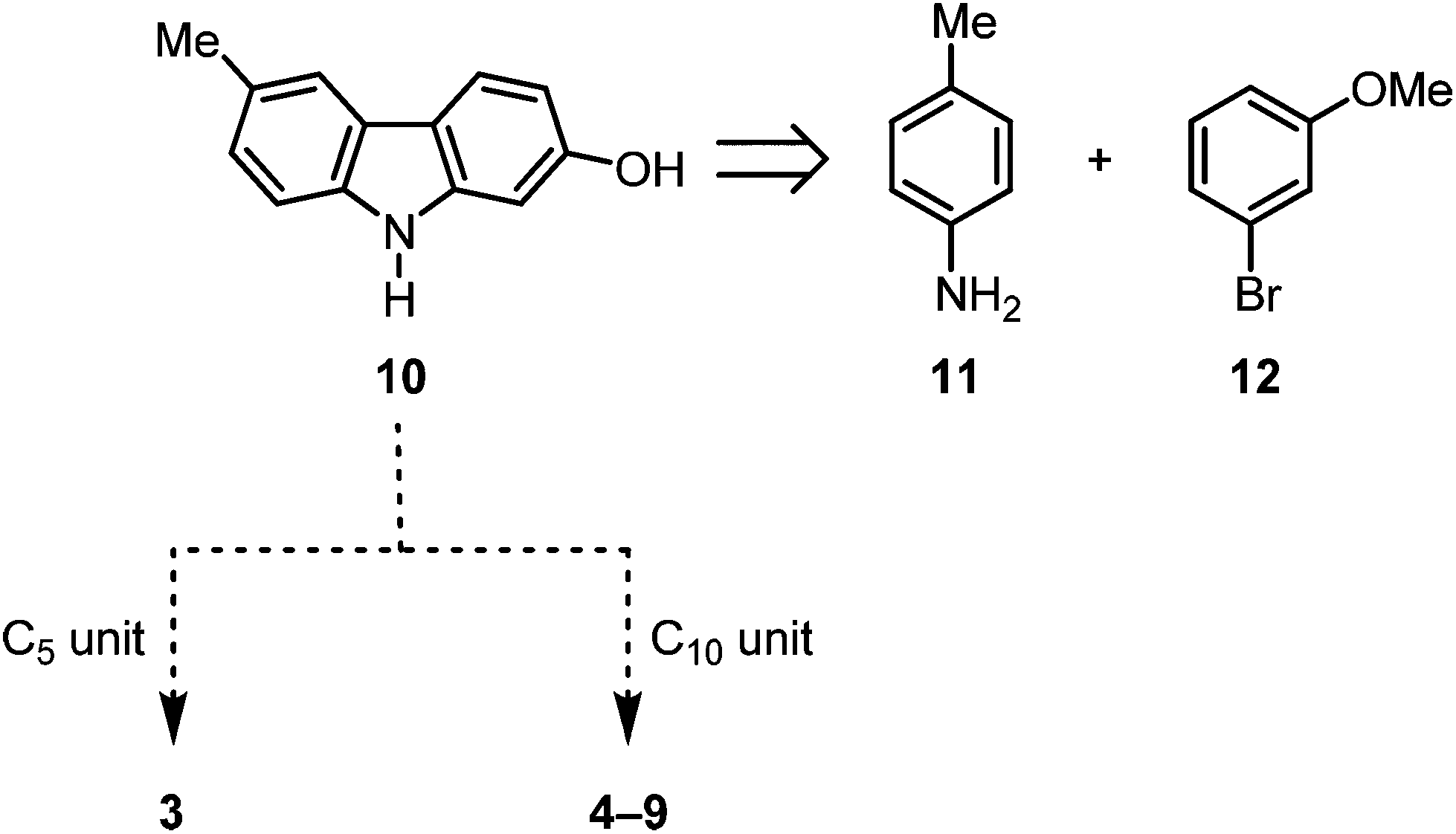 | ||
| Scheme 1 Synthetic route to the pyrano[3,2-a]carbazoles 3–9. | ||
Wu et al. isolated murrayamine-J (5),8 murrayamine-M (7)8 and murrayamine-G (8),9 from the leaves of Murraya euchrestifolia. The hexacyclic pyrano[3,2-a]carbazole alkaloid isomurrayazoline (9) was obtained in 1982 by Chakraborty et al. from the stem bark of Murraya koenigii.10
We have developed diverse synthetic approaches to pyrano[3,2-a]carbazoles including girinimbine (1), mahanimbine (2), pyrayafoline A–E and monoterpenoid pyrano[3,2-a]carbazole alkaloids.11–14 Herein, we describe the synthesis of isogirinimbine (3), (±)-mahanimbicine [(±)-isomahanimbine] [(±)-4], murrayamine-J (5) and the cyclic monoterpenoid pyrano[3,2-a]carbazole alkaloids 6–9, which biogenetically derive from 2-hydroxy-6-methylcarbazole (10). Key steps of our approach are an efficient construction of the carbazole 10 based on our palladium-catalyzed route15 and a subsequent annulation of either a C5 or a C10 building block (Scheme 1). The substitution pattern present in compound 10 has been generated previously in our synthesis of 7-oxygenated carbazole alkaloids.16
Results and discussion
Buchwald–Hartwig coupling of p-toluidine (11) and m-bromoanisole (12) in the presence of SPhos (2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl)17 afforded the diarylamine 13 (Scheme 2). Alternatively, compound 13 has been prepared quantitatively on a 25 g scale by Buchwald–Hartwig coupling of m-anisidine and p-bromotoluene (see Experimental section).16 Palladium(II)-catalyzed oxidative cyclization of 13 provided the desired 2-methoxy-6-methylcarbazole (14)16 as major product (89% yield) and up to 5% of glycoborine (15)18 as by-product. Cleavage of the methyl ether led to 2-hydroxy-6-methylcarbazole (10). Formation of the dimethylpropargyl ether via Godfrey's method,19 followed by a thermally induced sequence of Claisen rearrangement, 1,5-hydrogen shift and electrocyclic ring closure20 provided isogirinimbine (3) in 63% yield and as a by-product the furo[3,2-a]carbazole 17 in up to 3% yield. The structure of isogirinimbine (3) has been fully supported by its spectroscopic data which confirm it as an isomer of the natural product girinimbine (1). The 3-methyl-regioisomer of 17 was obtained previously as by-product in our synthesis of girinimbine (1).12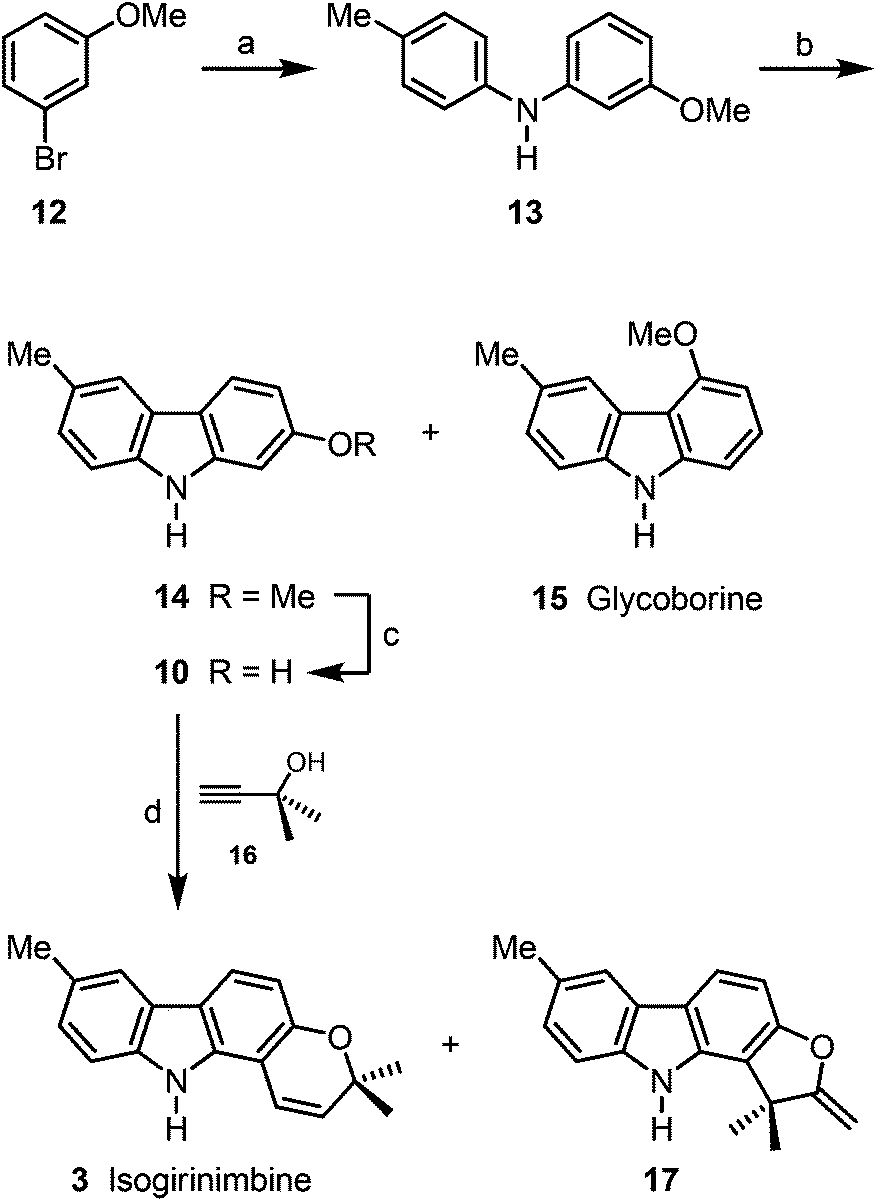 | ||
| Scheme 2 Synthesis of isogirinimbine (3). Reagents and conditions: (a) 1.3 equiv. 11, 5 mol% Pd(OAc)2, 10 mol% SPhos, 1.4 equiv. Cs2CO3, toluene, reflux, 17.5 h, 98%; (b) 3 mol% Pd(OAc)2, 10 mol% K2CO3, PivOH, 100 °C, 20.5 h, 89% 14 and ≤5% 15; (c) 1.9 equiv. BBr3, −78 °C to rt, 2 h, 91%; (d) 1. 1.1 equiv. 16, 1.1 equiv. TFAA, 2.8 equiv. DBU, 0.2 mol% CuI, MeCN, −15 °C to rt, 6.5 h; 2. toluene, reflux, 23 h, 63% 3 and ≤3% 17. | ||
We envisaged (±)-mahanimbicine [(±)-isomahanimbine] [(±)-4] as crucial intermediate for the synthesis of the formyl derivative murrayamine-J (5) and the cyclic monoterpenoid pyrano[3,2-a]carbazole alkaloids 6–9. Thus, we have developed two alternative synthetic routes for the synthesis of (±)-4. The first approach requires no protecting group (Scheme 3). Reaction of 2-hydroxy-6-methylcarbazole (10) with the carbonate 1821 in the presence of catalytic amounts of copper(I) iodide and subsequent thermally induced rearrangement provided (±)-mahanimbicine [(±)-4] in 49% yield along with the furo[3,2-a]carbazole 19 in up to 5% yield as by-product.
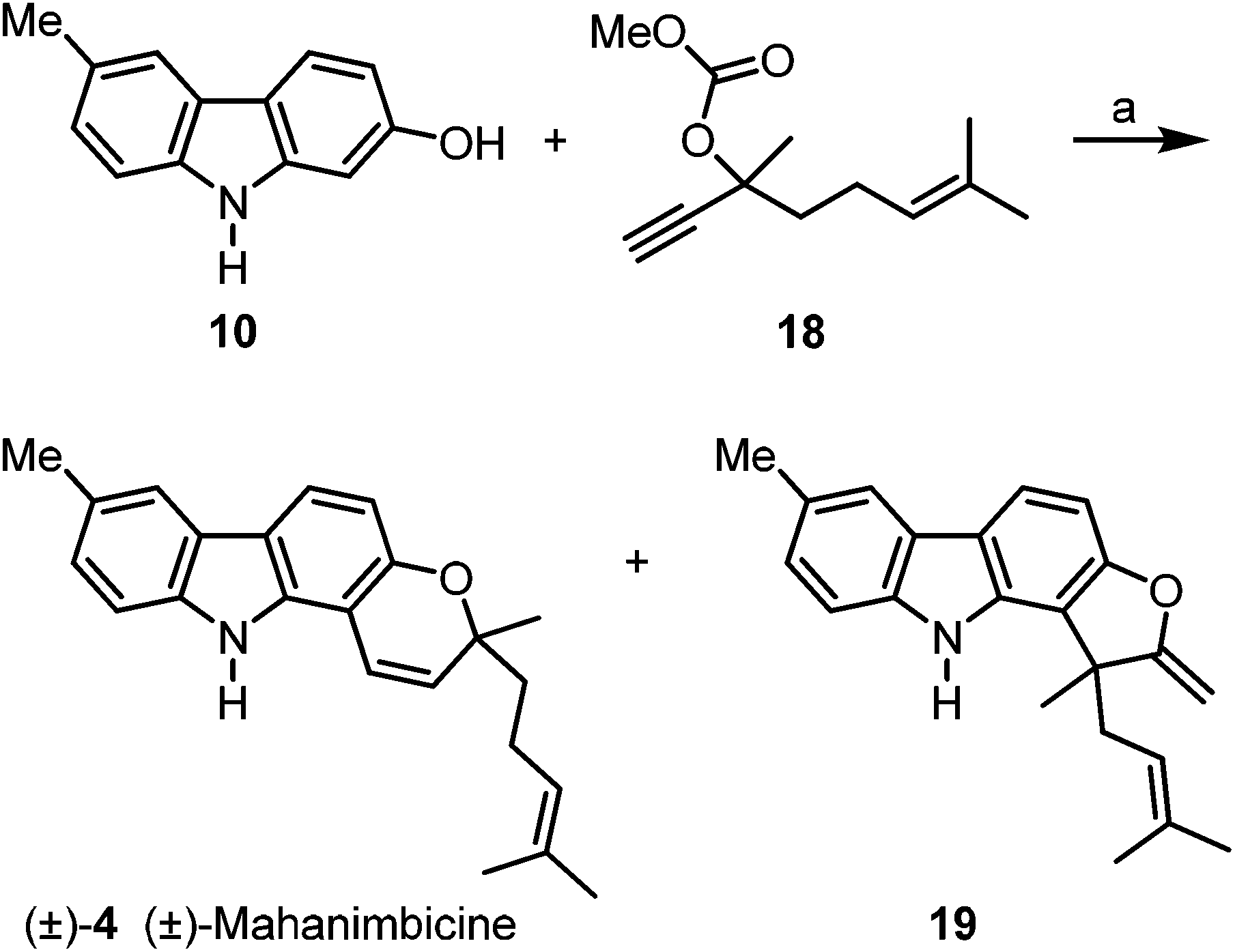 | ||
| Scheme 3 Synthesis of (±)-mahanimbicine [(±)-4]. Reagents and conditions: (a) 1. 1.5 equiv. 18, 2.0 equiv. DBU, 0.5 mol% CuI, MeCN, rt, 22 h; 2. toluene, reflux, 22.5 h, 49% (±)-4 and ≤5% 19. | ||
Alternatively, 2-methoxy-6-methylcarbazole (14) was initially protected by transformation to the N-tosylcarbazole 20 (Scheme 4). Cleavage of the methyl ether, copper-catalyzed reaction with the carbonate 18 and thermal rearrangement led in 82% yield to a mixture of the pyrano[3,2-a]carbazole 21 and the pyrano[2,3-b]carbazole 22 in a ratio of 7.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Finally, removal of the tosyl group by treatment with tetrabutylammonium fluoride22 at elevated temperature provided (±)-mahanimbicine [(±)-4].
:1. Finally, removal of the tosyl group by treatment with tetrabutylammonium fluoride22 at elevated temperature provided (±)-mahanimbicine [(±)-4].
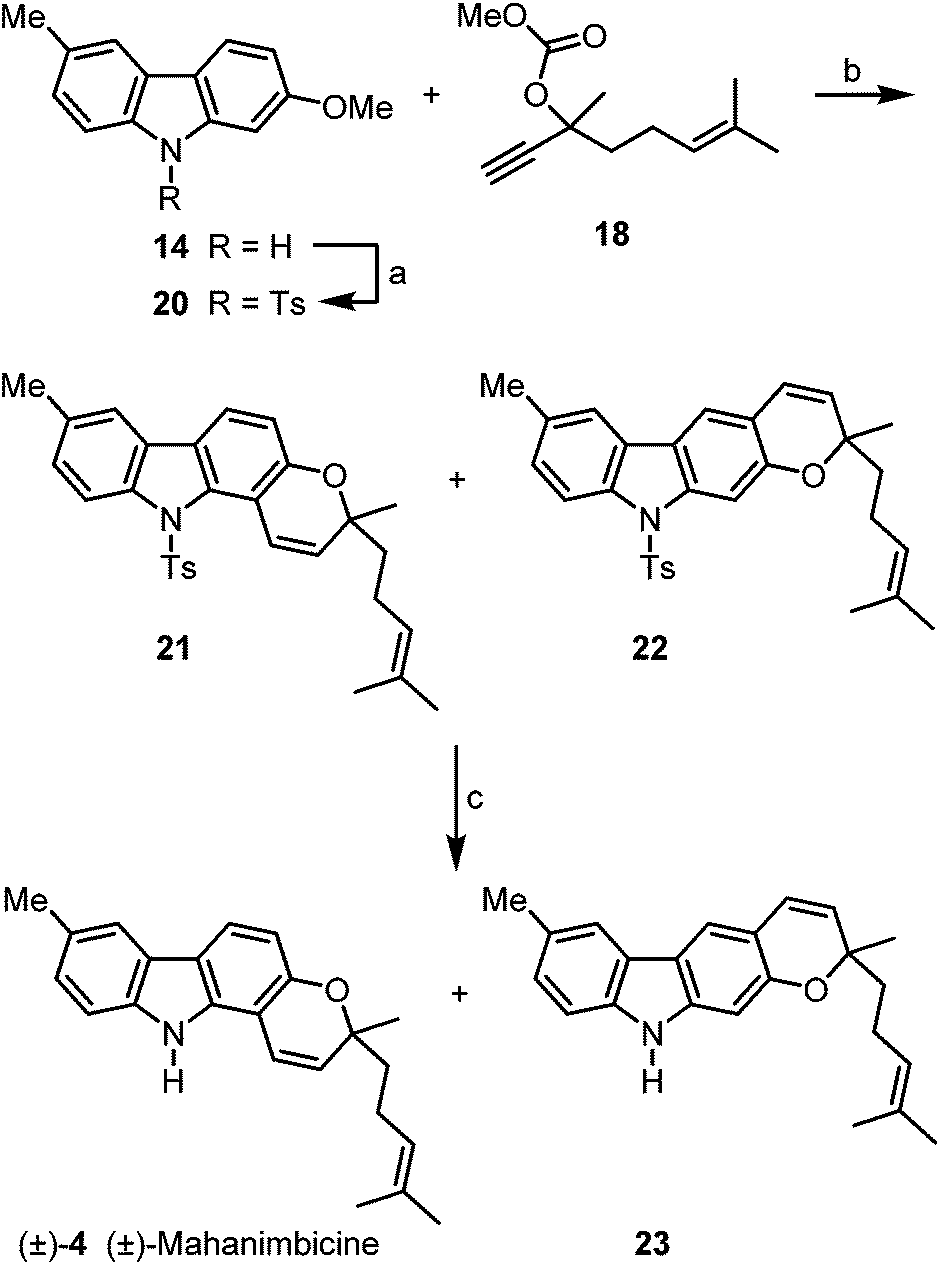 | ||
| Scheme 4 Alternative route to (±)-mahanimbicine [(±)-4]. Reagents and conditions: (a) 4.1 equiv. NaH, 1.5 equiv. TsCl, THF, 0 °C to rt, 16.25 h, 80%; (b) 1. 3.0 equiv. BBr3, CH2Cl2, −78 °C to rt, 2.5 h; 2. 2.0 equiv. 18, 3.0 equiv. DBU, 0.5 mol% CuI, MeCN, rt, 22 h; 3. xylene, reflux, 27.5 h, 82% (3 steps, ratio 21/22 = 7.7:1); (c) 4.0 equiv. TBAF, THF, 75 °C, 6 h, 79% (±)-4 and 9% 23. | ||
Using the first approach (Scheme 3), (±)-mahanimbicine [(±)-4] is available in 5 steps and 40% overall yield based on p-bromotoluene. Our second route (Scheme 4) leads to (±)-4 in 7 steps and 46% overall yield based on the same starting material. It is interesting to note, that annulation of the pyran ring with the carbonate 18 at 2-hydroxy-6-methylcarbazole (10) provides the furo[3,2-a]carbazole 19 as by-product, whereas annulation at the corresponding N-tosylcarbazole gives the pyrano[2,3-b]carbazole 22 as by-product. This outcome is explained by the steric demand of the tosyl group which suppresses the formation of N-tosyl-19 with the quaternary carbon center in close proximity to the protecting group; instead linear pyran annulation and thus formation of compound 22 is observed.
Using (±)-mahanimbicine [(±)-4] as relay compound the carbazole alkaloids 5–9 are accessible, following putative biogenetic routes. Oxidation of (±)-mahanimbicine [(±)-4] with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) afforded murrayamine-J (5) (Scheme 5). Intramolecular [2 + 2] cycloaddition of [(±)-4] led to bicyclomahanimbicine (6). Oxidation of 6 with DDQ gave murrayamine-M (7).
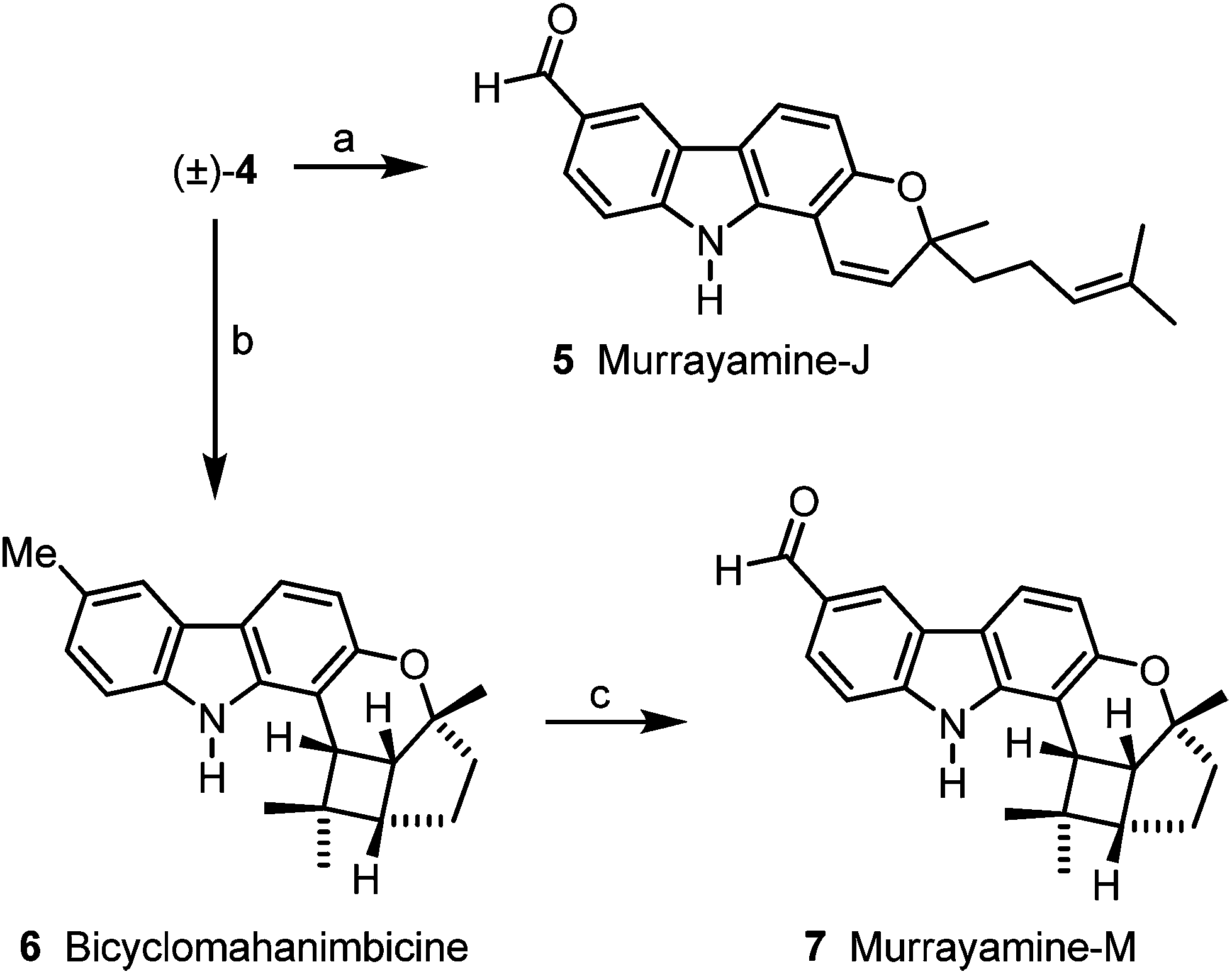 | ||
| Scheme 5 Synthesis of murrayamine-J (5), bicyclomahanimbicine (6) and murrayamine-M (7). Reagents and conditions: (a) 8.0 equiv. DDQ, MeOH–THF–water (10:1:1), rt, 6.25 h, 67%; (b) hν, toluene, rt, 14 d, 39%; (c) 6.6 equiv. DDQ, MeOH–THF–water (10:1:2), rt, 5 h, 51%. | ||
For the Brønsted acid promoted cycloisomerization of (±)-mahanimbicine [(±)-4], we took advantage of our previous study on the conversion of mahanimbine (2) into cyclomahanimbine and mahanimbidine.12 On treatment of (±)-4 with one equivalent camphor-10-sulfonic acid (CSA) at room temperature to 70 °C for 16 d, (±)-4 and rapidly formed 9 were both completely converted into 8. Thus, murrayamine-G (8) was obtained in 65% yield (Scheme 6, Table 1). Cycloisomerization of (±)-mahanimbicine [(±)-4] in the presence of catalytic amounts of CSA in hexane at room temperature afforded in 70% yield a 1:1 mixture of 8 and 9 which after separation by preparative HPLC led to pure isomurrayazoline (9). Our synthetic route leads to isomurrayazoline (9) in 8 steps and 16% overall yield based on p-bromotoluene.
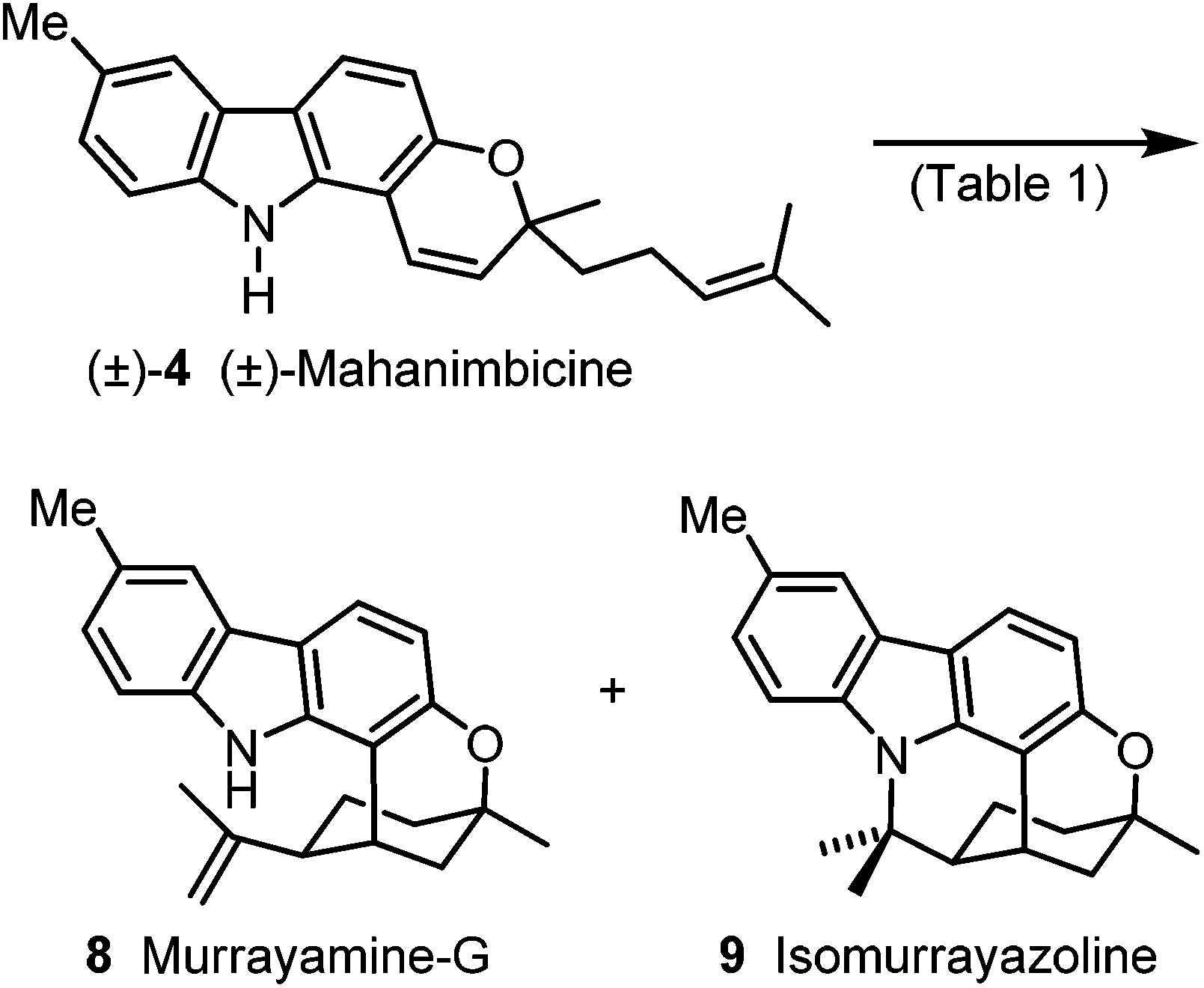 | ||
| Scheme 6 Synthesis of murrayamine-G (8) and isomurrayazoline (9). Reagents and conditions: see Table 1. | ||
| Reaction conditions | Yield | Ratio, 8:9 |
|---|---|---|
| 1.0 equiv. CSA, PhMe, rt to 70 °C, 16 d | 65% 8 | — |
| 8 mol% CSA, hexane, rt, 11.5 d | 70% 8, 9 | 1:1 |
Conclusions
We have developed a highly efficient palladium-catalyzed route to 2-hydroxy-6-methylcarbazole (10) (3 steps, 81% overall yield). Using appropriate C5 or C10 building blocks, compound 10 has been transformed into isogirinimbine (3) and (±)-mahanimbicine [(±)-4], respectively. (±)-Mahanimbicine [(±)-4] served as central intermediate for the synthesis of cyclic monoterpenoid pyrano[3,2-a]carbazole alkaloids and could be converted into murrayamine-J (5), bicyclomahanimbicine (6), murrayamine-M (7), murrayamine-G (8) and isomurrayazoline (9). The spectroscopic data of the alkaloids were in full agreement with those reported for the corresponding natural products. For the compounds 3, 5 and 7–9 we have achieved the first total synthesis. The present results emphasize the utility of our synthetic methodology and demonstrate that for the first time also the whole series of carbazole alkaloids which is isomeric to girinimbine (1) and mahanimbine (2) is available by synthesis on large scale. The biological properties of 3–9, e.g. their potential anti-TB activity,23 are under further investigation.Experimental
General methods
All reactions were carried out in oven-dried glassware using dry solvents under an argon atmosphere unless stated otherwise. Acetonitrile, dichloromethane, tetrahydrofuran and toluene were dried using a solvent purification system (MBraun-SPS). Palladium(II) acetate was recrystallized from glacial acetic acid. All other chemicals were used as received from commercial sources. Flash chromatography was performed on a Büchi Sepacore system equipped with an UV monitor using silica gel from Acros Organics (0.035–0.070 mm). Thin layer chromatography was performed with TLC plates from Merck (60 F254) using UV-light for visualization. Melting points were measured on a Gallenkamp MPD 350 melting point apparatus. Ultraviolet spectra were recorded on a Perkin Elmer 25 UV/VIS spectrometer. Infrared spectra were recorded on a Thermo Nicolet Avatar 360 FT-IR spectrometer using the ATR method (Attenuated Total Reflectance). NMR spectra were recorded on Bruker Avance II 300, DRX 500 and Avance III 600 spectrometers. Chemical shifts δ are reported in parts per million with the non-deuterated solvent as internal standard.24 The following abbreviations have been used: s: singlet, d: doublet, t: triplet, m: multiplet and br: broad. Mass spectra were recorded on a Finnigan MAT-95 spectrometer (electron impact, 70 eV) or by GC/MS-coupling using an Agilent Technologies 6890 N GC System equipped with a 5973 Mass Selective Detector (electron impact, 70 eV). ESI-MS spectra were recorded on an Esquire LC with an ion trap detector from Bruker. Positive and negative ions were detected. Elemental analyses were measured on an EuroVector EuroEA3000 elemental analyzer.:5:1) provided 3-methoxy-N-(4-methylphenyl)aniline (13) as colourless solid, yield: 2.24 g (98%), m.p. 68–70 °C. UV (MeOH): λ = 283 nm. IR (ATR): ν = 3365, 3000, 1596, 1512, 1492, 1463, 1438, 1389, 1324, 1302, 1283, 1256, 1237, 1198, 1158, 1107, 1032, 992, 951, 832, 774, 753, 686, 649, 632 cm−1. 1H NMR (500 MHz, CDCl3): δ = 2.31 (s, 3 H), 3.77 (s, 3 H), 5.62 (br s, 1 H), 6.43–6.45 (m, 1 H), 6.58–6.60 (m, 2 H), 7.01–7.04 (m, 2 H), 7.09–7.11 (m, 2 H), 7.13–7.16 (m, 1 H). 13C NMR and DEPT (125 MHz, CDCl3): δ = 20.68 (CH3), 55.16 (CH3), 102.33 (CH), 105.43 (CH), 109.32 (CH), 119.36 (2 CH), 129.83 (2 CH), 130.03 (CH), 131.18 (C), 139.91 (C), 145.39 (C), 160.67 (C). EI-MS: m/z (%) = 213 (100) [M+], 197 (4), 182 (4), 168 (4), 154 (5). HRMS: m/z calcd for C14H15NO [M+]: 213.1154; found: 213.1158. Elemental analysis calcd for C14H15NO: C 78.84, H 7.09, N 6.57; found: C 78.97, H 7.08, N 6.40%.
Crystal data for 13: C14H15NO, M = 213.27 g mol−1, crystal size: 0.50 × 0.40 × 0.10 mm3, monoclinic, space group P21/c, a = 8.856(1) Å, b = 13.861(1) Å, c = 10.868(1) Å, β = 92.41(1)°, V = 1332.9(2) Å3, Z = 4, ρcalcd = 1.063 g cm−3, μ = 0.067 mm−1, T = 293(2) K, λ = 0.71073 Å, θ range 3.26–25.37°, 18034 reflections collected, 2165 independent reflections (Rint = 0.0280), 151 parameters. The structure was solved by direct methods and refined by full-matrix least-squares on F2; final R indices [I > 2σ(I)]: R1 = 0.0436, wR2 = 0.1205; maximal residual electron density: 0.179 e Å−3 (Fig. S1‡) CCDC 1000251.
Method B: m-Anisidine (18.9 g, 153 mmol) was added portionwise over a period of 3 h to a solution of p-bromotoluene (20.0 g, 117 mol), caesium carbonate (45.7 g, 140 mmol), rac-BINAP (3.61 g, 5.80 mmol) and palladium(II) acetate (1.53 g, 6.82 mmol) in toluene (80 mL) at reflux. The mixture was heated at reflux for 13 h (total reaction time 16 h), then cooled to room temperature, filtered over a short pad of silica gel and Celite (diethyl ether), and the solvent was removed. Purification of the residue by column chromatography on silica gel (petroleum ether–acetone, 15:1) provided the diarylamine 13 as light yellow solid, yield: 25.4 g (100%). Spectroscopic data, see above.
:1:0.2 to 29:1:0.2) provided 2-methoxy-6-methylcarbazole (14) as colourless solid, yield: 3.52 g (89%), m.p. 230–233 °C (ref. 16: 227–228 °C). For spectroscopic data, see ref. 16. Glycoborine (5-methoxy-3-methylcarbazole) (15) was obtained from the less polar fraction as by-product in up to 5% yield, m.p. 138–140 °C (ref. 18a: 155–156 °C). UV (MeOH): λ = 226, 238, 244, 254 (sh), 277 (sh), 287, 323, 337 mm. Fluorescence (MeOH): λex = 287 nm, λem = 345, 359 nm. IR (ATR): ν = 3398, 3048, 3012, 2913, 2846, 1624, 1604, 1585, 1554, 1506, 1473, 1453, 1438, 1388, 1345, 1314, 1294, 1258, 1225, 1178, 1101, 1058, 973, 915, 879, 802, 781, 745, 716, 698, 620, 590, 535 cm−1. 1H NMR (500 MHz, CDCl3): δ = 2.54 (s, 3 H), 4.09 (s, 3 H), 6.67 (d, J = 8.0 Hz, 1 H), 7.02 (d, J = 8.0 Hz, 1 H), 7.21 (dd, J = 8.2, 1.3 Hz, 1 H), 7.29 (d, J = 8.2 Hz, 1 H), 7.32 (t, J = 8.0 Hz, 1 H), 7.94 (br s, 1 H), 8.12 (d, J = 0.8 Hz, 1 H). 13C NMR and DEPT (125 MHz, CDCl3): δ = 21.43 (CH3), 55.37 (CH3), 100.10 (CH), 103.48 (CH), 109.52 (CH), 112.40 (C), 122.77 (C), 122.89 (CH), 126.16 (CH), 126.42 (CH), 128.86 (C), 136.82 (C), 141.16 (C), 156.19 (C). EI-MS (70 eV): m/z (%) = 211 (100) [M+], 196 (27), 168 (68), 167 (19). Elemental analysis calcd for C14H13NO: C 79.59, H 6.20, N 6.63; found: C 79.86, H 6.36, N 6.69%.
:1) provided 2-hydroxy-6-methylcarbazole (10) as colourless solid, yield: 93 mg (91%), m.p. 262–264 °C (ref. 5: 245 °C, decomp.). UV (MeOH): λ = 260, 305, 322 (sh), 335 (sh) nm. IR (ATR): ν = 3399, 3286, 2913, 2855, 1617, 1508, 1488, 1461, 1416, 1343, 1308, 1295, 1275, 1218, 1158, 1134, 1106, 1036, 955, 937, 885, 836, 824, 805, 766, 730, 690, 652, 627 cm−1. 1H NMR (500 MHz, acetone-d6): δ = 2.45 (s, 3 H), 6.71 (dd, J = 8.4, 2.1 Hz, 1 H), 6.90 (d, J = 2.1 Hz, 1 H), 7.08 (dd, J = 8.2, 1.0 Hz, 1 H), 7.28 (d, J = 8.2 Hz, 1 H), 7.74 (s, 1 H), 7.85 (d, J = 8.4 Hz, 1 H), 8.32 (s, 1 H), 9.94 (br s, 1 H). 13C NMR and DEPT (125 MHz, acetone-d6): δ = 21.48 (CH3), 97.34 (CH), 109.06 (CH), 110.92 (CH), 117.03 (C), 119.78 (CH), 121.46 (CH), 124.65 (C), 125.99 (CH), 128.42 (C), 139.17 (C), 142.93 (C), 157.38 (C). EI-MS (70 eV): m/z (%) = 197 (100) [M+], 196 (55), 167 (7). HRMS: m/z calcd for C13H11NO [M+]: 197.0841; found: 197.0855.
:1 to 3:1) provided isogirinimbine (3) as colourless solid, yield: 84.4 mg (63%), m.p. 186–187 °C. UV (MeOH): λ = 221 (sh), 237, 278 (sh), 289, 332, 337, 353 nm. Fluorescence (MeOH): λex = 289 nm, λem = 362, 378 nm. IR (ATR): ν = 3415, 2969, 2919, 2853, 1640, 1606, 1519, 1474, 1460, 1418, 1400, 1373, 1359, 1339, 1295, 1207, 1157, 1114, 1071, 1036, 898, 882, 859, 800, 746, 721, 704, 652, 585, 578, 562, 540 cm−1. 1H NMR (500 MHz, acetone-d6): δ = 1.44 (s, 6 H), 2.45 (s, 3 H), 5.76 (d, J = 9.8 Hz, 1 H), 6.63 (d, J = 8.4 Hz, 1 H), 6.90 (d, J = 9.8 Hz, 1 H), 7.11 (dd, J = 8.2, 1.1 Hz, 1 H), 7.30 (d, J = 8.2 Hz, 1 H), 7.76 (d, J = 0.6 Hz, 1 H), 7.79 (d, J = 8.4 Hz, 1 H), 10.25 (br s, 1 H). 13C NMR and DEPT (125 MHz, acetone-d6): δ = 21.48 (CH3), 27.84 (2 CH3), 76.48 (C), 105.66 (C), 109.71 (CH), 111.18 (CH), 118.18 (C), 118.34 (CH), 119.98 (CH), 121.01 (CH), 124.70 (C), 126.42 (CH), 128.88 (C), 129.99 (CH), 137.99 (C), 139.34 (C), 152.32 (C). EI-MS: m/z (%) = 263 (28) [M+], 248 (100), 233 (5), 217 (4), 204 (9), 124 (14). HRMS: m/z calcd for C18H17NO [M+]: 263.1310; found: 263.1302.
1,1,7-Trimethyl-2-methylene-1,10-dihydro-2H-furo[3,2-a]carbazole (17) was obtained as a by-product in up to 3% yield as colourless oil. 1H NMR (300 MHz, acetone-d6): δ = 1.68 (s, 6 H), 2.46 (s, 3 H), 4.35 (d, J = 2.6 Hz, 1 H), 4.61 (d, J = 2.6 Hz, 1 H), 6.80 (d, J = 8.3 Hz, 1 H), 7.14 (dd, J = 8.1, 1.0 Hz, 1 H), 7.32 (d, J = 8.1 Hz, 1 H), 7.82 (d, J = 1.0 Hz, 1 H), 7.92, (d, J = 8.3 Hz, 1 H), 10.21 (br s, 1 H). 13C NMR and DEPT (75 MHz, acetone-d6): δ = 21.46 (CH3), 28.69 (2 CH3), 44.78 (C), 82.07 (CH2), 105.52 (CH), 111.39 (CH), 116.22 (C), 120.02 (CH), 120.25 (C), 120.48 (CH), 124.56 (C), 126.83 (CH), 129.07 (C), 136.56 (C), 139.62 (C), 155.55 (C), 173.56 (C). ESI-MS (−25 V): m/z = 262 [(M − H)−].
:1 to 20:1) provided carbonate 18 as colourless liquid, yield: 3.68 g (88%). IR (ATR): ν = 3287, 2959, 2923, 2853, 1754, 1699, 1684, 1651, 1635, 1441, 1376, 1338, 1257, 1165, 1114, 1074, 1022, 940, 884, 834, 790, 666, 631 cm−1. 1H NMR (500 MHz, CDCl3): δ = 1.60 (s, 3 H), 1.66 (d, J = 0.8 Hz, 3 H), 1.70 (s, 3 H), 1.82 (ddd, J = 13.6, 11.0, 5.9 Hz, 1 H), 1.96 (ddd, J = 13.6, 10.6, 6.0 Hz, 1 H), 2.11–2.23 (m, 2 H), 2.58 (s, 1 H), 3.75 (s, 3 H), 5.09 (m, 1 H). 13C NMR and DEPT (125 MHz, CDCl3): δ (ppm) = 17.57 (CH3), 22.79 (CH2), 25.59 (CH3), 26.17 (CH3), 41.17 (CH2), 54.29 (CH3), 73.85 (CH), 76.78 (C), 83.03 (C), 122.85 (CH), 132.43 (C), 153.49 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). EI-MS (70 eV): m/z (%) = 210 (0.4) [M+], 151 (7), 119 (100), 105 (13), 91 (50), 69 (44). Elemental analysis calcd for C12H18O3: C 68.54, H 8.63; found: C 68.38, H 8.93%.
:5:1 to 69:5:1) provided 10 (154 mg, 34%) and (±)-mahanimbicine [(±)-4] as colourless solid, yield: 372 mg (49%), m.p. 137–139 °C (ref. 5 and 7: 142 °C). UV (MeOH): λ = 238, 289, 335, 353 nm. Fluorescence (MeOH): λex = 289 nm, λem = 361 nm. IR (ATR): ν = 3428, 3409, 3012, 2968, 2916, 2856, 2727, 1639, 1608, 1517, 1473, 1454, 1419, 1402, 1374, 1338, 1297, 1223, 1208, 1190, 1164, 1126, 1090, 1036, 945, 909, 882, 816, 798, 748, 719, 673, 584 cm−1. 1H NMR (500 MHz, CDCl3): δ = 1.45 (s, 3 H), 1.58 (s, 3 H), 1.66 (d, J = 0.8 Hz, 3 H), 1.70–1.82 (m, 2 H), 2.11–2.21 (m, 2 H), 2.50 (s, 3 H), 5.11 (m, 1 H), 5.66 (d, J = 9.8 Hz, 1 H), 6.64 (d, J = 9.8 Hz, 1 H), 6.72 (dd, J = 8.3, 0.5 Hz, 1 H), 7.14 (dd, J = 8.2, 1.1 Hz, 1 H), 7.28 (d, J = 8.2 Hz, 1 H), 7.73 (s, 1 H), 7.74 (d, J = 8.3 Hz, 1 H), 7.84 (br s, 1 H). 13C NMR and DEPT (125 MHz, CDCl3): δ = 17.63 (CH3), 21.44 (CH3), 22.73 (CH2), 25.66 (CH3), 25.95 (CH3), 40.80 (CH2), 78.30 (C), 104.51 (C), 109.44 (CH), 110.06 (CH), 117.30 (CH), 117.39 (C), 119.45 (CH), 120.39 (CH), 124.11 (CH), 124.14 (C), 125.73 (CH), 128.65 (CH), 129.01 (C), 131.71 (C), 136.56 (C), 137.70 (C), 151.70 (C). EI-MS (70 eV): m/z (%) = 331 (45) [M+], 288 (11), 248 (100), (10), 210 (51), 209 (36), 180 (17). Elemental analysis calcd for C23H25NO: C 83.34, H 7.60, N 4.23; found: C 83.59, H 7.88, N 4.23%.
O). EI-MS (70 eV): m/z (%) = 210 (0.4) [M+], 151 (7), 119 (100), 105 (13), 91 (50), 69 (44). Elemental analysis calcd for C12H18O3: C 68.54, H 8.63; found: C 68.38, H 8.93%.
:5:1 to 69:5:1) provided 10 (154 mg, 34%) and (±)-mahanimbicine [(±)-4] as colourless solid, yield: 372 mg (49%), m.p. 137–139 °C (ref. 5 and 7: 142 °C). UV (MeOH): λ = 238, 289, 335, 353 nm. Fluorescence (MeOH): λex = 289 nm, λem = 361 nm. IR (ATR): ν = 3428, 3409, 3012, 2968, 2916, 2856, 2727, 1639, 1608, 1517, 1473, 1454, 1419, 1402, 1374, 1338, 1297, 1223, 1208, 1190, 1164, 1126, 1090, 1036, 945, 909, 882, 816, 798, 748, 719, 673, 584 cm−1. 1H NMR (500 MHz, CDCl3): δ = 1.45 (s, 3 H), 1.58 (s, 3 H), 1.66 (d, J = 0.8 Hz, 3 H), 1.70–1.82 (m, 2 H), 2.11–2.21 (m, 2 H), 2.50 (s, 3 H), 5.11 (m, 1 H), 5.66 (d, J = 9.8 Hz, 1 H), 6.64 (d, J = 9.8 Hz, 1 H), 6.72 (dd, J = 8.3, 0.5 Hz, 1 H), 7.14 (dd, J = 8.2, 1.1 Hz, 1 H), 7.28 (d, J = 8.2 Hz, 1 H), 7.73 (s, 1 H), 7.74 (d, J = 8.3 Hz, 1 H), 7.84 (br s, 1 H). 13C NMR and DEPT (125 MHz, CDCl3): δ = 17.63 (CH3), 21.44 (CH3), 22.73 (CH2), 25.66 (CH3), 25.95 (CH3), 40.80 (CH2), 78.30 (C), 104.51 (C), 109.44 (CH), 110.06 (CH), 117.30 (CH), 117.39 (C), 119.45 (CH), 120.39 (CH), 124.11 (CH), 124.14 (C), 125.73 (CH), 128.65 (CH), 129.01 (C), 131.71 (C), 136.56 (C), 137.70 (C), 151.70 (C). EI-MS (70 eV): m/z (%) = 331 (45) [M+], 288 (11), 248 (100), (10), 210 (51), 209 (36), 180 (17). Elemental analysis calcd for C23H25NO: C 83.34, H 7.60, N 4.23; found: C 83.59, H 7.88, N 4.23%.
The furo[3,2-a]carbazole 19 was obtained as by-product in up to 5% yield. 1H NMR (500 MHz, CDCl3): δ = 1.35 (s, 3 H), 1.47–1.53 (m, 1 H), 1.53 (s, 3 H), 1.64 (s, 3 H), 1.78 (ddd, J = 13.4, 11.9, 4.6 Hz, 1 H), 1.90–1.98 (m, 1 H), 2.12 (ddd, J = 13.4, 11.9, 4.8 Hz, 1 H), 2.51 (s, 3 H), 4.24 (d, J = 2.8 Hz, 1 H), 4.77 (d, J = 2.8 Hz, 1 H), 4.97 (m, 1 H), 6.84 (d, J = 8.3 Hz, 1 H), 7.17 (dd, J = 8.2, 1.0 Hz, 1 H), 7.30 (d, J = 8.2 Hz, 1 H), 7.74 (m, 1 H), 7.77 (d, J = 1.0 Hz, 1 H), 7.83 (d, J = 8.3 Hz, 1 H). 13C NMR and DEPT (125 MHz, CDCl3): δ (ppm) = 17.61 (CH3), 21.58 (CH3), 24.21 (CH2), 25.70 (CH3), 28.65 (CH3), 41.95 (CH2), 48.39 (C), 82.67 (CH2), 102.61 (CH), 110.29 (CH), 113.24 (C), 119.47 (C), 119.67 (CH), 119.99 (CH), 123.52 (CH), 124.10 (C), 126.29 (CH), 129.40 (C), 132.30 (C), 135.74 (C), 137.95 (C), 155.63 (C), 170.22 (C).
:5:1) provided 2-methoxy-6-methyl-9-tosylcarbazole (20) as colourless solid, yield: 969 mg (80%), m.p. 145–148 °C. UV (MeOH): λ = 224, 268, 296, 309, 326 nm. Fluorescence (MeOH): λex = 268 nm, λem = 396 nm. IR (ATR): ν = 3091, 2992, 2949, 2917, 2831, 1622, 1580, 1493, 1477, 1453, 1361, 1299, 1280, 1253, 1190, 1167, 1147, 1130, 1090, 1044, 985, 938, 869, 842, 801, 777, 736, 705, 675 cm−1. 1H NMR (500 MHz, acetone-d6): δ = 2.27 (s, 3 H), 2.43 (s, 3 H), 3.95 (s, 3 H), 7.01 (dd, J = 8.6, 2.3 Hz, 1 H), 7.25–7.29 (m, 3 H), 7.73–7.77 (m, 3 H), 7.86 (d, J = 2.3 Hz, 1 H), 7.90 (d, J = 8.6 Hz, 1 H), 8.14 (d, J = 8.5 Hz, 1 H). 13C NMR and DEPT (125 MHz, acetone-d6): δ = 21.21 (CH3), 21.35 (CH3), 56.07 (CH3), 100.72 (CH), 112.87 (CH), 115.55 (CH), 120.43 (CH), 120.60 (C), 121.76 (CH), 127.38 (2 CH), 127.63 (C), 128.10 (CH), 130.73 (2 CH), 134.78 (C), 135.54 (C), 137.25 (C), 140.76 (C), 146.38 (C), 160.86 (C). EI-MS (70 eV): m/z (%) = 365 (65) [M+], 210 (100), 167 (26). HRMS: m/z calcd for C21H19NO3S [M+]: 365.1086; found: 365.1099. Elemental analysis calcd for C21H19NO3S: C 69.02, H 5.24, N 3.83, S 8.77; found: C 69.14, H 5.20, N 3.82, S 9.31%.
2-Hydroxy-6-methyl-9-tosylcarbazole: UV (MeOH): λ = 222, 244 (sh), 260, 267, 275, 297, 308 nm. Fluorescence (MeOH): λex = 260 nm, λem = 381 nm. IR (ATR): ν = 3482, 3035, 2919, 1612, 1558, 1499, 1442, 1398, 1346, 1296, 1268, 1215, 1186, 1161, 1114, 1088, 1027, 991, 952, 854, 811, 790, 739, 718, 701, 679, 660, 604 cm−1. 1H NMR (500 MHz, acetone-d6): δ = 2.28 (s, 3 H), 2.42 (s, 3 H), 6.92 (dd, J = 8.5, 2.1 Hz, 1 H), 7.23–7.27 (m, 3 H), 7.69–7.74 (m, 3 H), 7.81 (d, J = 2.4 Hz, 1 H), 7.82 (d, J = 8.2 Hz, 1 H), 8.11 (d, J = 8.5 Hz, 1 H), 8.80 (s, 1 H). 13C NMR and DEPT (125 MHz, acetone-d6): δ = 21.17 (CH3), 21.32 (CH3), 102.55 (CH), 113.72 (CH), 115.44 (CH), 119.70 (C), 120.16 (CH), 121.77 (CH), 127.28 (2 CH), 127.72 (CH), 127.87 (C), 130.66 (2 CH), 134.62 (C), 135.64 (C), 137.09 (C), 140.90 (C), 146.26 (C), 158.55 (C). EI-MS (70 eV): m/z (%) = 351 (79) [M+], 196 (100), 167 (12), 91 (5). HRMS: m/z calcd for C20H17NO3S [M+]: 351.0929; found: 351.0926. Elemental analysis calcd for C20H17NO3S: C 68.36, H 4.88, N 3.99, S 9.12; found: C 68.55, H 4.86, N 3.94, S 9.78%.
A solution of 3,7-dimethyloct-6-en-1-yn-3-yl methyl carbonate (18) (577 mg, 2.74 mmol) in acetonitrile (12 mL) was added over a period of 12 h at room temperature to a solution of the crude 2-hydroxy-6-methyl-9-tosylcarbazole (539 mg), DBU (0.62 mL, 0.63 g, 4.1 mmol) and copper(I) iodide (1.3 mg, 7 μmol) in acetonitrile (35 mL) and the mixture was stirred at room temperature for 10 h (total reaction time 22 h). The mixture was diluted with diethyl ether and washed with water, 10% aqueous HCl and brine. The aqueous layers were extracted with diethyl ether, the combined organic layers were dried over sodium sulfate, the solvent was evaporated and the residue was dried in vacuum. The crude product was dissolved in xylenes (25 mL), the solution was heated at reflux for 27.5 h and the solvent was evaporated. Purification of the residue by column chromatography on silica gel (pentane–dichloromethane–ethyl acetate, gradient elution, 99:1 to 15:1) provided a mixture of N-tosylmahanimbicine (21) and compound 22, combined yield: 546 mg (82%), ratio of 21:22 = 7.7:1 (determined by 1H NMR integration). UV (MeOH): λ = 230, 266, 313 nm. Fluorescence (MeOH): λex = 266 nm, λem = 359, 419 nm. IR (ATR): ν = 2967, 2921, 2853, 1725, 1631, 1596, 1476, 1451, 1398, 1368, 1272, 1172, 1086, 1035, 960, 918, 890, 810, 782, 749, 726, 703, 672, 662, 629, 575, 542 cm−1. 1H NMR (500 MHz, acetone-d6): δ (major isomer) = 1.52 (s, 3 H), 1.58 (s, 3 H), 1.67 (s, 3 H), 1.80–1.90 (m, 2 H), 2.18–2.24 (m, 2 H), 2.21 (s, 3 H), 2.39 (s, 3 H), 5.18 (m, 1 H), 5.80 (d, J = 10.0 Hz, 1 H), 6.88 (dd, J = 8.5, 0.7 Hz, 1 H), 7.00 (d, J = 8.2 Hz, 2 H), 7.08 (m, 2 H), 7.24 (d, J = 10.0 Hz, 1 H), 7.24 (m, 1 H), 7.52 (d, J = 0.7 Hz, 1 H), 7.61 (d, J = 8.3 Hz, 1 H), 8.04 (d, J = 8.3 Hz, 1 H). 13C NMR and DEPT (125 MHz, acetone-d6): δ (major isomer) = 17.66 (CH3), 21.23 (CH3), 21.27 (CH3), 23.37 (CH2), 25.83 (2 CH3), 40.99 (CH2), 78.40 (C), 115.19 (C), 115.89 (CH), 119.61 (CH), 120.02 (CH), 121.02 (CH), 122.80 (CH), 124.49 (C), 124.97 (CH), 127.53 (CH), 127.82 (2 CH), 127.83 (CH), 129.73 (2 CH), 130.55 (C), 132.08 (C), 133.15 (C), 136.34 (C), 138.19 (C), 139.75 (C), 145.67 (C), 154.82 (C). ESI-MS (+25 V): m/z (%) = 486.3 [(M + H)+], 988.7 [(2M + NH4)+]. Elemental analysis calcd for C30H31NO3S: C 74.20, H 6.43, N 2.88, S 6.60; found: C 74.46, H 6.68, N 2.97, S 6.49%.
:22 = 7.7:1, see above). The mixture was irradiated in the microwave reactor at 300 W and 70 °C for 3 h and then at 75 °C for 2 h. A 1 M solution of TBAF in THF (0.28 mL, 0.28 mmol) was added and the mixture was irradiated in the microwave reactor at 300 W and 75 °C for 1 h. The mixture was diluted with diethyl ether, and washed first with an aqueous solution of ammonium chloride and then with brine. The aqueous layers were extracted with diethyl ether, the combined organic layers were dried over sodium sulfate and the solvent was evaporated. Purification of the residue by column chromatography on silica gel (pentane–dichloromethane–diethyl ether, gradient elution, 119:1:0.2 to 14:1:0.2) provided (±)-mahanimbicine [(±)-4] as colourless solid (yield: 37.1 mg, 79%; spectroscopic data, see above) and 23 as light yellow solid, yield: 4.0 mg (9%). 1H-NMR (500 MHz, CDCl3): δ = 1.43 (s, 3 H), 1.57 (s, 3 H), 1.65 (s, 3 H), 1.67–1.80 (m, 2 H), 2.14–2.17 (m, 2 H), 2.49 (s, 3 H), 5.10 (br t, J = 7.1 Hz, 1 H), 5.55 (d, J = 9.8 Hz, 1 H), 6.53 (d, J = 9.8 Hz, 1 H), 6.78 (s, 1 H), 7.12 (dd, J = 8.1, 1.1 Hz, 1 H), 7.23 (d, J = 8.1 Hz, 1 H), 7.57 (s, 1 H), 7.71 (br s, 1 H), 7.82 (br s, 1 H).
:0:0 to 13:5:1) provided murrayamine-J (5) as colourless solid, yield: 27.9 mg (67%), m.p. 93–97 °C (ref. 8: oil). UV (MeOH): λ = 246, 267, 311, 347 (sh) nm. Fluorescence (MeOH): λex = 311 nm, λem = 350 nm. IR (ATR): ν = 3322, 3045, 2966, 2918, 2848, 2757, 1771, 1735, 1717, 1698, 1662, 1604, 1580, 1508, 1474, 1462, 1415, 1393, 1343, 1320, 1278, 1224, 1176, 1121, 1085, 1031, 962, 895, 813, 784, 754, 721, 662, 629 cm−1. 1H NMR (600 MHz, CDCl3): δ = 1.46 (s, 3 H), 1.58 (s, 3 H), 1.66 (s, 3 H), 1.71–1.83 (m, 2 H), 2.11–2.21 (m, 2 H), 5.10 (m, 1 H), 5.71 (d, J = 9.8 Hz, 1 H), 6.67 (d, J = 9.8 Hz, 1 H), 6.81 (d, J = 8.4 Hz, 1 H), 7.47 (d, J = 8.4 Hz, 1 H), 7.83 (d, J = 8.4 Hz, 1 H), 7.89 (dd, J = 8.4, 1.4 Hz, 1 H), 8.35 (br s, 1 H), 8.46 (s, 1 H), 10.07 (s, 1 H). 13C NMR and DEPT (150 MHz, CDCl3): δ = 17.62 (CH3), 22.70 (CH2), 25.65 (CH3), 26.05 (CH3), 40.88 (CH2), 78.69 (C), 104.98 (C), 110.69 (CH), 110.91 (CH), 116.75 (CH), 117.07 (C), 120.85 (CH), 122.77 (CH), 123.93 (CH), 124.23 (C), 126.33 (CH), 129.35 (C), 129.43 (CH), 131.85 (C), 136.75 (C), 143.42 (C), 152.65 (C), 192.01 (CHO). EI-MS (70 eV): m/z (%) = 345 (33) [M+], 330 (4), 262 (100), 234 (4). HRMS: m/z calcd for C23H23NO2 [M+]: 345.1729; found: 345.1731. Elemental analysis calcd for C23H23NO2: C 79.97, H 6.71, N 4.05; found: C 79.22, H 6.94, N 4.14%.
:1:0.2 to 23:1:0.2) provided bicyclomahanimbicine (6) as colourless solid, yield: 15.7 mg (39%), m.p. 224–225 °C (ref. 5: 218 °C, decomp.). UV (MeOH): λ = 223, 242, 256, 261, 288 (sh), 304, 322 (sh), 335 (sh) nm. Fluorescence (MeOH): λex = 242 nm, λem = 344, 359 nm. IR (ATR): ν = 3452, 3021, 2964, 2944, 2913, 2856, 1711, 1609, 1508, 1483, 1454, 1415, 1381, 1365, 1339, 1293, 1217, 1194, 1177, 1142, 1085, 1022, 976, 918, 873, 802, 766, 740, 672, 645, 589 cm−1. 1H NMR (500 MHz, CDCl3): δ = 0.77 (s, 3 H), 1.45 (s, 3 H), 1.57 (s, 3 H), 1.63–1.76 (m, 3 H), 2.07–2.14 (m, 1 H), 2.49–2.54 (m, 1 H), 2.50 (s, 3 H), 2.72 (dd, J = 9.4, 7.7 Hz, 1 H), 3.28 (d, J = 9.4 Hz, 1 H), 6.77 (d, J = 8.4 Hz, 1 H), 7.14 (dd, J = 8.2, 1.2 Hz, 1 H), 7.28 (d, J = 8.2 Hz, 1 H), 7.40 (br s, 1 H), 7.75 (s, 1 H), 7.76 (d, J = 8.4 Hz, 1 H). 13C NMR and DEPT (125 MHz, CDCl3): δ = 18.53 (CH3), 21.42 (CH3), 25.61 (CH2), 27.57 (CH3), 35.08 (CH3), 37.12 (CH), 37.65 (CH), 38.00 (CH2), 39.22 (C), 46.39 (CH), 83.54 (C), 106.48 (C), 109.99 (CH), 111.14 (CH), 116.62 (C), 118.94 (CH), 119.46 (CH), 124.35 (C), 125.41 (CH), 128.88 (C), 137.46 (C), 139.64 (C), 151.85 (C). EI-MS (70 eV): m/z (%) = 331 (30) [M+], 248 (100), 234 (3), 218 (2). HRMS: m/z calcd for C23H25NO [M+]: 331.1936; found: 331.1937.
:0:0 to 14:5:1) provided murrayamine-M (7) as light yellow solid, yield: 15 mg (51%), m.p. 216–218 °C (ref. 8: oil). UV (MeOH): λ = 243, 255 (sh), 293, 329 (sh) nm. Fluorescence (MeOH): λex = 293 nm, λem = 368 nm. IR (ATR): ν = 3416, 3385, 2947, 2863, 1735, 1698, 1673, 1654, 1605, 1572, 1508, 1474, 1458, 1414, 1318, 1220, 1182, 1151, 1114, 1088, 1021, 926, 891, 818, 787, 729, 686, 632, 610 cm−1. 1H NMR (500 MHz, CDCl3): δ = 0.78 (s, 3 H), 1.45 (s, 3 H), 1.59 (s, 3 H), 1.66–1.78 (m, 3 H), 2.04–2.12 (m, 1 H), 2.54 (m, 1 H), 2.74 (dd, J = 9.5, 7.7 Hz, 1 H), 3.30 (d, J = 9.5 Hz, 1 H), 6.88 (d, J = 8.5 Hz, 1 H), 7.48 (d, J = 8.4 Hz, 1 H), 7.82 (br s, 1 H), 7.85 (d, J = 8.5 Hz, 1 H), 7.88 (dd, J = 8.4, 1.6 Hz, 1 H), 8.48 (m, 1 H), 10.07 (s, 1 H). 13C NMR and DEPT (125 MHz, CDCl3): δ = 18.72 (CH3), 25.59 (CH2), 27.42 (CH3), 35.10 (CH3), 36.96 (CH), 37.66 (CH), 38.19 (CH2), 39.30 (C), 46.40 (CH), 83.88 (C), 107.17 (C), 110.68 (CH), 112.77 (CH), 116.48 (C), 119.44 (CH), 122.88 (CH), 124.46 (C), 126.01 (CH), 129.32 (C), 139.84 (C), 143.10 (C), 152.92 (C), 192.03 (CHO). EI-MS (70 eV): m/z (%) = 345 (24) [M+], 262 (100), 233 (3), 204 (4). HRMS: m/z calcd for C23H23NO2 [M+]: 345.1729; found: 345.1746.
:1:0.2 to 17:1:0.2) provided murrayamine-G (8) as colourless crystals, yield: 41.4 mg (65%), m.p. 166–169 °C (ref. 9: 173–176 °C). (MeOH): λ = 219 (sh), 241, 257 (sh), 262, 307, 323 (sh) nm. Fluorescence (MeOH): λex = 307 nm, λem = 358 nm. IR (ATR): ν = 3463, 3074, 2963, 2920, 2862, 1613, 1519, 1481, 1445, 1420, 1378, 1312, 1297, 1217, 1172, 1159, 1100, 1027, 989, 963, 915, 888, 816, 798, 744, 658, 621, 584, 559 cm−1. 1H NMR (500 MHz, CDCl3): δ = 1.43 (s, 3 H), 1.48–1.50 (m, 1 H), 1.50 (s, 3 H), 1.61–1.67 (m, 2 H), 1.94 (dt, J = 12.9, 3.1 Hz, 1 H), 2.03 (dd, J = 12.9, 2.7 Hz, 1 H), 2.11 (dt, J = 9.3, 2.3 Hz, 1 H), 2.49 (s, 3 H), 2.58 (m, 1 H), 3.41 (m, 1 H), 4.73 (s, 1 H), 4.81 (t, J = 1.5 Hz, 1 H), 6.72 (d, J = 8.5 Hz, 1 H), 7.11 (dd, J = 8.1, 1.2 Hz, 1 H), 7.22 (d, J = 8.1 Hz, 1 H), 7.70 (br s, 1 H), 7.71 (s, 1 H), 7.73 (d, J = 8.5 Hz, 1 H). 13C NMR and DEPT (125 MHz, CDCl3): δ = 21.41 (CH3), 21.57 (CH3), 23.00 (CH2), 28.89 (CH3), 35.95 (CH), 37.40 (CH2), 39.67 (CH2), 48.54 (CH), 74.08 (C), 105.67 (C), 108.70 (CH), 109.89 (CH), 112.09 (CH2), 115.36 (C), 119.08 (CH), 119.22 (CH), 124.40 (C), 125.05 (CH), 128.65 (C), 137.60 (C), 140.08 (C), 149.89 (C), 155.17 (C). EI-MS (70 eV): m/z (%) = 331 (80) [M+], 316 (11), 248 (100), 210 (9). HRMS: m/z calcd for C23H25NO [M+]: 331.1936; found: 331.1936. Elemental analysis calcd for C23H25NO: C 83.34, H 7.60, N 4.23; found: C 83.03, H 7.79, N 4.18%.
:1:0.2 to 16:1:0.2) provided a mixture of murrayamine G (8) and isomurrayazoline (9) as colourless solid, yield: 28.4 mg (70%), ratio of 8:9 = 1:1 (determined by the 1H NMR spectrum). The two isomers were separated by preparative HPLC on a Grace Vydac C8 column (208TP1050, 50 × 250 mm), gradient elution with 55 mL min−1 (THF–H2O, 20–70% THF in 25 min) to afford murrayamine-G (8) as colourless crystals (spectroscopic data: see above) and isomurrayazoline (9) as colourless solid, m.p. 224–227 °C (ref. 10: 269–270 °C). UV (MeOH): λ = 239 (sh), 246, 262 (sh), 307, 327 (sh), 340 (sh) nm. Fluorescence (MeOH): λex = 246 nm, λem = 366 nm. IR (ATR): ν = 3047, 2965, 2922, 2900, 1631, 1592, 1446, 1416, 1347, 1307, 1288, 1221, 1204, 1180, 1152, 1134, 1087, 1068, 1037, 985, 941, 914, 882, 855, 801, 787, 771, 744, 680, 625, 602, 593, 556 cm−1. 1H NMR (500 MHz, CDCl3): δ = 0.13–0.21 (m, 1 H), 1.26–1.33 (m, 1 H), 1.28 (s, 3 H), 1.44 (s, 3 H), 1.47–1.54 (m, 1 H), 1.68 (ddd, J = 15.3, 6.8, 3.2 Hz, 1 H), 1.90 (s, 3 H), 1.90–1.93 (m, 1 H), 1.98 (ddd, J = 11.1, 5.5, 2.4 Hz, 1 H), 2.41 (ddd, J = 13.3, 5.3, 3.4 Hz, 1 H), 2.49 (s, 3 H), 3.29 (d, J = 4.6 Hz, 1 H), 6.64 (d, J = 8.2 Hz, 1 H), 7.09 (dd, J = 8.3, 1.3 Hz, 1 H), 7.38 (d, J = 8.3 Hz, 1 H), 7.58 (d, J = 8.2 Hz, 1 H), 7.72 (m, 1 H). 13C-NMR and DEPT (125 MHz, CDCl3): δ = 21.31 (CH3), 21.70 (CH2), 22.91 (CH3), 28.12 (CH), 29.15 (CH3), 30.14 (CH3), 36.06 (CH2), 36.56 (CH2), 48.35 (CH), 60.47 (C), 76.26 (C), 107.73 (C), 108.87 (CH), 113.16 (CH), 114.51 (C), 119.00 (CH), 120.16 (CH), 124.10 (CH), 127.50 (C), 128.82 (C), 138.92 (C), 144.32 (C), 156.37 (C). EI-MS (70 eV): m/z (%) = 331 (99) [M+], 316 (100), 288 (15), 248 (63). Elemental analysis calcd for C23H25NO: C 83.34, H 7.60, N 4.23; found: C 83.00, H 7.89, N 4.26%.
Acknowledgements
We thank Micha P. Krahl for experimental support.Notes and references
- (a) D. P. Chakraborty and S. Roy, in Progress in the Chemistry of Organic Natural Products, ed. W. Herz, H. Grisebach, G. W. Kirby, W. Steglich and C. Tamm, Springer-Verlag, Wien, 1991, vol. 57, p. 71 Search PubMed; (b) H.-J. Knölker and K. R. Reddy, Chem. Rev., 2002, 102, 4303 CrossRef PubMed.
- (a) H.-J. Knölker and K. R. Reddy, in The Alkaloids, ed. G. A. Cordell, Academic Press, Amsterdam, 2008, vol. 65, p. 1 Search PubMed; (b) A. W. Schmidt, K. R. Reddy and H.-J. Knölker, Chem. Rev., 2012, 112, 3193 CrossRef CAS PubMed.
- (a) D. P. Chakraborty, B. K. Barman and P. K. Bose, Sci. Cult., 1964, 30, 445 CAS; (b) N. L. Dutta and C. Quasim, Indian J. Chem., 1969, 7, 307 CAS; (c) S. P. Kureel, R. S. Kapil and S. P. Popli, Chem. Ind., 1970, 1262 CAS; (d) B. S. Joshi, V. N. Kamat, D. H. Gawad and T. R. Govindachari, Phytochemistry, 1972, 11, 2065 CrossRef CAS.
- (a) D. P. Chakraborty, K. C. Das and P. K. Bose, Sci. Cult., 1966, 32, 83 Search PubMed; (b) N. S. Narasimhan, M. V. Paradkar and V. P. Chitguppi, Tetrahedron Lett., 1968, 9, 5501 CrossRef; (c) N. S. Narasimhan, M. V. Paradkar, V. P. Chitguppi and S. L. Kelkar, Indian J. Chem., 1975, 13, 993 CAS; (d) N. S. Narasimhan, M. V. Paradkar and A. M. Gokhale, Indian J. Chem., 1976, 14B, 329 CAS; (e) H. Furukawa, T. S. Wu, T. Ohta and C.-S. Kuoh, Chem. Pharm. Bull., 1985, 33, 4132 CrossRef CAS; (f) K. M. Meragelman, T. C. McKee and M. R. Boyd, J. Nat. Prod., 2000, 63, 427 CrossRef CAS PubMed.
- S. P. Kureel, R. S. Kapil and S. P. Popli, Chem. Ind., 1970, 958 CAS.
- W. M. Bandaranayake, M. J. Begley, B. O. Brown, D. G. Clarke, L. Crombie and D. A. Whiting, J. Chem. Soc., Perkin Trans. 1, 1974, 998 RSC.
- B. S. Joshi, V. N. Kamat and D. H. Gawad, Tetrahedron, 1970, 26, 1475 CrossRef CAS.
- T.-S. Wu, M.-L. Wang, P.-L. Wu, C. Ito and H. Furukawa, Phytochemistry, 1996, 41, 1433 CrossRef CAS.
- T.-S. Wu, M.-L. Wang and P.-L. Wu, Phytochemistry, 1996, 43, 785 CrossRef CAS.
- L. Bhattacharya, S. K. Roy and D. P. Chakraborty, Phytochemistry, 1982, 21, 2432 CrossRef CAS.
- (a) H.-J. Knölker and C. Hofmann, Tetrahedron Lett., 1996, 37, 7947 CrossRef; (b) K. K. Gruner and H.-J. Knölker, Org. Biomol. Chem., 2008, 6, 3902 RSC; (c) K. K. Gruner, T. Hopfmann, K. Matsumoto, A. Jäger, T. Katsuki and H.-J. Knölker, Org. Biomol. Chem., 2011, 9, 2057 RSC.
- R. Hesse, K. K. Gruner, O. Kataeva, A. W. Schmidt and H.-J. Knölker, Chem. – Eur. J., 2013, 19, 14098 CrossRef CAS PubMed.
- V. P. Kumar, K. K. Gruner, O. Kataeva and H.-J. Knölker, Angew. Chem., Int. Ed., 2013, 52, 11073 CrossRef CAS PubMed.
- (a) R. Hesse, A. Jäger, A. W. Schmidt and H.-J. Knölker, Org. Biomol. Chem., 2014, 12, 3866 RSC; (b) K. K. Gruner, O. Kataeva, A. W. Schmidt and H.-J. Knölker, Chem. – Eur. J., 2014, 20, 8536 CrossRef PubMed; (c) R. Hesse, O. Kataeva, A. W. Schmidt and H.-J. Knölker, Chem. – Eur. J., 2014, 20 DOI:10.1002/chem.201403645.
- (a) H.-J. Knölker and N. O'Sullivan, Tetrahedron, 1994, 50, 10893 CrossRef; (b) H.-J. Knölker, Curr. Org. Synth., 2004, 1, 309 CrossRef; (c) R. Forke, A. Jäger and H.-J. Knölker, Org. Biomol. Chem., 2008, 6, 2481 RSC; (d) R. Forke, M. P. Krahl, F. Däbritz, A. Jäger and H.-J. Knölker, Synlett, 2008, 1870 CAS; (e) H.-J. Knölker, Chem. Lett., 2009, 38, 8 CrossRef; (f) I. Bauer and H.-J. Knölker, Top. Curr. Chem., 2012, 309, 203 CrossRef CAS; (g) T. Gensch, M. Rönnefahrt, R. Czerwonka, A. Jäger, O. Kataeva, I. Bauer and H.-J. Knölker, Chem. – Eur. J., 2012, 18, 770 CrossRef CAS PubMed; (h) L. Huet, R. Forke, A. Jäger and H.-J. Knölker, Synlett, 2012, 1230 CAS.
- M. P. Krahl, A. Jäger, T. Krause and H.-J. Knölker, Org. Biomol. Chem., 2006, 4, 3215 CAS.
- (a) J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046 CrossRef CAS; (b) M. D. Charles, P. Schultz and S. L. Buchwald, Org. Lett., 2005, 7, 3965 CrossRef CAS PubMed; (c) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338 CrossRef CAS PubMed.
- (a) A. K. Chakravarty, T. Sarkar, K. Masuda, T. Takey, H. Doi, E. Kotani and K. Shiojima, Indian J. Chem., 2001, 40B, 484 CAS; (b) S. Cheenpracha and S. Laphookhieo, Phytochem. Lett., 2011, 4, 187 CrossRef CAS PubMed; (c) J. T. Kuethe and K. G. Childers, Adv. Synth. Catal., 2008, 350, 1577 CrossRef CAS.
- J. D. Godfrey Jr., R. H. Mueller, T. C. Sedergran, N. Soundararajan and V. J. Colandrea, Tetrahedron Lett., 1994, 35, 6405 CrossRef.
- (a) I. Iwai and J. Ide, Chem. Pharm. Bull., 1962, 10, 926 CrossRef CAS; (b) I. Iwai and J. Ide, Chem. Pharm. Bull., 1963, 11, 1042 CrossRef CAS; (c) J. Zsindely and H. Schmid, Helv. Chim. Acta, 1968, 51, 1510 CrossRef CAS; (d) J. Hlubucek, E. Ritchie and W. C. Taylor, Tetrahedron Lett., 1969, 10, 1369 CrossRef; (e) P. E. Brown and R. A. Lewis, J. Chem. Soc., Perkin Trans. 1, 1992, 573 RSC.
- (a) J. Tsuji, T. Sugiura and I. Minami, Synthesis, 1987, 603 CrossRef CAS; (b) S. Yamaguchi, M. Maekawa, Y. Murayama, M. Miyazawa and Y. Hirai, Tetrahedron Lett., 2004, 45, 6971 CrossRef CAS PubMed; (c) S. Yamaguchi, M. Nedachi, M. Maekawa, Y. Murayama, M. Miyazawa and Y. Hirai, J. Heterocycl. Chem., 2006, 43, 29 CrossRef CAS.
- A. Yasuhara and T. Sakamoto, Tetrahedron Lett., 1998, 39, 595 CrossRef CAS.
- (a) T. A. Choi, R. Czerwonka, W. Fröhner, M. P. Krahl, K. R. Reddy, S. G. Franzblau and H.-J. Knölker, ChemMedChem, 2006, 1, 812 CrossRef CAS PubMed; (b) T. A. Choi, R. Czerwonka, R. Forke, A. Jäger, J. Knöll, M. P. Krahl, T. Krause, K. R. Reddy, S. G. Franzblau and H.-J. Knölker, Med. Chem. Res., 2008, 17, 374 CrossRef CAS.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512 CrossRef CAS.
Footnotes |
| † Part 121 of Transition Metals in Organic Synthesis; for Part 120, see ref. 14c. |
| ‡ Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for all compounds. CCDC 1000251. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob01151a |
| This journal is © The Royal Society of Chemistry 2014 |